The Slowly Aggregating Salmon Calcitonin: A Useful Tool for the Study of the Amyloid Oligomers Structure and Activity


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Calcitonins
3. Our Contribution to the Study of sCT
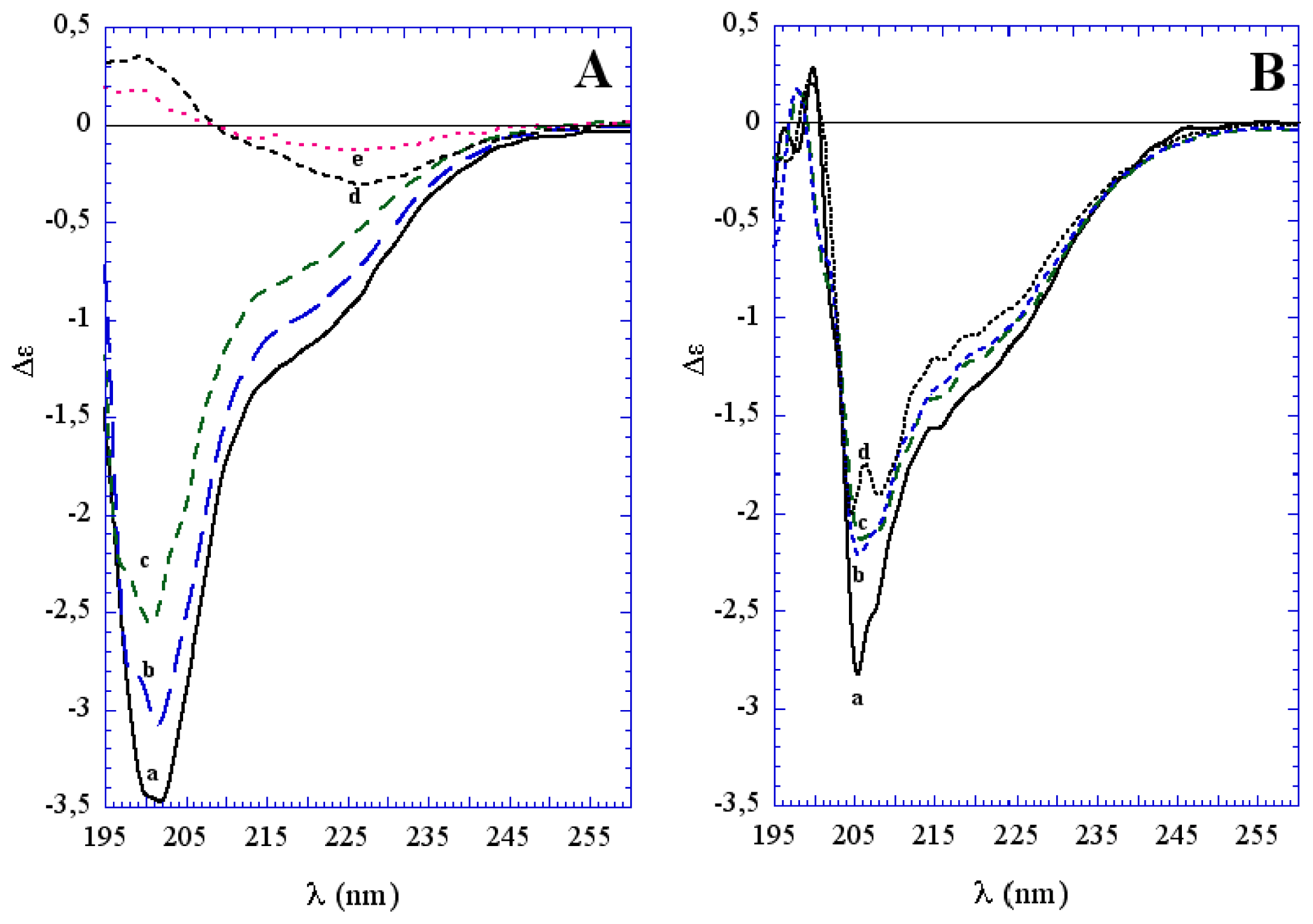
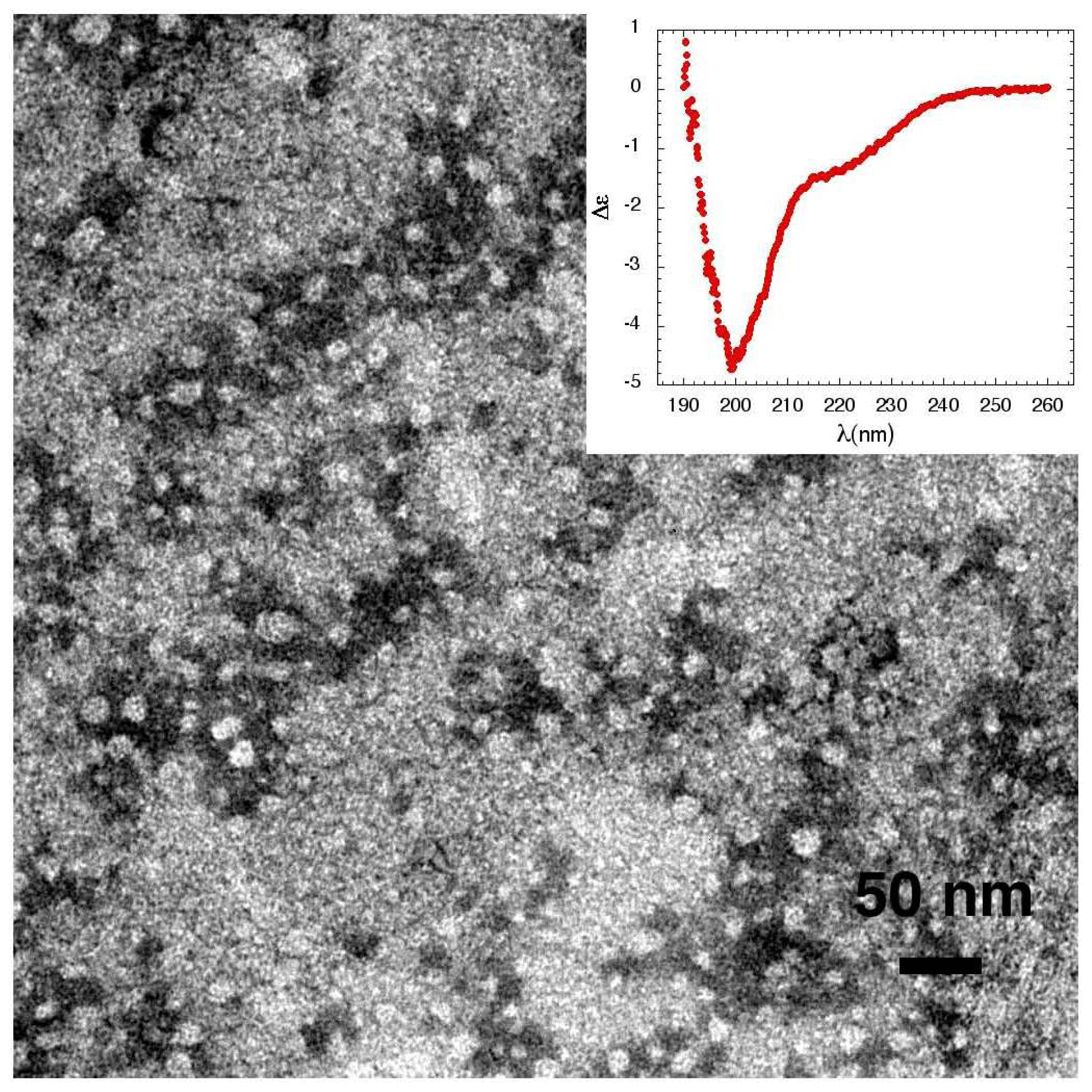
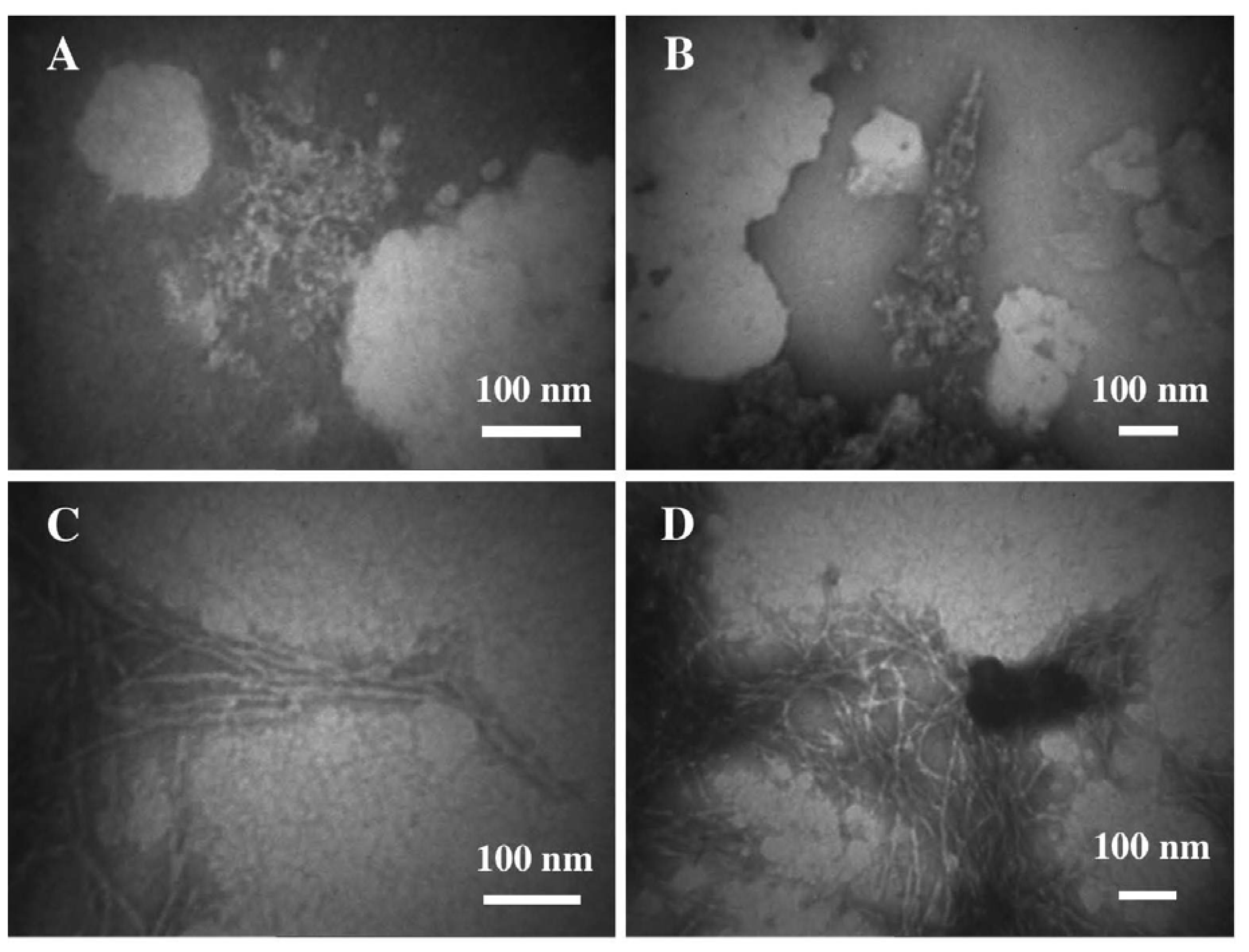
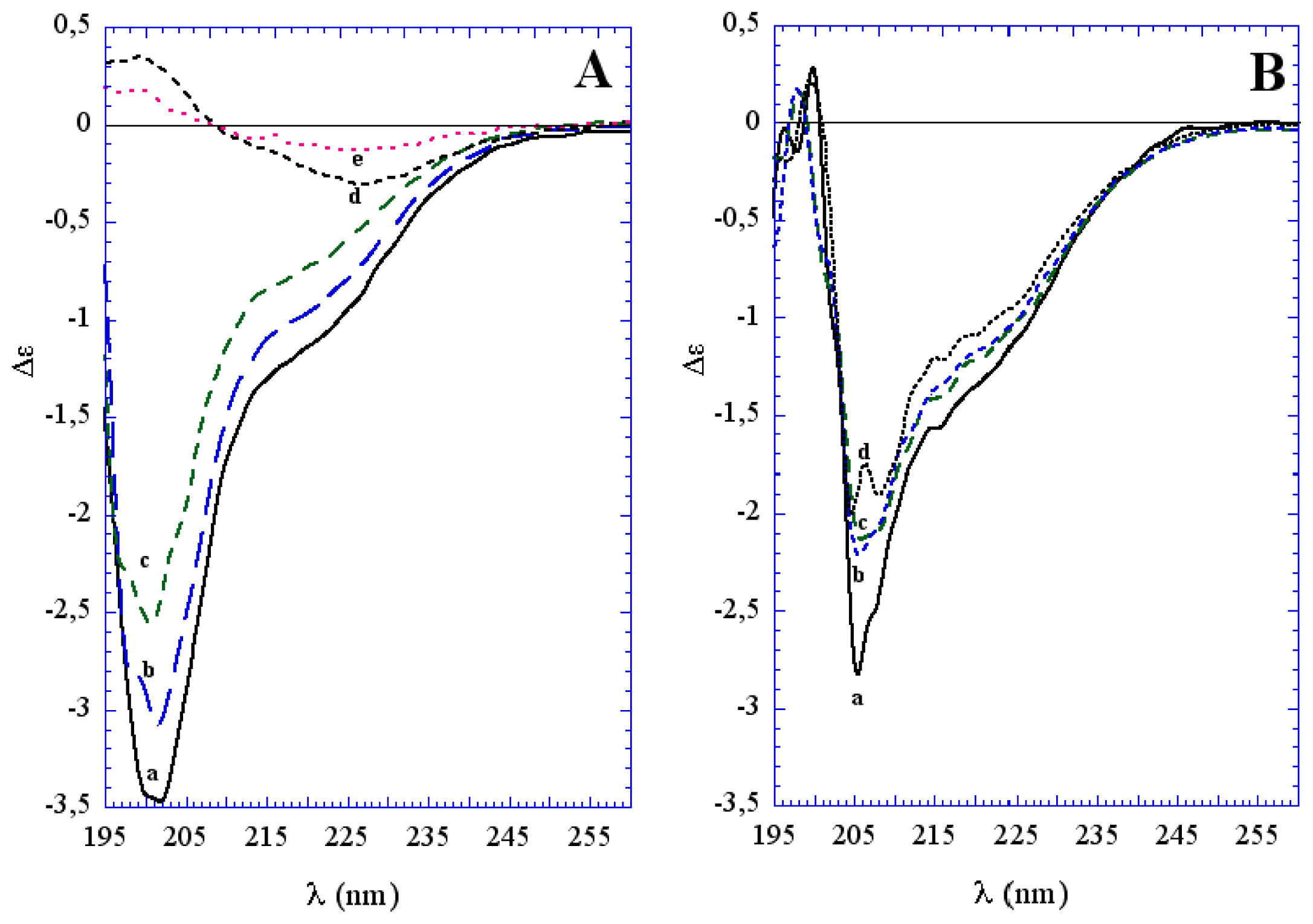
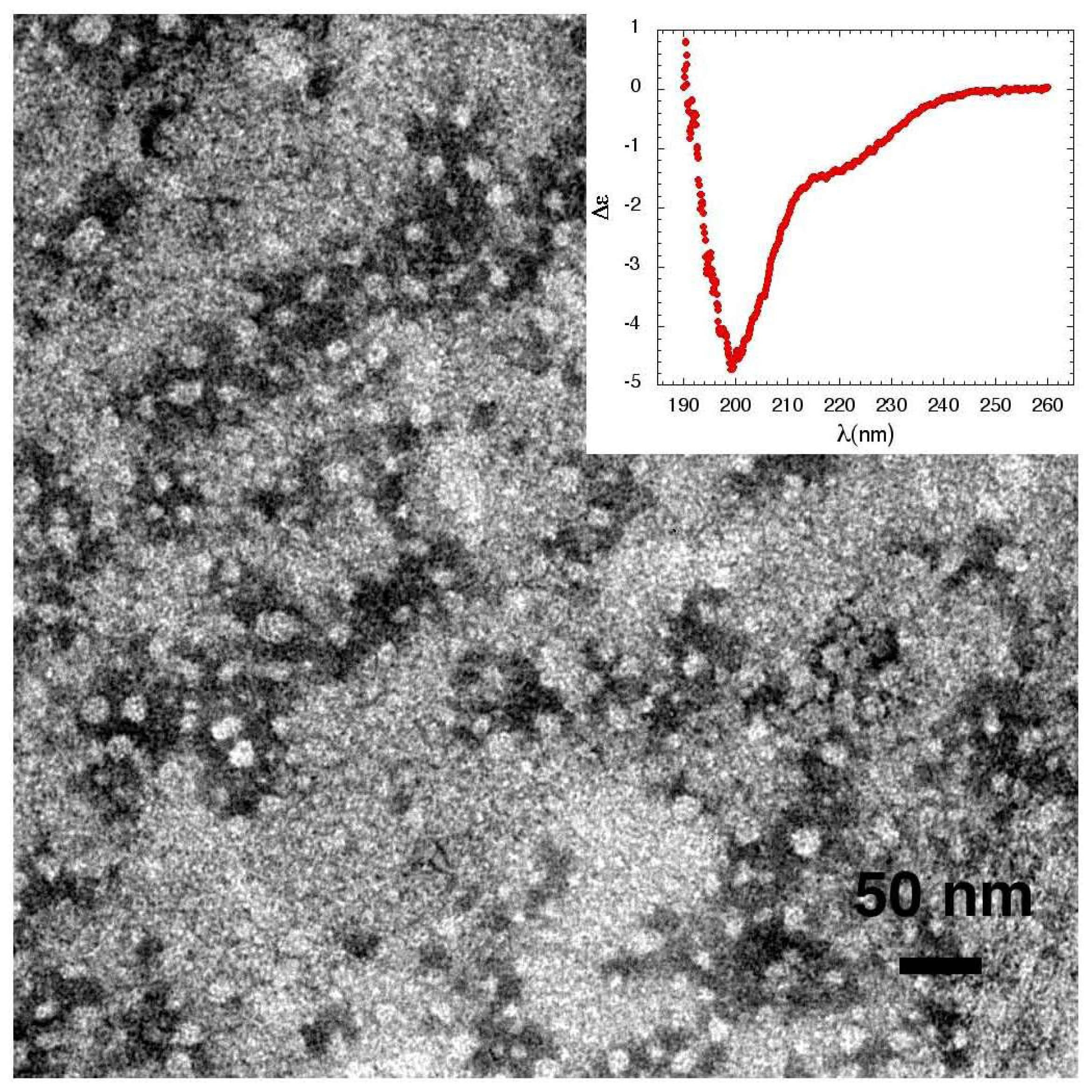
3.1. The Effect of a Mild Oxidation on sCT Aggregation
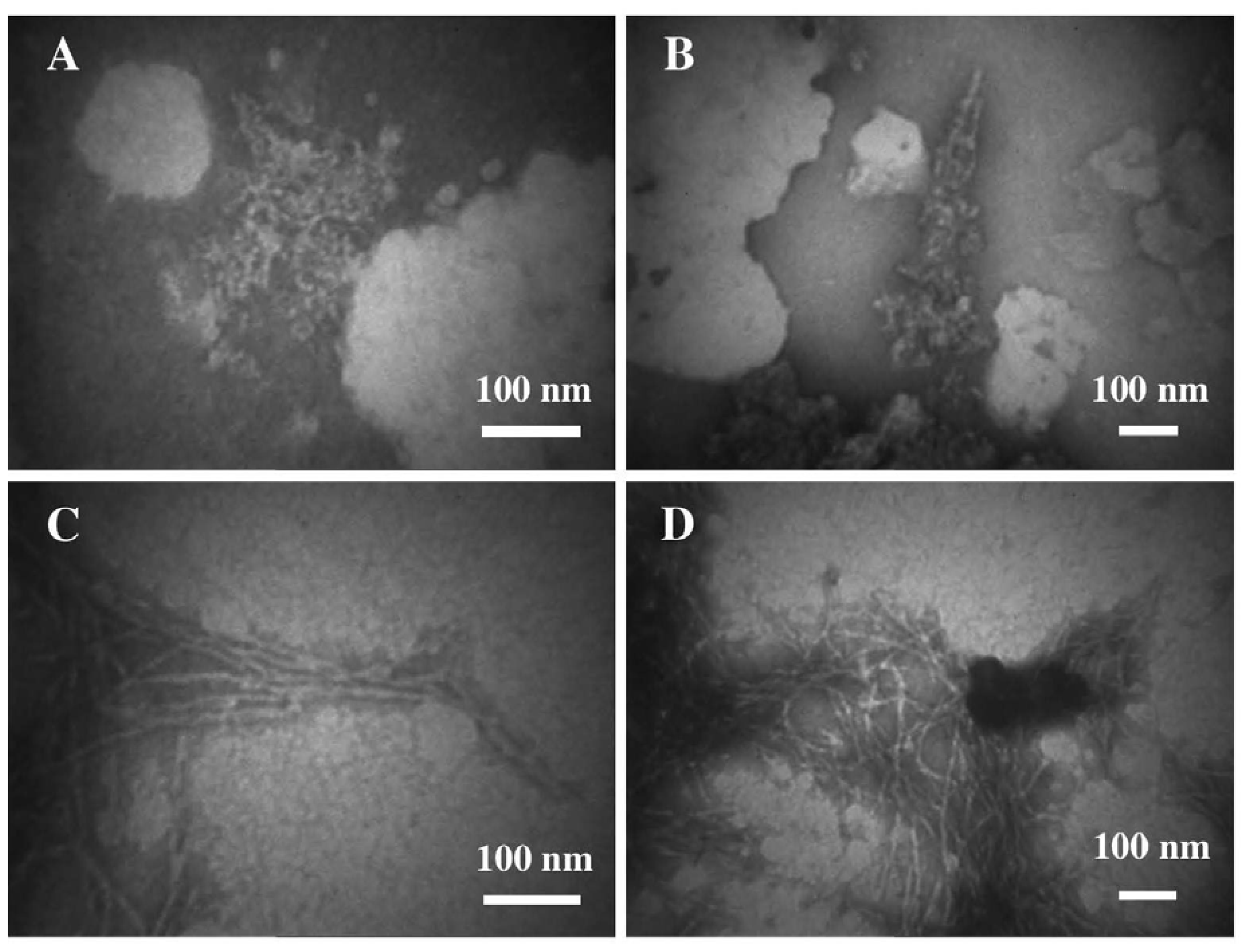
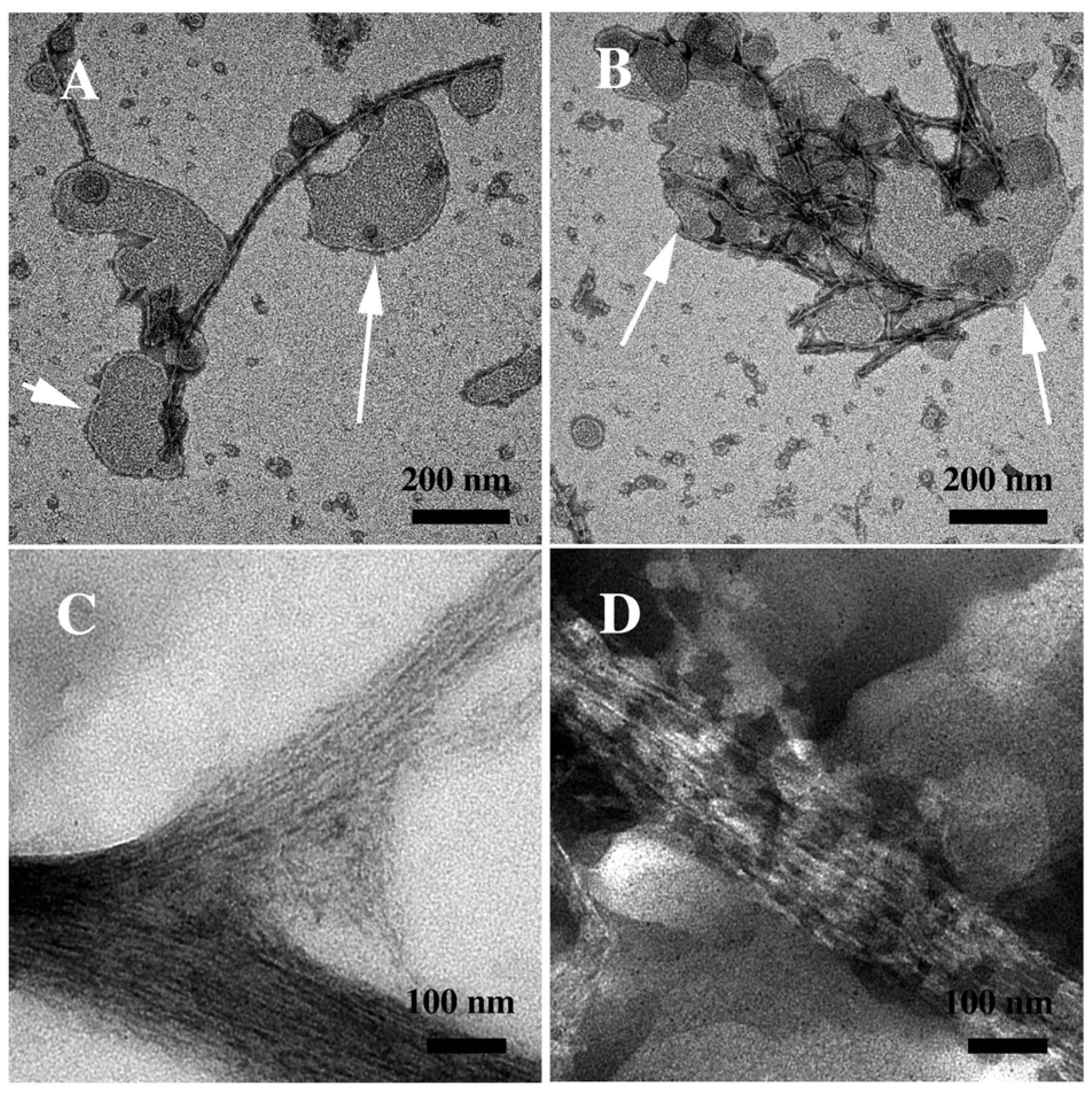
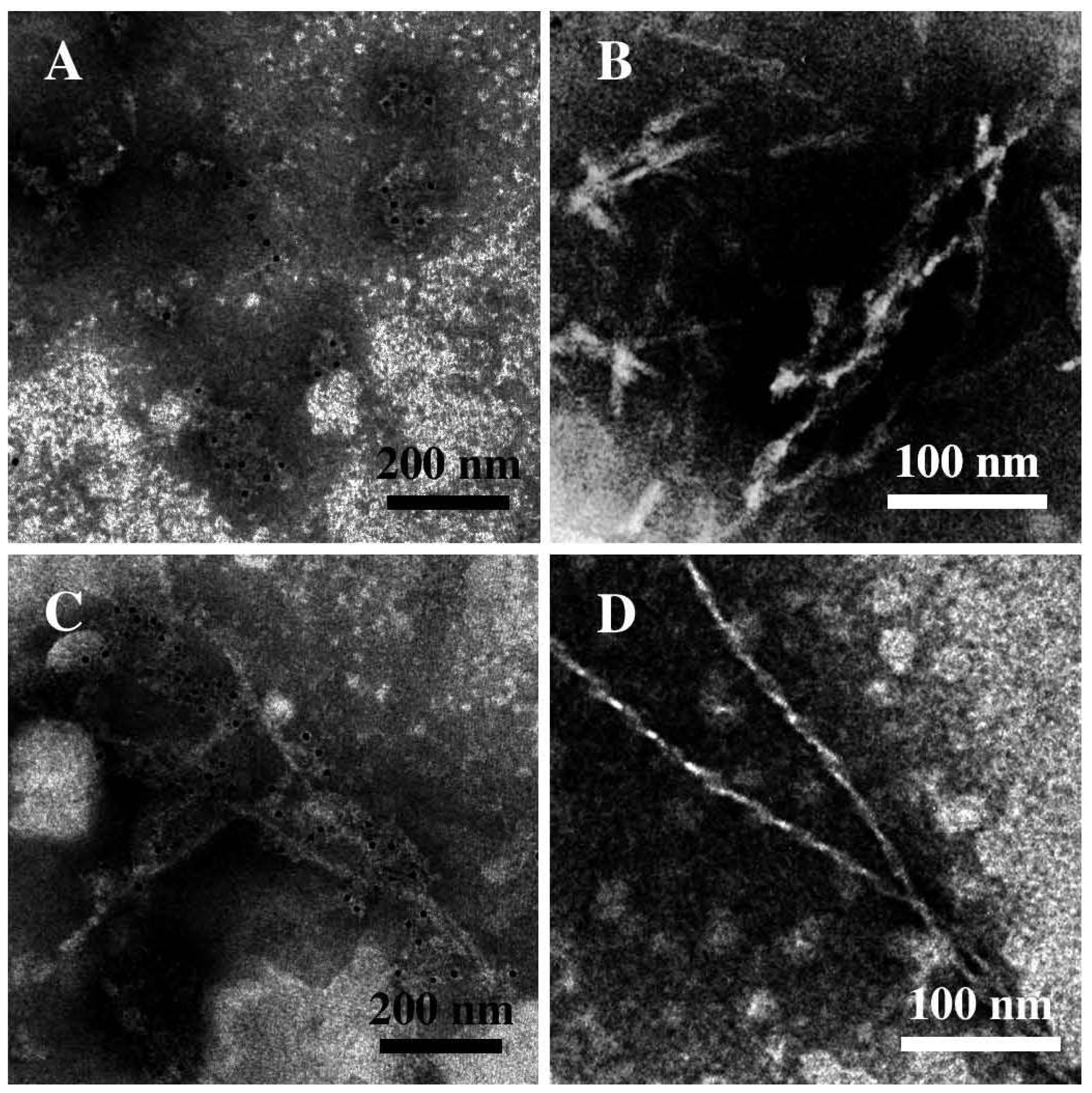
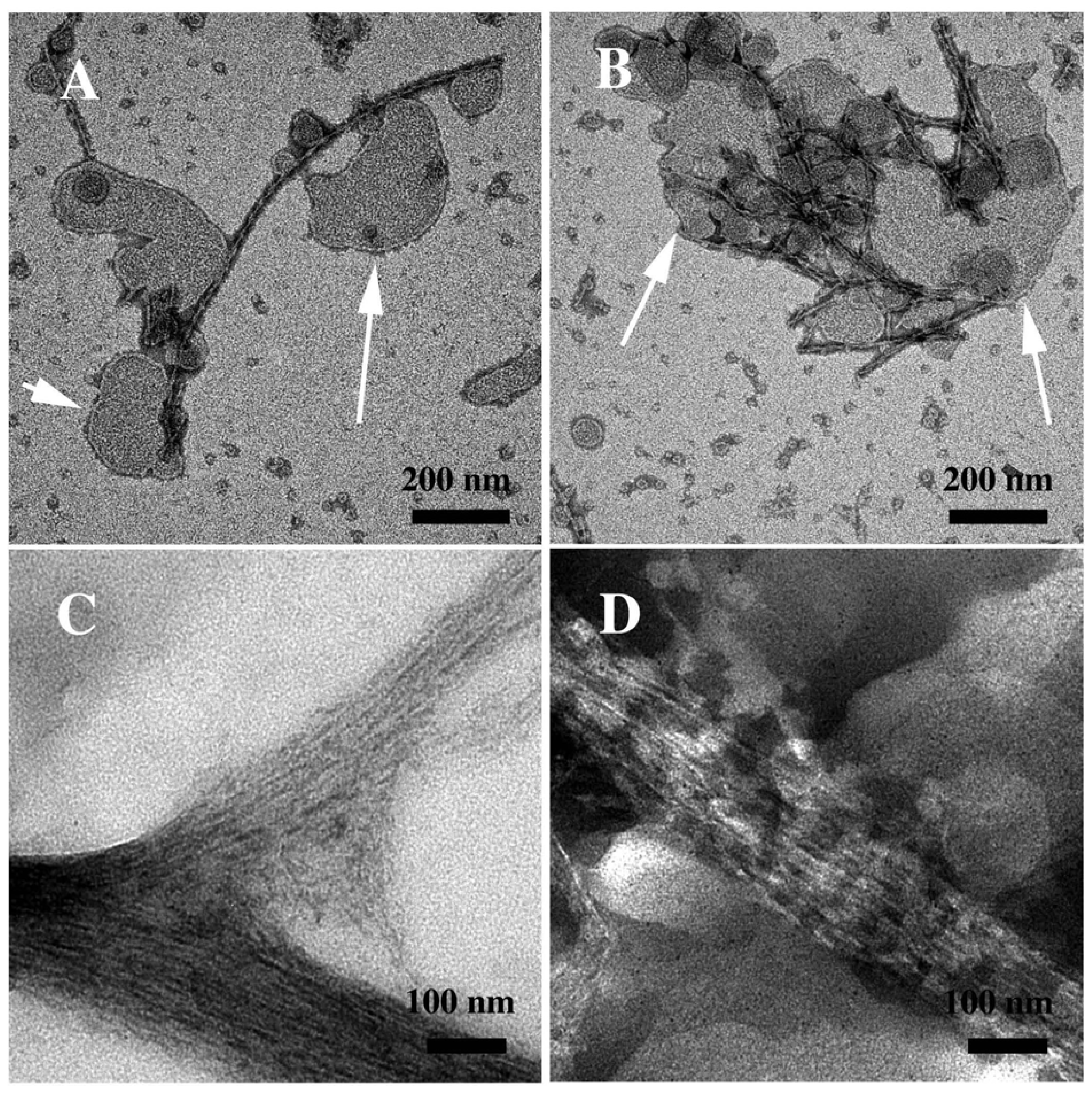
3.2. The Effect of a Strong Oxidation and the Presence of Liposomes on sCT Aggregation
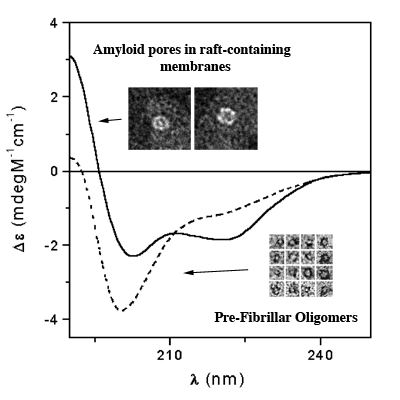
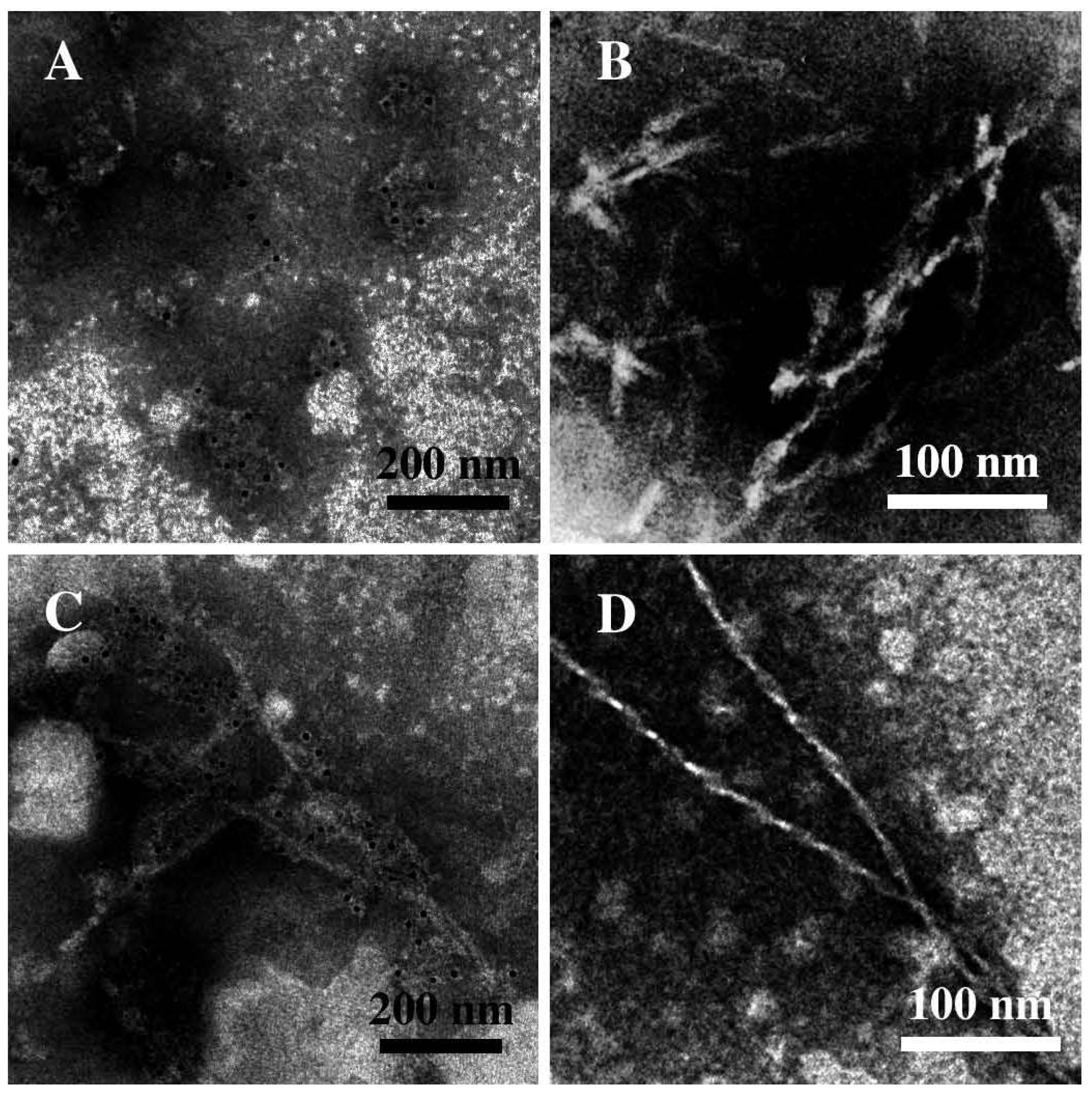
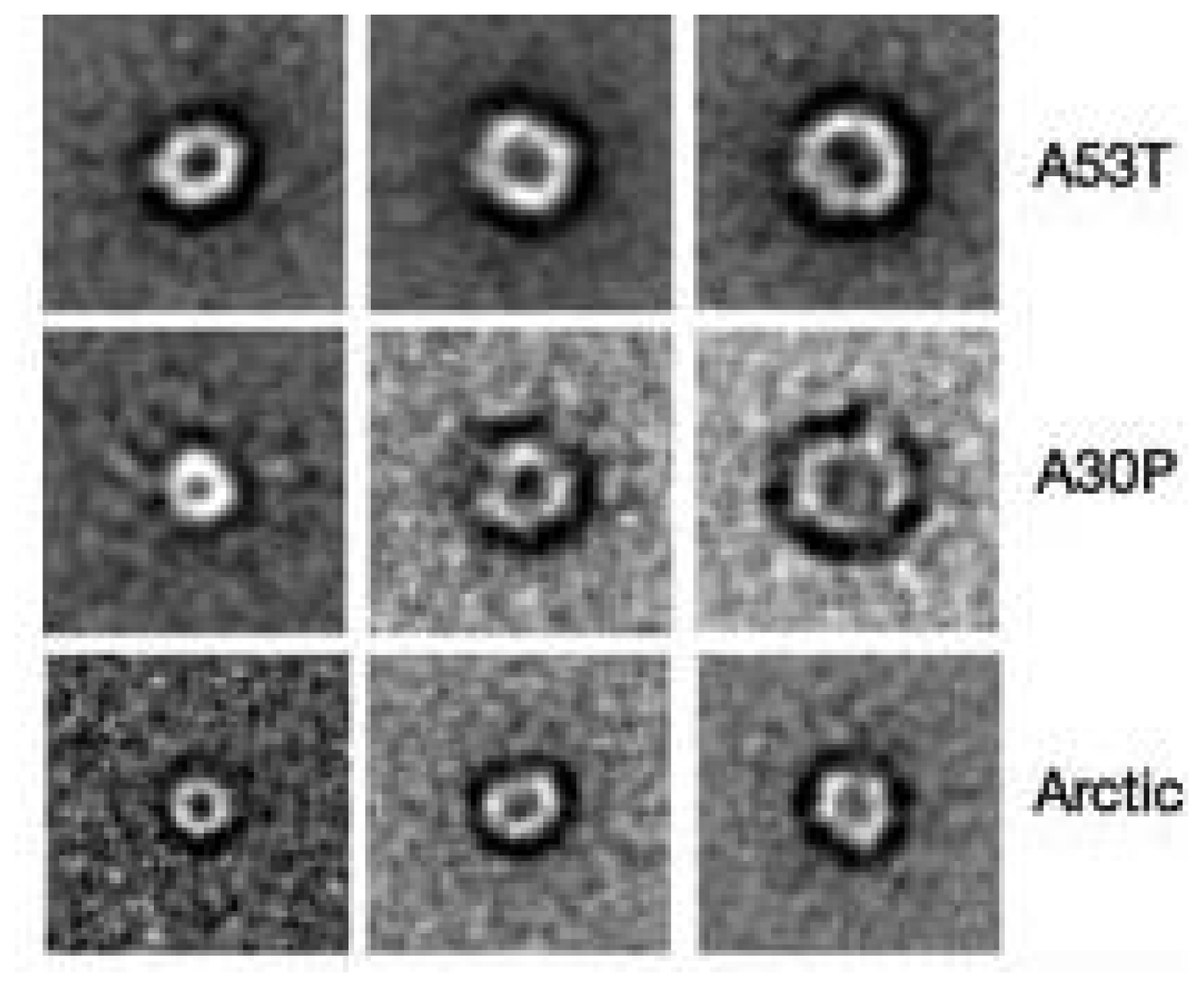
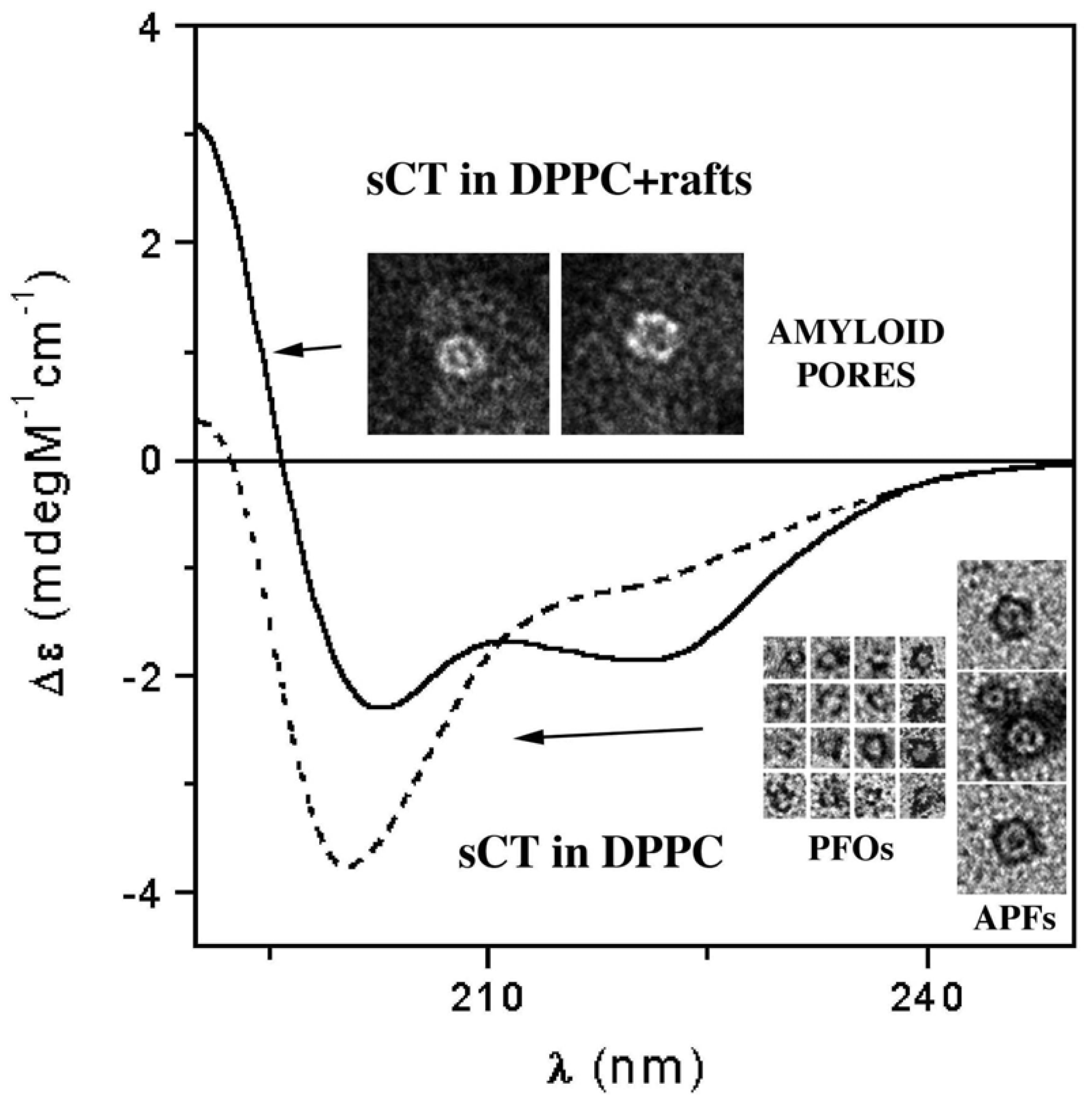
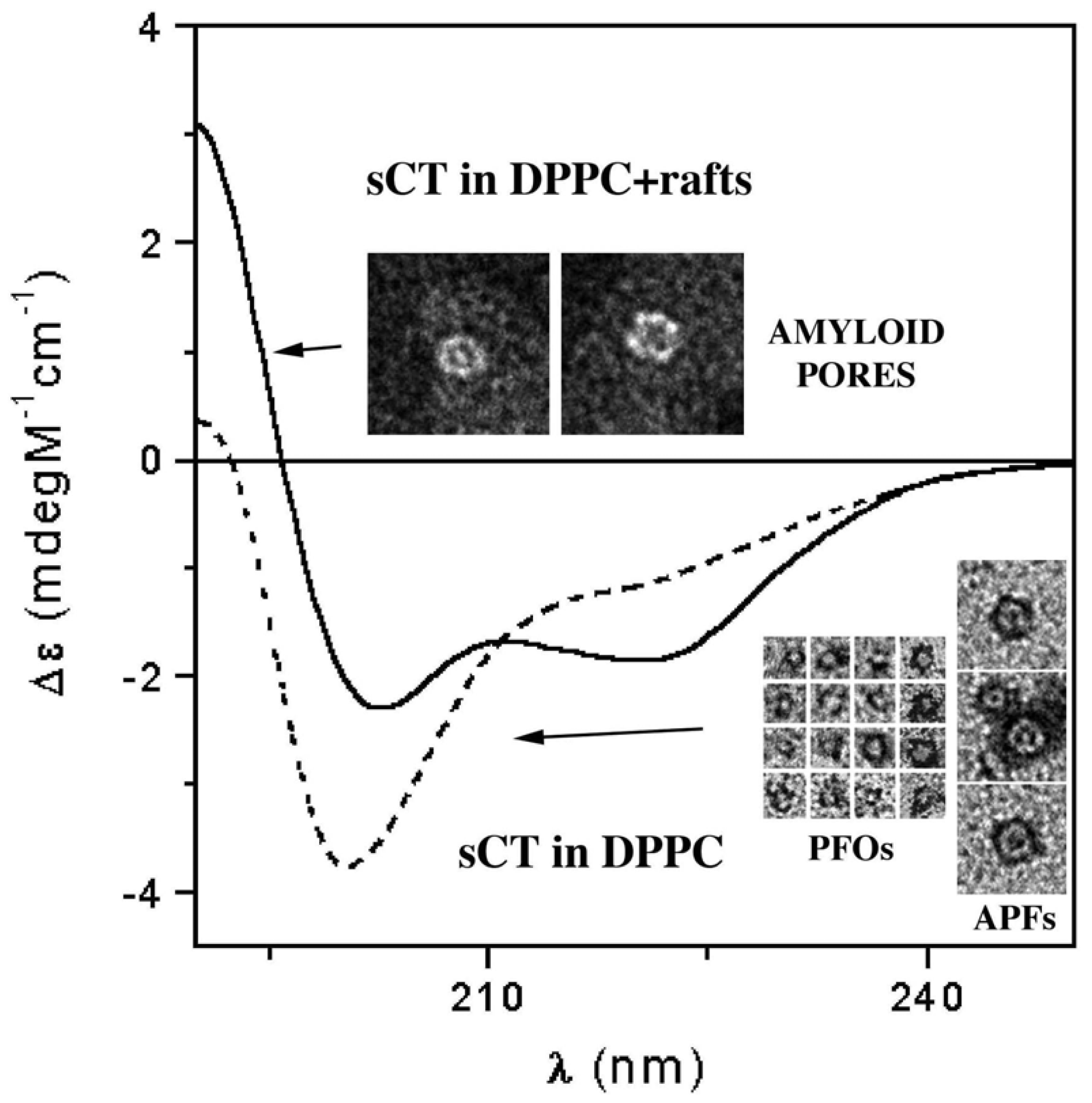
3.3. The Effect of Lipid Rafts on sCT Aggregation
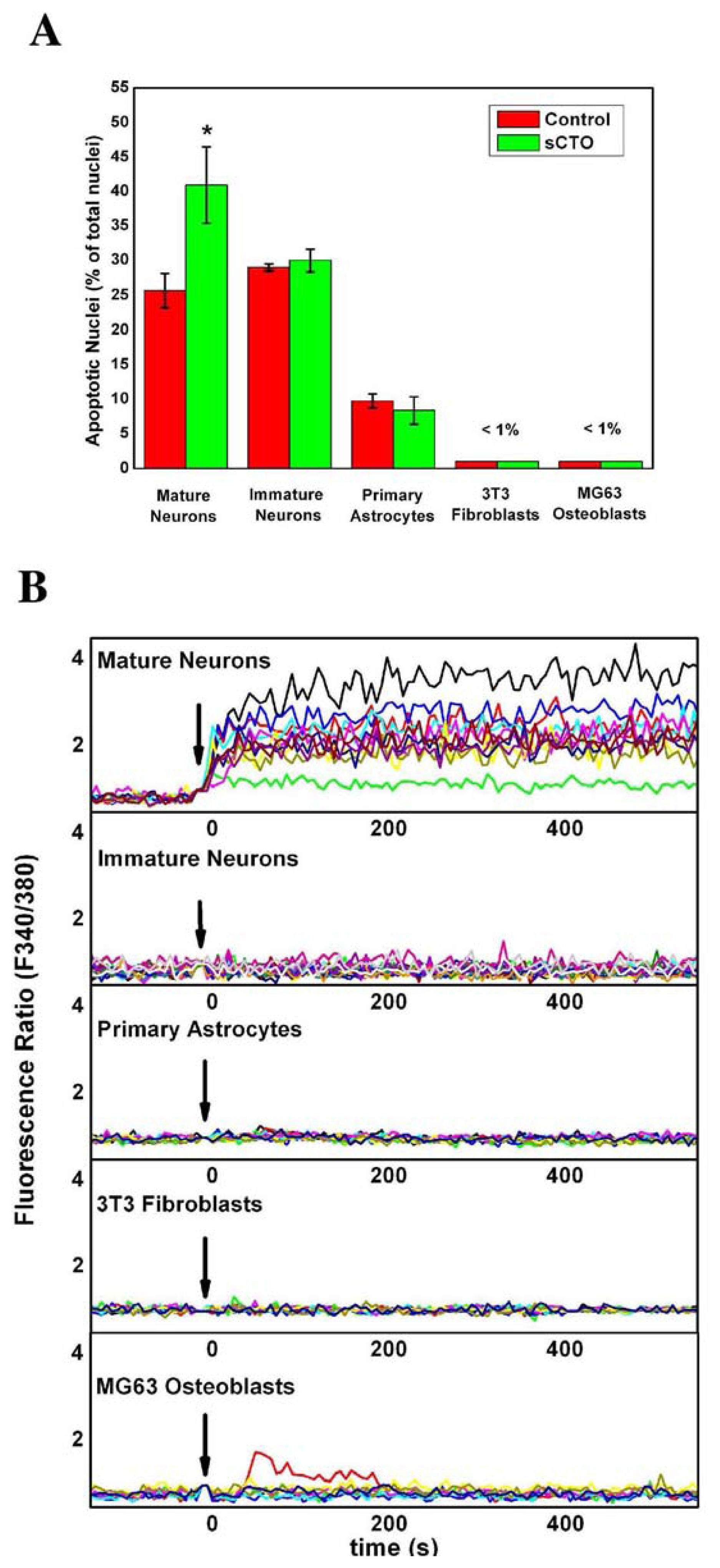
3.4. The Effect of sCT Aggregates on Ca2+ Flow
3.5. The Mechanism of Ca2+-permeabilization Induced by sCTOs Applied in Cell Cultures
4. Concluding Remarks
References
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun 1984, 122, 1131–1135. [Google Scholar]
- DeArmond, S.J.; McKinley, M.P.; Barry, R.A. Identification of prion amyloid filaments in scrapieinfected brain. Cell 1985, 41, 221–235. [Google Scholar]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar]
- Schnabel, J. Protein folding: The dark side of proteins. Nature 2010, 464, 828–829. [Google Scholar]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–553. [Google Scholar]
- Lashuel, H.A.; Grillo-Bosch, D. In vitro preparation of prefibrillar intermediates of amyloid-beta and alfa-synuclein. Methods Mol. Biol 2005, 299, 19–33. [Google Scholar]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T., Jr. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar]
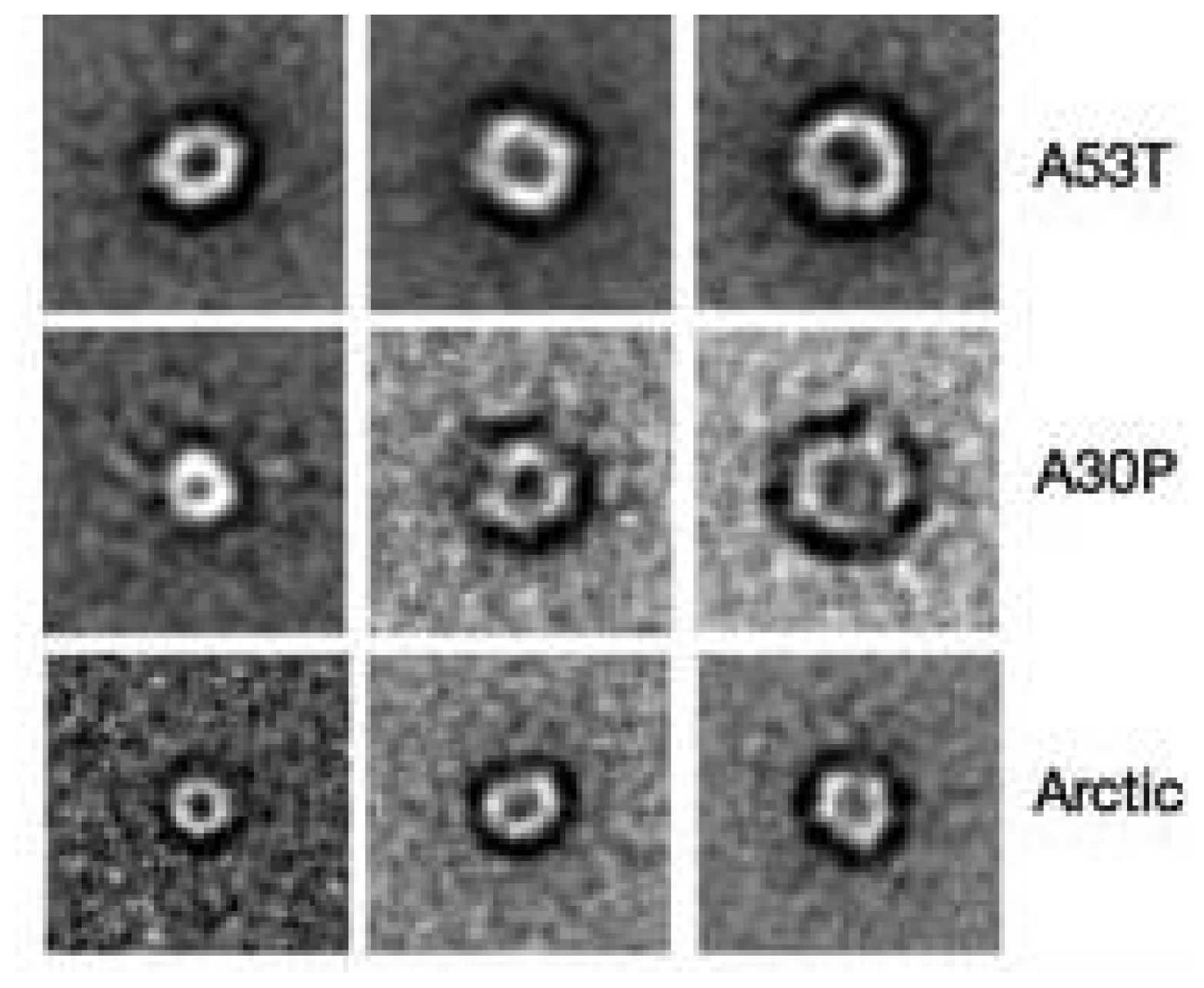
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar]
- Lashuel, H.A.; Lansbury, P.T., Jr. Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q. Rev. Biophys. 2006, 39, 167–201. [Google Scholar]
- Glabe, C.G. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 2006, 27, 570–575. [Google Scholar]
- Kagan, B.L.; Azimov, R.; Azimova, R. Amyloid peptide channels. J. Membr. Biol 2004, 202, 1–10. [Google Scholar]
- Volles, M.J.; Lansbury, P.T., Jr. Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson’s disease. Biochemistry 2003, 42, 7871–7878. [Google Scholar]
- Rochet, J.C.; Outeiro, T.F.; Conway, K.A.; Ding, T.T.; Volles, M.J.; Lashuel, H.A.; Bieganski, R.M.; Lindquist, S.L.; Lansbury, P.T. Interactions among alpha-synuclein, dopamine, and biomembranes: Some clues for understanding neurodegeneration in Parkinson’s disease. J. Mol. Neurosci 2004, 23, 23–34. [Google Scholar]
- Lin, H.; Bhatia, R.; Lal, R. Amyloid beta protein forms ion channels: Implications for Alzheimer’s disease pathophysiology. FASEB J 2001, 15, 2433–2444. [Google Scholar]
- Durell, S.R.; Guy, H.R.; Arispe, N.; Rojas, E.; Pollard, H.B. Theoretical models of the ion channel structure of amyloid beta-protein. Biophys. J 1994, 67, 2137–2145. [Google Scholar]
- Shafrir, Y.; Durell, S.R.; Anishkin, A.; Guy, H.R. Beta-barrel models of soluble amyloid beta oligomers and annular protofibrils. Proteins 2010, 78, 3458–3472. [Google Scholar]
- Shafrir, Y.; Durell, S.; Arispe, N.; Guy, H.R. Models of membrane-bound Alzheimer’s abeta peptide assemblies. Proteins 2010, 78, 3473–3487. [Google Scholar]
- Jang, H.; Zheng, J.; Nussinov, R. Models of beta-amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys. J 2007, 93, 1938–1949. [Google Scholar]
- Kakio, A.; Nishimoto, S.I.; Yanagisawa, K.; Kozutsumi, Y.; Matsuzaki, K. Cholesterol-dependent formation of gm1 ganglioside-bound amyloid beta-protein, an endogenous seed for Alzheimer amyloid. J. Biol. Chem 2001, 276, 24985–24990. [Google Scholar]
- Kakio, A.; Nishimoto, S.; Yanagisawa, K.; Kozutsumi, Y.; Matsuzaki, K. Interactions of amyloid beta-protein with various gangliosides in raft-like membranes: Importance of gm1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry 2002, 41, 7385–7390. [Google Scholar]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol 2000, 1, 31–39. [Google Scholar]
- Hancock, J.F. Lipid rafts: Contentious only from simplistic standpoints. Nat. Rev. Mol. Cell Biol 2006, 7, 456–462. [Google Scholar]
- Cordy, J.M.; Hooper, N.M.; Turner, A.J. The involvement of lipid rafts in Alzheimer’s disease. Mol. Membr. Biol 2006, 23, 111–122. [Google Scholar]
- Kayed, R.; Pensalfini, A.; Margol, L.; Sokolov, Y.; Sarsoza, F.; Head, E.; Hall, J.; Glabe, C. Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J. Biol. Chem 2009, 284, 4230–4237. [Google Scholar]
- Chesnut, C.H., 3rd; Azria, M.; Silverman, S.; Engelhardt, M.; Olson, M.; Mindeholm, L. Salmon calcitonin: A review of current and future therapeutic indications. Osteoporos. Int. 2008, 19, 479–491. [Google Scholar]
- Breimer, L.H.; MacIntyre, I.; Zaidi, M. Peptides from the calcitonin genes: Molecular genetics, structure and function. Biochem. J 1988, 255, 377–390. [Google Scholar]
- Cafforio, P.; de Matteo, M.; Brunetti, A.E.; Dammacco, F.; Silvestris, F. Functional expression of the calcitonin receptor by human T and B cells. Hum. Immunol 2009, 70, 678–685. [Google Scholar]
- Bauer, H.H.; Aebi, U.; Haner, M.; Hermann, R.; Muller, M.; Merkle, H.P. Architecture and polymorphism of fibrillar supramolecular assemblies produced by in vitro aggregation of human calcitonin. J. Struct. Biol 1995, 115, 1–15. [Google Scholar]
- Arvinte, T.; Cudd, A.; Drake, A.F. The structure and mechanism of formation of human calcitonin fibrils. J. Biol. Chem 1993, 268, 6415–6422. [Google Scholar]
- Arvinte, T.; Drake, A.F. Comparative study of human and salmon calcitonin secondary structure in solutions with low dielectric constants. J. Biol. Chem 1993, 268, 6408–6414. [Google Scholar]
- Ferrone, F.A.; Hofrichter, J.; Sunshine, H.R.; Eaton, W.A. Kinetic studies on photolysis-induced gelation of sickle cell hemoglobin suggest a new mechanism. Biophys. J 1980, 32, 361–380. [Google Scholar]
- Meyer, J.P.; Pelton, J.T.; Hoflack, J.; Saudek, V. Solution structure of salmon calcitonin. Biopolymers 1991, 31, 233–241. [Google Scholar]
- Epand, R.M.; Epand, R.F.; Orlowski, R.C.; Schlueter, R.J.; Boni, L.T.; Hui, S.W. Amphipathic helix and its relationship to the interaction of calcitonin with phospholipids. Biochemistry 1983, 22, 5074–5084. [Google Scholar]
- Gilchrist, P.J.; Bradshaw, J.P. Amyloid formation by salmon calcitonin. Biochim. Biophys. Acta 1993, 1182, 111–114. [Google Scholar]
- Gaudiano, M.C.; Diociaiuti, M.; Bertocchi, P.; Valvo, L. Effects induced by hydroxyl radicals on salmon calcitonin: A RP-HPLC, CD and TEM study. Biochim. Biophys. Acta 2003, 1623, 33–40. [Google Scholar]
- Siligardi, G.; Samori, B.; Melandri, S.; Visconti, M.; Drake, A.F. Correlations between biological activities and conformational properties for human, salmon, eel, porcine calcitonins and elcatonin elucidated by CD spectroscopy. Eur. J. Biochem 1994, 221, 1117–1125. [Google Scholar]
- van der Zee, J.; Krootjes, B.B.; Chignell, C.F.; Dubbelman, T.M.; Van Steveninck, J. Hydroxyl radical generation by a light-dependent Fenton reaction. Free Radic. Biol. Med 1993, 14, 105–113. [Google Scholar]
- Gaudiano, M.C.; Colone, M.; Bombelli, C.; Chistolini, P.; Valvo, L.; Diociaiuti, M. Early stages of salmon calcitonin aggregation: Effect induced by ageing and oxidation processes in water and in the presence of model membranes. Biochim. Biophys. Acta 2005, 1750, 134–145. [Google Scholar]
- Halliwell, B. Free radicals, antioxidants, and human disease: Curiosity, cause, or consequence? Lancet 1994, 344, 721–724. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M.; Cross, C.E. Free radicals, antioxidants, and human disease: Where are we now? J. Lab. Clin. Med 1992, 119, 598–620. [Google Scholar]
- Diociaiuti, M.; Molinari, A.; Ruspantini, I.; Gaudiano, M.C.; Ippoliti, R.; Lendaro, E.; Bordi, F.; Chistolini, P.; Arancia, G. P-glycoprotein inserted in planar lipid bilayers formed by liposomes opened on amorphous carbon and langmuir-blodgett monolayer. Biochim. Biophys. Acta 2002, 1559, 21–31. [Google Scholar]
- Bradshaw, J.P. Phosphatydylglycerol promotes bilayer insertion of salmon calcitonin. Biophys. J 1997, 72, 2180–2186. [Google Scholar]
- Diociaiuti, M.; Polzi, L.Z.; Valvo, L.; Malchiodi-Albedi, F.; Bombelli, C.; Gaudiano, M.C. Calcitonin forms oligomeric pore-like structures in lipid membranes. Biophys. J 2006, 91, 2275–2281. [Google Scholar]
- Malchiodi-Albedi, F.; Contrusciere, V.; Raggi, C.; Fecchi, K.; Rainaldi, G.; Paradisi, S.; Matteucci, A.; Santini, M.T.; Sargiacomo, M.; Frank, C.; et al. Lipid raft disruption protects mature neurons against amyloid oligomer toxicity. Biochim. Biophys. Acta 2010, 1802, 406–415. [Google Scholar]
- Ledesma, M.D.; Brugger, B.; Bunning, C.; Wieland, F.T.; Dotti, C.G. Maturation of the axonal plasma membrane requires upregulation of sphingomyelin synthesis and formation of protein-lipid complexes. EMBO J 1999, 18, 1761–1771. [Google Scholar]

© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Diociaiuti, M.; Gaudiano, M.C.; Malchiodi-Albedi, F. The Slowly Aggregating Salmon Calcitonin: A Useful Tool for the Study of the Amyloid Oligomers Structure and Activity. Int. J. Mol. Sci. 2011, 12, 9277-9295. https://doi.org/10.3390/ijms12129277
Diociaiuti M, Gaudiano MC, Malchiodi-Albedi F. The Slowly Aggregating Salmon Calcitonin: A Useful Tool for the Study of the Amyloid Oligomers Structure and Activity. International Journal of Molecular Sciences. 2011; 12(12):9277-9295. https://doi.org/10.3390/ijms12129277
Chicago/Turabian StyleDiociaiuti, Marco, Maria Cristina Gaudiano, and Fiorella Malchiodi-Albedi. 2011. "The Slowly Aggregating Salmon Calcitonin: A Useful Tool for the Study of the Amyloid Oligomers Structure and Activity" International Journal of Molecular Sciences 12, no. 12: 9277-9295. https://doi.org/10.3390/ijms12129277