Anchoring Intrinsically Disordered Proteins to Multiple Targets: Lessons from N-Terminus of the p53 Protein

Abstract
:
1. Introduction
2. Results and Discussion
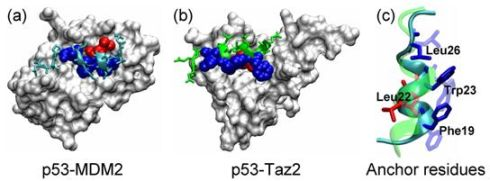
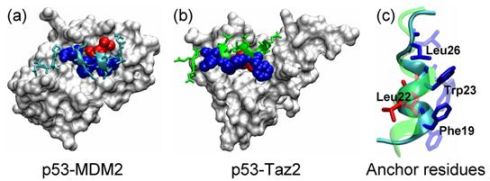
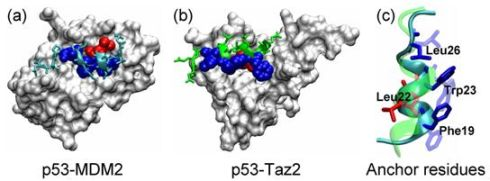
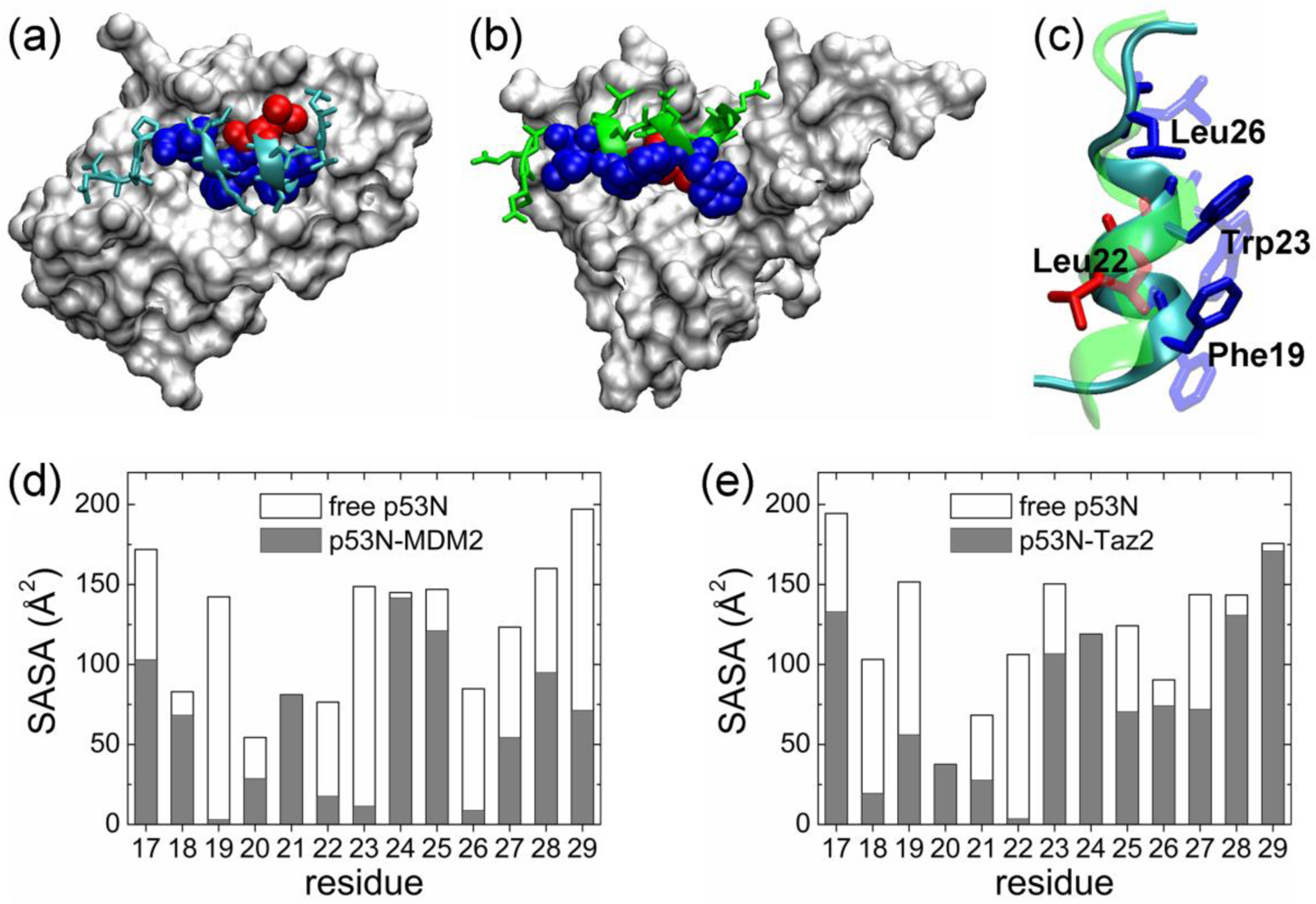
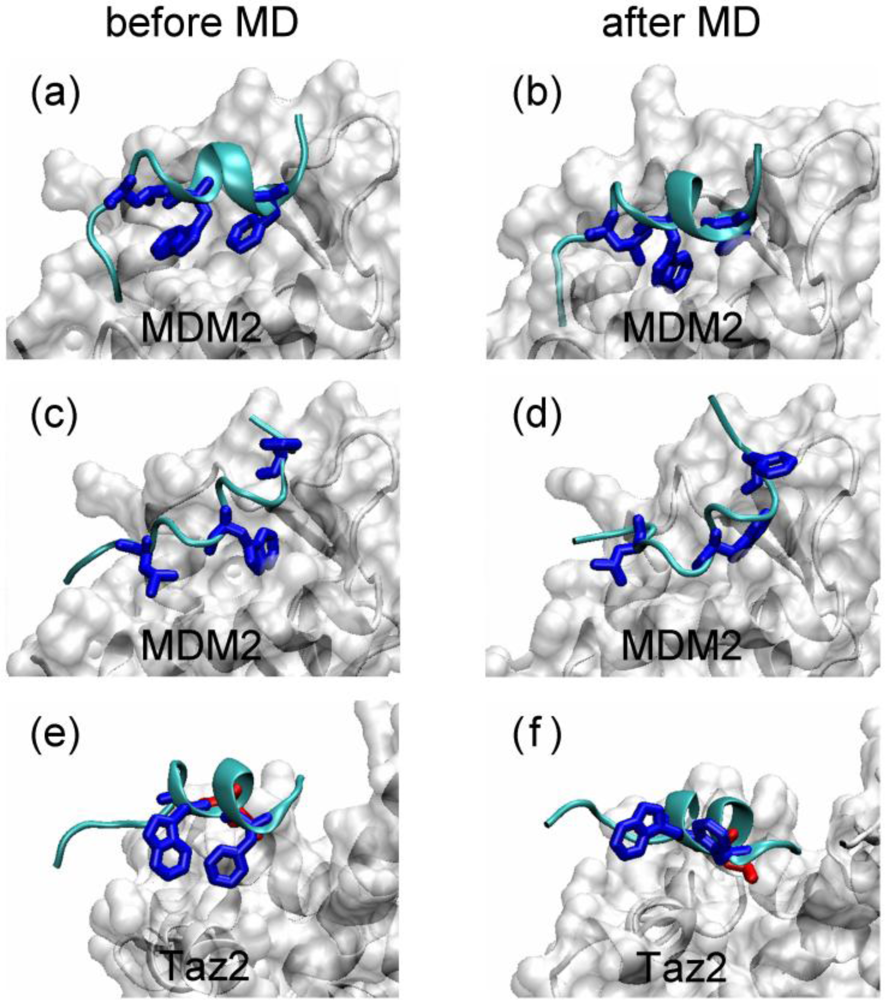
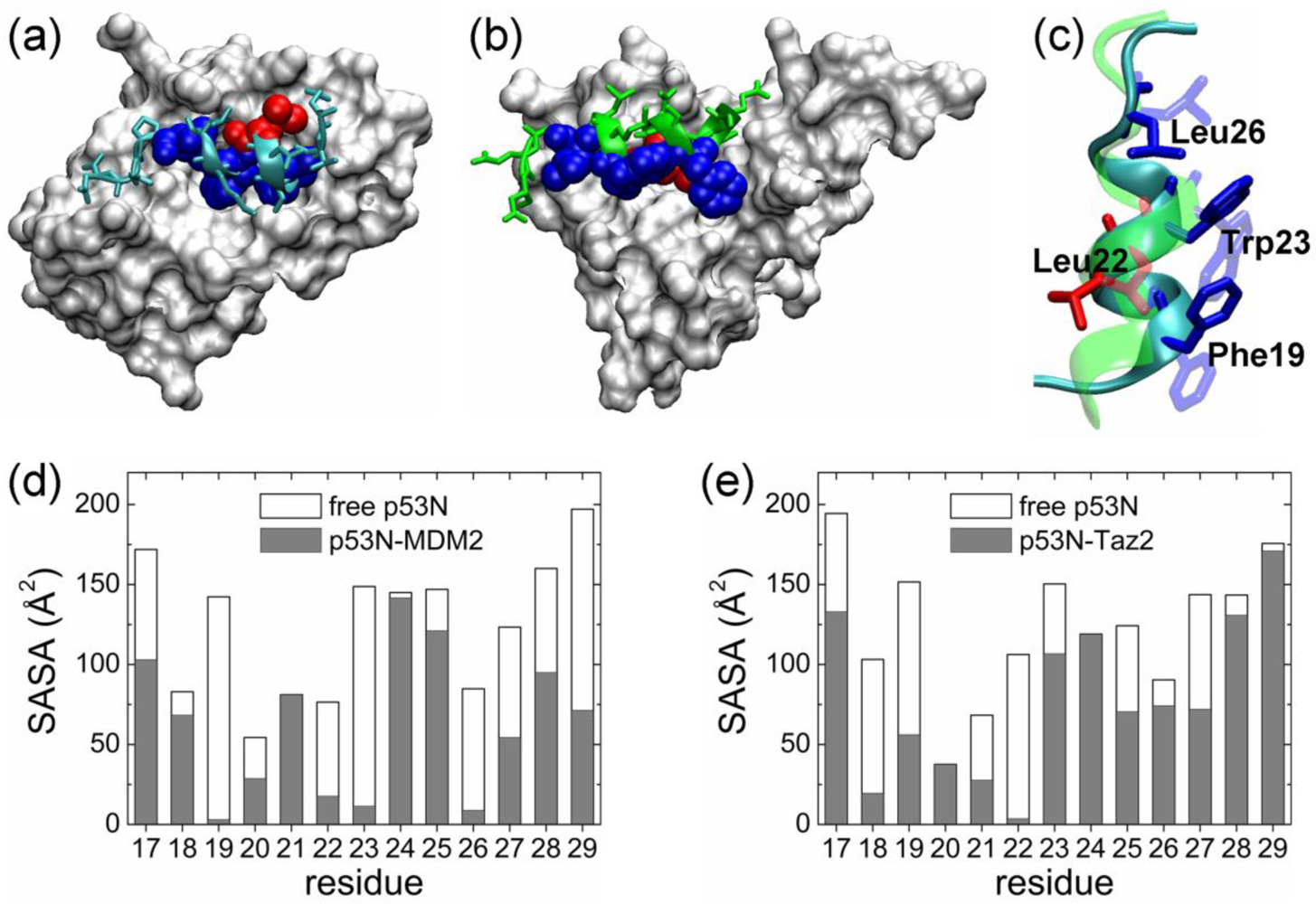
2.1. Anchor Residues in the p53N Complexes
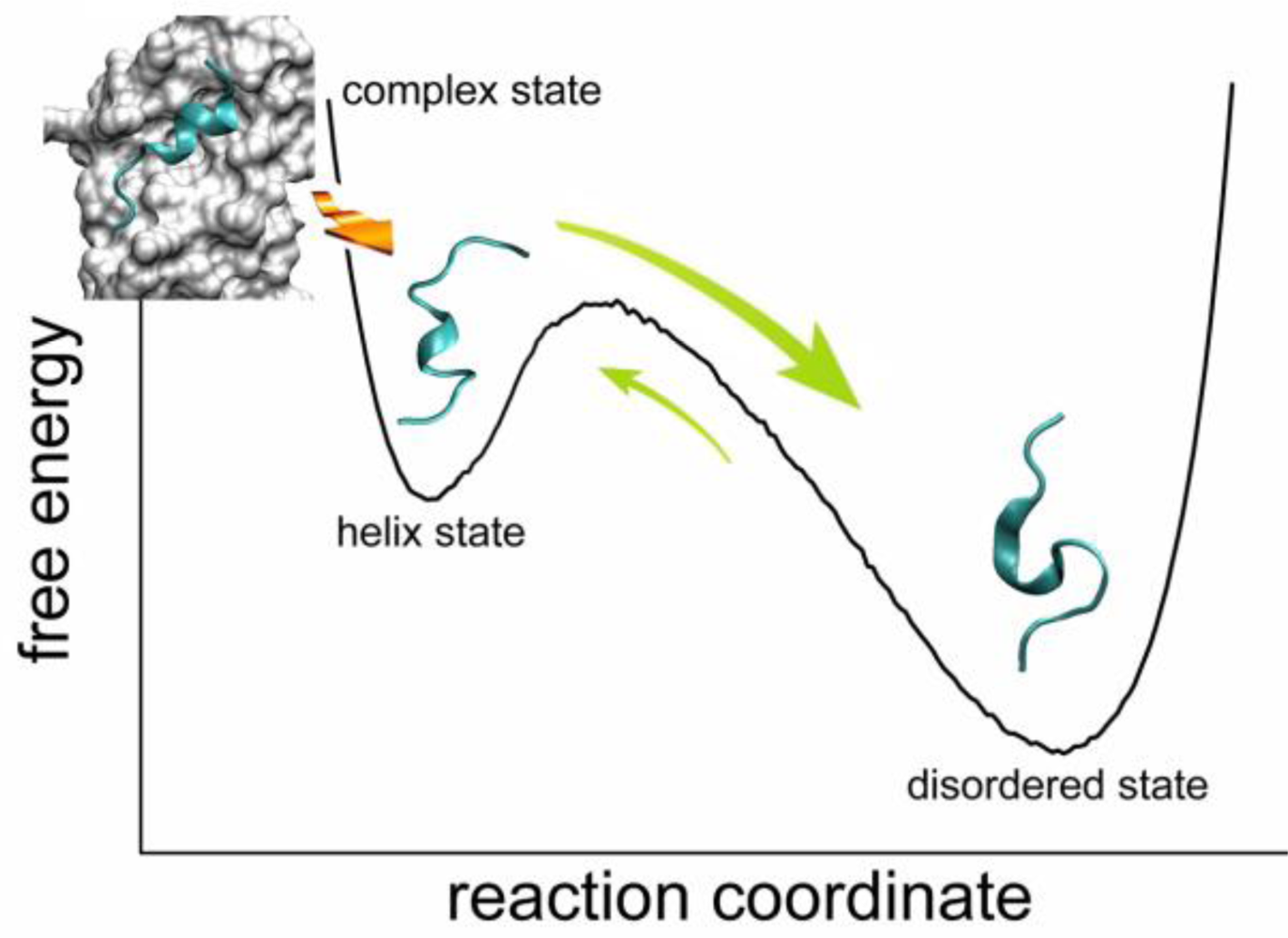
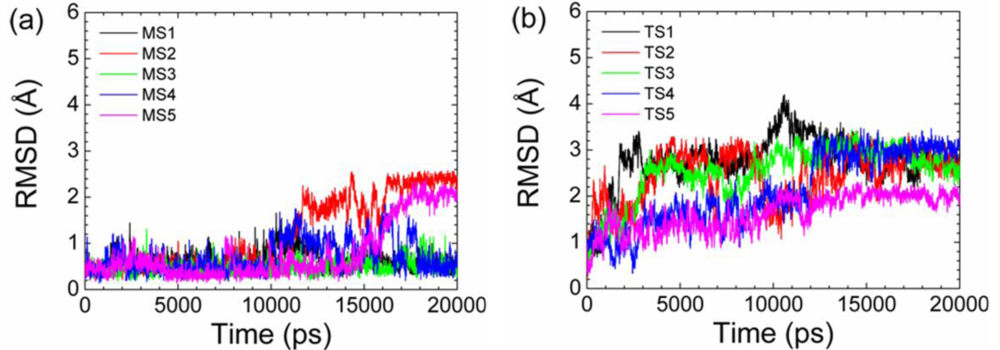
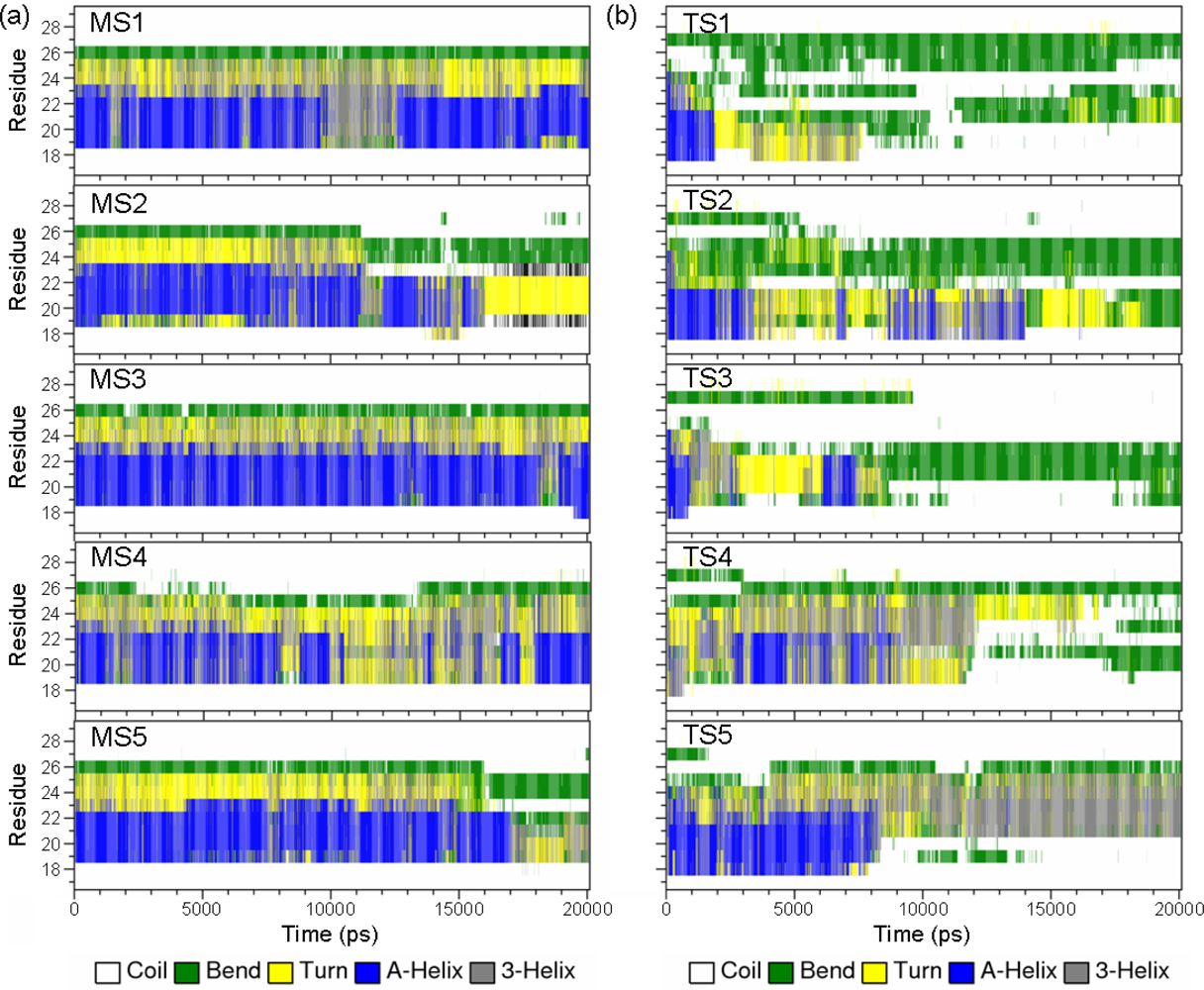
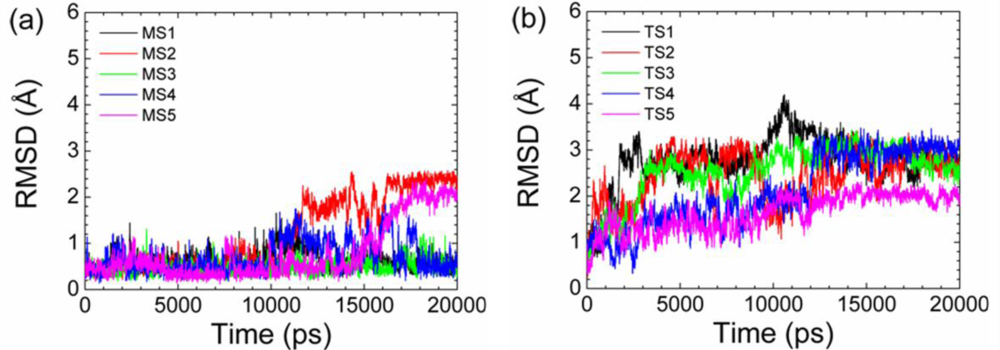
2.2. Transient Stable Helix of the p53N
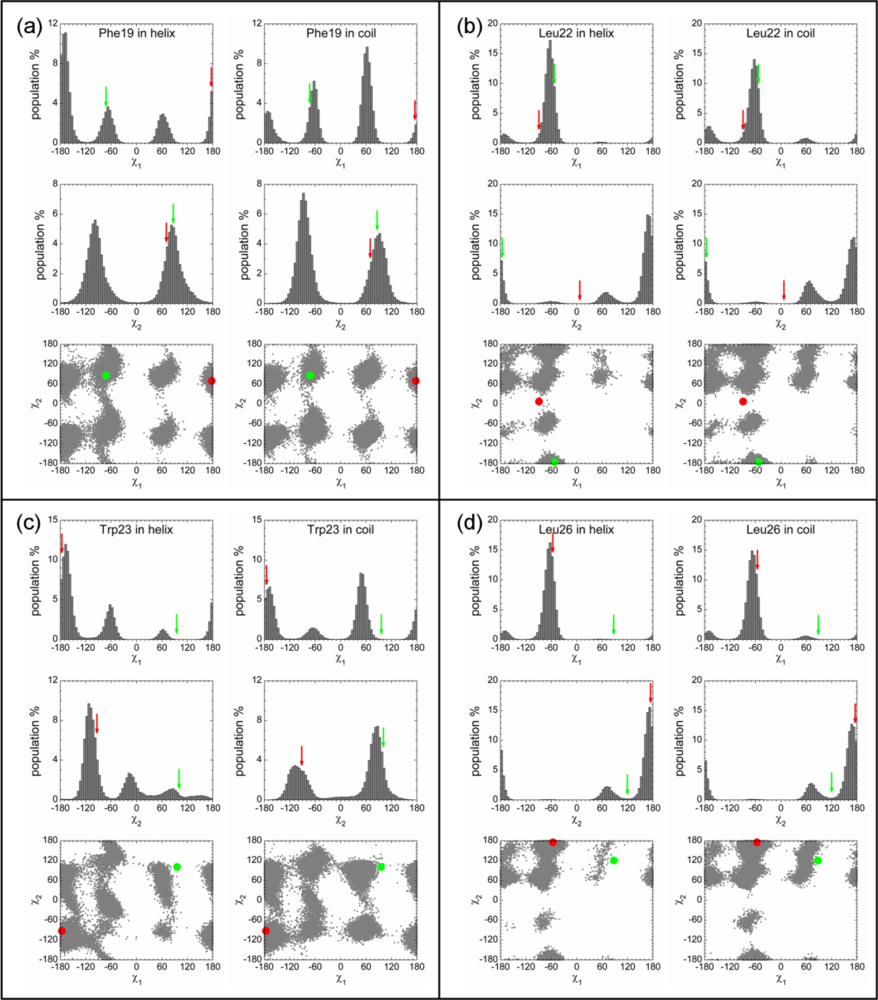
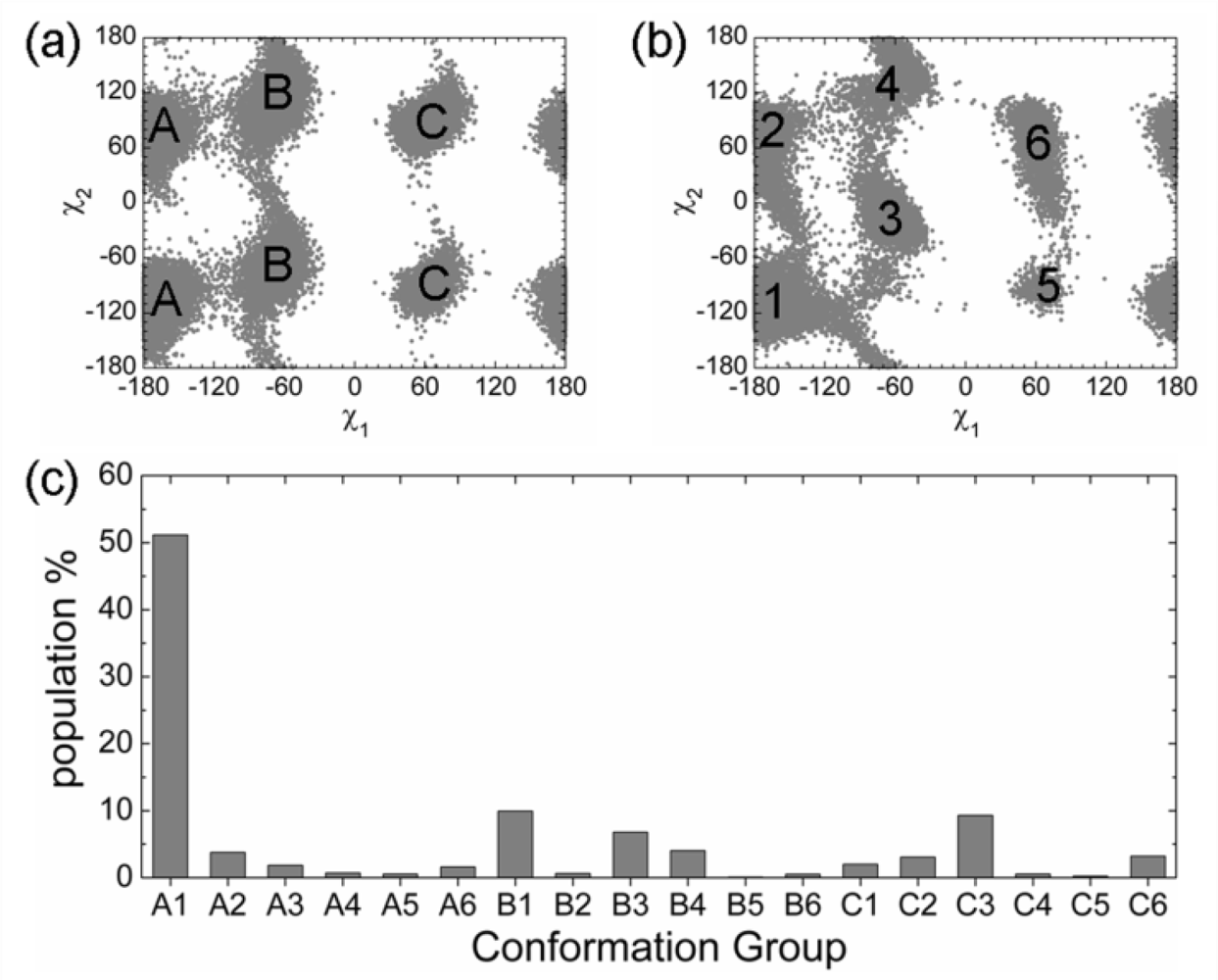
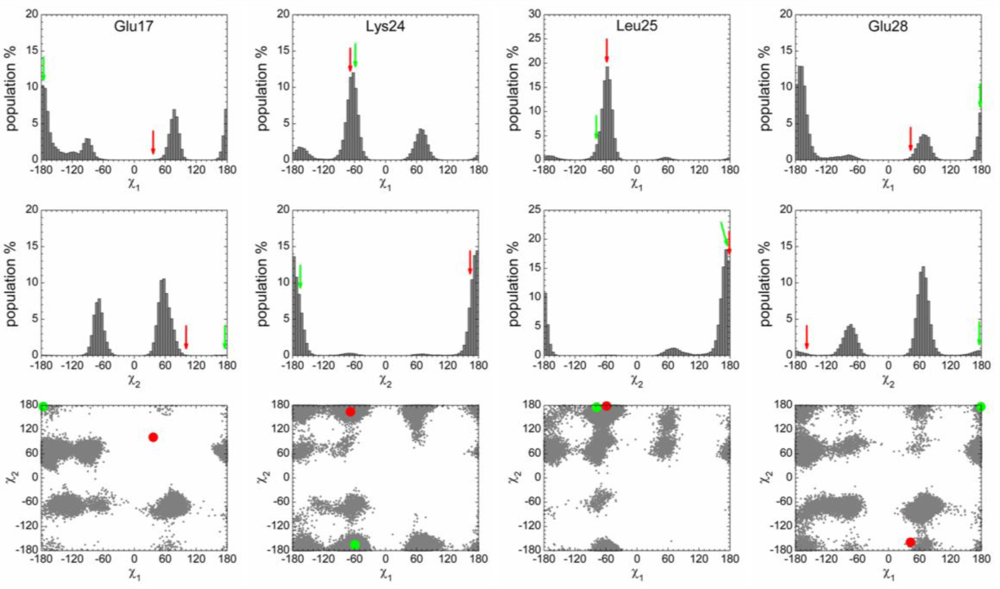
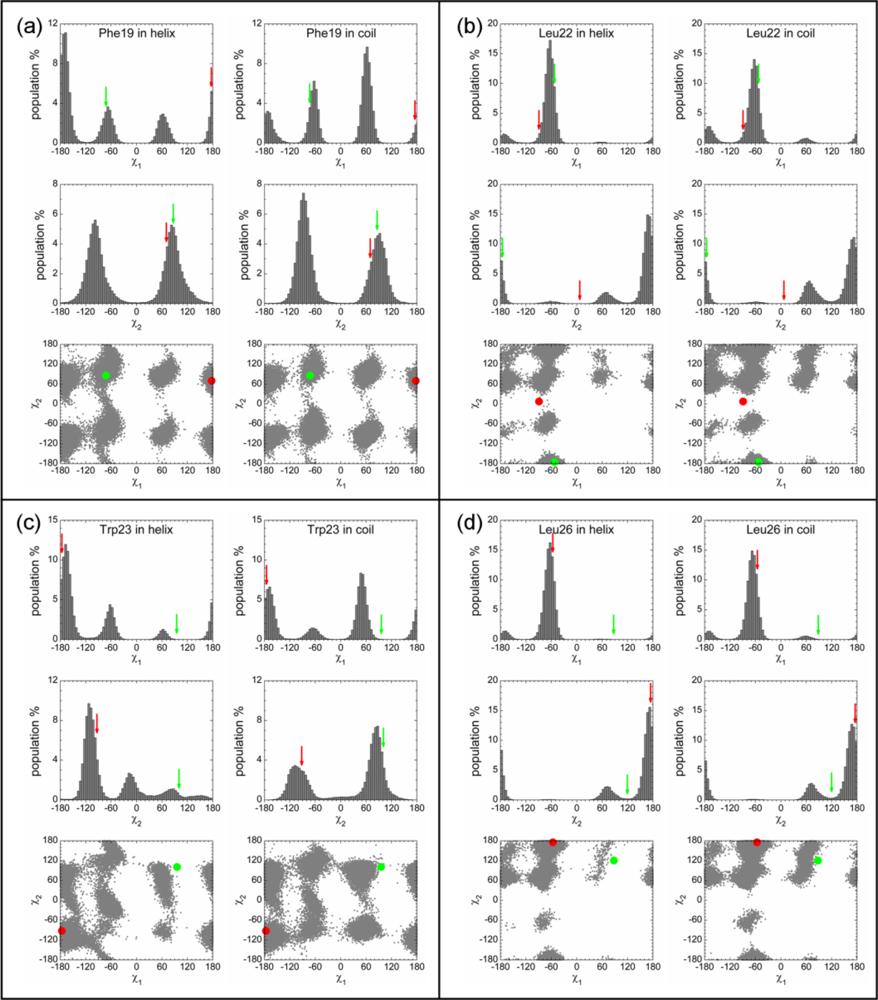
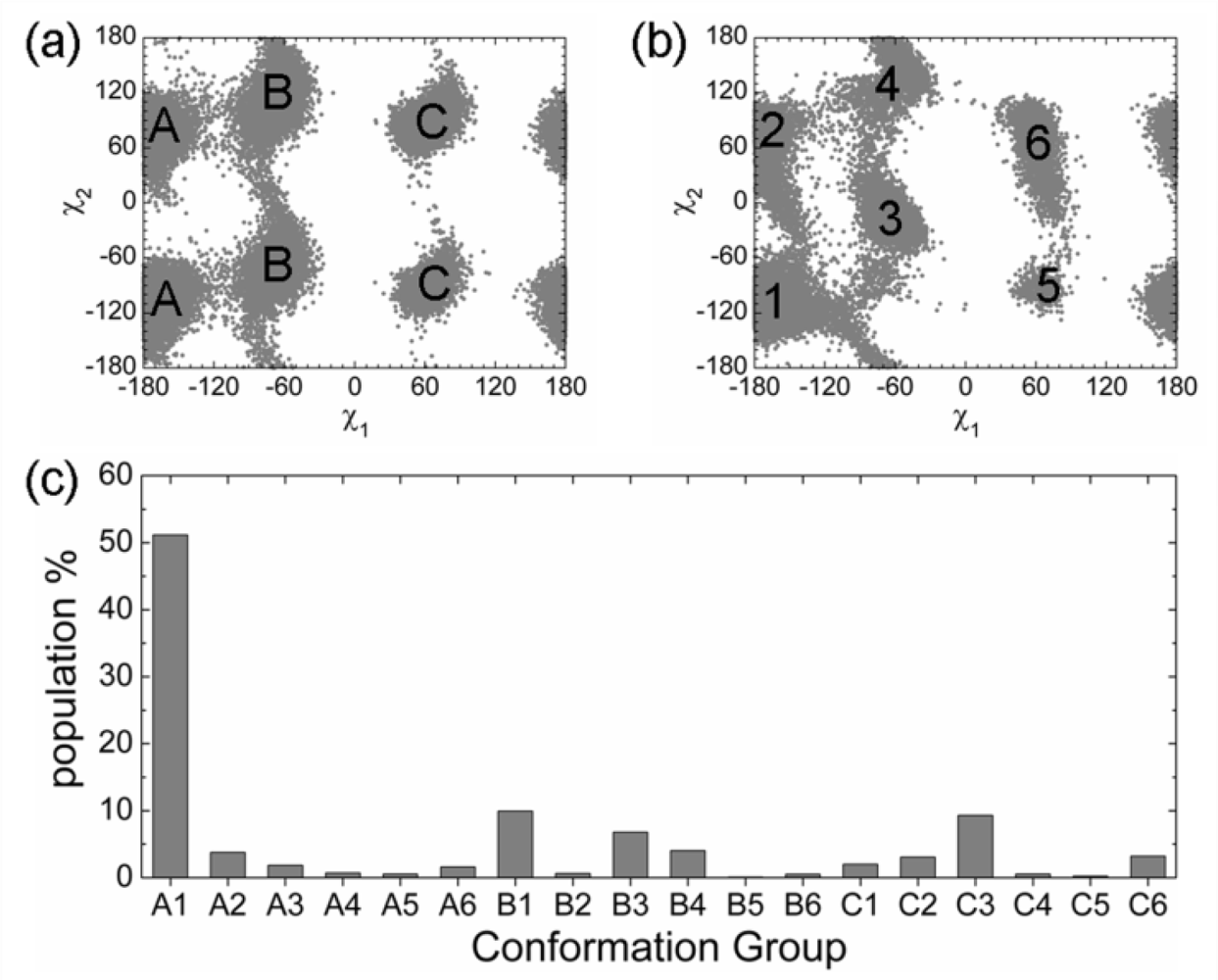
2.3. Analysis of the Side Chain Conformations of the Anchor Residues
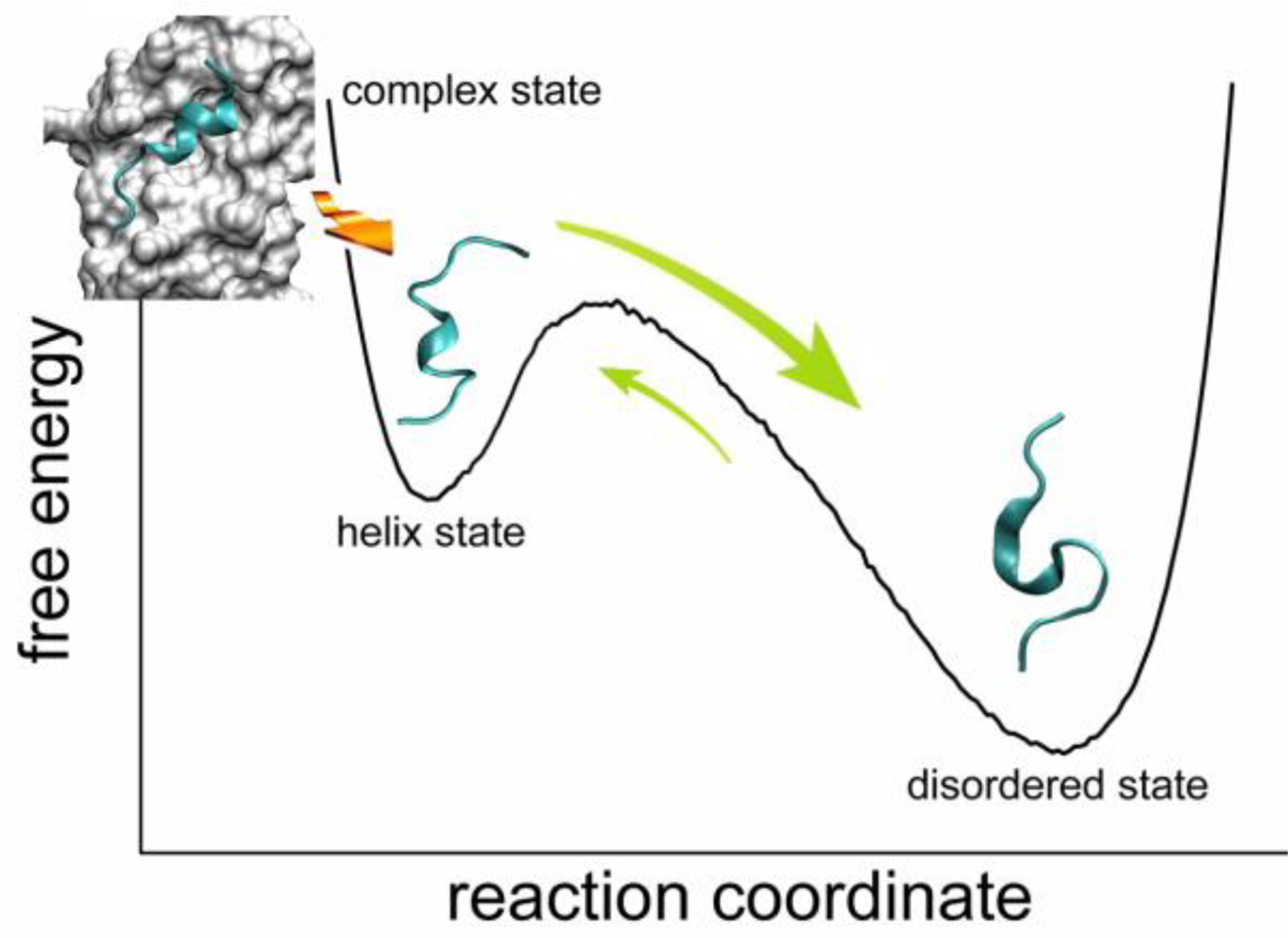
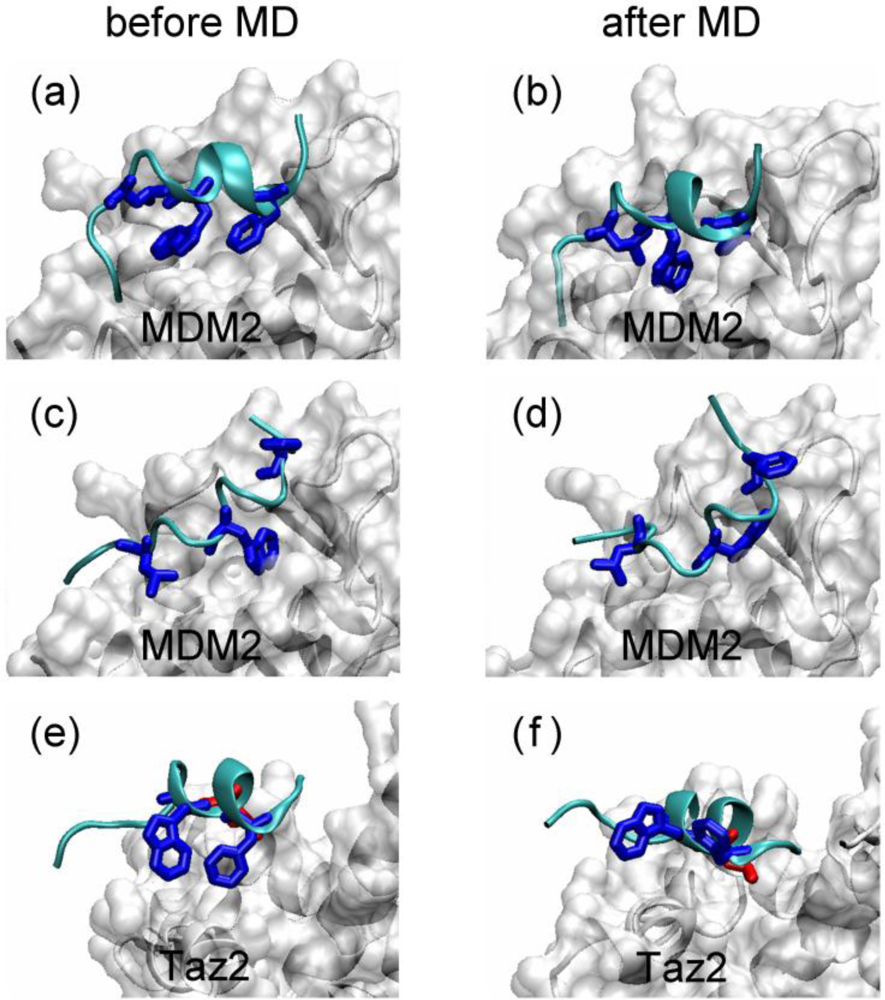
2.4. Transient Formation of Helical Structures Promotes the Binding Process
2.5. Discussions: Roles of Anchor Residues in Molecular Recognition
3. Method Section
3.1. Systems Setup
3.2. Molecular Dynamics Simulations
3.3. Analysis
4. Conclusions
Supplementary Materials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Simulations | Initial Conformation* | Simulation Duration (ns) | Unfolding Time (ns) |
|---|---|---|---|
| MS1 | M | 100 | 43 |
| MS2 | M | 20 | 11 |
| MS3 | M | 100 | 42 |
| MS4 | M | 100 | 25 |
| MS5 | M | 20 | 17 |
| MS6 | M | 60 | 28 |
| MS7 | M | 20 | 10 |
| MS8 | M | 20 | 15 |
| MS9 | M | 60 | 23 |
| MS10 | M | 60 | 31 |
| TS1 | T | 20 | 2 |
| TS2 | T | 20 | 4 |
| TS3 | T | 20 | 3 |
| TS4 | T | 20 | 9 |
| TS5 | T | 20 | 8 |
| DS1 | D | 50 | - |
| DS2 | D | 50 | - |
| DS3 | D | 50 | - |
| DS4 | D | 50 | - |
| DS5 | D | 50 | - |
Acknowledgments
References
- Larsen, TA; Olson, AJ; Goodsell, DS. Morphology of protein-protein interfaces. Structure 1998, 6, 421–427. [Google Scholar]
- Lo Conte, L; Chothia, C; Janin, J. The atomic structure of protein-protein recognition sites. J. Mol. Biol 1999, 285, 2177–2198. [Google Scholar]
- Ma, BY; Elkayam, T; Wolfson, H; Nussinov, R. Protein-protein interactions: Structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Natl. Acad. Sci. USA 2003, 100, 5772–5777. [Google Scholar]
- Bai, HJ; Ma, WZ; Liu, SY; Lai, LH. Dynamic property is a key determinant for protein-protein interactions. Proteins 2008, 70, 1323–1331. [Google Scholar]
- Zhang, QC; Petrey, D; Norel, R; Honig, BH. Protein interface conservation across structure space. Proc. Natl. Acad. Sci. USA 2010, 107, 10896–10901. [Google Scholar]
- Rajamani, D; Thiel, S; Vajda, S; Camacho, CJ. Anchor residues in protein-rotein interactions. Proc. Natl. Acad. Sci. USA 2004, 101, 11287–11292. [Google Scholar]
- Kimura, SR; Brower, RC; Vajda, S; Camacho, CJ. Dynamical view of the positions of key side chains in protein-protein recognition. Biophys. J 2001, 80, 635–642. [Google Scholar]
- Li, X; Keskin, O; Ma, B; Nussinov, R; Liang, J. Protein-protein interactions: Hot spots and structurally conserved residues often locate in complemented pockets that pre-organized in the unbound states: Implications for docking. J. Mol. Biol 2004, 344, 781–795. [Google Scholar]
- Camacho, CJ. Modeling side-chains using molecular dynamics improve recognition of binding region in CAPRI targets. Proteins 2005, 60, 245–251. [Google Scholar]
- Smith, GR; Sternberg, MJE; Bates, PA. The relationship between the flexibility of proteins and their conformational states on forming protein-protein complexes with an application to protein-protein docking. J. Mol. Biol 2005, 347, 1077–1101. [Google Scholar]
- Yogurtcu, ON; Erdemli, SB; Nussinov, R; Turkay, M; Keskin, O. Restricted mobility of conserved residues in protein-protein interfaces in molecular simulations. Biophys. J 2008, 94, 3475–3485. [Google Scholar]
- Ben-Shimon, A; Eisenstein, M. Computational mapping of anchoring spots on protein surfaces. J. Mol. Biol 2010, 402, 259–277. [Google Scholar]
- Csermely, P; Palotai, R; Nussinov, R. Induced fit, conformational selection and independent dynamic segments: An extended view of binding events. Trends Biochem. Sci 2010, 35, 539–546. [Google Scholar]
- Wlodarski, T; Zagrovic, B. Conformational selection and induced fit mechanism underlie specificity in noncovalent interactions with ubiquitin. Proc. Natl. Acad. Sci. USA 2009, 106, 19346–19351. [Google Scholar]
- Fink, AL. Compact intermediate states in protein folding. Annu. Rev. Biophys. Biomol. Struct 1995, 24, 495–522. [Google Scholar]
- Wright, PE; Dyson, HJ. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol 1999, 293, 321–331. [Google Scholar]
- Uversky, VN; Gillespie, JR; Fink, AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar]
- Dunker, AK; Lawson, JD; Brown, CJ; Williams, RM; Romero, P; Oh, JS; Oldfield, CJ; Campen, AM; Ratliff, CM; Hipps, KW; Ausio, J; Nissen, MS; Reeves, R; Kang, C; Kissinger, CR; Bailey, RW; Griswold, MD; Chiu, W; Garner, EC; Obradovic, Z. Intrinsically disordered protein. J. Mol. Graphics Modell 2001, 19, 26–59. [Google Scholar]
- Dunker, AK; Obradovic, Z. The protein trinity–linking function and disorder. Nat. Biotechnol 2001, 19, 805–806. [Google Scholar]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci 2002, 27, 527–533. [Google Scholar]
- Uversky, VN. Natively unfolded proteins: A point where biology waits for physics. Protein Sci 2002, 11, 739–756. [Google Scholar]
- Uversky, VN. What does it mean to be natively unfolded? Eur. J. Biochem 2002, 269, 2–12. [Google Scholar]
- Dunker, AK; Brown, CJ; Lawson, JD; Iakoucheva, LM; Obradović, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar]
- Dunker, AK; Brown, CJ; Obradović, Z. Identification and functions of usefully disordered proteins. Adv. Protein Chem 2002, 62, 25–49. [Google Scholar]
- Tompa, P; Csermely, P. The role of structural disorder in the function of RNA and protein chaperones. FASEB J 2004, 18, 1169–1175. [Google Scholar]
- Dyson, HJ; Wright, PE. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol 2005, 6, 197–208. [Google Scholar]
- Radivojac, P; Iakoucheva, LM; Oldfield, CJ; Obradovic, Z; Uversky, VN; Dunker, AK. Intrinsic disorder and functional proteomics. Biophys. J 2007, 92, 1439–1456. [Google Scholar]
- Huang, YQ; Liu, ZR. Intrinsically disordered proteins: The new sequence-structure-function relations. Acta Phys. Chim. Sin 2010, 26, 2061–2072. [Google Scholar]
- Uversky, VN; Dunker, AK. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar]
- Turoverov, KK; Kuznetsova, IM; Uversky, VN. The protein kingdom extended: Ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog. Biophys. Mol. Biol 2010, 102, 73–84. [Google Scholar]
- He, B; Wang, KJ; Liu, YL; Xue, B; Uversky, VN; Dunker, AK. Predicting intrinsic disorder in proteins: An overview. Cell Res 2009, 19, 929–949. [Google Scholar]
- Tompa, P; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci 2008, 33, 2–8. [Google Scholar]
- Galea, CA; Wang, Y; Sivakolundu, SG; Kriwacki, RW. Regulation of cell division by intrinsically unstructured proteins: Intrinsic flexibility, modularity, and signaling conduits. Biochemistry 2008, 47, 7598–7609. [Google Scholar]
- Shoemaker, BA; Portman, JJ; Wolynes, PG. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. USA 2000, 97, 8868–8873. [Google Scholar]
- Huang, YQ; Liu, ZR. Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: A critical assessment of the “fly-casting” mechanism. J. Mol. Biol 2009, 393, 1143–1159. [Google Scholar]
- Mittag, T; Kay, LE; Forman-Kay, JD. Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit 2010, 23, 105–116. [Google Scholar]
- Huang, YQ; Liu, ZR. Nonnative interactions in coupled folding and binding processes of intrinsically disordered proteins. PLoS ONE 2010, e15375. [Google Scholar]
- Patil, A; Kinoshita, K; Nakamura, H. Hub promiscuity in protein-protein interaction networks. Int. J. Mol. Sci 2010, 11, 1930–1943. [Google Scholar]
- Receveur-Bréchot, V; Bourhis, J-M; Uversky, VN; Canard, B; Longhi, S. Assessing protein disorder and induced folding. Proteins 2006, 62, 24–45. [Google Scholar]
- Mittag, T; Forman-Kay, JD. Atomic-level characterization of disordered protein ensembles. Curr. Opin. Struct. Biol 2007, 17, 3–14. [Google Scholar]
- Chen, HM; Rhoades, E. Fluorescence characterization of denatured proteins. Curr. Opin. Struct. Biol 2008, 18, 516–524. [Google Scholar]
- Eliezer, D. Biophysical characterization of intrinsically disordered proteins. Curr. Opin. Struct. Biol 2009, 19, 23–30. [Google Scholar]
- Uversky, V; Longhi, S. Instrumental Analysis of Intrinsically Disordered Proteins: Assessing Structure and Conformation; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2010. [Google Scholar]
- Csizmók, V; Bokor, M; Bánki, P; Klement, T; Medzihradszky, KF; Friedrich, P; Tompa, KA; Tompa, P. Primary contact sites in intrinsically unstructured proteins: The case of calpastatin and microtubule-associated protein 2. Biochemistry 2005, 44, 3955–3964. [Google Scholar]
- Oldfield, CJ; Cheng, Y; Cortese, MS; Brown, CJ; Uversky, VN; Dunker, AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar]
- Mohan, A; Oldfield, CJ; Radivojac, P; Vacic, V; Cortese, MS; Dunker, AK; Uversky, VN. Analysis of molecular recognition features (MoRFs). J. Mol. Biol 2006, 362, 1043–1059. [Google Scholar]
- Vacic, V; Oldfield, CJ; Mohan, A; Radivojac, P; Cortese, MS; Uversky, VN; Dunker, AK. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res 2007, 6, 2351–2366. [Google Scholar]
- Cheng, YG; Oldfield, CJ; Meng, JW; Romero, P; Uversky, VN; Dunker, AK. Mining α-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry 2007, 46, 13468–13477. [Google Scholar]
- Fuxreiter, M; Simon, I; Friedrich, P; Tompa, P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol 2004, 338, 1015–1026. [Google Scholar]
- Dancheck, B; Naim, AC; Peti, W. Detailed structural characterization of unbound protein phosphatase 1 inhibitors. Biochemistry 2008, 47, 12346–12356. [Google Scholar]
- Yoon, M-K; Venkatachalam, V; Huang, A; Choi, B-S; Stultz, CM; Chou, JJ. Residual structure within the disordered C-terminal segment of p21(Waf1/Cip1/Sdi1) and its implications for molecular recognition. Protein Sci 2009, 18, 337–347. [Google Scholar]
- Marsh, JA; Dancheck, B; Ragusa, MJ; Allaire, M; Forman-Kay, JD; Peti, W. Structural diversity in free and bound states of intrinsically disordered protein phosphatase 1 regulators. Structure 2010, 18, 1094–1103. [Google Scholar]
- Gunasekaran, K; Tsai, C-J; Nussinov, R. Analysis of ordered and disordered protein complexes reveals structural features discriminating between stable and unstable monomers. J. Mol. Biol 2004, 341, 1327–1341. [Google Scholar]
- Mészáros, B; Tompa, P; Simon, I; Dosztányi, Z. Molecular principles of the interactions of disordered proteins. J. Mol. Biol 2007, 372, 549–561. [Google Scholar]
- Oldfield, CJ; Meng, JW; Yang, JY; Yang, MQ; Uversky, VN; Dunker, AK. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 2008, 9, S1. [Google Scholar]
- Solt, I; Magyar, C; Simon, I; Tompa, P; Fuxreiter, M. Phosphorylation-induced transient intrinsic structure in the kinase-inducible domain of CREB facilitates its recognition by the KIX domain of CBP. Proteins 2006, 64, 749–757. [Google Scholar]
- Ganguly, D; Chen, JH. Atomistic details of the disordered states of KID and pKID. Implications in coupled binding and folding. J. Am. Chem. Soc 2009, 131, 5214–5223. [Google Scholar]
- Joerger, AC; Fersht, AR. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem 2008, 77, 557–582. [Google Scholar]
- Lee, H; Mok, KH; Muhandiram, R; Park, K-H; Suk, J-E; Kim, D-H; Chang, J; Sung, YC; Choi, KY; Han, K-H. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J. Biol. Chem 2000, 275, 29426–29432. [Google Scholar]
- Espinoza-Fonseca, LM; Trujillo-Ferrara, JG. Transient stability of the helical pattern of region F19-L22 of the N-terminal domain of p53: A molecular dynamics simulation study. Biochem. Biophys. Res. Comm 2006, 343, 110–116. [Google Scholar]
- Xu, HB; Ye, H; Osman, NE; Sadler, K; Won, E-Y; Chi, S-W; Yoon, HS. The MDM2-binding region in the transactivation domain of p53 also acts as a Bcl-XL-binding motif. Biochemistry 2009, 48, 12159–12168. [Google Scholar]
- Kussie, PH; Gorina, S; Marechal, V; Elenbaas, B; Moreau, J; Levine, AJ; Pavletich, NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar]
- Chen, H-F; Luo, R. Binding induced folding in p53-MDM2 complex. J. Am. Chem. Soc 2007, 129, 2930–2937. [Google Scholar]
- Lowry, DF; Stancik, A; Shrestha, RM; Daughdrill, GW. Modeling the accessible conformations of the intrinsically unstructured transactivation domain of p53. Proteins 2008, 71, 587–598. [Google Scholar]
- Dunbrack, RL; Cohen, FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci 1997, 6, 1661–1681. [Google Scholar]
- Dastidar, SG; Lane, DP; Verma, CS. Modulation of p53 binding to MDM2: Computational studies reveal important roles of Tyr100. BMC Bioinformatics 2009, 10, S6. [Google Scholar]
- Dastidar, SG; Madhumalar, A; Fuentes, G; Lane, DP; Verma, CS. Forces mediating protein-protein interactions: A computational study of p53 “approaching” MDM2. Theor. Chem. Acc 2010, 125, 621–635. [Google Scholar]
- Uhrinova, S; Uhrin, D; Powers, H; Watt, K; Zheleva, D; Fischer, P; McInnes, C; Barlow, PN. Structure of free MDM2 N-terminal domain reveals conformational adjustments that accompany p53-binding. J. Mol. Biol 2005, 350, 587–598. [Google Scholar]
- Li, C; Pazgier, M; Li, CQ; Yuan, WR; Liu, M; Wei, G; Lu, W-Y; Lu, W. Systematic mutational analysis of peptide inhibition of the p53-MDM2/MDMX interactions. J. Mol. Biol 2010, 398, 200–213. [Google Scholar]
- Zondlo, SC; Lee, AE; Zondlo, NJ. Determinants of specificity of MDM2 for the activation domains of p53 and p65: Proline27 disrupts the MDM2-binding motif of p53. Biochemistry 2006, 45, 11945–11957. [Google Scholar]
- Guharoy, M; Janin, J; Robert, CH. Side-chain rotamer transitions at protein-protein interfaces. Proteins 2010, 78, 3219–3225. [Google Scholar]
- Sivakolundu, SG; Bashford, D; Kriwacki, RW. Disordered p27Kip1 exhibits intrinsic structure resembling the Cdk2/Cyclin A-bound conformation. J. Mol. Biol 2005, 353, 1118–1128. [Google Scholar]
- Jensen, MR; Houben, K; Lescop, E; Blanchard, L; Ruigrok, RWH; Blackledge, M. Quantitative conformational analysis of partially folded proteins from residual dipolar couplings: application to the molecular recognition element of Sendai virus nucleoprotein. J. Am. Chem. Soc 2008, 130, 8055–8061. [Google Scholar]
- Ghosh, RP; Nikitina, T; Horowitz-Scherer, RA; Gierasch, LM; Uversky, VN; Hite, K; Hansen, JC; Woodcock, CL. Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry 2010, 49, 4395–4410. [Google Scholar]
- Gely, S; Lowry, DF; Bernard, C; Jensen, MR; Blackledge, M; Costanzo, S; Bourhis, J-M; Darbon, H; Daughdrill, G; Longhi, S. Solution structure of the C-terminal X domain of the measles virus phosphoprotein and interaction with the intrinsically disordered C-terminal domain of the nucleoprotein. J. Mol. Recognit 2010, 23, 435–447. [Google Scholar]
- Jiao, WT; McDonald, DQ; Coxon, JM; Parker, EJ. Molecular modeling studies of peptide inhibitors highlight the importance of conformational prearrangement for inhibition of calpain. Biochemistry 2010, 49, 5533–5539. [Google Scholar]
- Kjaergaard, M; Teilum, K; Poulsen, FM. Conformational selection in the molten globule state of the nuclear coactivator binding domain of CBP. Proc. Natl. Acad. Sci. USA 2010, 107, 12535–12540. [Google Scholar]
- Ahmad, M; Gu, W; Helms, V. Mechanism of fast peptide recognition by SH3 domains. Angew. Chem. Int. Ed 2008, 47, 7626–7630. [Google Scholar]
- Fischer, E. Einfluss der configuration auf die wirkung der enzyme. Ber. Dtsch. Chem. Ges 1894, 27, 2985–2993. [Google Scholar]
- Koshland, DE. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar]
- Monod, J; Wyman, J; Changeux, J-P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol 1965, 12, 88–118. [Google Scholar]
- Tsai, CJ; Kumar, S; Ma, BY; Nussinov, R. Folding funnels, binding funnels, and protein function. Protein Sci 1999, 8, 1181–1190. [Google Scholar]
- Boehr, DD; Nussinov, R; Wright, PE. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol 2009, 5, 789–796. [Google Scholar]
- Huang, YQ; Liu, ZR. Smoothing molecular interactions: The “kinetic buffer” effect of intrinsically disordered proteins. Proteins 2010, 78, 3251–3259. [Google Scholar]
- Dunker, AK; Garner, E; Guilliot, S; Romero, P; Albrecht, K; Hart, J; Obradovic, Z; Kissinger, C; Villafranca, JE. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput 1998, 3, 473–484. [Google Scholar]
- Dunker, AK; Cortese, MS; Romero, P; Iakoucheva, LM; Uversky, VN. Flexible nets-The roles of intrinsic disorder in protein interaction networks. FEBS J 2005, 272, 5129–5148. [Google Scholar]
- Ngan, C-H; Beglov, D; Rudnitskaya, AN; Kozakov, D; Waxman, DJ; Vajda, S. The structural basis of pregnane X receptor binding promiscuity. Biochemistry 2009, 48, 11572–11581. [Google Scholar]
- Feng, H; Jenkins, LMM; Durell, SR; Hayashi, R; Mazur, SJ; Cherry, S; Tropea, JE; Miller, M; Wlodawer, A; Appella, E; Bai, Y. Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation. Structure 2009, 17, 202–210. [Google Scholar]
- Berendsen, HJC; van der Spoel, D; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun 1995, 91, 43–56. [Google Scholar]
- Hess, B; Kutzner, C; Spoel, Dvd; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput 2008, 4, 435–447. [Google Scholar]
- Kaminski, GA; Friesner, RA; Tirado-Rives, J; Jorgensen, WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar]
- Berendsen, HJC; Grigera, JR; Straatsma, TP. The missing term in effective pair potentials. J. Phys. Chem 1987, 91, 6269–6271. [Google Scholar]
- Darden, T; York, D; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- Hess, B; Bekker, H; Berendsen, HJC; Fraaije, JGEM. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 1997, 18, 1463–1472. [Google Scholar]
- Bussi, G; Donadio, D; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys 2007, 126, 014101. [Google Scholar]
- Parrinello, M; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys 1981, 52, 7182–7190. [Google Scholar]
- Kabsch, W; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar]
- Humphrey, W; Dalke, A; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar]
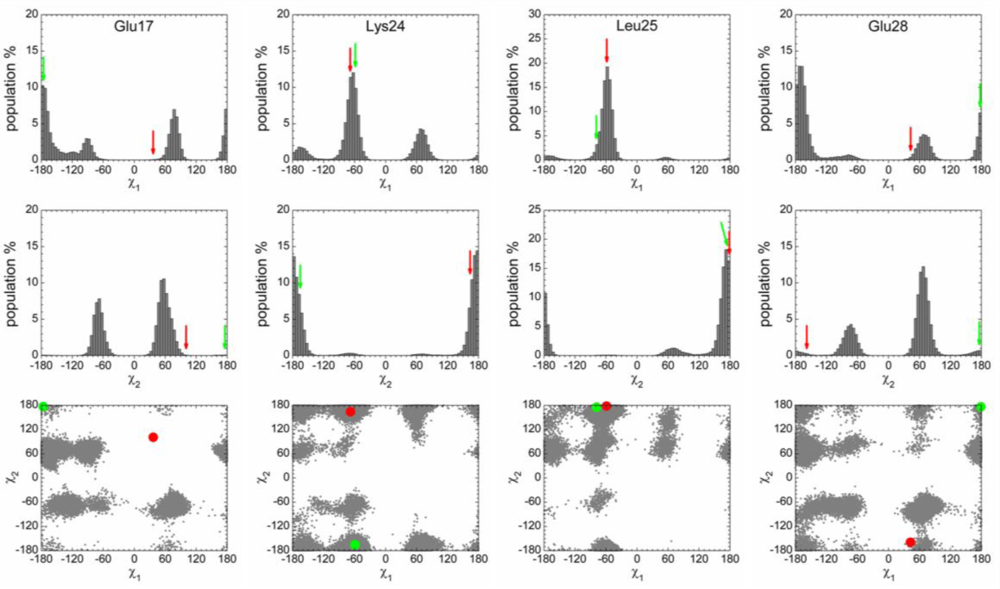
| Complex compared | State of p53N | Population of bound-like conformation * | |||
|---|---|---|---|---|---|
| Phe19 | Leu22 | Trp23 | Leu26 | ||
| p53N-MDM2 | Helix | 59.5% | – | 62.4% | 82.7% |
| Disordered | 19.2% | – | 18.7% | 76.2% | |
| Rotamer library | 31.71% | 3.65% | 16.21% | 62.52% | |
| p53N-Taz2 | Helix | 12.2% | 81.8% | – | – |
| Disordered | 6.0% | 67.1% | – | – | |
| Rotamer library | 47.08% | 62.52% | 5.32% | <1% | |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, Y.; Liu, Z. Anchoring Intrinsically Disordered Proteins to Multiple Targets: Lessons from N-Terminus of the p53 Protein. Int. J. Mol. Sci. 2011, 12, 1410-1430. https://doi.org/10.3390/ijms12021410
Huang Y, Liu Z. Anchoring Intrinsically Disordered Proteins to Multiple Targets: Lessons from N-Terminus of the p53 Protein. International Journal of Molecular Sciences. 2011; 12(2):1410-1430. https://doi.org/10.3390/ijms12021410
Chicago/Turabian StyleHuang, Yongqi, and Zhirong Liu. 2011. "Anchoring Intrinsically Disordered Proteins to Multiple Targets: Lessons from N-Terminus of the p53 Protein" International Journal of Molecular Sciences 12, no. 2: 1410-1430. https://doi.org/10.3390/ijms12021410
APA StyleHuang, Y., & Liu, Z. (2011). Anchoring Intrinsically Disordered Proteins to Multiple Targets: Lessons from N-Terminus of the p53 Protein. International Journal of Molecular Sciences, 12(2), 1410-1430. https://doi.org/10.3390/ijms12021410