Coupled Folding and Specific Binding: Fishing for Amphiphilicity

Abstract
:1. Introduction
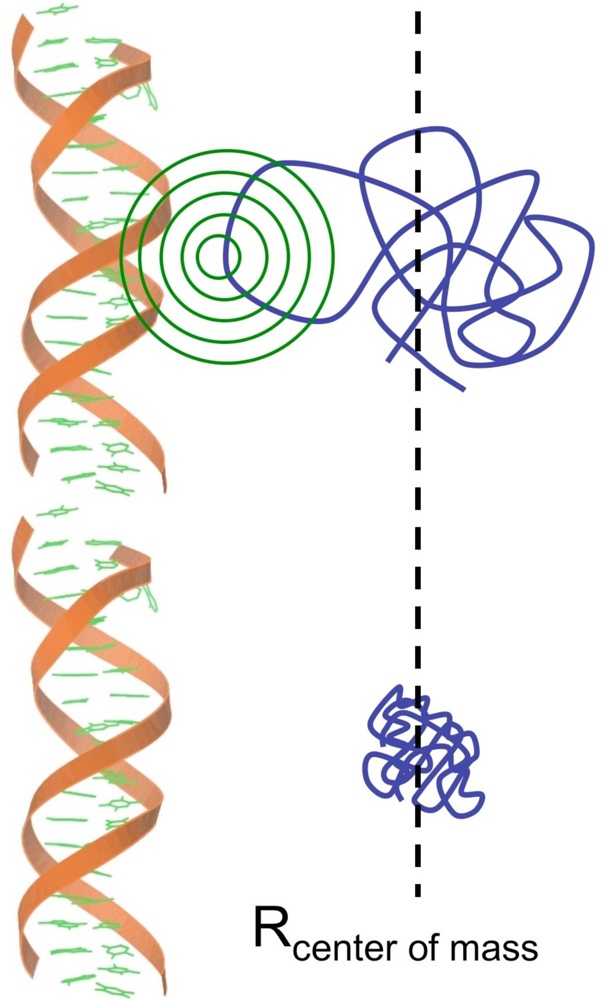
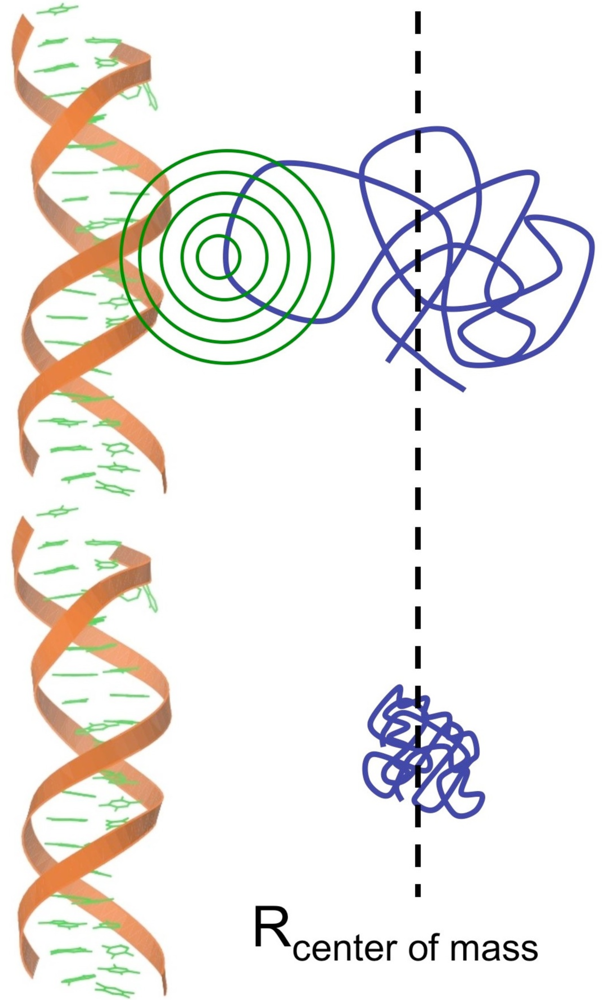
2. DNA/RNA Binding Intrinsically Disordered Proteins
2.1. DNA Binding Proteins
2.2. RNA Binding Proteins
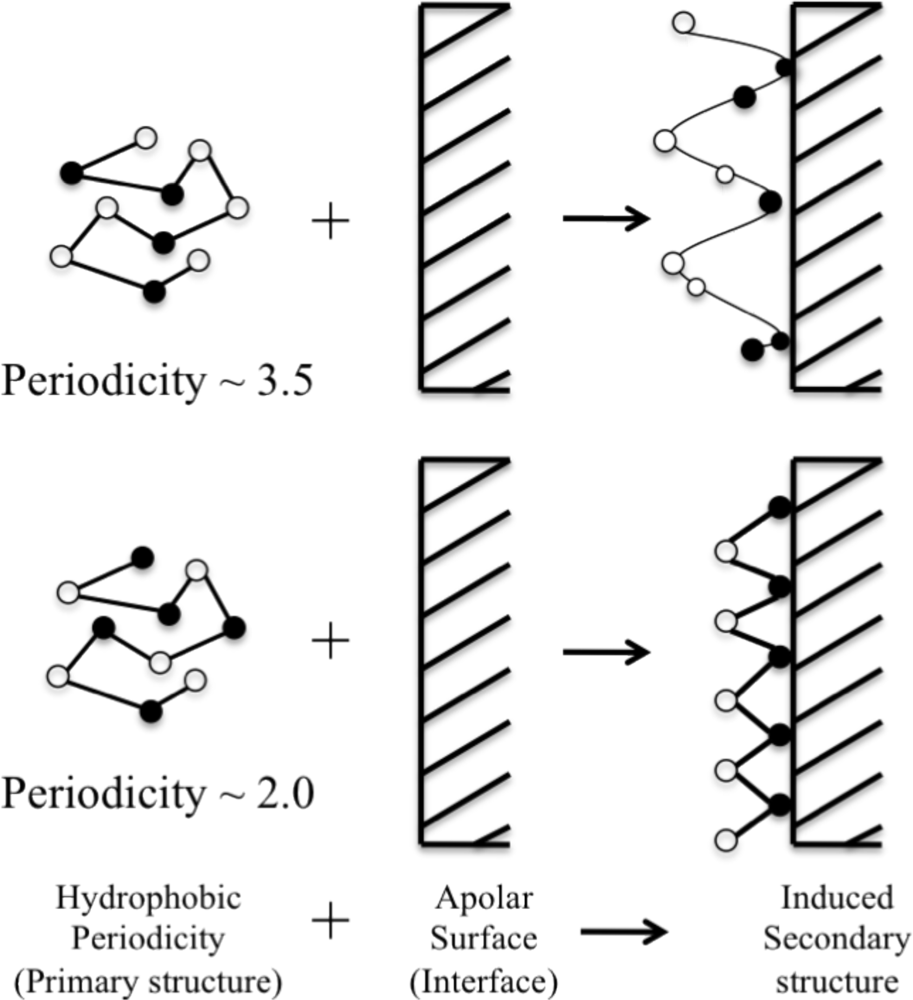
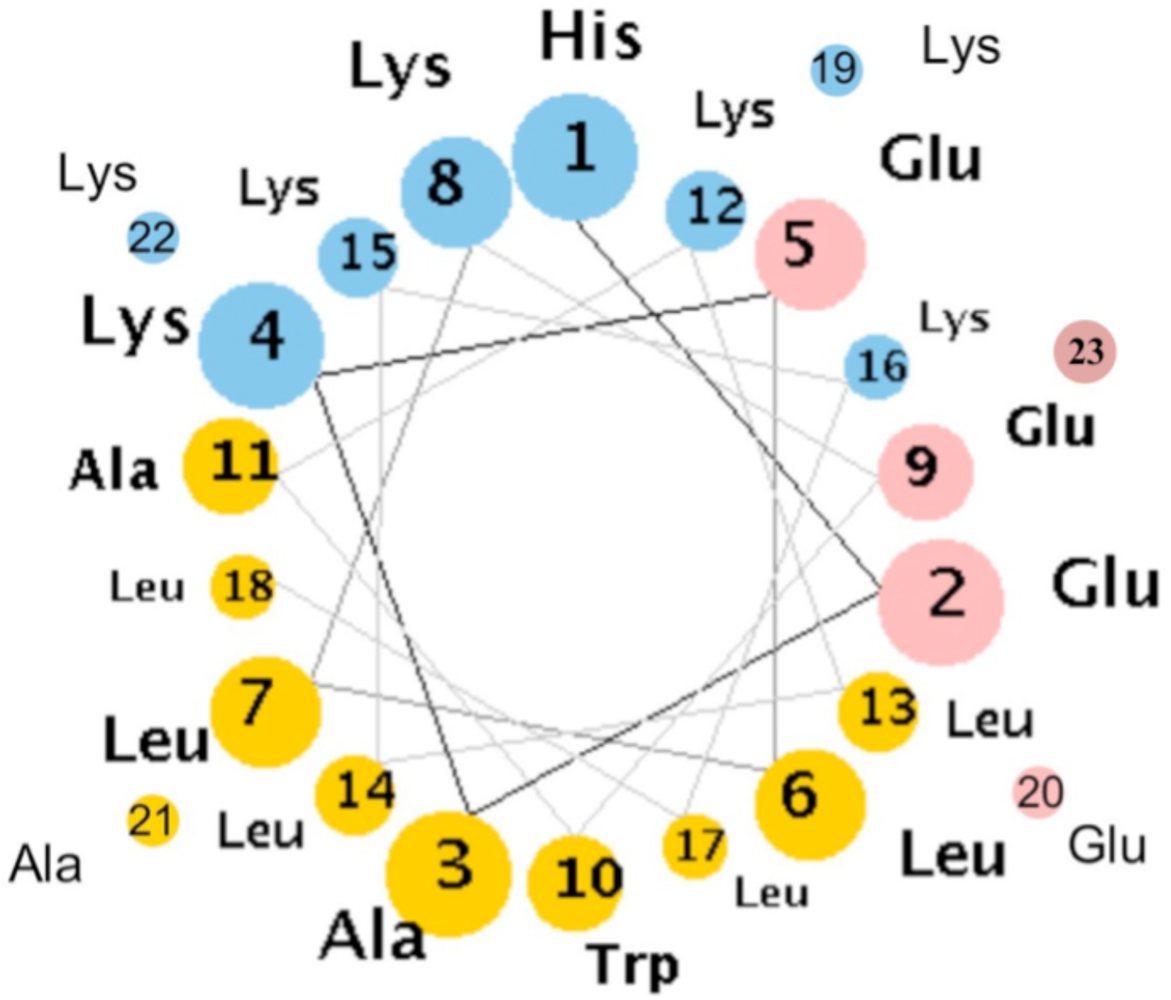
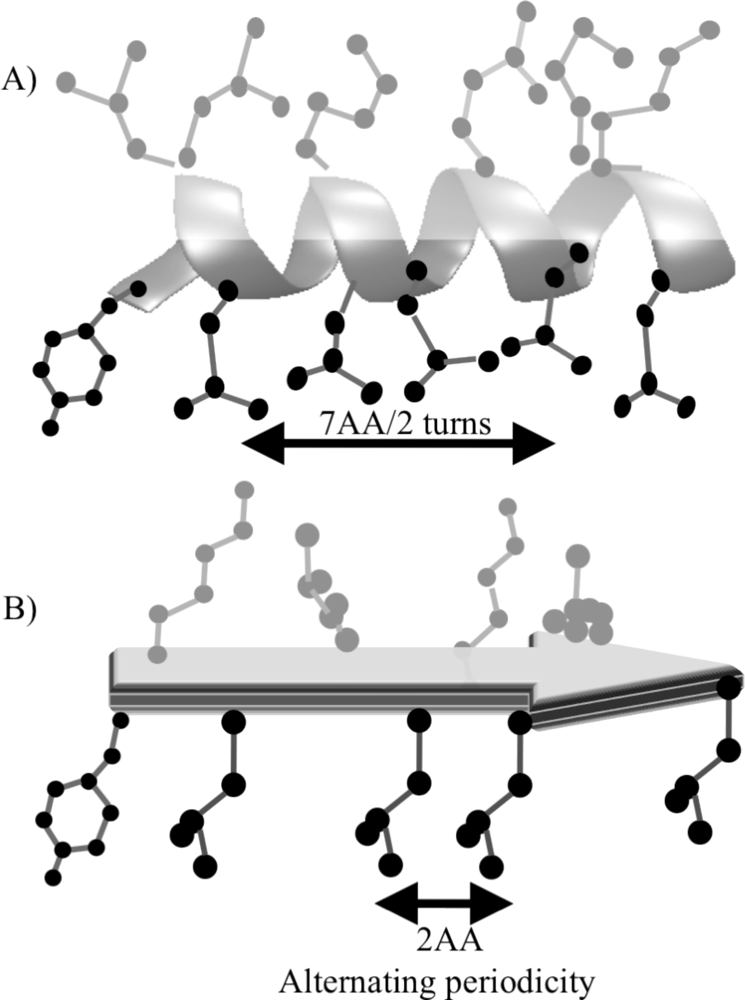
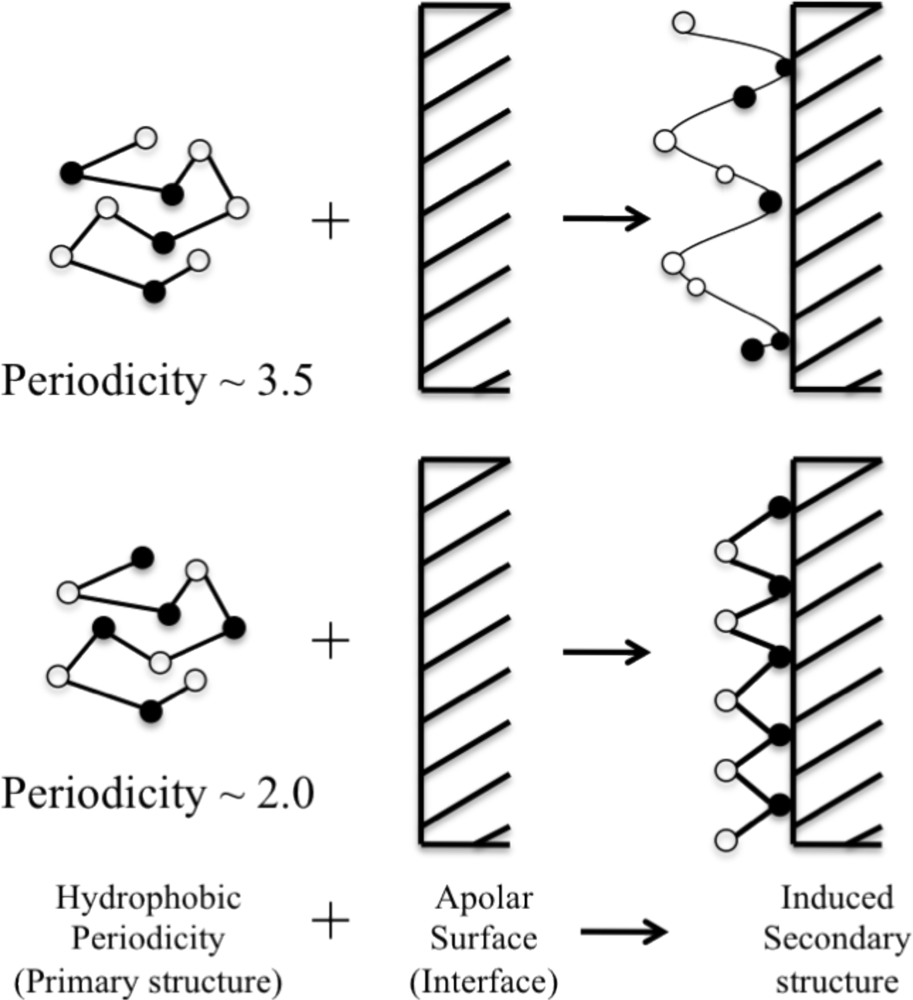
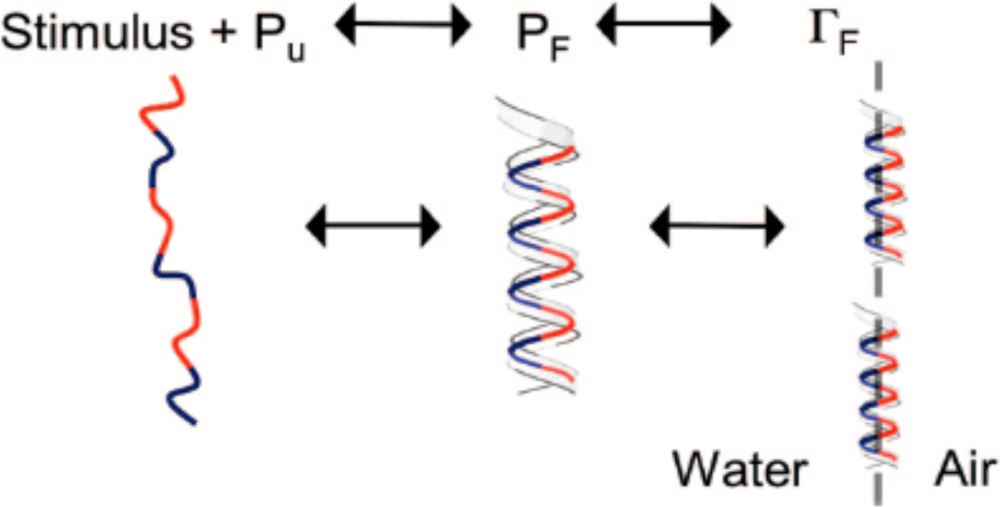
3. Designing Amphiphilic α-Helix Peptide
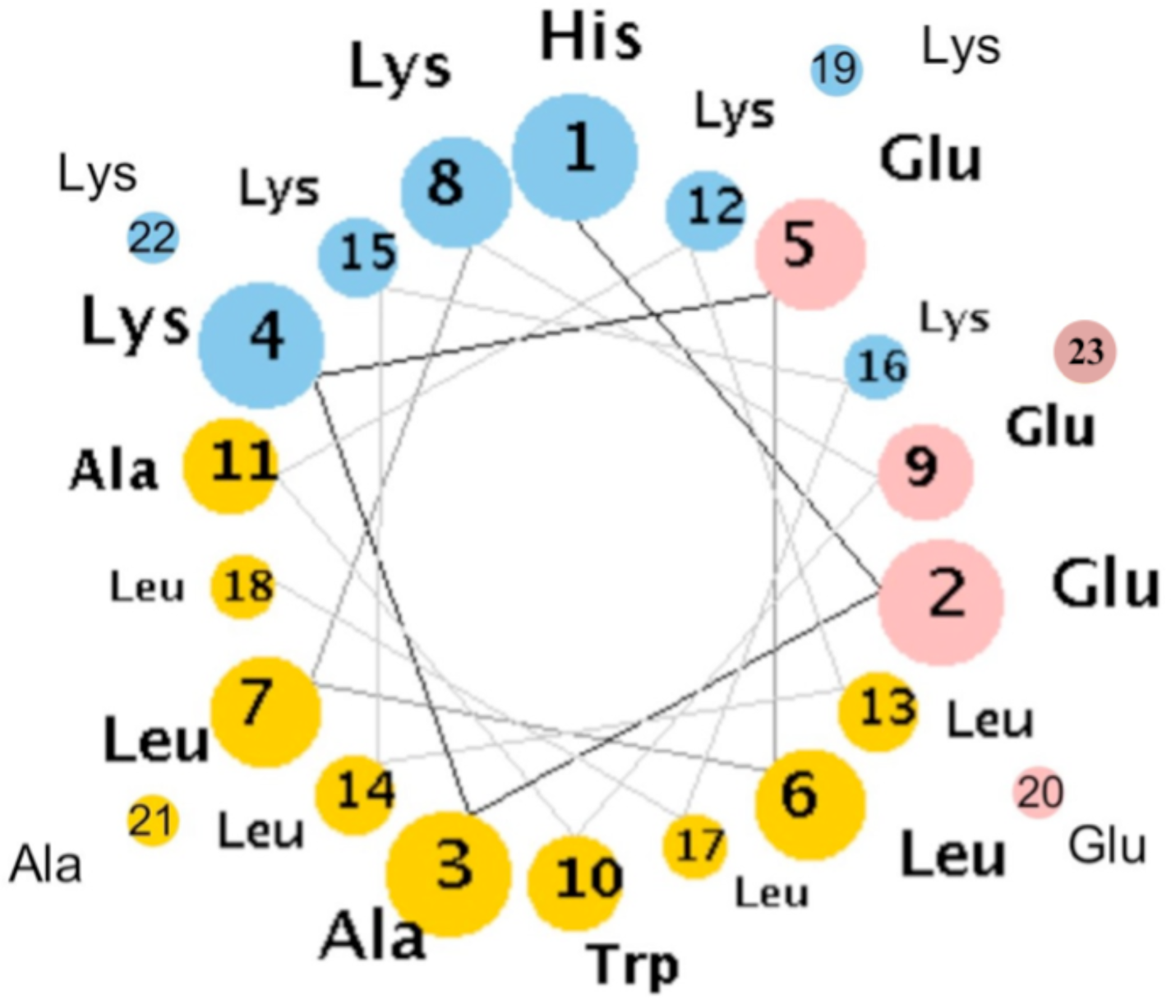
4. Engineered Synthetic Peptides
4.1. Membrane Mimics
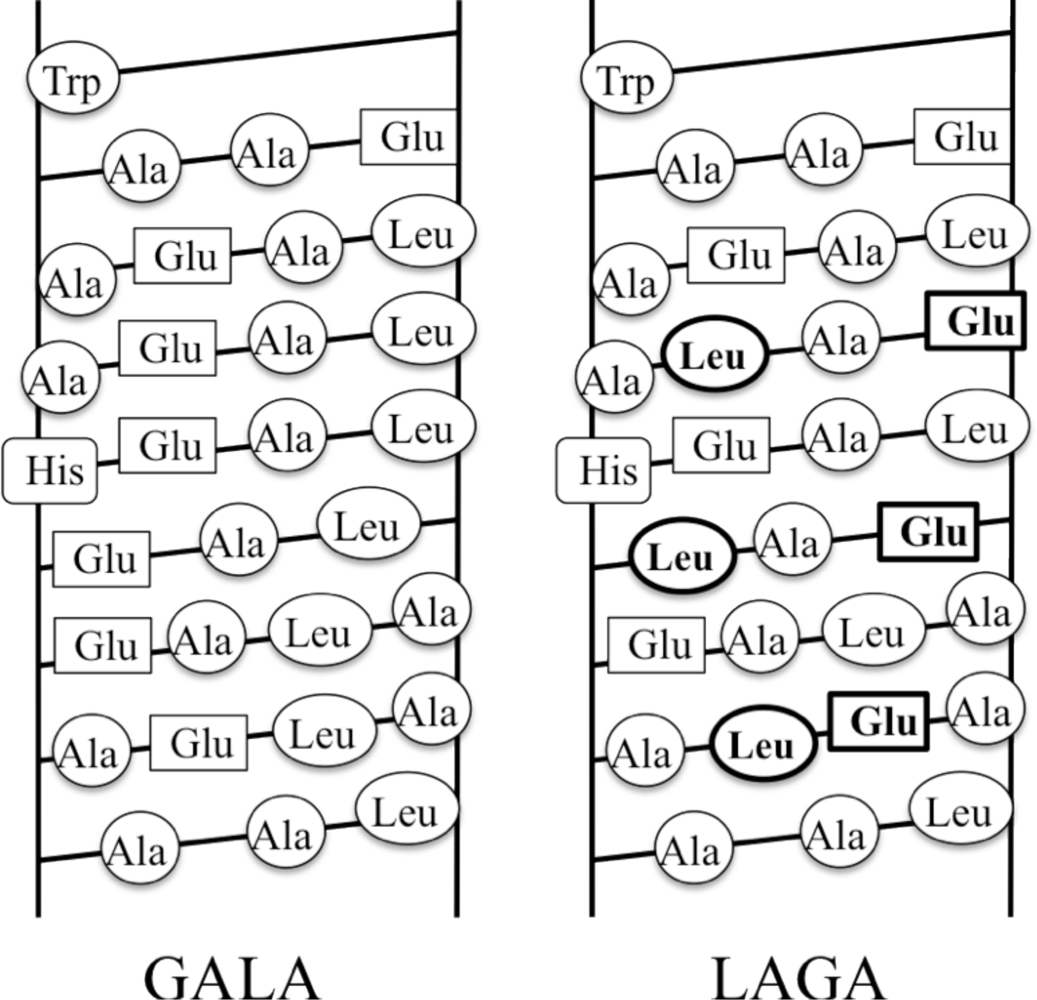
4.2. de Novo Designs
5. Conclusions
Acknowledgments
References
- Dunker, AK; Brown, CJ; Lawson, JD; Iakoucheva, LM; Obradović, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar]
- Dyson, HJ; Wright, PE. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol 2005, 6, 197–208. [Google Scholar]
- Fuxreiter, M; Tompa, P; Simon, I; Uversky, VN; Hansen, JC; Asturias, FJ. Malleable machines take shape in eukaryotic transcriptional regulation. Nat. Chem. Biol 2008, 4, 728–737. [Google Scholar]
- Iakoucheva, LM; Brown, CJ; Lawson, JD; Obradovic, Z; Dunker, AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol 2002, 323, 573–584. [Google Scholar]
- Iakoucheva, LM; Radivojac, P; Brown, CJ; O’Connor, TR; Sikes, JG; Obradovic, Z; Dunker, AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res 2004, 32, 1037–1049. [Google Scholar]
- Dames, SA; Martinez-Yamout, M; de Guzman, RN; Dyson, HJ; Wright, PE. Structural basis for Hif-1α/CBP recognition in the cellular hypoxic response. Proc. Nat. Acad. Sci. USA 2002, 99, 5271–5276. [Google Scholar]
- Creighton, TE. Protein folding. Biochem. J 1990, 270, 1–16. [Google Scholar]
- Pérez-Payà, E; Houghten, R; Blondelle, S. The role of amphipathicity in the folding, self-association and biological activity of multiple subunit small proteins. J. Biol. Chem 1995, 270, 1048. [Google Scholar]
- Kaiser, E; Kezdy, F. Amphiphilic secondary structure: Design of peptide hormones. Science 1984, 223, 249–255. [Google Scholar]
- Kaiser, ET; Kezdy, FJ. Secondary structures of proteins and peptides in amphiphilic environments: A review. Proc. Nat. Acad. Sci. USA 1983, 80, 1137–1143. [Google Scholar]
- Wright, PE; Dyson, HJ. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol 1999, 293, 321–331. [Google Scholar]
- Dunker, AK; Lawson, JD; Brown, CJ; Williams, RM; Romero, P; Oh, JS; Oldfield, CJ; Campen, AM; Ratliff, CM; Hipps, KW; Ausio, J; Nissen, MS; Reeves, R; Kang, C; Kissinger, CR; Bailey, RW; Griswold, MD; Chiu, W; Garner, EC; Obradovic, Z. Intrinsically disordered protein. J. Mol. Graphics Modell 2001, 19, 26–59. [Google Scholar]
- Dyson, HJ; Wright, PE. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol 2002, 12, 54–60. [Google Scholar]
- von Hippel, PH. From “simple” DNA-protein interactions to the macromolecular machines of gene expression. Annu. Rev. Biophys. Biomol. Struct 2007, 36, 79–105. [Google Scholar]
- Verkhivker, GM; Bouzida, D; Gehlhaar, DK; Rejto, PA; Freer, ST; Rose, PW. Simulating disorder-order transitions in molecular recognition of unstructured proteins: Where folding meets binding. Proc. Nat. Acad. Sci. USA 2003, 100, 5148–5153. [Google Scholar]
- Slutsky, M; Mirny, LA. Kinetics of protein-DNA interaction: Facilitated target location in sequence-dependent potential. Biophys. J 2004, 87, 4021–4035. [Google Scholar]
- Spolar, RS; Record, MT, Jr. Coupling of local folding to site-specific binding of proteins to DNA. Science 1994, 263, 777–784. [Google Scholar]
- Wright, PE; Dyson, HJ. Linking folding and binding. Curr. Opin. Struct. Biol 2009, 19, 31–38. [Google Scholar]
- Sugase, K; Dyson, HJ; Wright, PE. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 2007, 447, 1021–1025. [Google Scholar]
- Huang, Y; Liu, Z. Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: A critical assessment of the “Fly-Casting” mechanism. J. Mol. Biol 2009, 393, 1143–1159. [Google Scholar]
- Shoemaker, BA; Portman, JJ; Wolynes, PG. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Nat. Acad. Sci. USA 2000, 97, 8868–8873. [Google Scholar]
- Tsai, C-J; Kumar, S; Ma, B; Nussinov, R. Folding funnels, binding funnels, and protein function. Protein Sci 1999, 8, 1181–1190. [Google Scholar]
- Landschulz, WH; Johnson, PF; McKnight, SL. The leucine zipper: A hypothetical structure common to a new class of DNA binding proteins. Science 1988, 240, 1759–1764. [Google Scholar]
- O’Shea, EK; Rutkowski, R; Kim, PS. Evidence that the leucine zipper is a coiled coil. Science 1989, 243, 538–542. [Google Scholar]
- O’Neil, KT; Shuman, JD; Ampe, C; DeGrado, WF. DNA-induced increase in the alphahelical content of C/EBP and GCN4. Biochemistry 1991, 30, 9030–9034. [Google Scholar]
- O’Neil, K; Hoess, R; DeGrado, W. Design of DNA-binding peptides based on the leucine zipper motif. Science 1990, 249, 774–778. [Google Scholar]
- Talanian, R; McKnight, C; Kim, P. Sequence-specific DNA binding by a short peptide dimer. Science 1990, 249, 769–771. [Google Scholar]
- Talanian, RV; McKnight, CJ; Rutkowski, R; Kim, PS. Minimum length of a sequence-specific DNA binding peptide. Biochemistry 1992, 31, 6871–6875. [Google Scholar]
- Kim, Y-G; Park, H-J; Kim, KK; Lowenhaupt, K; Rich, A. A peptide with alternating lysines can act as a highly specific Z-DNA binding domain. Nucl. Acids Res 2006, 34, 4937–4942. [Google Scholar]
- Tu, RS; Marullo, R; Pynn, R; Bitton, R; Bianco-Peled, H; Tirrell, MV. Cooperative DNA binding and assembly by a bZip peptide-amphiphile. Soft Matter 2010, 6, 1035–1044. [Google Scholar]
- Cui, H; Webber, MJ; Stupp, SI. Self-Assembly of peptide amphiphiles: From molecules to nanostructures to biomaterials. Biopolymers 2010, 94, 1–18. [Google Scholar]
- Kokkoli, E; Mardilovich, A; Wedekind, A; Rexeisen, EL; Garg, A; Craig, JA. Self-assembly and applications of biomimetic and bioactive peptide-amphiphiles. Soft Matter 2006, 2, 1015–1024. [Google Scholar]
- Leulliot, N; Varani, G. Current topics in RNA-protein recognition: Control of specificity and biological function through induced fit and conformational capture. Biochemistry 2001, 40, 7947–7956. [Google Scholar]
- Williamson, JR. Proteins that bind RNA and the labs who love them. Nat. Struct. Mol. Biol 2001, 8, 390–391. [Google Scholar]
- Agalarov, SC; Prasad, GS; Funke, PM; Stout, CD; Williamson, JR. Structure of the S15,S6,S18-rRNA complex: Assembly of the 30S ribosome central domain. Science 2000, 288, 107–112. [Google Scholar]
- Henkels, CH; Kurz, JC; Fierke, CA; Oas, TG. Linked folding and anion binding of the bacillus subtilis ribonuclease P protein. Biochemistry 2001, 40, 2777–2789. [Google Scholar]
- Bouvet, P; Allain, FHT; Finger, LD; Dieckmann, T; Feigon, J. Recognition of pre-formed and flexible elements of an RNA stem-loop by nucleolin. J. Mol. Biol 2001, 309, 763–775. [Google Scholar]
- Allain, FHT; Bouvet, P; Dieckmann, T; Feigon, J. Molecular basis of sequence-specific recognition of pre-ribosomal RNA by nucleolin. EMBO J 2000, 19, 6870–6881. [Google Scholar]
- Mogridge, J; Legault, P; Li, J; van Oene, MD; Kay, LE; Greenblatt, J. Independent ligand-induced folding of the RNA-binding domain and two functionally distinct antitermination regions in the phage lambda N protein. Mol. Cell 1998, 1, 265–275. [Google Scholar]
- Bloomer, AC; Champness, JN; Bricogne, G; Staden, R; Klug, A. Protein disk of tobacco mosaic virus at 2.8Å resolution showing the interactions within and between subunits. Nature 1978, 276, 362–368. [Google Scholar]
- Champness, JN; Bloomer, AC; Bricogne, G; Butler, PJG; Klug, A. The structure of the protein disk of tobacco mosaic virus to 5Å resolution. Nature 1976, 259, 20–24. [Google Scholar]
- Jardetzky, O; Akasaka, K; Vogel, D; Morris, S; Holmes, KC. Unusual segmental flexibility in a region of tobacco mosaic virus coat protein. Nature 1978, 273, 564–566. [Google Scholar]
- Stubbs, G; Warren, S; Holmes, K. Structure of RNA and RNA binding site in tobacco mosaic virus from 4Å map calculated from X-ray fibre diagrams. Nature 1977, 267, 216–221. [Google Scholar]
- Mattaj, IW. RNA recognition: A family matter? Cell 1993, 73, 837–840. [Google Scholar]
- Burd, CG; Dreyfuss, G. Conserved structures and diversity of functions of RNA-binding proteins. Science 1994, 265, 615–621. [Google Scholar]
- Lazinski, D; Grzadzielska, E; Das, A. Sequence-specific recognition of RNA hairpins by bacteriophage antiterminators requires a conserved arginine-rich motif. Cell 1989, 59, 207–218. [Google Scholar]
- Daly, TJ; Rusche, JR; Maione, TE; Frankel, AD. Circular dichroism studies of the HIV-1 Rev protein and its specific RNA binding site. Biochemistry 1990, 29, 9791–9795. [Google Scholar]
- Tan, R; Frankel, AD. Costabilization of peptide and RNA structure in an HIV Rev peptide-RRE complex. Biochemistry 1994, 33, 14579–14585. [Google Scholar]
- Tan, R; Chen, L; Buettner, JA; Hudson, D; Frankel, AD. RNA recognition by an isolated alpha helix. Cell 1993, 73, 1031–1040. [Google Scholar]
- Kjems, J; Calnan, BJ; Frankel, AD; Sharp, PA. Specific binding of a basic peptide from HIV-1 REV. EMBO J 1992, 11, 1119–1129. [Google Scholar]
- DeGrado, W; Wasserman, Z; Lear, J. Protein design, a minimalist approach. Science 1989, 243, 622–628. [Google Scholar]
- Kamtekar, S; Schiffer, J; Xiong, H; Babik, J; Hecht, M. Protein design by binary patterning of polar and nonpolar amino acids. Science 1993, 262, 1680–1685. [Google Scholar]
- Bryson, JW; Betz, SF; Lu, HS; Suich, DJ; Zhou, HX; O’Neil, KT; DeGrado, WF. Protein design: A hierarchic approach. Science 1995, 270, 935–941. [Google Scholar]
- Beasley, J; Hecht, M. Protein design: The choice of de novo sequences. J. Biol. Chem 1997, 272, 2031. [Google Scholar]
- Tu, RS; Tirrell, M. Bottom-up design of biomimetic assemblies. Adv. Drug Delivery Rev 2004, 56, 1537–1563. [Google Scholar]
- Zhou, N; Kay, C; Hodges, R. Synthetic model proteins. Positional effects of interchain hydrophobic interactions on stability of two-stranded alpha-helical coiled-coils. J. Biol. Chem 1992, 267, 2664. [Google Scholar]
- Lear, J; Wasserman, Z; DeGrado, W. Synthetic amphiphilic peptide models for protein ion channels. Science 1988, 240, 1177–1181. [Google Scholar]
- Aurora, R; Creamer, T; Srinivasan, R; Rose, G. Local interactions in protein folding: Lessons from the alpha-helix. J. Biol. Chem 1997, 272, 1413. [Google Scholar]
- Chou, PY; Fasman, GD. Conformational parameters for amino acids in helical, beta-sheet, and random coil regions calculated from proteins. Biochemistry 1974, 13, 211–222. [Google Scholar]
- DeGrado, WF; Lear, JD. Induction of peptide conformation at apolar water interfaces. 1. A study with model peptides of defined hydrophobic periodicity. J. Am. Chem. Soc 1985, 107, 7684–7689. [Google Scholar]
- Xiong, H; Buckwalter, BL; Shieh, HM; Hecht, MH. Periodicity of polar and nonpolar amino acids is the major determinant of secondary structure in self-assembling oligomeric peptides. Proc. Nat. Acad. Sci. USA 1995, 92, 6349–6353. [Google Scholar]
- Monera, OD; Sereda, TJ; Zhou, NE; Kay, CM; Hodges, RS. Relationship of sidechain hydrophobicity and alpha-helical propensity on the stability of the single-stranded amphipathic alpha-helix. J. Pept. Sci 1995, 1, 319–329. [Google Scholar]
- O’Neil, K; DeGrado, W. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science 1990, 250, 646–651. [Google Scholar]
- Lyu, P; Liff, M; Marky, L; Kallenbach, N. Side chain contributions to the stability of alpha-helical structure in peptides. Science 1990, 250, 669–673. [Google Scholar]
- Narita, M; Tomotake, Y; Isokawa, S; Matsuzawa, T; Miyauchi, T. Syntheses and properties of resin-bound oligopeptides. 2. Infrared spectroscopic conformational analysis of cross-linked polystyrene resin bound oligoleucines in the swollen state. Macromolecules 1984, 17, 1903–1906. [Google Scholar]
- McLachlan, AD. Repeated helical pattern in apolipoprotein-A-I. Nature 1977, 267, 465–466. [Google Scholar]
- Fukushima, D; Kupferberg, JP; Yokoyama, S; Kroon, DJ; Kaiser, ET; Kezdy, FJ. A synthetic amphiphilic helical docosapeptide with the surface properties of plasma apolipoprotein A-I. J. Am. Chem. Soc 1979, 101, 3703–3704. [Google Scholar]
- Yokoyama, S; Fukushima, D; Kupferberg, J; Kezdy, F; Kaiser, E. The mechanism of activation of lecithin: Cholesterol acyltransferase by apolipoprotein AI and an amphiphilic peptide. J. Biol. Chem 1980, 255, 7333. [Google Scholar]
- Raghuraman, H; Chattopadhyay, A. Melittin: A membrane-active peptide with diverse functions. Biosci. Rep 2007, 27, 189–223. [Google Scholar]
- Blondelle, SE; Houghten, RA. Hemolytic and antimicrobial activities of the twenty-four individual omission analogs of melittin. Biochemistry 1991, 30, 4671–4678. [Google Scholar]
- DeGrado, WF; Kezdy, FJ; Kaiser, ET. Design, synthesis, and characterization of a cytotoxic peptide with melittin-like activity. J. Am. Chem. Soc 1981, 103, 679–681. [Google Scholar]
- DeGrado, WF; Musso, GF; Lieber, M; Kaiser, ET; Kezdy, FJ. Kinetics and mechanism of hemolysis induced by melittin and by a synthetic melittin analogue. Biophys. J 1982, 37, 329–338. [Google Scholar]
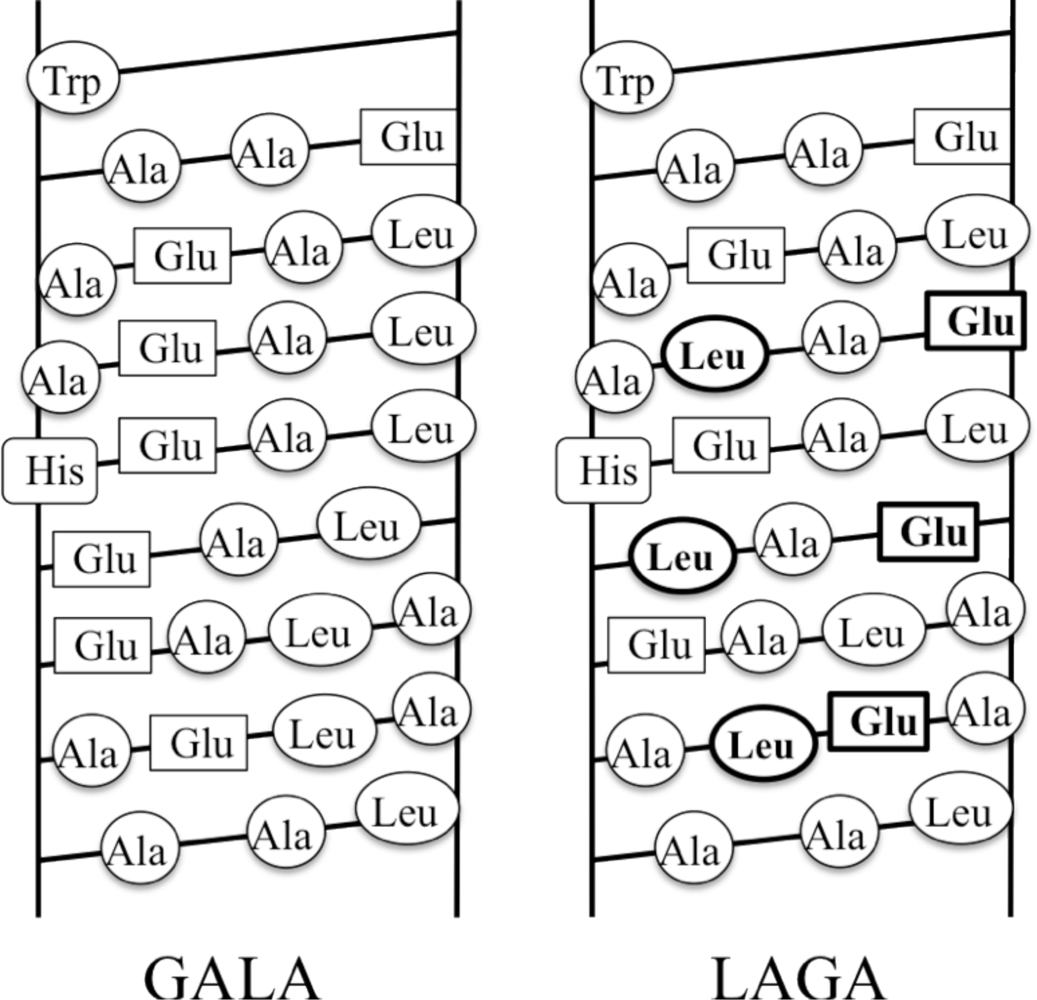
- Li, W; Nicol, F; Szoka, FC. GALA: A designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Adv. Drug Delivery Rev 2004, 56, 967–985. [Google Scholar]
- Subbarao, NK; Parente, RA; Szoka, FC; Nadasdi, L; Pongracz, K. The pH-dependent bilayer destabilization by an amphipathic peptide. Biochemistry 1987, 26, 2964–2972. [Google Scholar]
- Parente, R; Nir, S; Szoka, F. pH-dependent fusion of phosphatidylcholine small vesicles. Induction by a synthetic amphipathic peptide. J. Biol. Chem 1988, 263, 4724. [Google Scholar]
- Fattal, E; Nir, S; Parente, RA; Szoka, FC. Pore-forming peptides induce rapid phospholipid flip-flop in membranes. Biochemistry 1994, 33, 6721–6731. [Google Scholar]
- Nicol, F; Nir, S; Szoka, FC. Effect of cholesterol and charge on pore formation in bilayer vesicles by a pH-sensitive peptide. Biophys. J 1996, 71, 3288–3301. [Google Scholar]
- Nicol, F; Nir, S; Szoka, FC. Orientation of the pore-forming peptide GALA in POPC vesicles determined by a BODIPY-avidin/biotin binding assay. Biophys. J 1999, 76, 2121–2141. [Google Scholar]
- Shlomo Nir, FNFCS. Surface aggregation and membrane penetration by peptides: Relation to pore formation and fusion. Mol. Membr. Biol 1999, 16, 95–101. [Google Scholar]
- Parente, RA; Nir, S; Szoka, FC. Mechanism of leakage of phospholipid vesicle contents induced by the peptide GALA. Biochemistry 1990, 29, 8720–8728. [Google Scholar]
- Nicol, F; Nir, S; Szoka, FC. Effect of phospholipid composition on an amphipathic peptide-mediated pore formation in bilayer vesicles. Biophys. J 2000, 78, 818–829. [Google Scholar]
- Parente, RA; Nadasdi, L; Subbarao, NK; Szoka, FC. Association of a pH-sensitive peptide with membrane vesicles: Role of amino acid sequence. Biochemistry 1990, 29, 8713–8719. [Google Scholar]
- Plank, C; Oberhauser, B; Mechtler, K; Koch, C; Wagner, E. The influence of endosome-disruptive peptides on gene transfer using synthetic virus-like gene transfer systems. J. Biol. Chem 1994, 269, 12918. [Google Scholar]
- Haensler, J; Szoka, FC. Polyamidoamine cascade polymers mediate efficient transfection of cells in culture. Bioconjugate Chem 1993, 4, 372–379. [Google Scholar]
- Simoes, S; Slepushkin, V; Gaspar, R; de Lima, M; Duzgunes, N. Gene delivery by negatively charged ternary complexes of DNA, cationic liposomes and transferrin or fusigenic peptides. Gene Ther 1998, 5, 955. [Google Scholar]
- Simoes, S; Slepushkin, V; Pretzer, E; Dazin, P; Gaspar, R; Pedroso de Lima, MC; Duzgunes, N. Transfection of human macrophages by lipoplexes via the combined use of transferrin and pH-sensitive peptides. J. Leukocyte Biol 1999, 65, 270–279. [Google Scholar]
- Simoes, S; Slepushkin, V; Pires, P; Gaspar, R; de Lima, M; Dsuzgunes, N. Mechanisms of gene transfer mediated by lipoplexes associated with targeting ligands or pH-sensitive peptides. Gene Ther 1999, 6, 1798. [Google Scholar]
- Wyman, TB; Nicol, F; Zelphati, O; Scaria, PV; Plank, C; Szoka, FC. Design, synthesis, and characterization of a cationic peptide that binds to nucleic acids and permeabilizes bilayers. Biochemistry 1997, 36, 3008–3017. [Google Scholar]
- Plank, C; Tang, MX; Wolfe, AR; Szoka, FC. Branched cationic peptides for gene delivery: Role of type and number of cationic residues in formation and in vitro activity of DNA polyplexes. Hum. Gene Ther 2004, 10, 319–332. [Google Scholar]
- Jain, V; Jimenez, A; Maldarelli, C; Tu, RS. Dynamic surface activity by folding and unfolding an amphiphilic α-helix. Langmuir 2008, 24, 9923–9928. [Google Scholar]
- Shai, Y. Molecular recognition between membrane-spanning polypeptides. Trends in Biochem. Sci 1995, 20, 460–464. [Google Scholar]
- Han, X; Bushweller, JH; Cafiso, DS; Tamm, LK. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Mol. Biol 2001, 8, 715–720. [Google Scholar]
- Jain, VP; Maldarelli, C; Tu, RS. Modeling the dynamic folding and surface-activity of a helical peptide adsorbing to a pendant bubble interface. J. Colloid Interface Sci 2009, 331, 364–370. [Google Scholar]



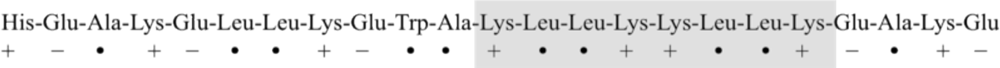


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
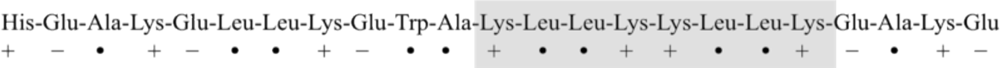{kind=link}
{kind=link}
{kind=link}
| Helical Residues | Pα | β-Sheet residues | Pβ |
|---|---|---|---|
| Glu(−) | 1.53 | Met | 1.67 |
| Ala | 1.45 | Val | 1.65 |
| Leu | 1.34 | Ile | 1.60 |
| His(+) | 1.24 | Cys | 1.30 |
| Met | 1.20 | Tyr | 1.29 |
| Gln | 1.17 | Phe | 1.28 |
| Trp | 1.14 | Gln | 1.23 |
| Val | 1.14 | Leu | 1.22 |
| Phe | 1.12 | Thr | 1.20 |
| Lys(+) | 1.07 | Trp | 1.19 |
| Ile | 1.00 | Ala | 0.97 |
| Asp(−) | 0.98 | Arg(+) | 0.90 |
| Thr | 0.82 | Gly | 0.81 |
| Ser | 0.79 | Asp(−) | 0.80 |
| Arg | 0.79 | Lys(+) | 0.74 |
| Cys | 0.77 | Ser | 0.72 |
| Asn | 0.73 | His(+) | 0.71 |
| Tyr | 0.61 | Asn | 0.65 |
| Pro | 0.59 | Pro | 0.62 |
| Gly | 0.53 | Glu(−) | 0.26 |
| Peptide | Hydrophobic repeat period | Intrinsic propensity | Result in bulk |
|---|---|---|---|
| LKKLLKL (A) | 3.5 | α (Pα = 1.19, Pβ = 1.06) | Sequence too short |
| (LKKLLKL)2 (B) | 3.5 | α (Pα = 1.19, Pβ = 1.06) | α |
| LKLKLKL (C) | 2.0 | α (Pα = 1.19, Pβ = 1.06) | β |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jain, V.P.; Tu, R.S. Coupled Folding and Specific Binding: Fishing for Amphiphilicity. Int. J. Mol. Sci. 2011, 12, 1431-1450. https://doi.org/10.3390/ijms12031431
Jain VP, Tu RS. Coupled Folding and Specific Binding: Fishing for Amphiphilicity. International Journal of Molecular Sciences. 2011; 12(3):1431-1450. https://doi.org/10.3390/ijms12031431
Chicago/Turabian StyleJain, Vikas P., and Raymond S. Tu. 2011. "Coupled Folding and Specific Binding: Fishing for Amphiphilicity" International Journal of Molecular Sciences 12, no. 3: 1431-1450. https://doi.org/10.3390/ijms12031431