3D QSAR Pharmacophore Modeling, in Silico Screening, and Density Functional Theory (DFT) Approaches for Identification of Human Chymase Inhibitors

Abstract
:
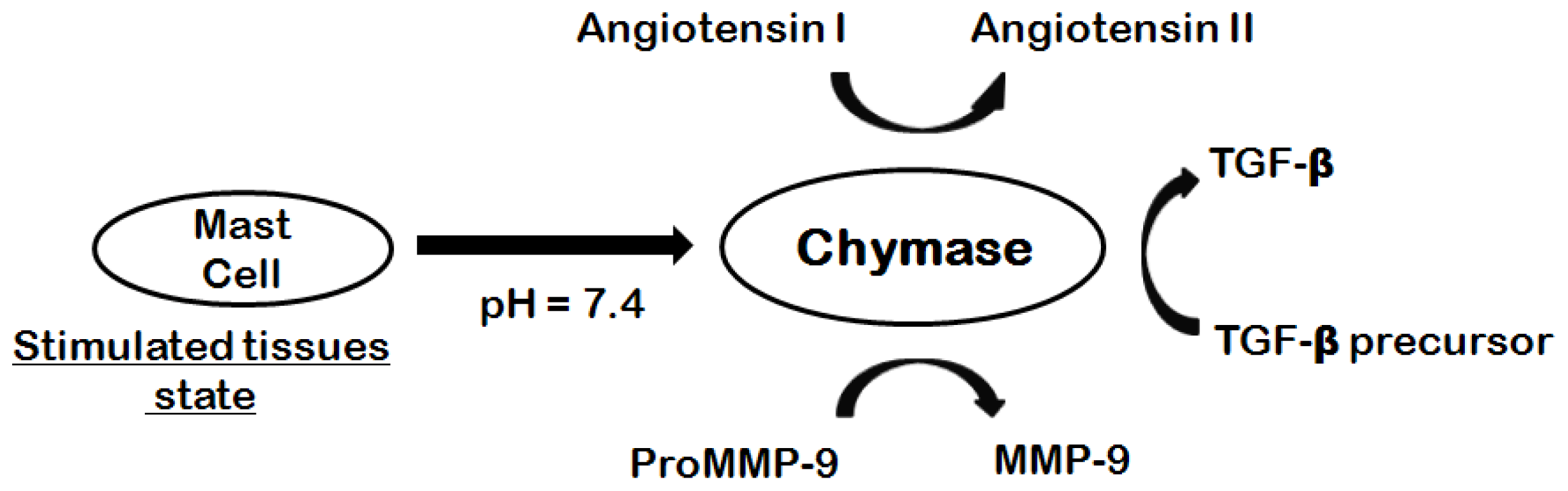
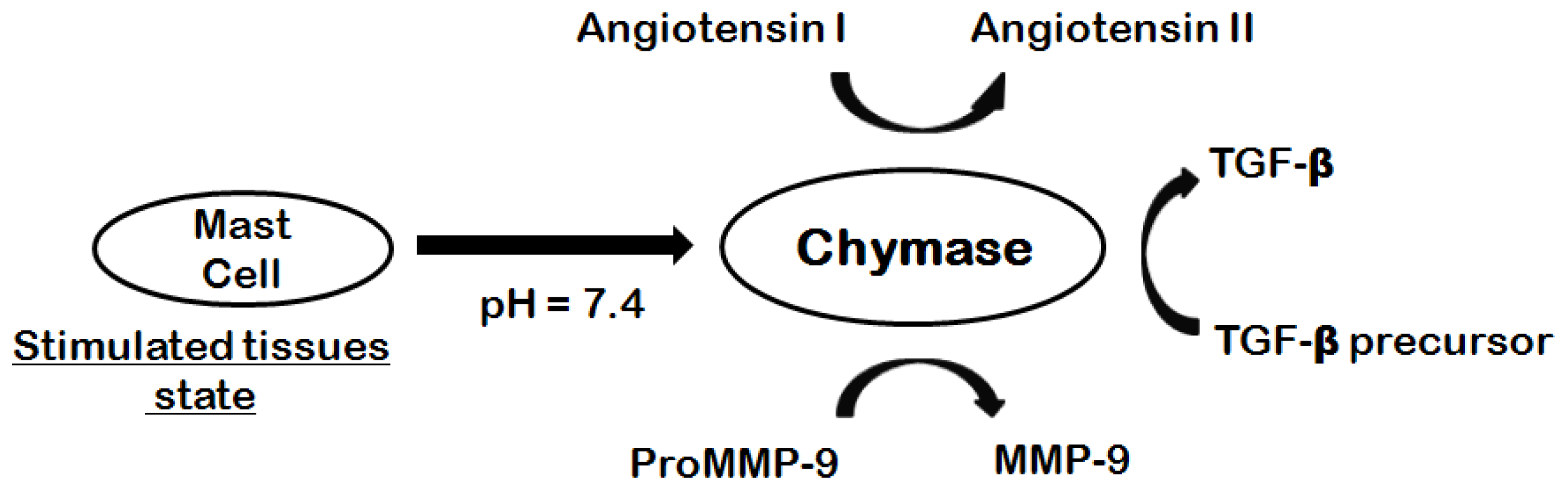
1. Introduction
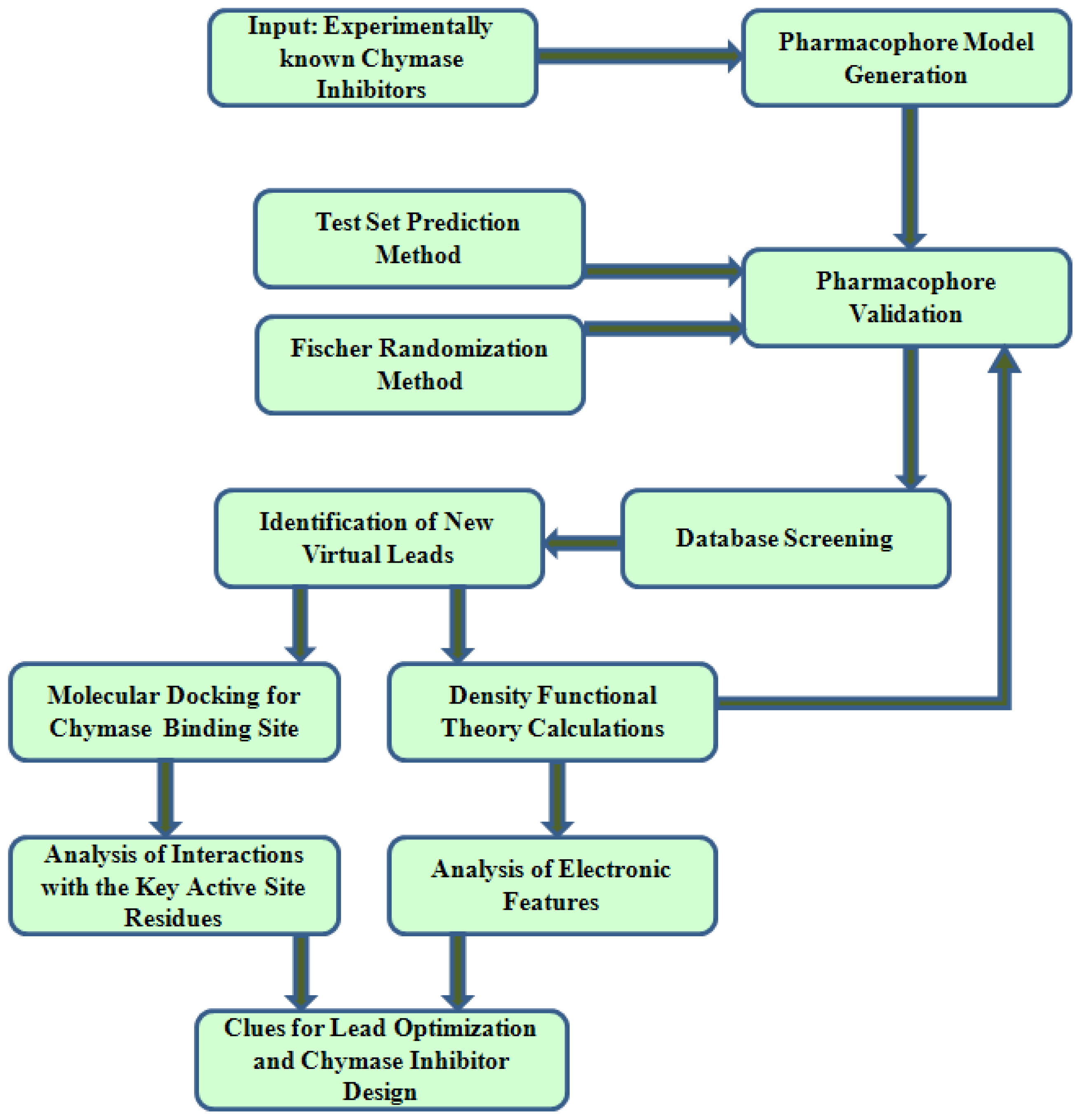
2. Results and Discussion
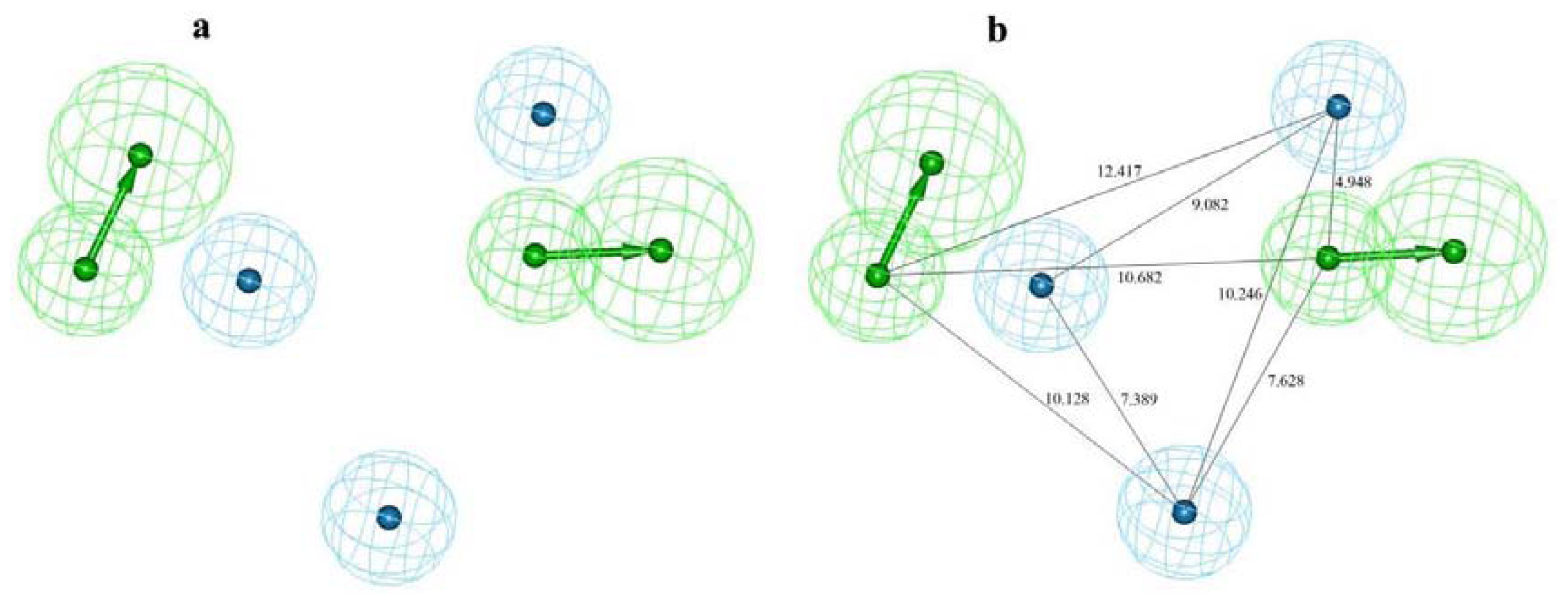
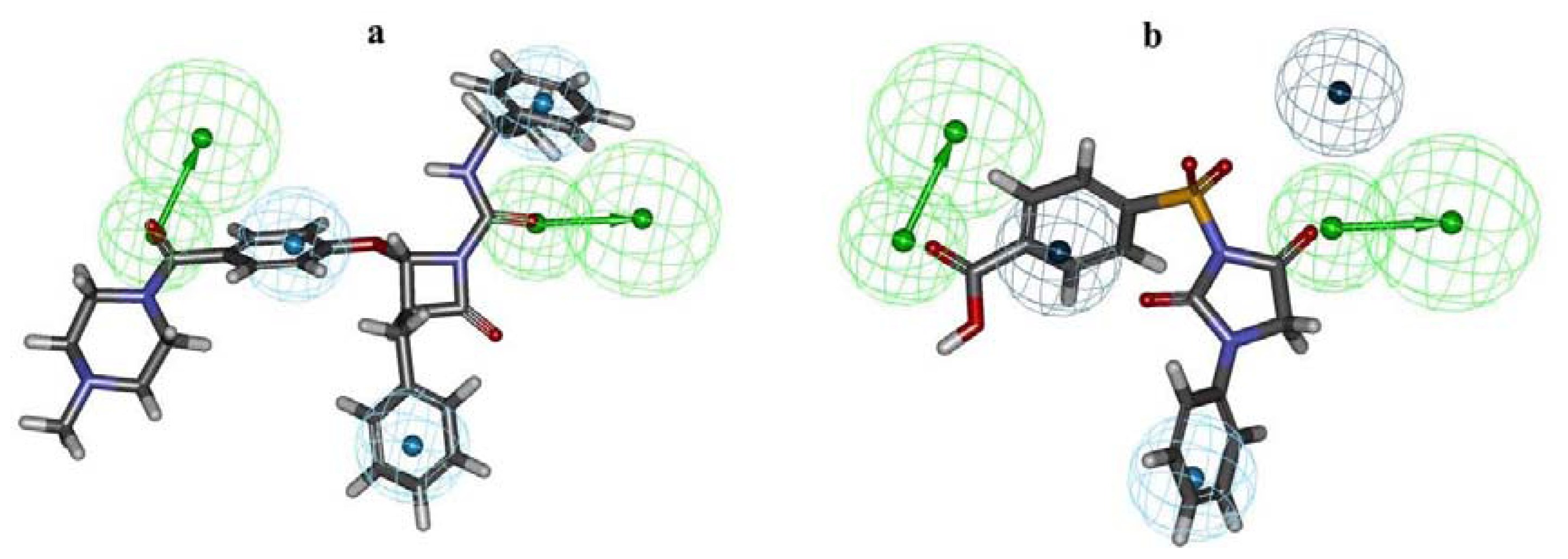
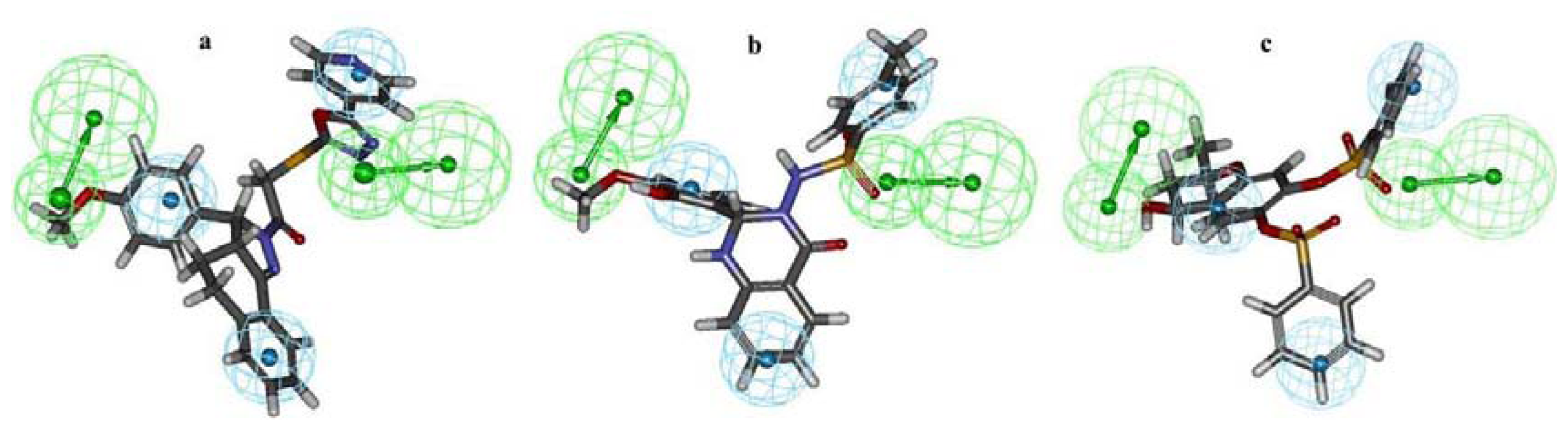
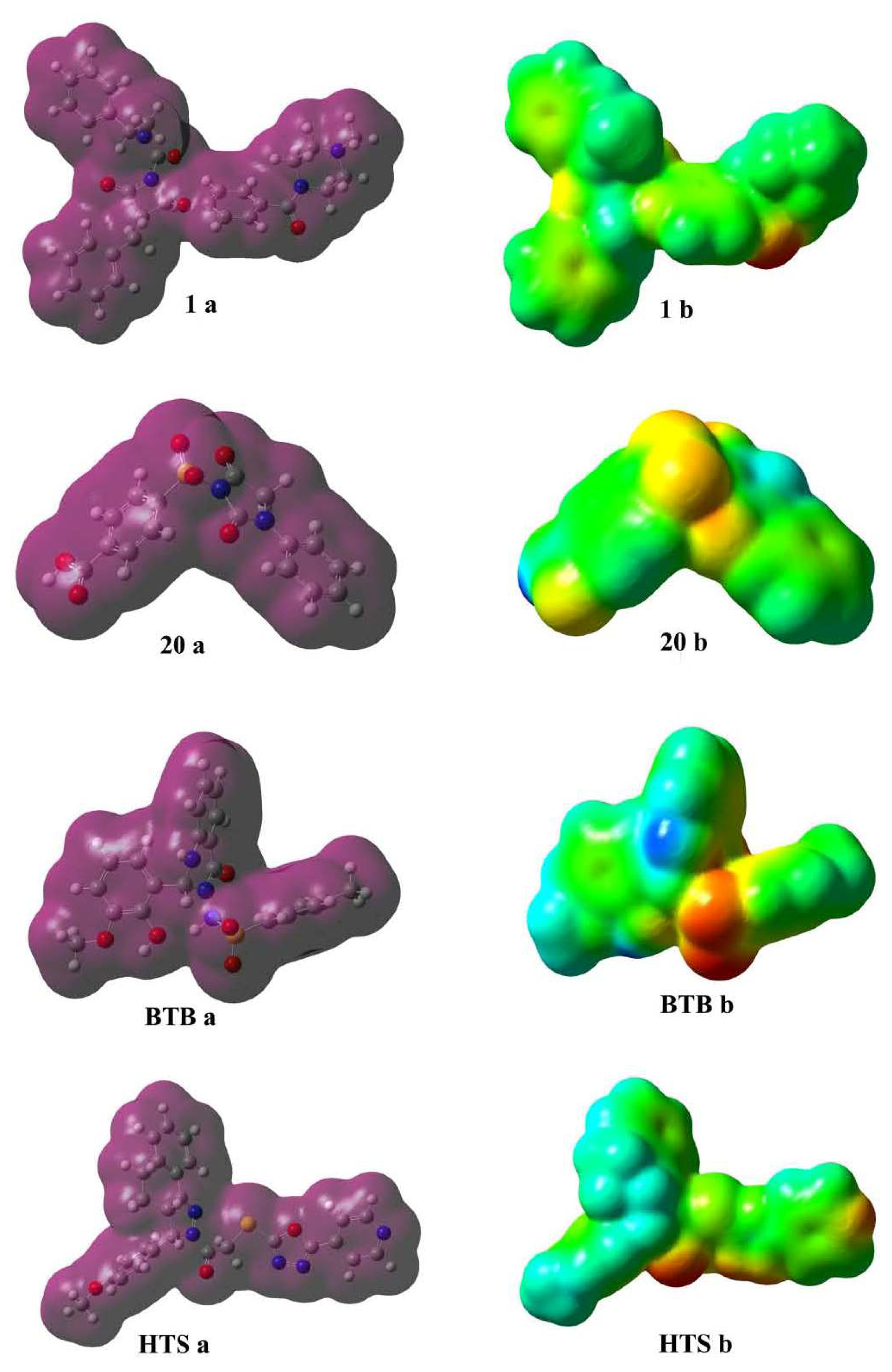
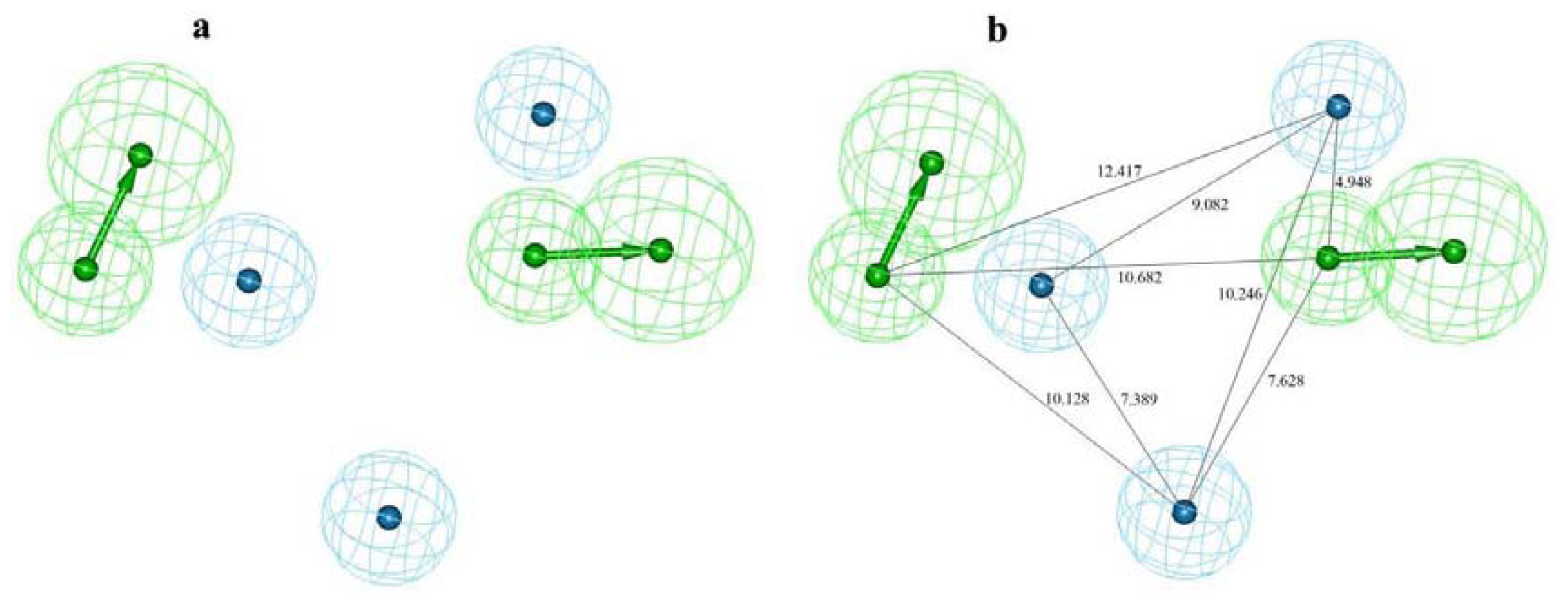
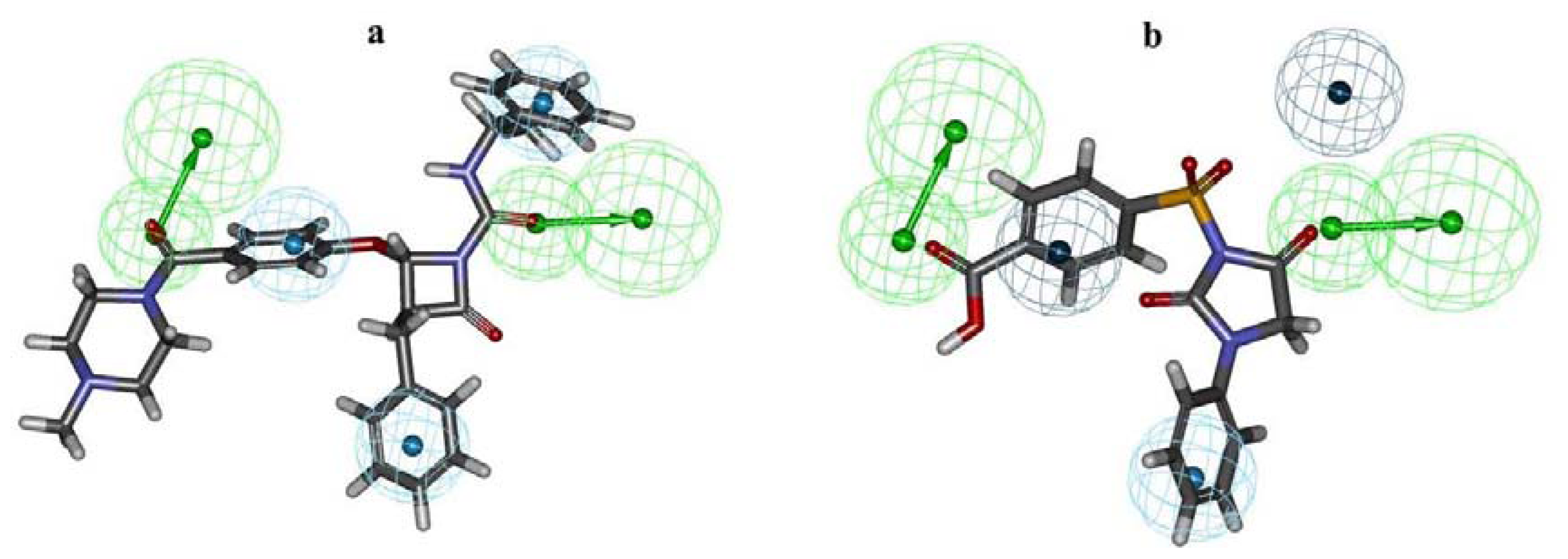
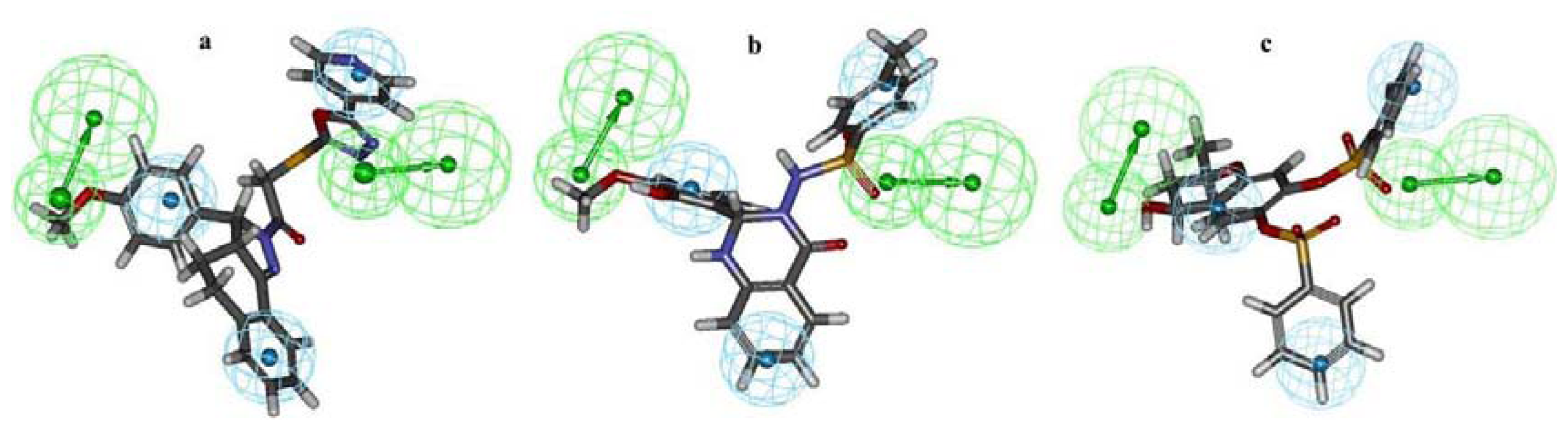
2.1. Pharmacophore Modeling
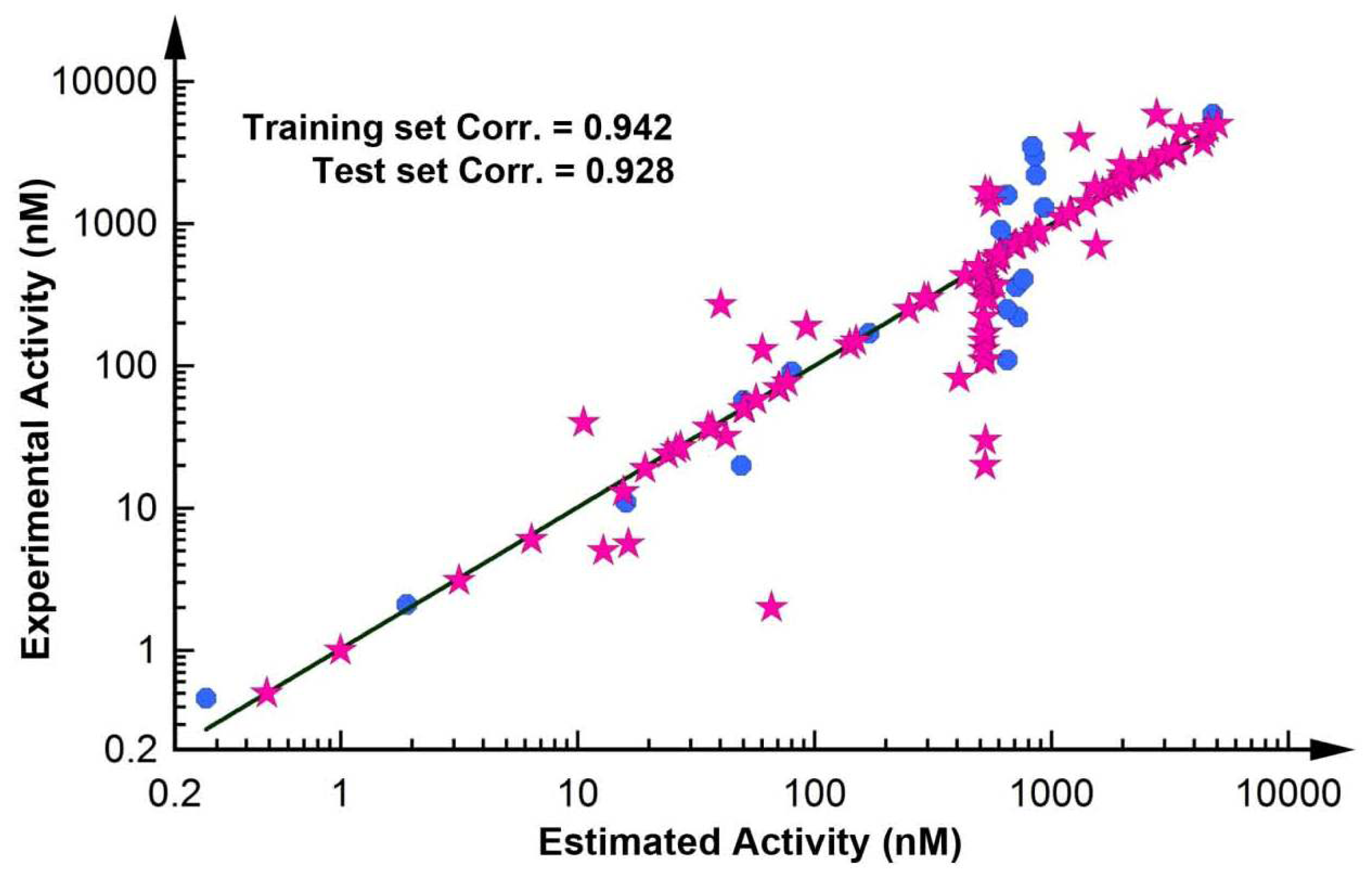
2.2. Pharmacophore Validation
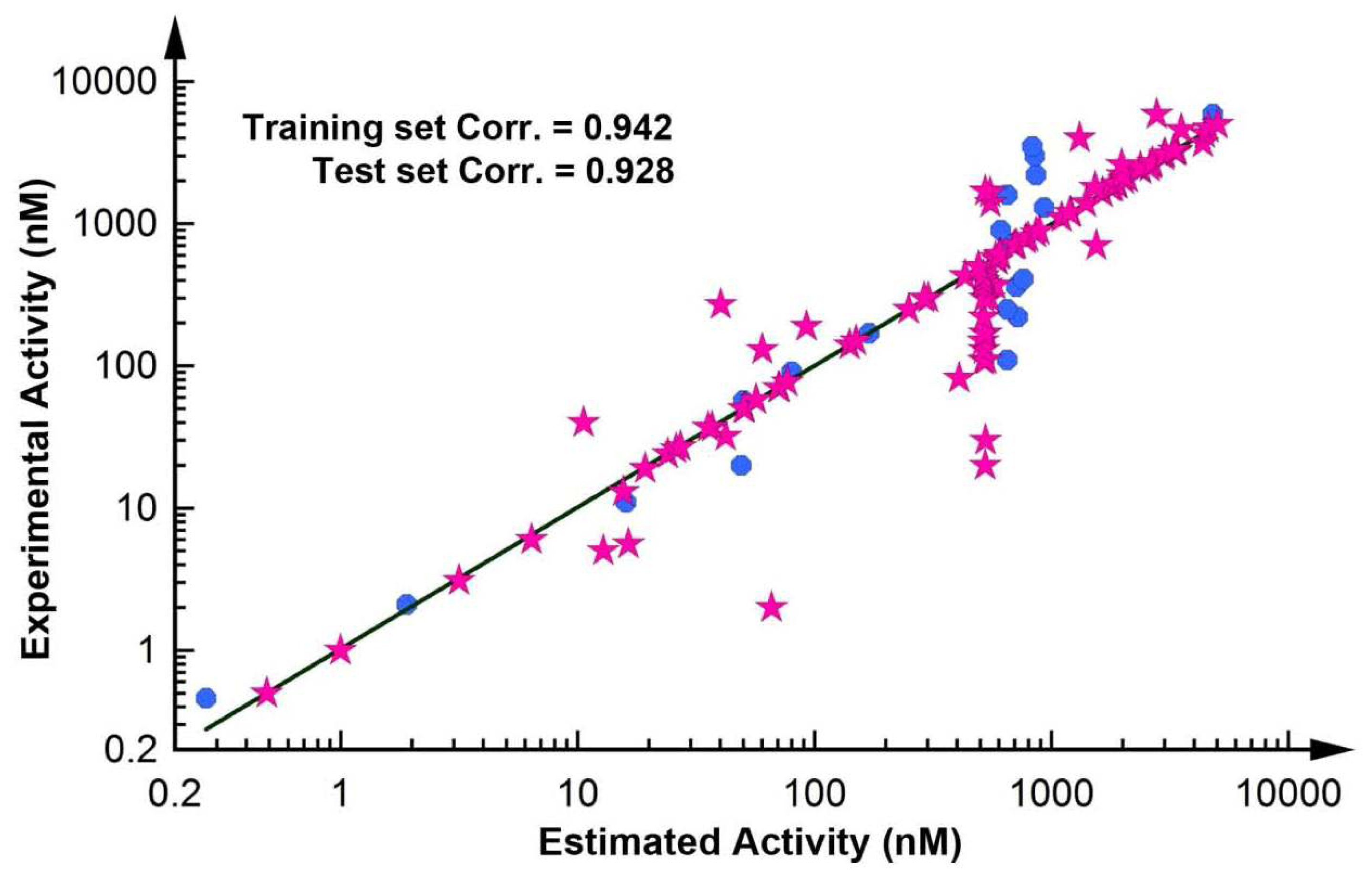
2.2.1. Test Set Prediction Method
2.2.2. Fischer Randomization Method
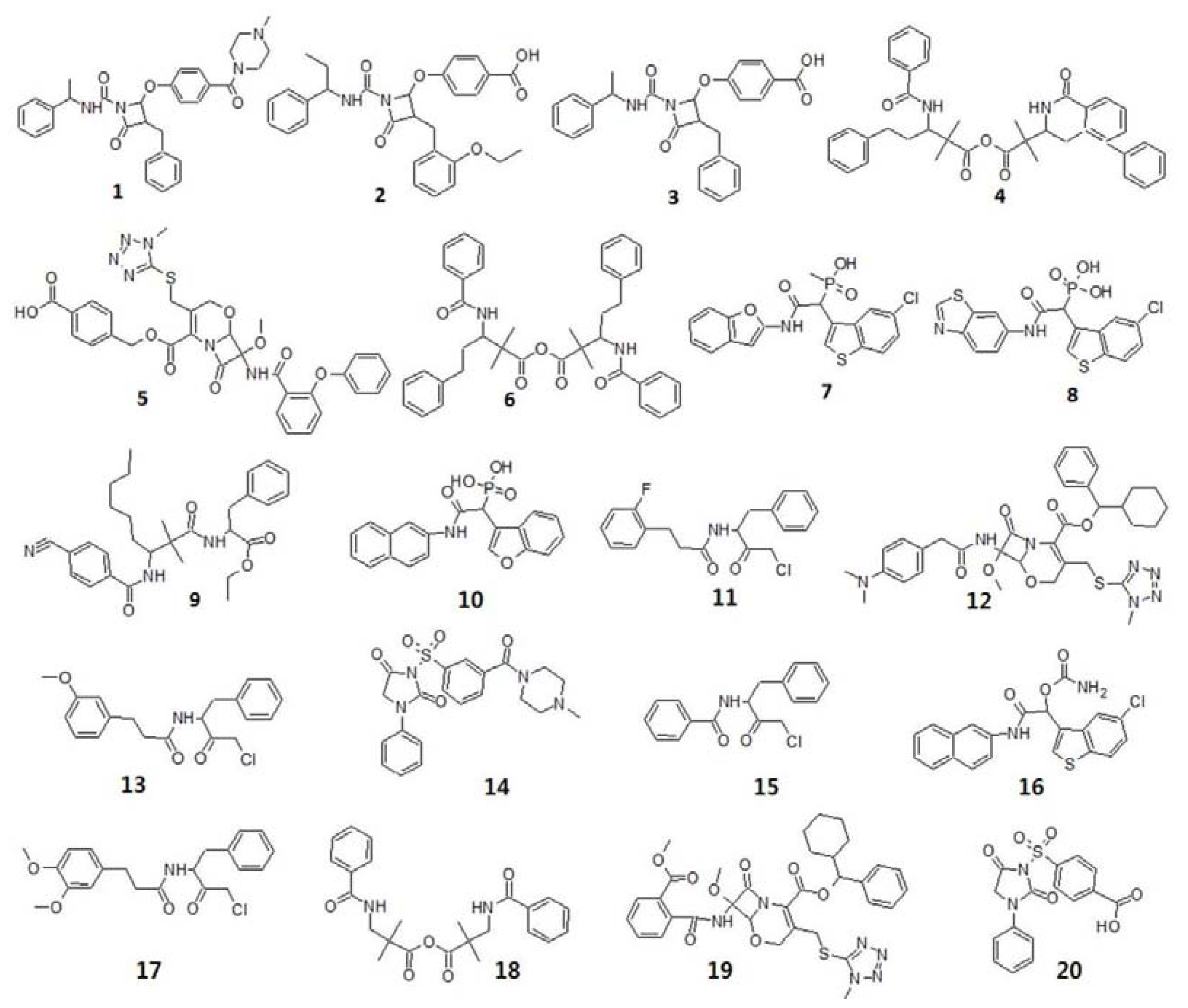
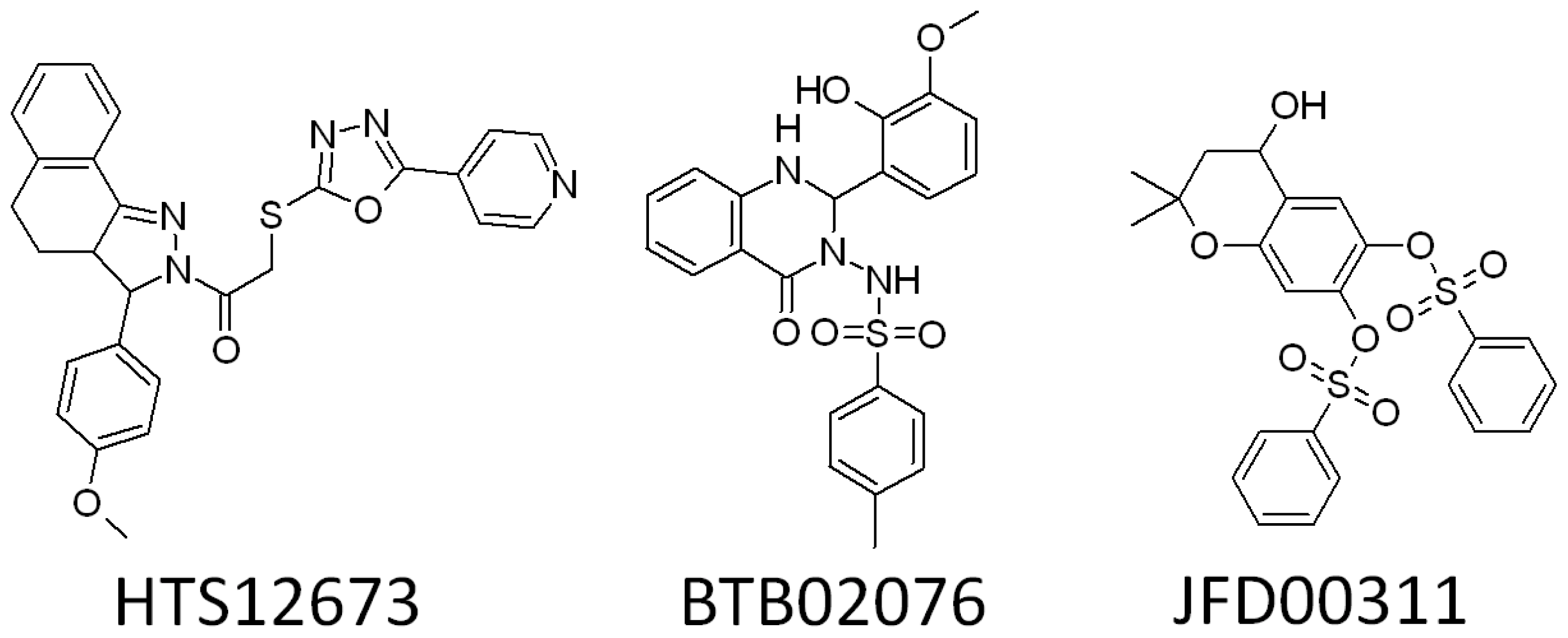
2.3. Search for New Potential Compounds Using Database Screening
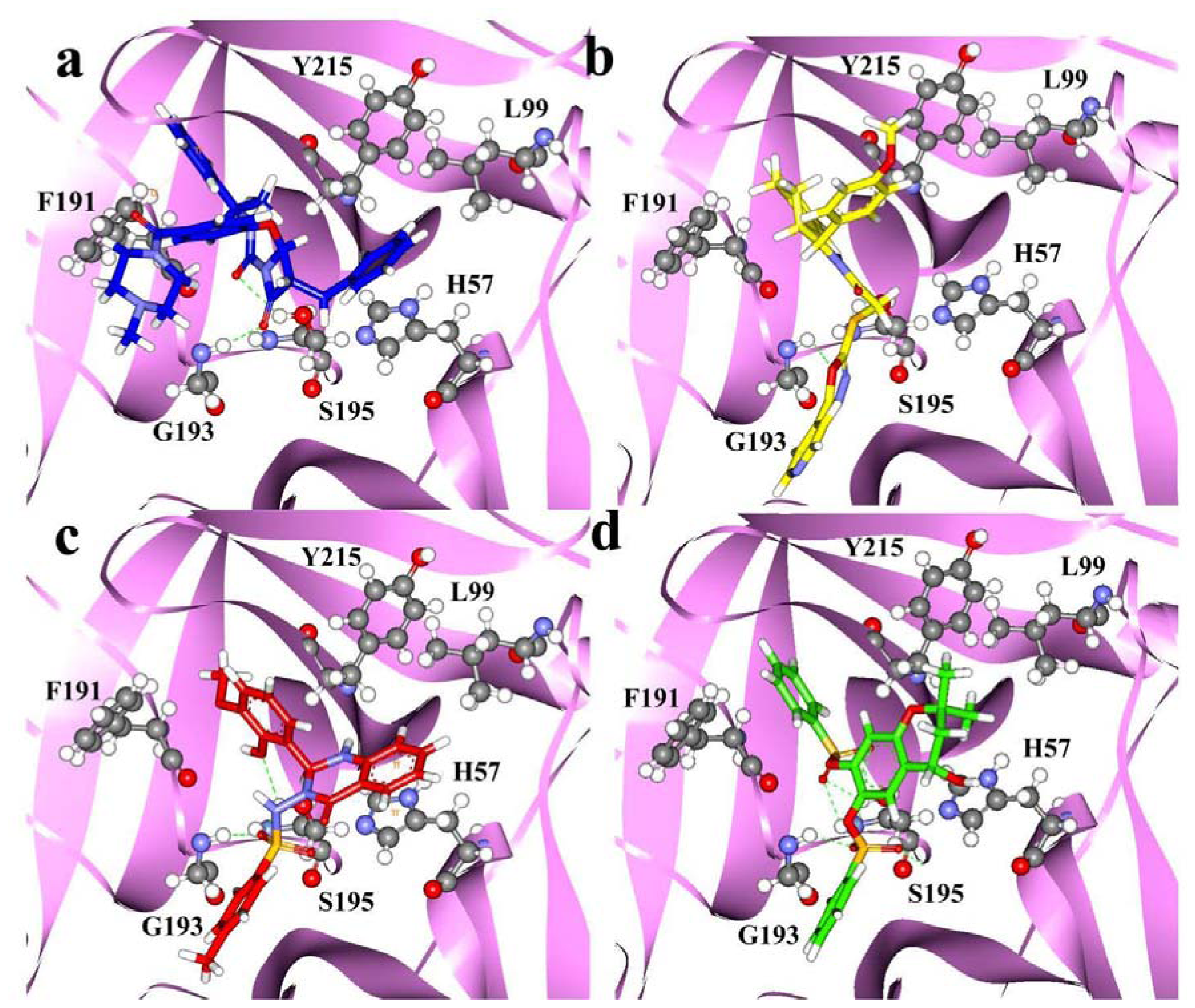
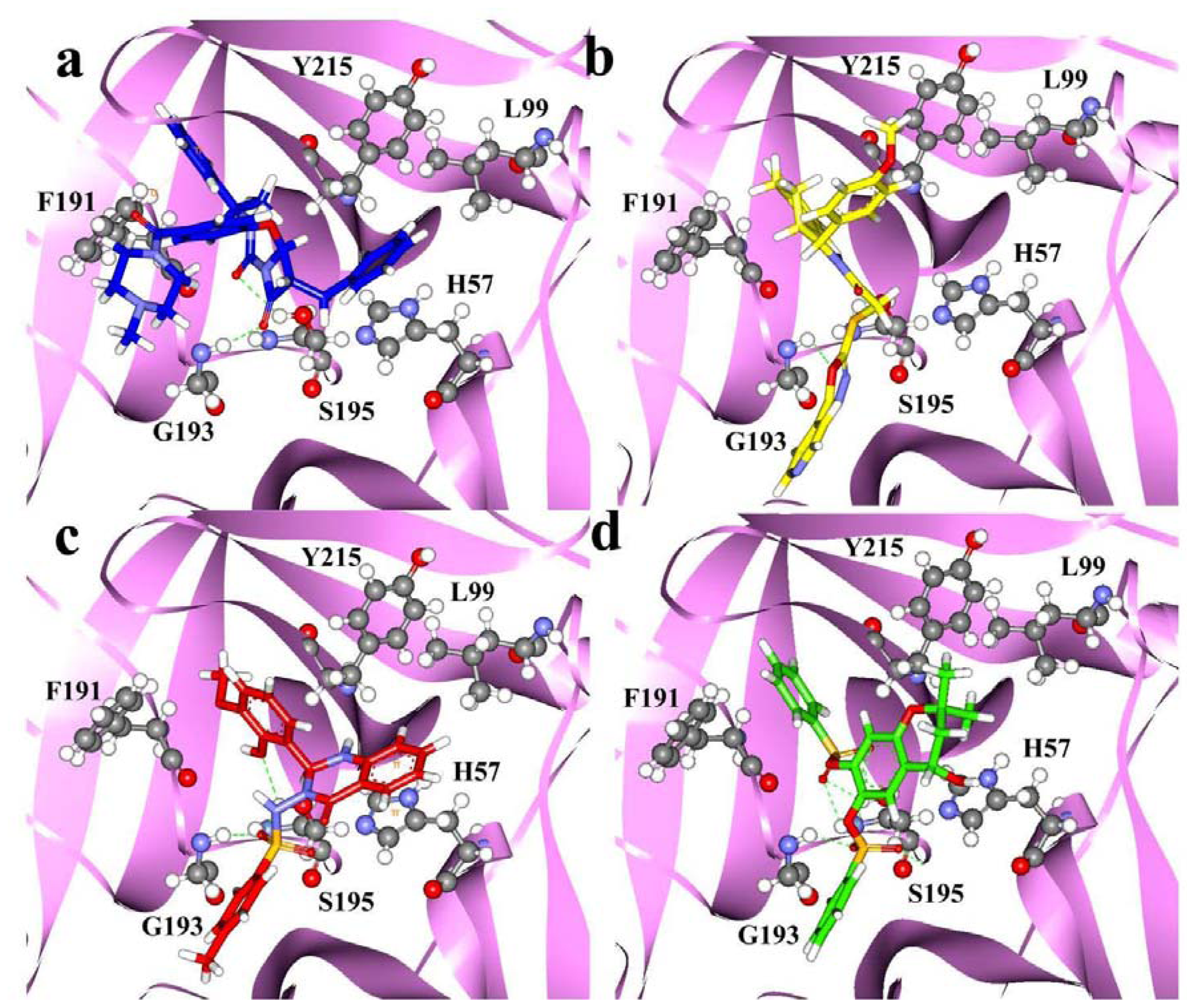
2.4. Molecular Docking
2.5. Density Functional Theory Calculations
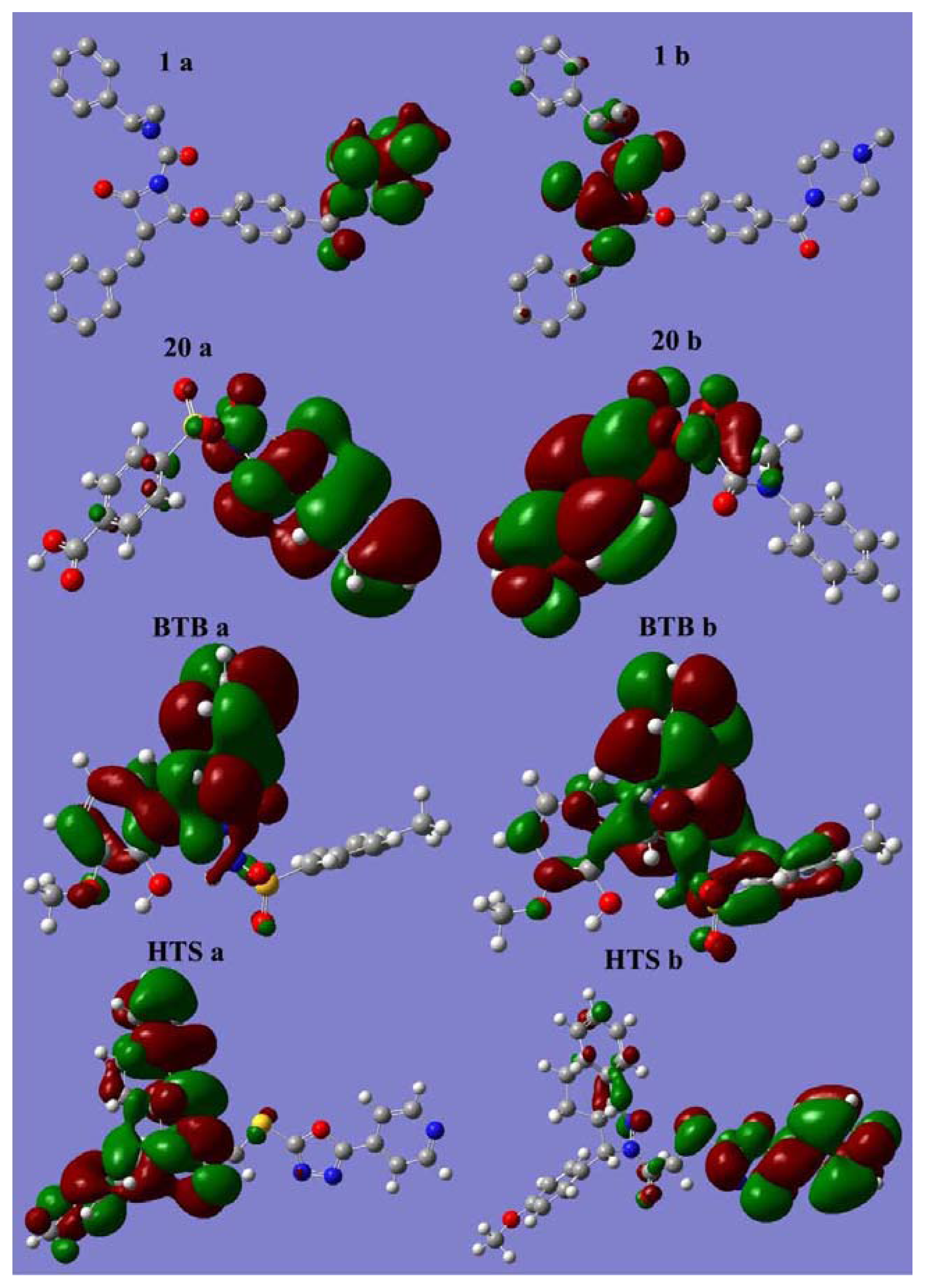
2.5.1. Analysis of Orbital Energies
2.5.2. Molecular Electrostatic Potential (MESP) Profiles
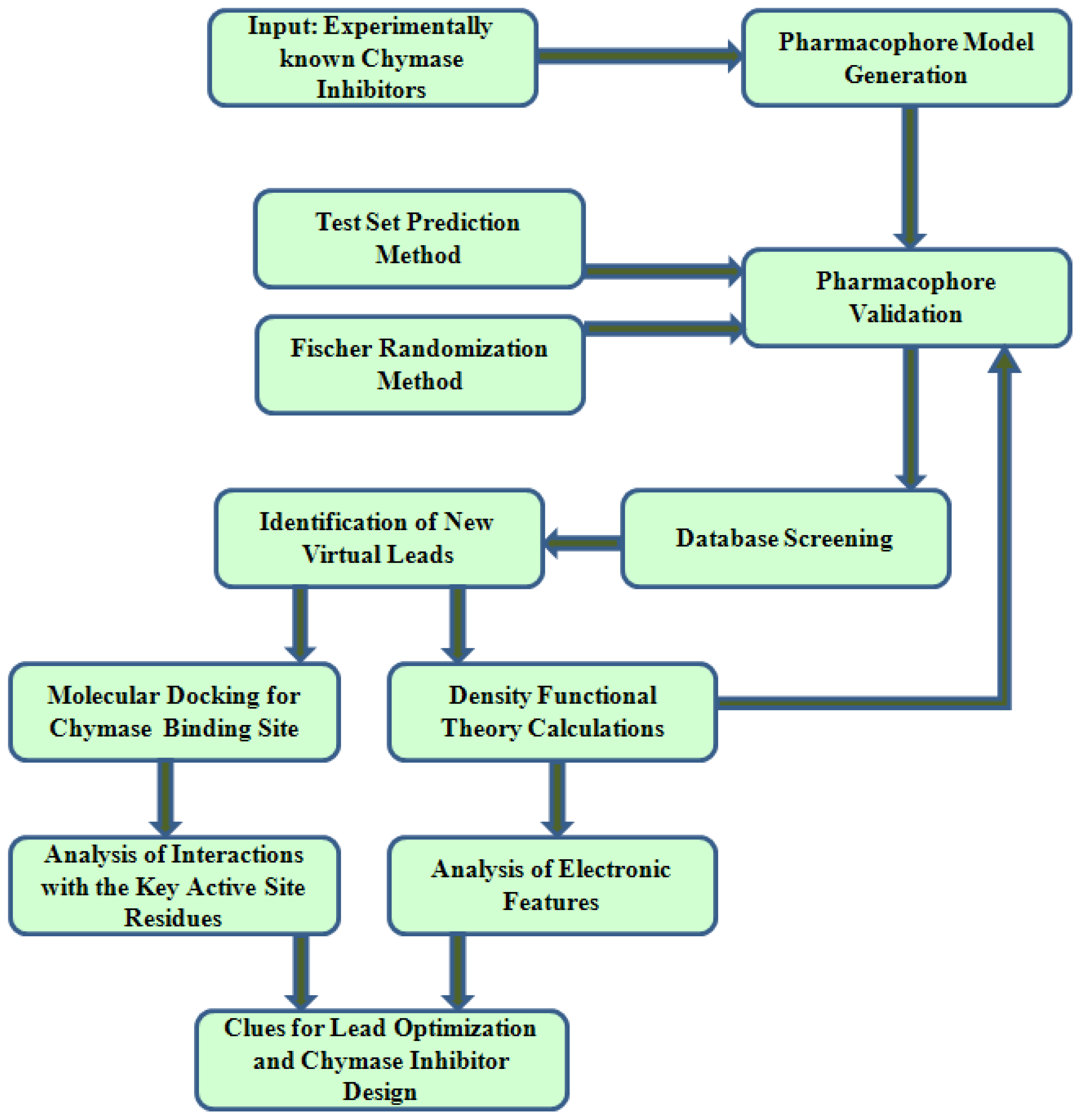
3. Materials and Methods
3.1. Pharmacophore Modeling
3.1.1. Selection of Training Set Compounds and Diverse Conformation Generation
3.1.2. Pharmacophore Model Generation
3.1.3. Pharmacophore Model Validation and Database Searching
3.2. Molecular Docking
3.3. Density Functional Theory (DFT) Calculations
3.3.1. Data Set of DFT Study
3.3.2. Calculation of Molecular Electrostatic Potential (MESP)
4. Conclusion
Acknowledgements
References
- Pierdomenico, S.D.; Nicola, D.M.; Esposito, A.L.; Mascio, D.R.; Ballone, E.; Lapenna, D.; Cuccurullo, F. Prognostic value of different indices of blood pressure variability in hypertensive patients. Am. J. Hypertens 2009, 22, 842–847. [Google Scholar]
- Flack, J.M.; Peters, R.; Shafi, T.; Alrefai, H.; Nasser, S.A.; Crook, E. Prevention of Hypertension and Its Complications: Theoretical Basis and Guidelines for Treatment. J. Am. Soc. Nephrol 2003, 14, S92–S98. [Google Scholar]
- Schmieder, R.E. Mechanisms for the Clinical Benefits of Angiotensin II Receptor Blockers. Am. J. Hypertens 2005, 18, 720–730. [Google Scholar]
- Caughey, G.H.; Raymond, W.W.; Wolters, P.J. Angiotensin II generation by mast cell α- and β-chymases. Biochim. Biophys. Acta 2000, 1480, 245–257. [Google Scholar]
- Amano, N.; Takai, S.; Jin, D.; Ueda, K.; Miyazaki, M. Possible roles of mast cell-derived chymase for skin rejuvenation. Lasers. Med. Sci 2009, 24, 223–229. [Google Scholar]
- Takai, S.; Miyazaki, M. Inhibition of transforming growth factor-β activation is a novel effect of chymase inactivation. Lett. Drug Des. Discov 2005, 2, 19–22. [Google Scholar]
- Omoto, Y.; Tokime, K.; Yamanaka, K.; Habe, K.; Morioka, T.; Kurokawa, I.; Tsutsui, H.; Yamanishi, K.; Nakanishi, K.; Mizutani, H. Human mast cell chymase cleaves Pro-IL-18 and generates a novel and biologically active IL-18 fragment. J. Immunol 2006, 177, 8315–8319. [Google Scholar]
- Huang, X.R.; Chen, W.Y.; Truong, L.D.; Lan, H.Y. Chymase is upregulated in diabetic nephropathy: implications for an alternative pathway of angiotensin II-mediated diabetic renal and vascular disease. J. Am. Soc. Nephrol 2003, 14, 1738–1747. [Google Scholar]
- Garavilla, L.D.; Greco, M.N.; Sukumar, N.; Chen, Z.; Pineda, A.O.; Mathews, F.S.; Cera, E.D.; Giardino, E.C.; Wells, G.I.; Haertlein, B.J.; et al. A novel, potent dual inhibitor of the leukocyte proteases cathepsin g and chymase. J. Biol. Chem 2005, 280, 18001–18007. [Google Scholar]
- Powers, J.C.; Tanaka, T.; Harper, J.W.; Minematsu, Y.; Baker, L.; Lincoln, D.; Crumley, K.V. Mammalian chymotrypsin-like enzymes. Comparative reactivities of rat mast cell proteases, human and dog skin chymases, and human cathepsin G with peptide 4-nitroanilide substrates and with peptide chloromethyl ketone and sulfonyl fluoride inhibitors. Biochemistry 1985, 24, 2048–2058. [Google Scholar]
- Burzycki, T.A.; Hoover, K.W.; Thomsen, D.L.; Sneddon, S.F.; Rauch, A.L.; Hoover, D.J. Beyond ACE Inhibitors and Calcium Antagonists. Presented at IBC Conference on Developmental Therapy for Hypertension, Philadelphia, PA, USA, 25 January 1993.
- Bastos, M.; Maeji, N.J.; Abeles, R.H. Inhibitors of human heart chymase based on a peptide library. Proc. Natl. Acad. Sci. USA 1995, 92, 6738–6742. [Google Scholar]
- Greco, M.N.; Hawkins, M.J.; Powell, E.T.; Almond, H.R.; Garavilla, L.; Hall, J.; Minor, L.K.; Wang, Y.; Corcoran, T.W.; Cera, E.D.; et al. Discovery of potent, selective, orally active, nonpeptide inhibitors of human mast cell chymase. J. Med. Chem 2007, 50, 1727–1730. [Google Scholar]
- Iijima, K.; Katada, J.; Yasuda, E.; Uno, I.; Hayashi, Y. N-[2,2-Dimethyl-3-(N-(4- Cyanobenzoyl)Amino)Nonanoyl]-L-Phenylalanine ethyl ester as a stable ester-type inhibitor of chymotrypsin-like serine proteases: Structural requirements for potent inhibition of A-chymotrypsin. J. Med. Chem 1999, 42, 312–323. [Google Scholar]
- Aoyama, Y.; Uenaka, M.; Kii, M.; Tanaka, M.; Konoike, T.; Hayasaki-Kajiwara, Y.; Naya, N.; Nakajima, M. Design, synthesis and pharmacological evaluation of 3-Benzylazetidine-2-one-based human chymase inhibitors. Bioorg. Med. Chem 2001, 9, 3065–3075. [Google Scholar]
- Aoyama, Y.; Uenaka, M.; Konoike, T.; Hayasaki-Kajiwara, Y.; Naya, N.; Nakajima, M. Inhibition of serine proteases: Activity of 1,3-Diazetidine-2,4-diones. Bioorg. Med. Chem. Lett 2001, 11, 1691–1694. [Google Scholar]
- Aoyama, Y.; Konoike, T.; Kanda, A.; Naya, N.; Nakajima, M. Total synthesis of human chymase inhibitor methyllinderone and structure-activity relationships of its derivatives. Bioorg. Med. Chem. Lett 2001, 11, 1695–1697. [Google Scholar]
- Hayashi, Y.; Iijima, K.; Katada, J.; Kiso, Y. Structure-activity relationship studies of chloromethyl ketone derivatives for selective human chymase inhibitors. Bioorg. Med. Chem. Lett 2000, 10, 199–201. [Google Scholar]
- Aoyama, Y.; Uenaka, M.; Konoike, T.; Iso, Y.; Nishitani, Y.; Kanda, A.; Naya, N.; Nakajima, M. Synthesis and structure-activity relationships of a new class of 1-oxacephem-based human chymase inhibitors. Bioorg. Med. Chem. Lett 2000, 10, 2397–2401. [Google Scholar]
- Niwata, S.; Fukami, H.; Sumida, M.; Ito, A.; Kakutani, S.; Saitoh, M.; Suzuki, K.; Imoto, M.; Shibata, H.; Imajo, S.; et al. Substituted 3-(Phenylsulfonyl)-1-phenylimidazolidine-2,4-dione derivatives as novel nonpeptide inhibitors of human heart chymase. J. Med. Chem 1997, 40, 2156–2163. [Google Scholar]
- He, Z.; Zhang, J.; Shi, X.H.; Hu, L.L.; Kong, X; Cai, Y.D.; Chou, K.C. Predicting drug-target interaction networks based on functional groups and biological features. PLoS One 2010, 5, e9603. [Google Scholar]
- Hu, L.L.; Huang, T.; Cai, Y.D.; Chou, K.C. Prediction of body fluids where proteins are secreted into based on protein interaction network. PLoS One 2011, 6, e22989. [Google Scholar]
- Chou, K.C. A vectorized sequence-coupling model for predicting HIV protease cleavage sites in proteins. J Biol. Chem 1993, 268, 16938–16948. [Google Scholar]
- Chou, K.C. Review: Prediction of HIV protease cleavage sites in proteins. Anal. Biochem 1996, 233, 1–14. [Google Scholar]
- Huang, T.; Shi, X.H.; Wang, P.; He, Z.; Feng, K.Y.; Hu, L.; Kong, X.; Li, Y.X.; Cai, Y.D.; Chou, K.C. Analysis and prediction of the metabolic stability of proteins based on their sequential features, subcellular locations and interaction networks. PLoS One 2010, 5, e10972. [Google Scholar]
- Chou, K.C.; Shen, H.B. Signal-CF: A subsite-coupled and window-fusing approach for predicting signal peptides. Biochem. Biophys. Res. Commun 2007, 357, 633–640. [Google Scholar]
- Lin, W.Z.; Fang, J.A.; Xiao, X.; Chou, K.C. iDNA-Prot: Identification of DNA binding proteins using random forest with grey model. PLoS One 2011, 6, e24756. [Google Scholar]
- Chen, L.; Feng, K.Y.; Cai, Y.D.; Chou, K.C.; Li, H.P. Predicting the network of substrate-enzyme-product triads by combining compound similarity and functional domain composition. BMC Bioinformatics 2010, 11, 293. [Google Scholar]
- Chou, K.C.; Shen, H.B. Cell-PLoc 2.0: An improved package of web-servers for predicting subcellular localization of proteins in various organisms. Nat. Sci 2010, 2, 1090–1103. [Google Scholar]
- Chou, K.C.; Wu, Z.C.; Xiao, X. iLoc-Euk: A multi-label classifier for predicting the subcellular localization of singleplex and multiplex eukaryotic proteins. PLoS One 2011, 6, e18258. [Google Scholar]
- Chou, K.C.; Shen, H.B. ProtIdent: A web server for identifying proteases and their types by fusing functional domain and sequential evolution information. Biochem. Biophys. Res. Commun 2008, 376, 321–325. [Google Scholar]
- Wang, P.; Hu, L.; Liu, G.; Jiang, N.; Chen, X.; Xu, J.; Zheng, W.; Li, L.; Tan, M.; Chen, Z.; et al. Prediction of antimicrobial peptides based on sequence alignment and feature selection methods. PLoS One 2011, 6, e18476. [Google Scholar]
- Chou, K.C.; Shen, H.B. MemType-2L: A Web server for predicting membrane proteins and their types by incorporating evolution information through Pse-PSSM. Biochem. Biophys. Res. Commun 2007, 360, 339–345. [Google Scholar]
- Xiao, X.; Wang, P.; Chou, K.C. GPCR-2L: Predicting G protein-coupled receptors and their types by hybridizing two different modes of pseudo amino acid compositions. Mol. Biosyst 2011, 7, 911–919. [Google Scholar]
- Wang, P.; Xiao, X.; Chou, K.C. NR-2L: A two-level predictor for identifying nuclear receptor subfamilies based on sequence-derived features. PLoS One 2011, 6, e23505. [Google Scholar]
- Xiao, X.; Wu, Z.C.; Chou, K.C. A multi-label classifier for predicting the subcellular localization of gram-negative bacterial proteins with both single and multiple sites. PLoS One 2011, 6, e20592. [Google Scholar]
- Huang, T.; Niu, S.; Xu, Z.; Huang, Y.; Kong, X.; Cai, Y.D.; Chou, K.C. Predicting transcriptional activity of multiple site p53 mutants based on hybrid properties. PLoS One 2011, 6, e22940. [Google Scholar]
- Chou, K.C. Review: Structural bioinformatics and its impact to biomedical science. Curr. Med. Chem 2004, 11, 2105–2134. [Google Scholar]
- Narkhede, S.S.; Degani, M.S. Pharmacophore refinement and 3D-QSAR studies of histamine H3 antagonists. QSAR Comb. Sci 2007, 26, 744–753. [Google Scholar]
- Geppert, H.; Vogt, M.; Bajorath, J. Current trends in ligandbased virtual screening: Molecular representations, data mining methods, new application areas, and performance evaluation. J. Chem. Inf. Model 2010, 50, 205–216. [Google Scholar]
- Du, Q.S.; Mezey, P.G.; Chou, K.C. Heuristic molecular lipophilicity potential (HMLP): A 2D-QSAR study to LADH of molecular family pyrazole and derivatives. J. Comput. Chem 2005, 26, 461–470. [Google Scholar]
- Du, Q.S.; Huang, R.B.; Wei, Y.T.; Du, L.Q.; Chou, K.C. multiple field three dimensional quantitative structure-activity relationship (MF-3D-QSAR). J. Comput. Chem 2008, 29, 211–219. [Google Scholar]
- Du, Q.S.; Huang, R.B.; Chou, K.C. Review: Recent advances in QSAR and their applications in predicting the activities of chemical molecules, peptides and proteins for drug design. Curr. Protein Pept. Sci 2008, 9, 248–259. [Google Scholar]
- Du, Q.S.; Huang, R.B.; Wei, Y.T.; Pang, Z.W.; Du, L.Q.; Chou, K.C. Fragment-Based quantitative structure-activity relationship (FB-QSAR) for fragment-based drug design. J. Comput. Chem 2009, 30, 295–304. [Google Scholar]
- Prado-Prado, F.J.; Gonzalez-Diaz, H.; de la Vega, O.M.; Ubeira, F.M.; Chou, K.C. Unified QSAR approach to antimicrobials. Part 3: First multi-tasking QSAR model for Input-Coded prediction, structural back-projection, and complex networks clustering of antiprotozoal compounds. Bioorg. Med. Chem 2008, 16, 5871–5880. [Google Scholar]
- Wei, H.; Wang, C.H.; Du, Q.S.; Meng, J.; Chou, K.C. Investigation into adamantane-based M2 inhibitors with FB-QSAR. Med. Chem 2009, 5, 305–317. [Google Scholar]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug. Des 2006, 67, 370–372. [Google Scholar]
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.L. Molecular docking and pharmacophore filtering in the discovery of dual-inhibitors for human leukotriene A4 hydrolase and leukotriene C4 synthase. J. Chem. Inf. Model 2011, 51, 33–44. [Google Scholar]
- Bag, S.; Tawari, N.R.; Degani, M.S. Insight into inhibitory activity of mycobacterial dihydrofolate reductase inhibitors by in-silico molecular modeling approaches. QSAR Comb. Sci 2009, 28, 296–311. [Google Scholar]
- Sirois, S.; Wei, D.Q.; Du, Q.S.; Chou, K.C. Virtual screening for SARS-CoV protease based on KZ7088 pharmacophore points. J. Chem. Inf. Comput. Sci 2004, 44, 1111–1122. [Google Scholar]
- Chou, K.C.; Wei, D.Q.; Du, Q.S.; Sirois, S.; Zhong, W.Z. Review: Progress in computational approach to drug development against SARS. Curr. Med. Chem 2006, 13, 3263–3270. [Google Scholar]
- Wan, J.; Zhang, L.; Yang, G.; Zhan, C. Quantitative structure-activity relationship for cyclic imide derivatives of protoporphyrinogen oxidase inhibitors: A study of quantum chemical descriptors from density functional theory. J. Chem. Inf. Comput. Sci 2004, 44, 2099–2105. [Google Scholar]
- Waller, C.L.; Marshall, G.R. Three-Dimensional quantitative structure-activity relationship of angiotesin-converting enzyme and thermolysin inhibitors. II. A comparison of CoMFA models incorporating molecular orbital fields and desolvation free energies based on active-analog and complementary-receptor-field alignment rules. J. Med. Chem 1993, 36, 2390–2403. [Google Scholar]
- Navajasra, C.; POSO, A.; Tuppurainenb, T.; Gynthe, J. Comparative molecular field analysis (CoMFA) of NIX compounds using different semi-empirical methods: LUMO field and its correlation with mutagenic activity. Quant. Struct. Act. Relat 1996, 5, 189–193. [Google Scholar]
- Iijima, K.; Katada, J.; Hayashi, Y. Symmetrical anhydride-type serine protease inhibitors: Structure-activity relationship studies of human chymase inhibitors. Bioorg. Med. Chem. Lett 1999, 9, 413–418. [Google Scholar]
- Koide, Y.; Tatsui, A.; Hasegawa, T.; Murakami, A.; Satoh, S.; Yamada, H.; Kazayama, S.; Takahashi, A. Identification of a stable chymase inhibitor using a pharmacophore-based database search. Bioorg. Med. Chem. Lett 2003, 13, 25–29. [Google Scholar]
- Gupta, A.K.; Chakroborty, S.; Srivastava, K.; Puri, S.K.; Saxena, A.K. Pharmacophore modeling of substituted 1,2,4-Trioxanes for quantitative prediction of their antimalarial activity. J. Chem. Inf. Model 2010, 50, 1510–1520. [Google Scholar]
- Kim, H.J.; Doddareddy, M.R.; Choo, H.; Cho, Y.S.; No, K.T.; Park, W.; Pae, A.N. New serotonin 5-HT6 ligands from common feature pharmacophore hypotheses. J. Chem. Inf. Model 2008, 48, 197–206. [Google Scholar]
- Nayana, R.S.; Bommisetty, S.K.; Singh, K.; Bairy, S.K.; Nunna, S.; Pramod, A.; Muttineni, R. Structural analysis of carboline derivatives as inhibitors of MAPKAP K2 using 3D QSAR and docking studies. J. Chem. Inf. Model 2009, 49, 53–67. [Google Scholar]
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.L. Pharmacophor-based virtual screening and bayesian model for the identification of potential human leukotriene A4 hydrolase inhibitors. Eur. J. Med. Chem 2011, 46, 1593–1603. [Google Scholar]
- Ruiz, J.; Pérez, C.; Pouplana, R. QSAR Study of dual cyclooxygenase and 5-Lipoxygenase inhibitors 2,6-di-tert-Butylphenol derivatives. Bioorg. Med. Chem 2003, 11, 4207–4216. [Google Scholar]
- Kenny, P.W. Hydrogen bonding, electrostatic potential, and molecular design. J. Chem. Inf. Model 2009, 49, 1234–1244. [Google Scholar]
- Nam, K.; Gao, J.L.; York, D.M. Electrostatic interactions in the hairpin ribozyme account for the majority of the rate acceleration without chemical participation by nucleobases. RNA 2008, 14, 1501–1507. [Google Scholar]
- Daga, P.R.; Doerksen, R.J. Stereoelectronic properties of spiroquinazolinones in differential PDE7 inhibitory activity. J. Comput. Chem 2008, 29, 1945–1954. [Google Scholar]
- Dehez, F.; Pebay-Peyroula, E.; Chipot, C. Binding of ADP in the mitochondrial ADP/ATP carrier is driven by an electrostatic funnel. J. Am. Chem. Soc 2008, 130, 12725–12733. [Google Scholar]
- Greco, M.; Hawkins, M.; Garavilla, L.D.; Powell, E.; Maryanoff, B.E. Novel inhibitors of chymase. U.S. Patent 20,080,096,844, 28 April 2008. [Google Scholar]
- Hawkins, M.J.; Greco, M.N.; Powell, E.; Garavilla, L.D.; Maryanoff, B.E. Novel inhibitors of chymase. U.S. Patent 2009. [Google Scholar]
- Aoyama, Y.; Uenaka, M.; Konoike, T.; Iso, Y.; Nishitani, Y.; Kanda, A.; Naya, N.; Nakajima, M. 1-Oxacephem-Based human chymase inhibitors: Discovery of stable inhibitors in human plasma. Bioorg. Med. Chem. Lett 2000, 10, 2403–2406. [Google Scholar]
- Chou, K.C.; Zhang, C.T. Review: Prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol 1995, 30, 275–349. [Google Scholar]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review). J. Theor. Biol 2011, 273, 236–247. [Google Scholar]
- Gu, Q.; Ding, Y.S.; Zhang, T.L. Prediction of G-Protein-Coupled receptor classes in low homology using Chou’s pseudo amino acid composition with approximate entropy and hydrophobicity patterns. Protein Pept. Lett 2010, 17, 559–567. [Google Scholar]
- Mohabatkar, H. Prediction of cyclin proteins using Chou’s pseudo amino acid composition. Protein Pept. Lett 2010, 17, 1207–1214. [Google Scholar]
- Chou, K.C.; Shen, H.B. Plant-mPLoc: A top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS One 2010, 5, e11335. [Google Scholar]
- Zeng, Y.H.; Guo, Y.Z.; Xiao, R.Q.; Yang, L.; Yu, L.Z.; Li, M.L. Using the augmented Chou’s pseudo amino acid composition for predicting protein submitochondria locations based on auto covariance approach. J. Theor. Biol 2009, 259, 366–372. [Google Scholar]
- Esmaeili, M.; Mohabatkar, H.; Mohsenzadeh, S. Using the concept of Chou’s pseudo amino acid composition for risk type prediction of human papillomaviruses. J. Theor. Biol 2010, 263, 203–209. [Google Scholar]
- Chou, K.C.; Wei, D.Q.; Zhong, W.Z. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Comm 2003, 308, 148–151. [Google Scholar]
- Du, Q.S.; Wang, S.; Wei, D.Q.; Sirois, S.; Chou, K.C. Molecular modelling and chemical modification for finding peptide inhibitor against SARS CoV Mpro. Anal. Biochem 2005, 337, 262–270. [Google Scholar]
- Huang, R.B.; Du, Q.S.; Wang, C.H.; Chou, K.C. An in-depth analysis of the biological functional studies based on the NMR M2 channel structure of influenza A virus. Biochem. Biophys. Res. Commun 2008, 377, 1243–1247. [Google Scholar]
- Du, Q.S.; Huang, R.B.; Wang, C.H.; Li, X.M.; Chou, K.C. Energetic analysis of the two controversial drug binding sites of the M2 proton channel in influenza A virus. J. Theor. Biol 2009, 259, 159–164. [Google Scholar]
- Du, Q.S.; Huang, R.B.; Wang, S.Q.; Chou, K.C. Designing inhibitors of M2 proton channel against H1N1 swine influenza virus. PLoS One 2010, 5, e9388. [Google Scholar]
- Wang, S.Q.; Du, Q.S.; Huang, R.B.; Zhang, D.W.; Chou, K.C. Insights from investigating the interaction of oseltamivir (Tamiflu) with neuraminidase of the 2009 H1N1 swine flu virus. Biochem. Biophys. Res. Commun 2009, 386, 432–436. [Google Scholar]
- Cai, L.; Wang, Y.; Wang, J.F.; Chou, K.C. Identification of proteins interacting with human SP110 during the process of viral infections. Med. Chem 2011, 7, 121–126. [Google Scholar]
- Liao, Q.H.; Gao, Q.Z.; Wei, J.; Chou, K.C. Docking and molecular dynamics study on the inhibitory activity of novel inhibitors on epidermal growth factor receptor (EGFR). Med. Chem 2011, 7, 24–31. [Google Scholar]
- Liu, X.Y.; Wang, S.Q.; Xu, W.R.; Tang, L.D.; Wang, R.L.; Chou, K.C. Docking and molecular dynamics simulations of peroxisome proliferator activated receptors interacting with pan agonist sodelglitazar. Protein Pept. Lett 2011, 18, 1021–1027. [Google Scholar]
- Raman, E.P.; Yu, W.; Guvench, O.; MacKerell, A.D. Reproducing crystal binding Modes of Ligand Functional Groups Using Site-Identification by Ligand Competitive Saturation (SILCS) Simulations. J. Chem. Inf. Model 2011, 51, 877–896. [Google Scholar]
- Lie, M.A.; Thomsen, R.; Pedersen, C.N.S.; Schiøtt, B.; Christensen, M.H. Molecular docking with ligand attached water molecules. J. Chem. Inf. Model 2011, 51, 909–917. [Google Scholar]
- Shah, F.; Mukherjee, P.; Gut, J.; Legac, J.; Rosenthal, P.J.; Tekwani, B.L.; Avery, M.A. Identification of novel malarial cysteine protease inhibitors using structure-based virtual screening of a focused cysteine protease inhibitor library. J. Chem. Inf. Model 2011, 51, 852–864. [Google Scholar]
- Díaz, L.; Bujons, J.; Delgado, A.; Guti, H.; Åqvist, J. Computational prediction of structure-activity relationships for the binding of aminocyclitols to β-Glucocerebrosidase. J. Chem. Inf. Model 2011, 51, 601–611. [Google Scholar]
- Helal, M.A.; Chittiboyina, A.G.; Avery, M.A. New insights into the binding mode of melanin concentrating hormone receptor-1 antagonists: Homology modeling and explicit membrane molecular dynamics simulation study. J. Chem. Inf. Model 2011, 51, 635–646. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev 1988, B37, 785–789. [Google Scholar]
- Chou, K.C.; Shen, H.B. Review: Recent advances in developing web-servers for predicting protein attributes. Nat. Sci 2009, 2, 63–92. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hypothesis | Total cost | ΔCost a | RMSD (Å) | Correlation (r) | Features |
|---|---|---|---|---|---|
| 1 | 89.663 | 92.703 | 1.176 | 0.942 | HBA, HBA, HY-AR, HY-AR, HY-AR |
| 2 | 91.454 | 90.912 | 1.25 | 0.934 | HBA, HBA, HY-AR, HY-AR, HY-AR |
| 3 | 94.811 | 87.555 | 1.348 | 0.924 | HBA, HY-AR, HY-AR, RA |
| 4 | 95.086 | 87.28 | 1.388 | 0.919 | HBA, HBA, HY-AR, HY-AR, HY-AR |
| 5 | 95.379 | 86.987 | 1.387 | 0.919 | HBA, HY-AR, RA, RA |
| 6 | 95.458 | 86.908 | 1.396 | 0.918 | HBA, HBA, HY-AR, HY-AR, HY-AR |
| 7 | 95.656 | 86.71 | 1.406 | 0.916 | HBA, HBA, HY-AR, HY-AR, HY-AR |
| 8 | 95.855 | 86.511 | 1.409 | 0.916 | HBA, HBA, HY-AR, HY-AR, HY-AR |
| 9 | 95.538 | 86.828 | 1.411 | 0.916 | HBD, HY-AR, RA, RA |
| 10 | 96.124 | 86.242 | 1.421 | 0.915 | HBA, HBA, HY-AR, HY-AR, HY-AR |
| Compound | Experimental activity (nM) | Estimated activity | Error | Activity scale a | Estimated activity scale |
|---|---|---|---|---|---|
| 1 | 0.46 | 0.27 | −1.7 | ++++ | ++++ |
| 2 | 2.1 | 1.9 | −1.1 | ++++ | ++++ |
| 3 | 11 | 16 | 1.4 | ++++ | ++++ |
| 4 | 20 | 49 | 2.5 | +++ | +++ |
| 5 | 57 | 50 | −1.1 | +++ | +++ |
| 6 | 91 | 80 | −1.1 | +++ | +++ |
| 7 | 110 | 650 | 5.9 | +++ | ++ |
| 8 | 170 | 170 | 1 | +++ | +++ |
| 9 | 220 | 720 | 3.3 | ++ | ++ |
| 10 | 250 | 650 | 2.6 | ++ | ++ |
| 11 | 360 | 710 | 2 | ++ | ++ |
| 12 | 410 | 760 | 1.9 | ++ | ++ |
| 13 | 730 | 660 | −1.1 | ++ | ++ |
| 14 | 900 | 610 | −1.5 | ++ | ++ |
| 15 | 1300 | 930 | −1.4 | ++ | ++ |
| 16 | 1600 | 650 | −2.6 | ++ | ++ |
| 17 | 2200 | 860 | −2.6 | + | ++ |
| 18 | 3000 | 850 | −3.5 | + | ++ |
| 19 | 3500 | 830 | −4.2 | + | ++ |
| 20 | 5900 | 4800 | −1.2 | + | + |
| Name | IC50 nM
| Error c | Activity scale d | Name | IC50 nM
| Error | Activity scale
| ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Exp. a | Est. b | Exp. | Est. | Exp. | Est. | Exp. | Est. | ||||
| 21 | 0.5 | 0.48 | −1.0 | ++++ | ++++ | 70 | 500 | 491.7 | −1.0 | ++ | ++ |
| 22 | 1 | 1.00 | −1.0 | ++++ | ++++ | 71 | 550 | 592.4 | 1.0 | ++ | ++ |
| 23 | 2 | 65.84 | 32.9 | ++++ | +++ | 72 | 580 | 579.668 | −1.0 | ++ | ++ |
| 24 | 3.1 | 3.16 | 1.0 | ++++ | ++++ | 73 | 600 | 601.005 | 1.0 | ++ | ++ |
| 25 | 5 | 12.84 | 2.5 | ++++ | ++++ | 74 | 609 | 609.043 | 1.0 | ++ | ++ |
| 26 | 5.6 | 16.38 | 2.9 | ++++ | ++++ | 75 | 700 | 1545.30 | 2.2 | ++ | ++ |
| 27 | 6 | 6.38 | 1.0 | ++++ | ++++ | 76 | 700 | 697.894 | −1.0 | ++ | ++ |
| 28 | 13 | 15.57 | 1.1 | ++++ | ++++ | 77 | 710 | 709.97 | −1.0 | ++ | ++ |
| 29 | 19 | 19.30 | 1.0 | ++++ | ++++ | 78 | 780 | 779.896 | −1.0 | ++ | ++ |
| 30 | 20 | 524.55 | 26.2 | +++ | ++ | 79 | 800 | 798.863 | −1.0 | ++ | ++ |
| 31 | 24 | 23.99 | −1.0 | +++ | +++ | 80 | 860 | 860.59 | 1.0 | ++ | ++ |
| 32 | 26 | 26.03 | 1.0 | +++ | +++ | 81 | 890 | 889.222 | −1.0 | ++ | ++ |
| 33 | 27 | 27.18 | 1.0 | +++ | +++ | 82 | 890 | 889.127 | −1.0 | ++ | ++ |
| 34 | 30 | 526.07 | 17.5 | +++ | ++ | 83 | 1100 | 1103.48 | 1.0 | ++ | ++ |
| 35 | 32 | 42.12 | 1.3 | +++ | +++ | 84 | 1200 | 1209.95 | 1.0 | ++ | ++ |
| 36 | 37 | 37.00 | −1.0 | +++ | +++ | 85 | 1200 | 1193.16 | −1.0 | ++ | ++ |
| 37 | 37 | 35.56 | −1.0 | +++ | +++ | 86 | 1400 | 1399.23 | −1.0 | ++ | ++ |
| 38 | 40 | 10.60 | −3.7 | +++ | +++ | 87 | 1400 | 552.461 | −2.5 | ++ | ++ |
| 39 | 50 | 50.85 | 1.0 | +++ | +++ | 88 | 1650 | 1645.71 | −1.0 | ++ | ++ |
| 40 | 50 | 50.17 | 1.0 | +++ | +++ | 89 | 1650 | 551.849 | −2.9 | ++ | ++ |
| 41 | 58 | 56.72 | −1.0 | +++ | +++ | 90 | 1700 | 524.94 | −3.2 | ++ | ++ |
| 42 | 70 | 71.11 | 1.0 | +++ | +++ | 91 | 1800 | 1821.04 | 1.0 | ++ | ++ |
| 43 | 70 | 70.25 | 1.0 | +++ | +++ | 92 | 1800 | 1776.13 | −1.0 | ++ | ++ |
| 44 | 77 | 76.65 | −1.0 | +++ | +++ | 93 | 1800 | 1530.73 | −1.1 | ++ | ++ |
| 45 | 82 | 408.48 | 4.9 | +++ | ++ | 94 | 1900 | 1901.07 | 1.0 | ++ | ++ |
| 46 | 109 | 517.17 | 4.7 | +++ | ++ | 95 | 1900 | 1876.01 | −1.0 | ++ | ++ |
| 47 | 110 | 532.07 | 4.8 | +++ | ++ | 96 | 2040 | 2022.46 | −1.0 | + | + |
| 48 | 130 | 518.66 | 3.9 | +++ | ++ | 97 | 2100 | 2095.67 | −1.0 | + | + |
| 49 | 130 | 60.13 | −2.1 | +++ | +++ | 98 | 2200 | 1991.66 | −1.1 | + | ++ |
| 50 | 140 | 140.46 | 1.0 | +++ | +++ | 99 | 2400 | 2386.02 | −1.0 | + | + |
| 51 | 150 | 514.80 | 3.4 | +++ | ++ | 100 | 2500 | 2594.48 | 1.0 | + | + |
| 52 | 150 | 149.81 | −1.0 | +++ | +++ | 101 | 2500 | 2373.56 | −1.0 | + | + |
| 53 | 170 | 531.86 | 3.1 | +++ | ++ | 102 | 2600 | 2653.51 | 1.0 | + | + |
| 54 | 170 | 520.35 | 3.0 | +++ | ++ | 103 | 2600 | 1980.42 | −1.3 | + | ++ |
| 55 | 190 | 92.38 | −2.0 | +++ | +++ | 104 | 2700 | 2668.93 | −1.0 | + | + |
| 56 | 220 | 519.50 | 2.3 | ++ | ++ | 105 | 3000 | 2980.04 | −1.0 | + | + |
| 57 | 250 | 249.79 | −1.0 | ++ | ++ | 106 | 3100 | 3021.26 | −1.0 | + | + |
| 58 | 270 | 40.16 | −6.7 | ++ | ++ | 107 | 3200 | 3351.54 | 1.0 | + | + |
| 59 | 300 | 546.21 | 1.8 | ++ | ++ | 108 | 3300 | 3370.61 | 1.0 | + | + |
| 60 | 300 | 521.04 | 1.7 | ++ | ++ | 109 | 3300 | 3333.56 | 1.0 | + | + |
| 61 | 300 | 301.67 | 1.0 | ++ | ++ | 110 | 3300 | 3277.62 | −1.0 | + | + |
| 62 | 300 | 289.88 | −1.0 | ++ | ++ | 111 | 3700 | 4354.53 | 1.1 | + | + |
| 63 | 370 | 580.52 | 1.5 | ++ | ++ | 112 | 4000 | 1316.33 | −3.0 | + | ++ |
| 64 | 380 | 531.50 | 1.3 | ++ | ++ | 113 | 4300 | 4440.06 | 1.0 | + | + |
| 65 | 400 | 517.75 | 1.2 | ++ | ++ | 114 | 4600 | 3529.94 | −1.3 | + | + |
| 66 | 400 | 513.35 | 1.2 | ++ | ++ | 115 | 4700 | 4603.40 | −1.0 | + | + |
| 67 | 430 | 515.92 | 1.2 | ++ | ++ | 116 | 5000 | 5036.50 | 1.0 | + | + |
| 68 | 430 | 515.00 | 1.1 | ++ | ++ | 117 | 5860 | 2779.37 | −2.1 | + | + |
| 69 | 430 | 431.38 | 1.0 | ++ | ++ | ||||||
| Trial No. | Total cost | Fixed cost | RMSD | Correlation (r) |
|---|---|---|---|---|
| Hypo1 | 89.663 | 75.791 | 1.176 | 0.942 |
| Results after randomization | ||||
| 1 | 114.486 | 77.911 | 1.821 | 0.858 |
| 2 | 108.259 | 72.031 | 1.796 | 0.863 |
| 3 | 98.26 | 74.85 | 1.529 | 0.9 |
| 4 | 113.25 | 77.605 | 1.851 | 0.851 |
| 5 | 112.27 | 77.909 | 1.729 | 0.874 |
| 6 | 108.84 | 75.77 | 1.749 | 0.869 |
| 7 | 141.304 | 78.861 | 2.463 | 0.717 |
| 8 | 109.86 | 72 | 1.857 | 0.852 |
| 9 | 112.265 | 77.915 | 1.849 | 0.851 |
| 10 | 113.941 | 77.584 | 1.774 | 0.867 |
| 11 | 101.143 | 72.068 | 1.593 | 0.894 |
| 12 | 116.666 | 74.077 | 1.959 | 0.834 |
| 13 | 114.356 | 69.48 | 1.97 | 0.834 |
| 14 | 98.277 | 77.638 | 1.433 | 0.913 |
| 15 | 108.878 | 72.047 | 1.753 | 0.872 |
| 16 | 117.228 | 78.041 | 1.961 | 0.831 |
| 17 | 102.183 | 78.085 | 1.359 | 0.926 |
| 18 | 113.597 | 77.563 | 1.891 | 0.843 |
| 19 | 106.121 | 74.044 | 1.706 | 0.876 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arooj, M.; Thangapandian, S.; John, S.; Hwang, S.; Park, J.K.; Lee, K.W. 3D QSAR Pharmacophore Modeling, in Silico Screening, and Density Functional Theory (DFT) Approaches for Identification of Human Chymase Inhibitors. Int. J. Mol. Sci. 2011, 12, 9236-9264. https://doi.org/10.3390/ijms12129236
Arooj M, Thangapandian S, John S, Hwang S, Park JK, Lee KW. 3D QSAR Pharmacophore Modeling, in Silico Screening, and Density Functional Theory (DFT) Approaches for Identification of Human Chymase Inhibitors. International Journal of Molecular Sciences. 2011; 12(12):9236-9264. https://doi.org/10.3390/ijms12129236
Chicago/Turabian StyleArooj, Mahreen, Sundarapandian Thangapandian, Shalini John, Swan Hwang, Jong Keun Park, and Keun Woo Lee. 2011. "3D QSAR Pharmacophore Modeling, in Silico Screening, and Density Functional Theory (DFT) Approaches for Identification of Human Chymase Inhibitors" International Journal of Molecular Sciences 12, no. 12: 9236-9264. https://doi.org/10.3390/ijms12129236
APA StyleArooj, M., Thangapandian, S., John, S., Hwang, S., Park, J. K., & Lee, K. W. (2011). 3D QSAR Pharmacophore Modeling, in Silico Screening, and Density Functional Theory (DFT) Approaches for Identification of Human Chymase Inhibitors. International Journal of Molecular Sciences, 12(12), 9236-9264. https://doi.org/10.3390/ijms12129236