QSAR Study and Molecular Design of Open-Chain Enaminones as Anticonvulsant Agents

Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Data
2.2. Geometry Optimization and Molecular Descriptors Calculation
2.3. Model Development
2.3.1. Linear Descriptors Search
2.3.2. Calculation of Flexible Descriptors
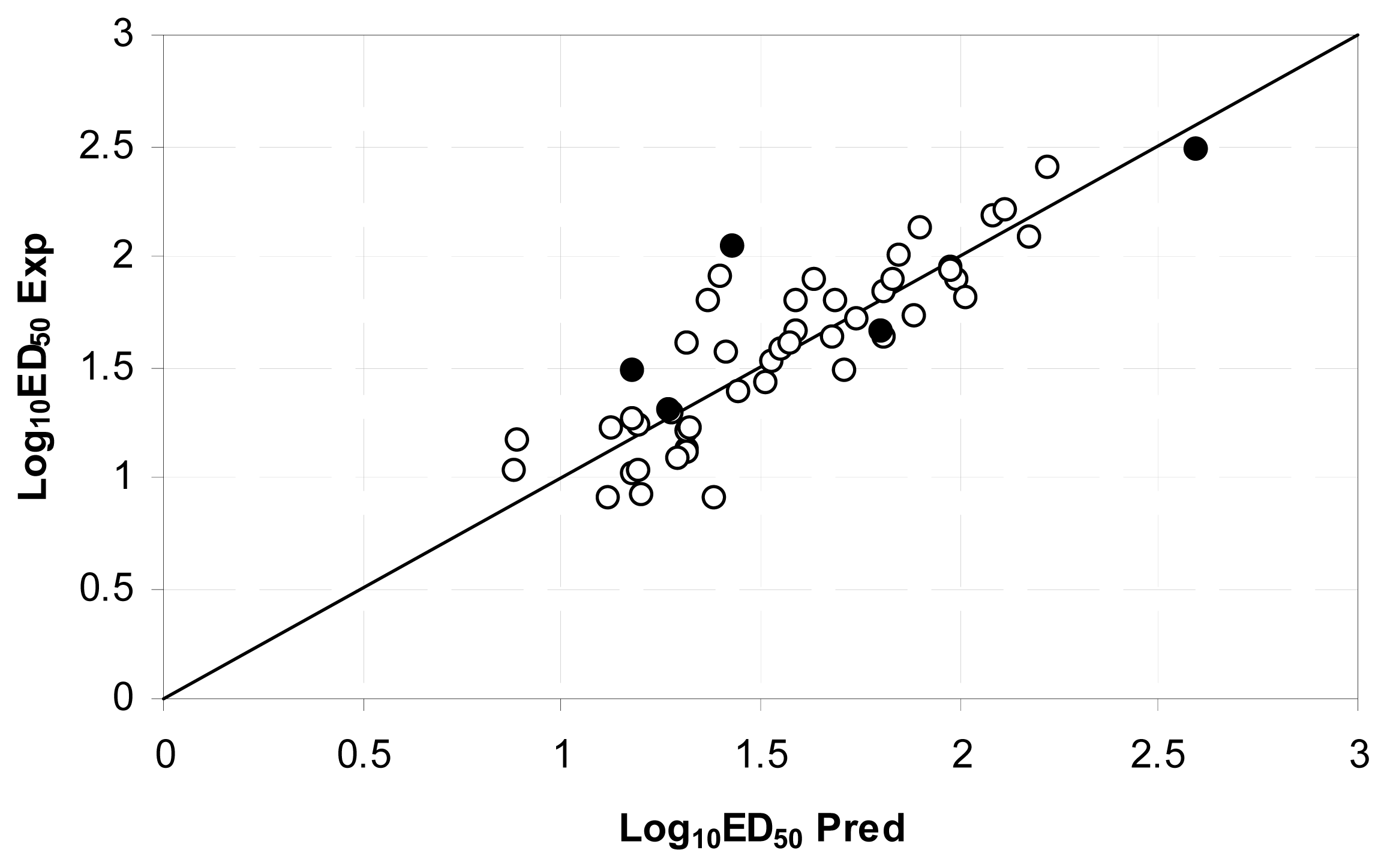
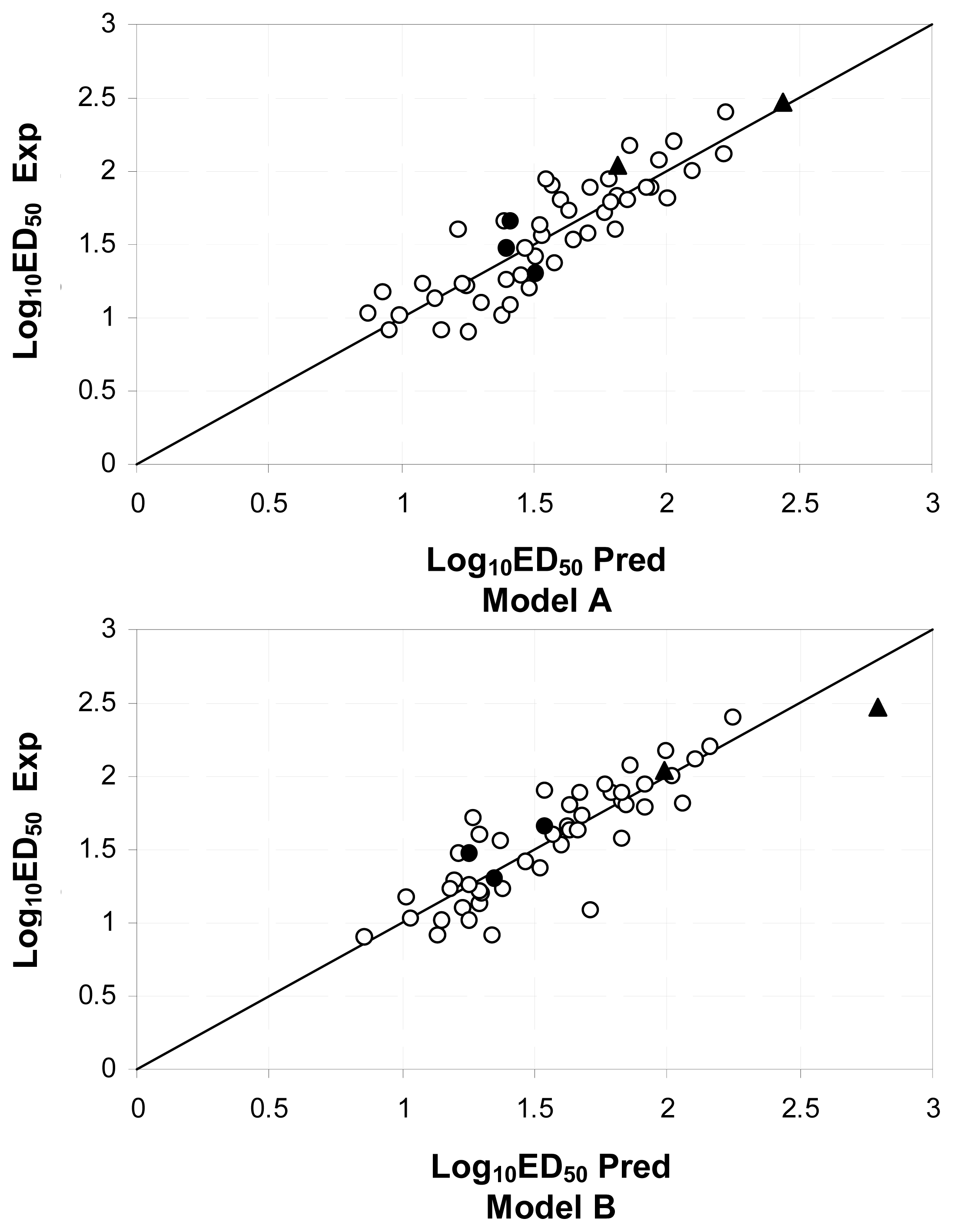
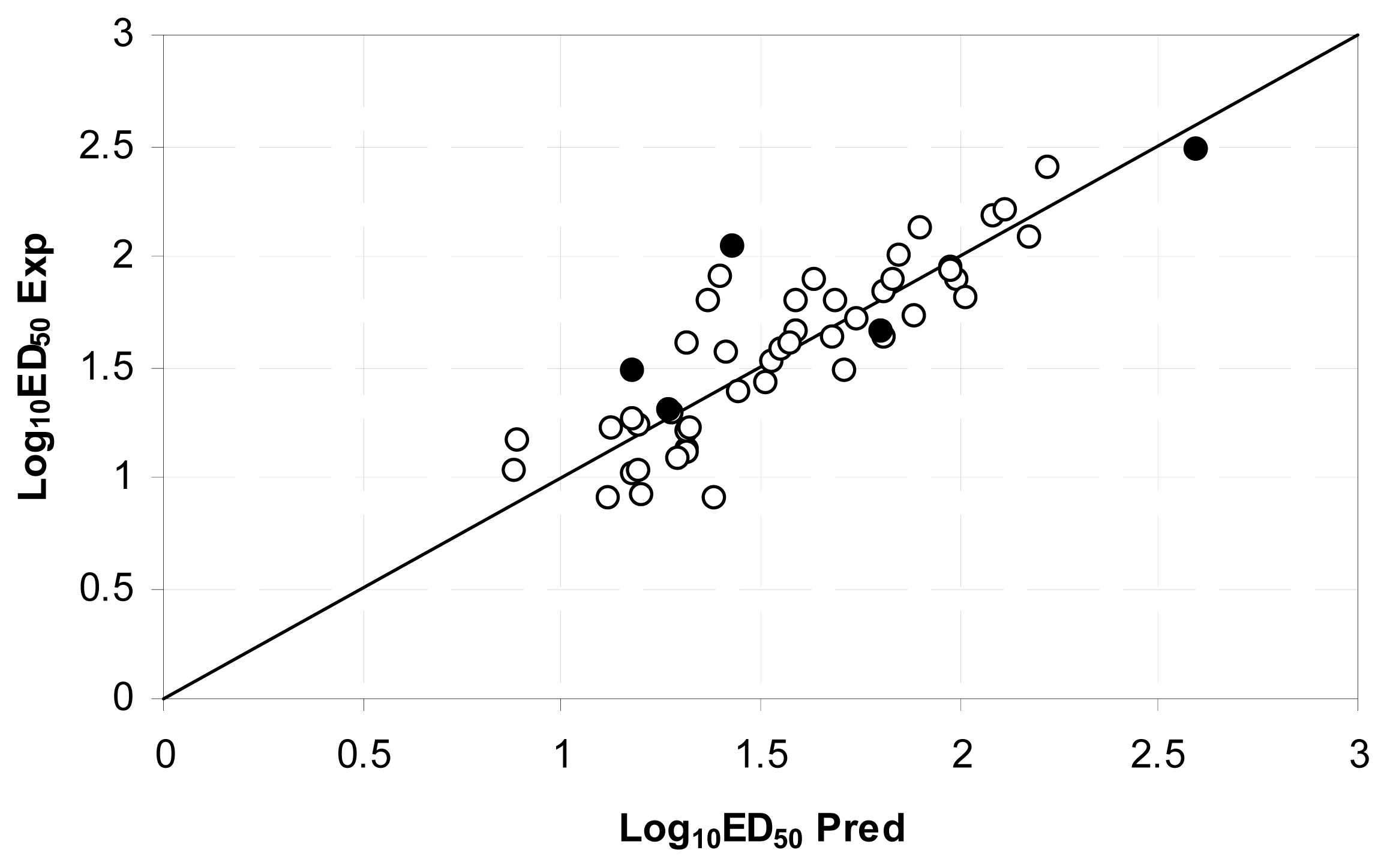
2.3.3. Model Validation
3. Results and Discussion
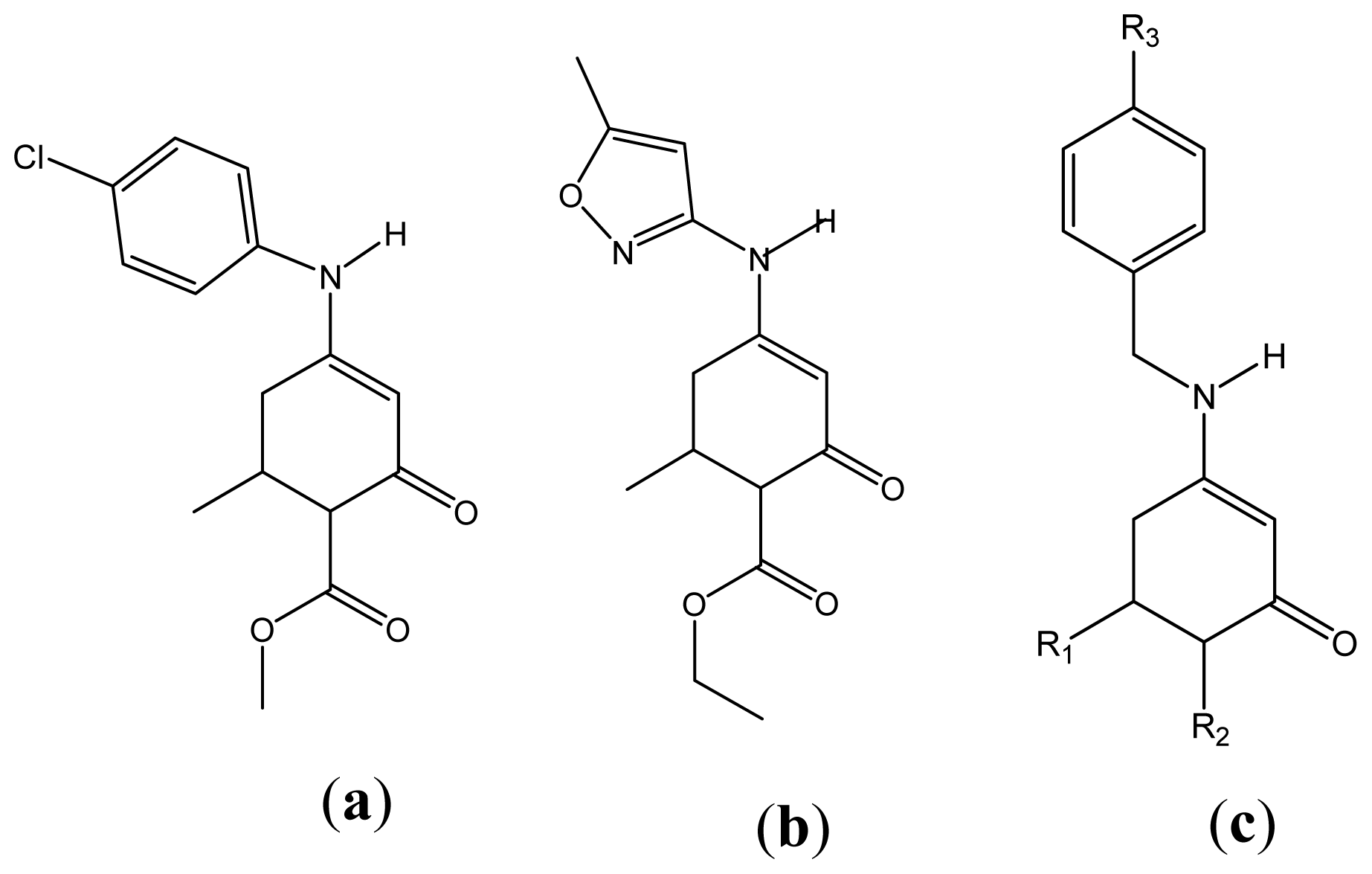
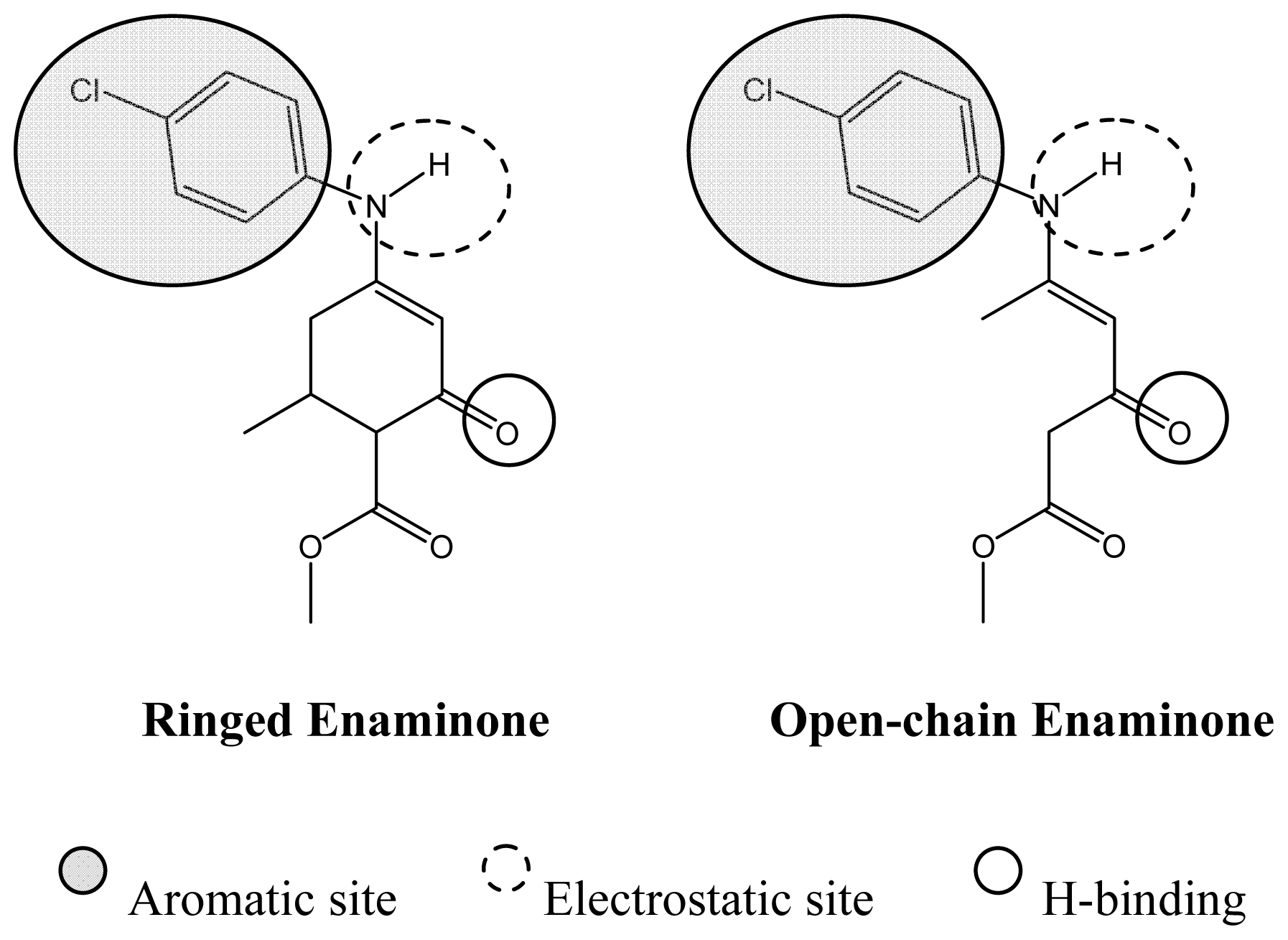
3.1. QSAR on Ringed-Enaminones
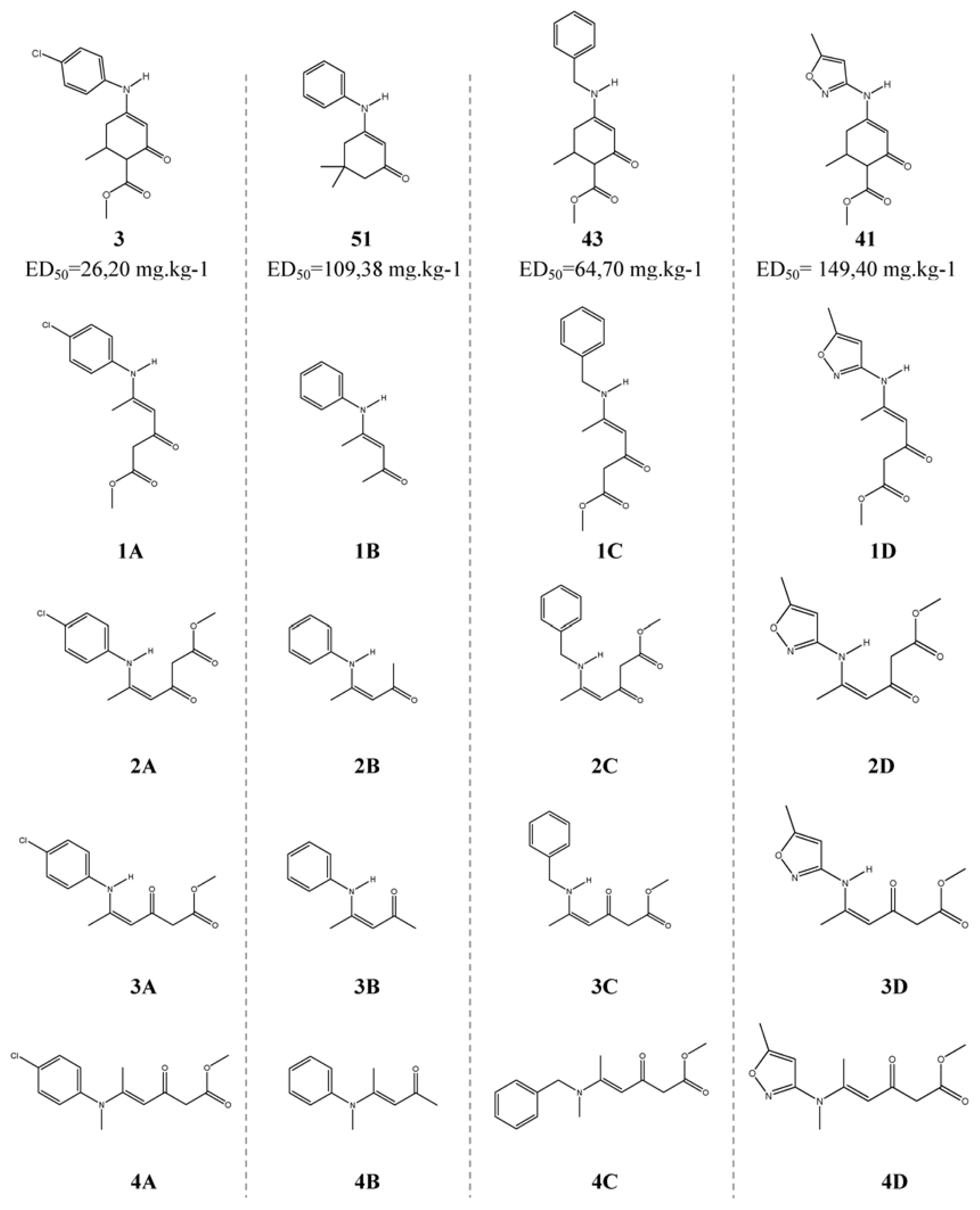
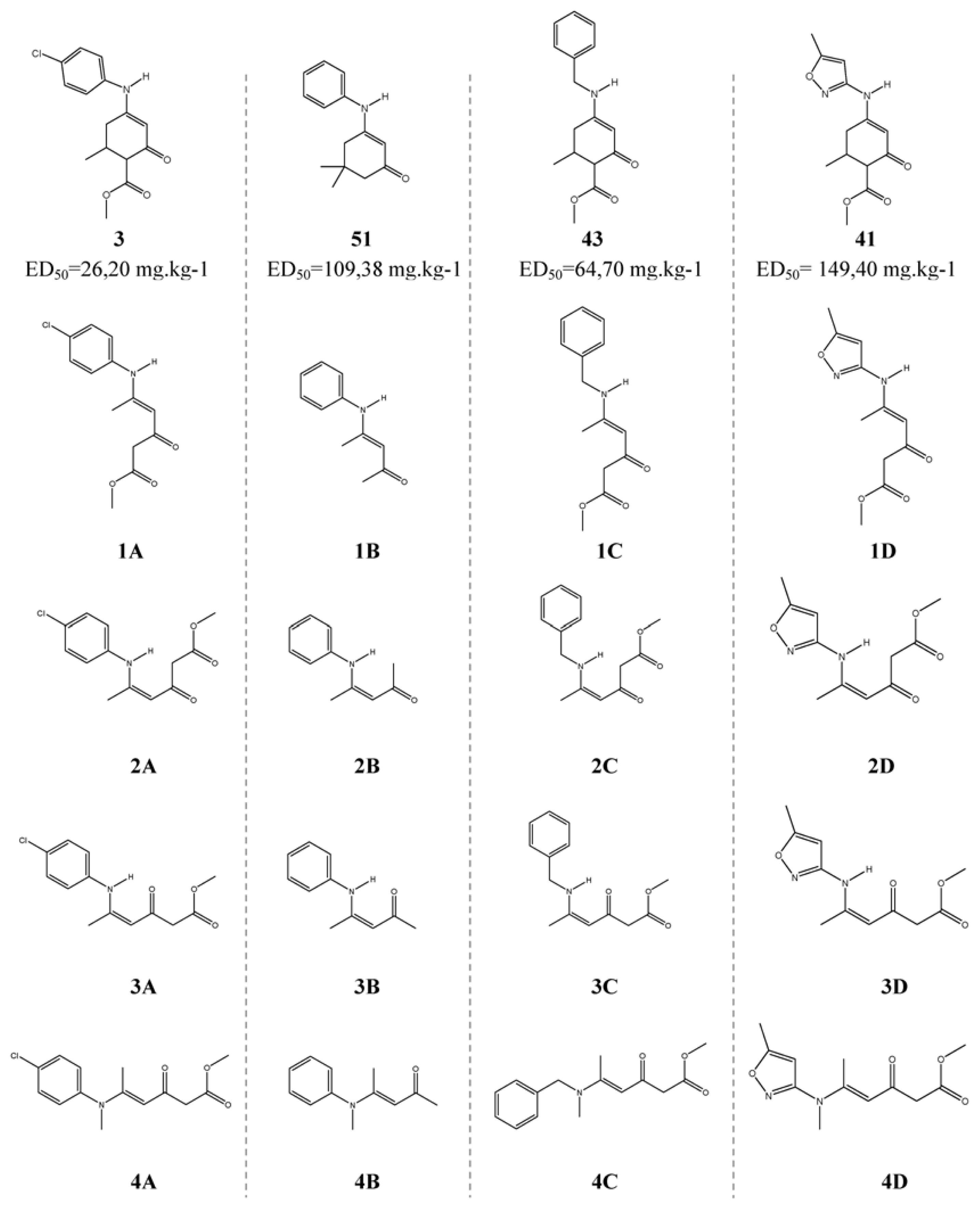
3.2. QSAR on Open-Chain Enaminones
4. Conclusions
Supplementary Material
ijms-12-09354-s001.pdfAcknowledgments
References
- Cook, A.G. Enaminas: Synthesis, Structure and Reaction; Cook, A.G., Ed.; Marcel Dekker: New York, NY, USA, 1969. [Google Scholar]
- Fraser, A.D. New drugs for the treatment of epilepsy. Clin. Biochem 1996, 29, 97–110. [Google Scholar]
- Porter, R.J.; Cereghino, J.J.; Gladding, G.D.; Hessie, B.J.; Kupferberg, H.J.; Scoville, B.; White, B.G. Antiepileptic drug development program. Clevel. Clin. Q 1984, 51, 293–305. [Google Scholar]
- Cox, D.S.; Gao, H.; Raje, S.; Scott, K.R.; Eddington, N.D. Enhancing the permeation of marker compounds and enaminone anticonvulsants across Caco-2 monolayers by modulating tight junctions using zonula occludens toxin. Eur. J. Pharm. Biopharm 2001, 52, 145–150. [Google Scholar]
- Cox, D.S.; Scott, K.R.; Gao, H.; Raje, S.; Eddington, N.D. Influence of multidrug resistance (MDR) proteins at the blood-brain barrier on the transport and brain distribution of enaminone anticonvulsants. J. Pharm. Sci 2001, 90, 1540–1552. [Google Scholar]
- Vamecq, J.; Lambert, D.; Poupaert, J.H.; Masereel, B.; Stables, J.P. Anticonvulsant activity and interactions with neuronal voltage-dependent sodium channel of analogues of ameltolide. J. Med. Chem 1998, 41, 3307–3313. [Google Scholar]
- Eddington, N.D.; Cox, D.S.; Khurana, M.; Salama, N.N.; Stables, J.P.; Harrison, S.J.; Negussie, A.; Taylor, R.S.; Tran, U.Q.; Moore, J.A.; et al. Synthesis and anticonvulsant activity of enaminones Part 7. Synthesis and anticonvulsant evaluation of ethyl 4-[(substituted phenyl)amino]-6-methyl-2-oxocyclohex-3-ene-1-carboxylates and their corresponding 5-methylcyclohex-2-enone derivatives. Eur. J. Med. Chem 2003, 38, 49–64. [Google Scholar]
- Eddington, N.D.; Cox, D.S.; Roberts, R.R.; Butcher, R.J.; Edafiogho, I.O.; Stables, J.P.; Cooke, N.; Goodwin, A.M.; Smith, C.A.; Scott, K.R. Synthesis and anticonvulsant activity of enaminones. 4. Investigations on isoxazole derivatives. Eur. J. Med. Chem 2002, 37, 635–648. [Google Scholar]
- Edafiogho, I.O.; Ananthalakshmi, K.V.; Kombian, S.B. Anticonvulsant evaluation and mechanism of action of benzylamino enaminones. Bioorgan. Med. Chem 2006, 14, 5266–5272. [Google Scholar]
- Kombian, S.B; Edafiogho, I.O.; Ananthalakshmi, K.V. Anticonvulsant enaminones depress excitatory synaptic transmission in the rat brain by enhancing extracellular GABA levels. Br. J. Pharm. 2005, 145, 945–953. [Google Scholar]
- Eberlin, M.N.; Takahata, Y.; Kascheres, C. The use of AM1 in structural analyses of primary and secondary enaminones. J. Mol. Struct. (Theochem) 1990, 207, 143–156. [Google Scholar]
- Kascheres, C.M. The chemistry of enaminones, diazocarbonyls and small rings: Our contribution. J. Braz. Chem. Soc 2003, 14, 945–969. [Google Scholar]
- Garro Martinez, J.C.; Manzanares, G.; Zamarbide, G.; Ponce, C.; Estrada, M.; Jáuregui, E. Geometrical Isomerism, tautomerism and conformational charges of 2-propenal-3-amine in its neutral and protonated forms. J. Mol. Struct. (Theochem.) 2001, 545, 17–27. [Google Scholar]
- Garro Martinez, J.C.; Zamarbide, G.N.; Ponce, C.; Estrada, M.R.; Tomás Vert, F.; Ponce, C. Theoretical study of a hydration mechanism in an enaminone pro-drug prototype. J. Mol. Struct. (Theochem.) 2003, 666–667, 617–627. [Google Scholar]
- Garro Martinez, J.C.; Zamarbide, G.N.; Estrada, M.R.; Castro, E.A. Geometrical isomerism and conformational charges of selected open-ring enaminones in its neutral and protonated forms. J. Mol. Struct. (Theochem. ) 2005, 725, 63–68. [Google Scholar]
- Carter, M.D.; Stephenson, V.C.; Weaver, D.F. Are anticonvulsants ‘two thirds’ of local anesthetics? A quantum pharmacology study. J. Mol. Struct. (Theochem. ) 2003, 638, 57–62. [Google Scholar]
- Garro Martinez, J.C.; Duchowicz, P.R.; Estrada, M.R.; Zamarbide, G.N.; Castro, E. Anticonvulsant activity of ringed enaminones: A QSAR study. QSAR Comb. Sci. 2009, 28, 1376–1385. [Google Scholar]
- Malawska, B.; Kulig, K.; Spiewak, A.; Stables, J.P. Investigation into new anticonvulsant derivatives of α-substituted N-benzylamides of γ-hydroxy- and γ-acetoxybutyric acid. Part 5: Search for new anticonvulsant compounds. Bioorg. Med. Chem 2004, 12, 625–632. [Google Scholar]
- Pandeya, S.N.; Raja, A.S. Synthesis of isatin semicarbazones as novel anticonvulsants—Role of hydrogen bonding. J. Pharm. Sci 2002, 5, 266–271. [Google Scholar]
- Aggarwal, N.; Mishra, P. Synthesis of 4-aryl substituted semicarbazones of some terpenes as novel anticonvulsants. Pharm. Pharmaceut. Sci 2004, 7, 260–264. [Google Scholar]
- Hansch, C.; Leo, A. Exploring QSAR. Fundamentals and Applications in Chemistry and Biology; American Chemical Society: Washington, DC, USA, 1995. [Google Scholar]
- Lacrãmã, A.M.; Putz, V.M.; Ostafe, V. A spectral-SAR model for the anionic-cationic interaction in ionic liquids: Application to vibrio fischeri ecotoxicity. Int. J. Mol. Sci 2007, 8, 842–863. [Google Scholar]
- Putz, V.M.; Lacrãmã, A.M. Introducing spectral structure activity relationship (S-SAR) analysis. Application to ecotoxicology. Int. J. Mol. Sci 2007, 8, 363–391. [Google Scholar]
- Chicu, S.A.; Putz, M.V. Köln-timişoara molecular activity combined models toward interspecies toxicity assessment. Int. J. Mol. Sci 2009, 10, 4474–4497. [Google Scholar]
- National Institutes of Health, Anticonvulsant Screening Project, Antiepileptic Drug Development Program; DHEW Publication No. (NIH) 78–1093; NIH: Bethesda, MD, USA, 1978.
- Hyperchem 7.5 (Hypercube). Available online: http://www.hyper.com/ accessed on 22 July 2010.
- Dragon 3.0 Evaluation Version. Available online: http://www.disat.unimib.it/chm accessed on 6 November 2008.
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Matlab 7.0; The MathWorks Inc: Natick, MA, USA, 2004.
- Duchowicz, P.R.; Castro, E.A.; Fernández, F.M.; González, M.P. A new search algorithm for QSPR/QSAR theories. Normal boiling points of some organic molecules. Chem. Phys. Lett 2005, 412, 376–380. [Google Scholar]
- Duchowicz, P.R.; Castro, E.A.; Fernández, F.M. Alternative algorithm for the search of an optimal set of descriptors in QSAR-QSPR studies. MATCH Commun. Math. Comput. Chem 2006, 55, 179–192. [Google Scholar]
- Duchowicz, P.R.; Mercader, A.G.; Fernández, F.M.; Castro, E.A. Prediction of aqueous toxicity for heterogeneous phenol derivatives by QSAR. Chemom. Intell. Lab. Syst 2008, 90, 97–107. [Google Scholar]
- Andrea, T.A.; Kalayeh, H. Applications of neural networks in quantitative structure-activity relationships of dihydrofolate reductase inhibitors. J. Med. Chem 1991, 34, 2824–2836. [Google Scholar]
- Kubinyi, H. Variable selection in QSAR studies. II. A Highly efficient combination of systematic search and evolution. Quant. Struct. Act. Relatsh 1994, 13, 393–401. [Google Scholar]
- Kubinyi, H. Variable selection in QSAR studies. I. An evolutionary algorithm. Quant. Struct. Act. Relatsh 1994, 13, 285–294. [Google Scholar]
- Coral 1.4. Available online: http://www.insilico.eu/coral accessed on 11 October 2010.
- ACD/ChemSketch Freeware, version 12.01; Advanced Chemistry Development, Inc: Toronto, ON, Canada, 2009. Available online: http://www.acdlabs.com accessed on 11 November 2010.
- Toropov, A.A.; Benfenati, E. SMILES in QSPR/QSAR Modeling: Results and perspectives. Curr. Drug Discov. Technol 2007, 4, 77–116. [Google Scholar]
- Toropov, A.A.; Benfenati, E. Additive SMILES-based optimal descriptors in QSAR modelling bee toxicity: Using rare SMILES attributes to define the applicability domain. Bioorg. Med. Chem 2008, 26, 4801–4809. [Google Scholar]
- Toropov, A.A.; Toropova, A.P.; Benfenati, E. Simplified molecular input line entry system-based optimal descriptors: Quantitative structure-activity relationship modeling mutagenicity of nitrated polycyclic aromatic hydrocarbons. Chem. Biol. Drug Des 2009, 73, 515–525. [Google Scholar]
- Toropov, A.A.; Toropova, A.P.; Benfenati, E.; Leszczynska, D.; Leszczynski, J. InChI-based optimal descriptors: QSAR analysis of fullerene [C60]-based HIV-1 PR inhibitors by correlation balance. Eur. J. Med. Chem 2010, 45, 1387–1394. [Google Scholar]
- Fernández, F.M.; Duchowicz, P.R.; Castro, E.A. Aplicación de los métodos QSAR/QSPR en fenómenos de adsorción de sustancias químicas sobre materiales sólidos. MATCH Commun. Math. Comput. Chem 2004, 51, 39–57. [Google Scholar]
- Fernández, F.M.; Castro, E.A.; Duchowicz, P.R. Los descriptores ortogonales en la. Teoría QSAR-QSPR. Afinidad 2004, 61, 476–495. [Google Scholar]
- Jaworska, J.; Nikolova-Jeliazkova, N.; Aldenberg, T. QSAR applicability domain estimation by projection of the training set in descriptor space: A review. Altern. Lab. Anim 2005, 33, 445–459. [Google Scholar]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci 2007, 26, 694–701. [Google Scholar]
- Weaver, S.; Gleeson, M.P. The importance of the domain of applicability in QSAR modeling. J. Mol. Graph. Model 2009, 26, 1315–1326. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | Type | Details |
|---|---|---|
| BELe6 | BCUT | Lowest eigenvalue n. 6 of Burden matrix/weighted by atomic Sanderson electronegativities |
| BELp8 | Lowest eigenvalue n. 8 of Burden matrix/weighted by atomic polarizabilities | |
| RDF025v | Radial Distribution Function | Radial Distribution Function—2.5/weighted by atomic van der Waals volumes |
| RDF025m | Radial Distribution Function—2.5/weighted by atomic masses | |
| RDF115m | Radial Distribution Function—11.5/weighted by atomic masses | |
| Mor15e | 3D-MoRSE | 3D-MoRSE—signal 15/weighted by atomic Sanderson electronegativities |
| R4e+ | GETAWAY | R maximal autocorrelation of lag 4/weighted by atomic Sanderson electronegativities |
| G(O..Cl) | Geometrical | Sum of geometrical distances between O..Cl |
| Homo-Lumo | Quantum Chemical | HOMO-LUMO energy gap |
| No. | Chemical name | ED50 (mg·Kg−1) | Exp. | Equation 3 | Equation 4 | Equation 5 |
|---|---|---|---|---|---|---|
| 1 | Ethyl 6-methyl-4-(5-methylisoxazol-3-ylamino)-2-oxocyclohex-3-enecarboxylate | 68.39 [4] | 1.835 | 1.815 | 1.831 | 1.813 |
| 2 | Methyl 4-(4-cyanophenylamino)-6-methyl-2-oxocyclohex-3-enecarboxylate | 248.31 [4] | 2.395 | 2.229 | 2.252 | 2.226 |
| 3 | Methyl 4-(4-chlorophenylamino)-6-methyl-2-oxocyclohex-3-enecarboxylate | 26.18 [4] | 1.418 | 1.509 | 1.466 | 1.517 |
| 4 | 2-acetamido-N-benzylpropanamide | 76.38 [5] | 1.883 | 1.716 | 1.677 | 1.634 |
| 5 | 2-acetamido-N-(3-fluorobenzyl)propanamide | 77.27 [5] | 1.888 | 1.945 | 1.791 | 1.994 |
| 6 | 2-acetamido-N-(2-fluorobenzyl)-2-(furan-2-yl)acetamide | 39.99 [5] | 1.602 | 1.215 | 1.294 | 1.315 |
| 7 | 2-acetamido-N-(3-fluorobenzyl)-2-(furan-2-yl)acetamide | 13.27 [5] | 1.123 | 1.132 | 1.291 | 1.315 |
| 8 | 2-acetamido-N-(4-fluorobenzyl)-2-(furan-2-yl)acetamide | 12.68 [5] | 1.103 | 1.302 | 1.228 | 1.315 |
| 9 | 2-acetamido-N-(2,5-difluorobenzyl)-2-(furan-2-yl)acetamida | 23.77 [5] | 1.376 | 1.577 | 1.522 | 1.448 |
| 10 | 2-acetamido-N-(2,6-difluorobenzyl)-2-(furan-2-yl)acetamide | 62.95 [5] | 1.799 | 1.604 | 1.631 | 1.687 |
| 11 | 2-acetamido-N-benzylpent-4-enamide | 33.57 [5] | 1.526 | 1.653 | 1.605 | 1.533 |
| 12 | 2-acetamido-N-benzyl-2-(tetrahydrofuran-2-yl)acetamide | 51.64 [5] | 1.713 | 1.770 | 1.272 | 1.746 |
| 13 | 2-acetamido-N-benzyl-2-(furan-2-yl)acetamide | 10.28 [5] | 1.012 | 1.383 | 1.252 | 1.182 |
| 14 | 2-acetamido-N-benzyl-2-(5-methylfuran-2-yl)acetamide | 19.19 [5] | 1.283 | 1.450 | 1.200 | 1.282 |
| 15 | 2-acetamido-N-benzyl-2-(1H-pyrrol-2-yl)acetamide | 16.07 [5] | 1.206 | 1.486 | 1.299 | 1.315 |
| 16 | 2-acetamido-N-benzyl-2-(5-methyl-1H-pyrrol-2-yl)acetamide | 36.48 [5] | 1.562 | 1.530 | 1.376 | 1.415 |
| 17 | 2-acetamido-N-benzyl-2-(thiophen-2-yl)acetamide | 44.77 [5] | 1.651 | 1.388 | 1.628 | 1.593 |
| 18 | 2-acetamido-N-benzyl-2-(thiophen-3-yl)acetamida | 87.70 [5] | 1.943 | 1.783 | 1.770 | 1.979 |
| 19 | 2-acetamido-N-benzyl-2-(1H-pyrrol-1-yl)acetamide | 80.17 [5] | 1.904 | 1.572 | 1.538 | 1.399 |
| 20 | 2-acetamido-N-benzyl-2-(1H-pyrazol-1-yl)acetamide | 16.48 [5] | 1.217 | 1.249 | 1.294 | 1.325 |
| 21 | 2-acetamido-N-benzyl-2-(pyridin-2-yl)acetamide | 10.79 [5] | 1.033 | 0.880 | 1.037 | 1.195 |
| 22 | 2-acetamido-3-amino-N-benzyl-3-thioxopropanamide | 86.50 [5] | 1.937 | 1.550 | 1.921 | 1.981 |
| 23 | 2-acetamido-N-benzyl-2-(ethylamino)acetamide | 42.36 [5] | 1.627 | 1.525 | 1.635 | 1.679 |
| 24 | 2-acetamido-N-benzyl-2-(hydroxy(methyl)amino)acetamide | 29.99 [5] | 1.477 | 1.465 | 1.215 | 1.712 |
| 25 | 2-acetamido-N-benzyl-2-(1-phenylhydrazinyl)acetamide | 42.76 [5] | 1.631 | 1.524 | 1.663 | 1.811 |
| 26 | 2-acetamido-N-benzyl-2-ethoxyacetamide | 61.94 [5] | 1.792 | 1.795 | 1.922 | 1.368 |
| 27 | 2-acetamido-N-benzyl-3-methoxypropanamide | 8.30 [5] | 0.919 | 0.954 | 1.135 | 1.201 |
| 28 | 2-acetamido-N-benzyl-3-ethoxypropanamide | 16.98 [5] | 1.230 | 1.232 | 1.385 | 1.197 |
| 29 | 2-acetamido-N-benzyl-2-(pyrazin-2-yl)acetamide | 14.79 [5] | 1.170 | 0.929 | 1.015 | 0.893 |
| 30 | 2-acetamido-N-benzyl-2-(pyrimidin-2-yl)acetamida | 8.09 [5] | 0.908 | 1.151 | 1.344 | 1.121 |
| 31 | 2-acetamido-N-benzyl-2-(oxazol-5-yl)acetamide | 10.50 [5] | 1.021 | 0.998 | 1.149 | 0.88 |
| 32 | 2-acetamido-N-benzyl-2-(thiazol-5-yl)acetamide | 11.99 [5] | 1.079 | 1.417 | 1.717 | 1.291 |
| 33 | 2-acetamido-2-(3-aminophenylamino)-N-benzylacetamide | 98.40 [5] | 1.993 | 2.102 | 2.023 | 1.85 |
| 34 | 2-acetamido-N-benzyl-2-(furan-2-yl)acetamide | 18.37 [5] | 1.264 | 1.396 | 1.255 | 1.182 |
| 35 | Ethyl 4-(4-chlorophenylamino)-6-methyl-2-oxo-3-cyclohexene-1-carboxylate | 16.67 [7] | 1.222 | 1.085 | 1.184 | 1.124 |
| 36 | Ethyl 4-(4-bromophenylamino)-6-methyl-2-oxo-3-cyclohexene-1-carboxylate | 7.89 [7] | 0.897 | 1.259 | 0.861 | 1.383 |
| 37 | Ethyl 6-methyl-2-oxo-4-(4-(trifluoromethoxy)phenylamino)cyclohex-3-enecarboxylate | 37.07 [7] | 1.569 | 1.708 | 1.831 | 1.553 |
| 38 | Ethyl 4-(4-cianophenylamino)-6-methyl-2-oxo-3-cyclohexene-1-carboxylate | 63.10 [7] | 1.800 | 1.852 | 1.847 | 1.595 |
| 39 | 3-(4-chlorophenylamino)-5-methyl-2-cyclohexenone | 40.36 [7] | 1.606 | 1.804 | 1.570 | 1.576 |
| 40 | 3-(4-iodophenylamino)-5-methyl-2-cyclohexenone | 76.91 [7] | 1.886 | 1.924 | 1.829 | 1.835 |
| 41 | Methyl 6-methyl-4-(5-methylisoxazol-3-ylamino)-2-oxocyclohex-3-cyclohexene-1-carboxylate | 149.28 [8] | 2.174 | 1.867 | 2.001 | 2.087 |
| 42 | Tert-butyl 6-methyl-4-(5-methylisoxazol-3-ylamino)-2-oxocyclohex-3-cyclohexene-1-carboxylate | 119.67 [8] | 2.078 | 1.974 | 1.861 | 2.181 |
| 43 | Methyl 4-(benzylamino)-6-methyl-2-oxocyclohex-3-cyclohexene-1-carboxylate | 64.57 [9] | 1.810 | 2.005 | 2.062 | 2.019 |
| 44 | Methyl 4-(4-fluorobenzylamino)-6-methyl-2-oxocyclohex-3-cyclohexene-1-carboxylate | 158.85 [9] | 2.201 | 2.030 | 2.164 | 2.118 |
| 45 | 3-(benzylamino)-5,5-dimethylcyclohex-2-cyclohexenone | 52.97 [9] | 1.724 | 1.633 | 1.678 | 1.892 |
| 46 | Methyl 4-(benzylamino)-6,6-dimethyl-2-oxocyclohex-3-enecarboxylate | 131.83 [10] | 2.120 | 2.219 | 2.107 | 1.904 |
| 47 * | Methyl 6-methyl-4-(4-nitrophenylamino)-2-oxocyclohex-3-enecarboxylate | 299.92 [4] | 2.477 | 2.441 | 2.793 | 2.599 |
| 48 * | 2-acetamido-N-benzyl-2-phenylacetamide | 20.28 [7] | 1.307 | 1.505 | 1.347 | 1.269 |
| 49 * | 2-acetamido-N-benzyl-2-(dimethylamino)acetamide | 45.29 [7] | 1.656 | 1.413 | 1.540 | 1.804 |
| 50 * | 2-acetamido-2-(furan-2-yl)-N-(pyridin-3-ylmethyl)acetamide | 29.99 [7] | 1.477 | 1.396 | 1.255 | 1.182 |
| 51 * | 5,5-dimethyl-3-(phenylamino)cyclohex-2-enone | 109.14 [10] | 2.038 | 1.812 | 1.990 | 1.434 |
| Molecule | Equation 3 | Equation 4 | PSA a |
|---|---|---|---|
| 1A | 2.051 | 2.041 | 2 |
| 2A | 2.426 | 2.093 | 2 |
| 3A | 2.456 | 2.229 | 2 |
| 4A | 2.323 | 2.037 | 2 |
| 1B | 0.956 | 0.245 | 1 |
| 2B | 1.529 | 0.903 | 1 |
| 3B | 1.103 | 0.667 | 1 |
| 4B | 1.393 | 0.758 | 1 |
| 1C | 1.329 | 1.241 | 1 |
| 2C | 1.462 | 1.556 | 1 |
| 3C | 1.794 | 1.446 | 1 |
| 4C | 1.505 | 1.177 | 1 |
| 1D | 1.295 | 1.416 | 1 |
| 2D | 1.583 | 1.375 | 1 |
| 3D | 2.172 | 1.980 | b |
| 4D | 1.291 | 1.183 | 1 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Garro Martinez, J.C.; Duchowicz, P.R.; Estrada, M.R.; Zamarbide, G.N.; Castro, E.A. QSAR Study and Molecular Design of Open-Chain Enaminones as Anticonvulsant Agents. Int. J. Mol. Sci. 2011, 12, 9354-9368. https://doi.org/10.3390/ijms12129354
Garro Martinez JC, Duchowicz PR, Estrada MR, Zamarbide GN, Castro EA. QSAR Study and Molecular Design of Open-Chain Enaminones as Anticonvulsant Agents. International Journal of Molecular Sciences. 2011; 12(12):9354-9368. https://doi.org/10.3390/ijms12129354
Chicago/Turabian StyleGarro Martinez, Juan C., Pablo R. Duchowicz, Mario R. Estrada, Graciela N. Zamarbide, and Eduardo A. Castro. 2011. "QSAR Study and Molecular Design of Open-Chain Enaminones as Anticonvulsant Agents" International Journal of Molecular Sciences 12, no. 12: 9354-9368. https://doi.org/10.3390/ijms12129354