Applications of Next-Generation Sequencing Technologies to Diagnostic Virology





Abstract
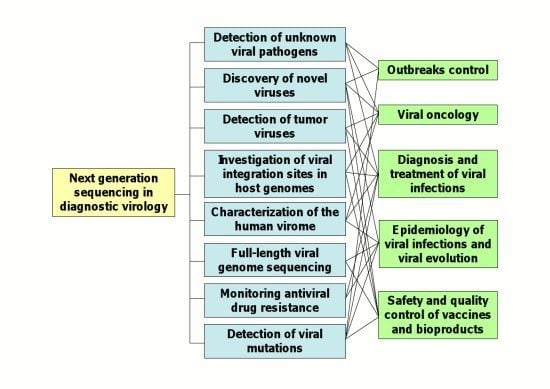
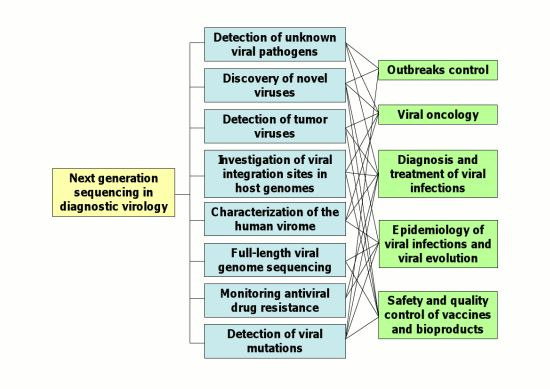
:
1. Introduction
2. Applications of NGS Technologies to Diagnostic Virology
2.1. Detection of Unknown Viral Pathogens and Discovery of Novel Viruses
2.2. Detection of Tumor Viruses
2.3. Characterization of the Human Virome
2.4. Full-Length Viral Genome Sequencing
2.5. Investigation of Viral Genome Variability and Characterization of Viral Quasispecies
2.7. Epidemiology of Viral Infections and Viral Evolution
2.8. Quality Control of Live-Attenuated Viral Vaccines
2.9. Data Analysis Issues
3. Conclusions
Acknowledgments
References
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet 2010, 19, R227–R240. [Google Scholar]
- Ansorge, W.J. Next-generation DNA sequencing techniques. N. Biotechnol 2009, 25, 195–203. [Google Scholar]
- Metzker, M.L. Sequencing technologies - the next generation. Nat. Rev. Genet 2010, 11, 31–46. [Google Scholar]
- Medini, D.; Serruto, D.; Parkhill, J.; Relman, D.A.; Donati, C.; Moxon, R.; Falkow, S.; Rappuoli, R. Microbiology in the post-genomic era. Nat. Rev. Microbiol 2008, 6, 419–430. [Google Scholar]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol 2008, 26, 1135–1145. [Google Scholar]
- MacLean, D.; Jones, J.D.; Studholme, D.J. Application of “next-generation” sequencing technologies to microbial genetics. Nat. Rev. Microbiol 2009, 7, 287–296. [Google Scholar]
- Morens, D.M.; Folkers, G.K.; Fauci, A.S. The challenge of emerging and re-emerging infectious diseases. Nature 2004, 430, 242–249. [Google Scholar]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar]
- Simons, J.N.; Pilot-Matias, T.J.; Leary, T.P.; Dawson, G.J.; Desai, S.M.; Schlauder, G.G.; Muerhoff, A.S.; Erker, J.C.; Buijk, S.L.; Chalmers, M.L.; et al. Identification of two flaviviruslike genomes in the GB hepatitis agent. Proc. Natl. Acad. Sci. USA 1995, 92, 3401–3405. [Google Scholar]
- Nishizawa, T.; Okamoto, H.; Konishi, K.; Yoshizawa, H.; Miyakawa, Y.; Mayumi, M. A novel DNA virus (TTV) associated with elevated transaminase levels in posttransfusion hepatitis of unknown etiology. Biochem. Biophys. Res. Commun 1997, 241, 92–97. [Google Scholar]
- Allander, T.; Tammi, M.T.; Eriksson, M.; Bjerkner, A.; Tiveljung-Lindell, A.; Andersson, B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc. Natl. Acad. Sci. USA 2005, 102, 12891–12896. [Google Scholar]
- Jones, M.S.; Kapoor, A.; Lukashov, V.V.; Simmonds, P.; Hecht, F.; Delwart, E. New DNA viruses identified in patients with acute viral infection syndrome. J. Virol 2005, 79, 8230–8236. [Google Scholar]
- Gaynor, A.M.; Nissen, M.D.; Whiley, D.M.; Mackay, I.M.; Lambert, S.B.; Wu, G.; Brennan, D.C.; Storch, G.A.; Sloots, T.P.; Wang, D. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog 2007, 3. [Google Scholar] [CrossRef]
- Allander, T.; Andreasson, K.; Gupta, S.; Bjerkner, A.; Bogdanovic, G.; Persson, M.A.; Dalianis, T.; Ramqvist, T.; Andersson, B. Identification of a third human polyomavirus. J. Virol 2007, 81, 4130–4136. [Google Scholar]
- Pyrc, K.; Jebbink, M.F.; Berkhout, B.; van der Hoek, L. Detection of new viruses by VIDISCA. Virus discovery based on cDNA-amplified fragment length polymorphism. Methods Mol. Biol 2008, 454, 73–89. [Google Scholar]
- van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.; Wolthers, K.C.; Wertheim-van Dillen, P.M.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med 2004, 10, 368–373. [Google Scholar]
- Wang, D.; Urisman, A.; Liu, Y.T.; Springer, M.; Ksiazek, T.G.; Erdman, D.D.; Mardis, E.R.; Hickenbotham, M.; Magrini, V.; Eldred, J.; et al. Viral discovery and sequence recovery using DNA microarrays. PLoS Biol 2003, 1, 257–260. [Google Scholar]
- Palacios, G.; Quan, P.L.; Jabado, O.J.; Conlan, S.; Hirschberg, D.L.; Liu, Y.; Zhai, J.; Renwick, N.; Hui, J.; Hegyi, H.; et al. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerg. Infect. Dis 2007, 13, 73–81. [Google Scholar]
- MacConaill, L.; Meyerson, M. Adding pathogens by genomic subtraction. Nat. Genet 2008, 40, 380–382. [Google Scholar]
- Tang, P.; Chiu, C. Metagenomics for the discovery of novel human viruses. Future Microbiol 2010, 5, 177–189. [Google Scholar]
- Palacios, G.; Druce, J.; Du, L.; Tran, T.; Birch, C.; Briese, T.; Conlan, S.; Quan, P.L.; Hui, J.; Marshall, J.; et al. A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med 2008, 358, 991–998. [Google Scholar]
- Nakamura, S.; Yang, C.S.; Sakon, N.; Ueda, M.; Tougan, T.; Yamashita, A.; Goto, N.; Takahashi, K.; Yasunaga, T.; Ikuta, K.; et al. Direct metagenomic detection of viral pathogens in nasal and fecal specimens using an unbiased high-throughput sequencing approach. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Quan, P.L.; Wagner, T.A.; Briese, T.; Torgerson, T.R.; Hornig, M.; Tashmukhamedova, A.; Firth, C.; Palacios, G.; Baisre-De-Leon, A.; Paddock, C.D.; et al. Astrovirus encephalitis in boy with X-linked agammaglobulinemia. Emerg. Infect. Dis 2010, 16, 918–925. [Google Scholar]
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; et al. Genetic detection and characterization of Lujo Virus, a new Hemorrhagic Fever-associated Arenavirus from Southern Africa. PLoS Pathog 2009, 5. [Google Scholar] [CrossRef]
- de Vries, M.; Deijs, M.; Canuti, M.; van Schaik, B.D.C.; Faria, N.R.; van de Garde, M.D.B.; Jachimowski, L.C.M.; Jebbink, M.F.; Jakobs, M.; Luyf, A.C.M.; et al. A sensitive assay for virus discovery in respiratory clinical samples. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Dean, F.B.; Nelson, J.R.; Giesler, T.L.; Lasken, R.S. Rapid amplification of plasmid and phage DNA using phi29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res 2001, 11, 1095–1099. [Google Scholar]
- Rector, A.; Tachezy, R.; Van Ranst, M. A sequence-independent strategy for detection and cloning of circular DNA virus genomes by using multiply primed rolling-circle amplification. J. Virol 2004, 78, 4993–4998. [Google Scholar]
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe 2010, 7, 509–515. [Google Scholar]
- van der Meijden, E.; Janssens, R.W.; Lauber, C.; Bouwes Bavinck, J.N.; Gorbalenya, A.E.; Feltkamp, M.C. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog 2010, 6. [Google Scholar] [CrossRef]
- Yongfeng, H.; Fan, Y.; Jie, D.; Jian, Y.; Ting, Z.; Lilian, S.; Jin, Q. Direct pathogen detection from swab samples using a new high-throughput sequencing technology. Clin. Microbiol. Infect 2011, 17, 241–244. [Google Scholar]
- Kuroda, M.; Katano, H.; Nakajima, N.; Tobiume, M.; Ainai, A.; Sekizuka, T.; Hasegawa, H.; Tashiro, M.; Sasaki, Y.; Arakawa, Y.; et al. Characterization of quasispecies of pandemic 2009 influenza A virus (A/H1N1/2009) by de novo sequencing using a next-generation DNA sequencer. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Greninger, A.L.; Chen, E.C.; Sittler, T.; Scheinerman, A.; Roubinian, N.; Yu, G.; Kim, E.; Pillai, D.R.; Guyard, C.; Mazzulli, T.; et al. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Yang, J.; Yang, F.; Ren, L.; Xiong, Z.; Wu, Z.; Dong, J.; Sun, L.; Zhang, T.; Hu, Y.; Du, J.; et al. Unbiased parallel detection of viral pathogens in clinical samples using a metagenomic approach. J. Clin. Microbiol 2011, 49, 3463–3469. [Google Scholar]
- Yozwiak, N.L.; Skewes-Cox, P.; Gordon, A.; Saborio, S.; Kuan, G.; Balmaseda, A.; Ganem, D.; Harris, E.; DeRisi, J.L. Human enterovirus 109: A novel interspecies recombinant enterovirus isolated from a case of acute pediatric respiratory illness in Nicaragua. J. Virol 2010, 84, 9047–9058. [Google Scholar]
- Cheval, J.; Sauvage, V.; Frangeul, L.; Dacheux, L.; Guigon, G.; Dumey, N.; Pariente, K.; Rousseaux, C.; Dorange, F.; Berthet, N.; et al. Evaluation of high throughput sequencing for identifying known and unknown viruses in biological samples. J. Clin. Microbiol 2011, 49, 3268–3275. [Google Scholar]
- Bishop-Lilly, K.A.; Turell, M.J.; Willner, K.M.; Butani, A.; Nolan, N.M.; Lentz, S.M.; Akmal, A.; Mateczun, A.; Brahmbhatt, T.N.; Sozhamannan, S.; et al. Arbovirus detection in insect vectors by rapid, high-throughput pyrosequencing. PLoS Negl. Trop. Dis 2010, 4, e878:1–e878:12. [Google Scholar]
- Wu, Q.; Luo, Y.; Lu, R.; Lau, N.; Lai, E.C.; Li, W.X.; Ding, S.W. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 1606–1611. [Google Scholar]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar]
- Quan, P.L.; Firth, C.; Street, C.; Henriquez, J.A.; Petrosov, A.; Tashmukhamedova, A.; Hutchison, S.K.; Egholm, M.; Osinubi, M.O.; Niezgoda, M.; et al. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. mBio 2010, 1, e00208–e00210. [Google Scholar]
- Weber, G.; Shendure, J.; Tanenbaum, D.M.; Church, G.M.; Meyerson, M. Identification of foreign gene sequences by transcript filtering against the human genome. Nat. Genet 2002, 30, 141–142. [Google Scholar]
- Xu, Y.; Stange-Thomann, N.; Weber, G.; Bo, R.; Dodge, S.; David, R.G.; Foley, K.; Beheshti, J.; Harris, N.L.; Birren, B.; et al. Pathogen discovery from human tissue by sequence-based computational subtraction. Genomics 2003, 81, 329–335. [Google Scholar]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar]
- Sauvage, V.; Foulongne, V.; Cheval, J.; Ar Gouilh, M.; Pariente, K.; Dereure, O.; Manuguerra, J.C.; Richardson, J.; Lecuit, M.; Burguiere, A.; Caro, V.; Eloit, M. Human polyomavirus related to african green monkey lymphotropic polyomavirus. Emerg. Infect. Dis 2011, 17, 1364–1370. [Google Scholar]
- Scuda, N.; Hofmann, J.; Calvignac-Spencer, S.; Ruprecht, K.; Liman, P.; Kuhn, J.; Hengel, H.; Ehlers, B. A novel human polyomavirus closely related to the african green monkey-derived lymphotropic polyomavirus. J. Virol 2011, 85, 4586–4590. [Google Scholar]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar]
- Barzon, L.; Militello, V.; Lavezzo, E.; Franchin, E.; Peta, E.; Squarzon, L.; Trevisan, M.; Pagni, S.; Dal Bello, F.; Toppo, S.; et al. Human papillomavirus genotyping by 454 next generation sequencing technology. J. Clin. Virol 2011, 52, 93–97. [Google Scholar]
- Ekstrom, J.; Bzhalava, D.; Svenback, D.; Forslund, O.; Dillner, J. High throughput sequencing reveals diversity of Human Papillomaviruses in cutaneous lesions. Int. J. Cancer 2011, 129, 2643–2650. [Google Scholar]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest 2008, 118, 3132–3142. [Google Scholar]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest 2008, 118, 3143–3150. [Google Scholar]
- Bushman, F.; Lewinski, M.; Ciuffi, A.; Barr, S.; Leipzig, J.; Hannenhalli, S.; Hoffmann, C. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol 2005, 3, 848–858. [Google Scholar]
- Wang, G.P.; Ciuffi, A.; Leipzig, J.; Berry, C.C.; Bushman, F.D. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res 2007, 17, 1186–1194. [Google Scholar]
- Varas, F.; Stadtfeld, M.; De Andres-Aguayo, L.; Maherali, N.; di Tullio, A.; Pantano, L.; Notredame, C.; Hochedlinger, K.; Graf, T. Fibroblast-derived induced pluripotent stem cells show no common retroviral vector insertions. Stem Cells 2009, 27, 300–306. [Google Scholar]
- Winkler, T.; Cantilena, A.; Metais, J.Y.; Xu, X.L.; Nguyen, A.D.; Borate, B.; Antosiewicz-Bourget, J.E.; Wolfsberg, T.G.; Thomson, J.A.; Dunbar, C.E. No evidence for clonal selection due to lentiviral integration sites in human induced pluripotent stem cells. Stem Cells 2010, 28, 687–694. [Google Scholar]
- Kane, N.M.; Nowrouzi, A.; Mukherjee, S.; Blundell, M.P.; Greig, J.A.; Lee, W.K.; Houslay, M.D.; Milligan, G.; Mountford, J.C.; von Kalle, C.; et al. Lentivirus-mediated reprogramming of somatic cells in the absence of transgenic transcription factors. Mol. Ther 2010, 18, 2139–2145. [Google Scholar]
- Cattoglio, C.; Pellin, D.; Rizzi, E.; Maruggi, G.; Corti, G.; Miselli, F.; Sartori, D.; Guffanti, A.; Di Serio, C.; Ambrosi, A.; et al. High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors. Blood 2010, 116, 5507–5517. [Google Scholar]
- Ciuffi, A.; Barr, S.D. Identification of HIV integration sites in infected host genomic DNA. Methods 2011, 53, 39–46. [Google Scholar]
- Schmidt, M.; Schwarzwaelder, K.; Bartholomae, C.; Zaoui, K.; Ball, C.; Pilz, I.; Braun, S.; Glimm, H.; von Kalle, C. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR). Nat. Methods 2007, 4, 1051–1057. [Google Scholar]
- Gabriel, R.; Eckenberg, R.; Paruzynski, A.; Bartholomae, C.C.; Nowrouzi, A.; Arens, A.; Howe, S.J.; Recchia, A.; Cattoglio, C.; Wang, W.; et al. Comprehensive genomic access to vector integration in clinical gene therapy. Nat. Med 2009, 15, 1431–1436. [Google Scholar]
- Brady, T.; Roth, S.L.; Malani, N.; Wang, G.P.; Berry, C.C.; Leboulch, P.; Hacein-Bey-Abina, S.; Cavazzana-Calvo, M.; Papapetrou, E.P.; Sadelain, M.; et al. A method to sequence and quantify DNA integration for monitoring outcome in gene therapy. Nucleic Acids Res 2011, 39. [Google Scholar] [CrossRef]
- Petrosino, J.F.; Highlander, S.; Luna, R.A.; Gibbs, R.A.; Versalovic, J. Metagenomic pyrosequencing and microbial identification. Clin. Chem 2009, 55, 856–866. [Google Scholar]
- Hamady, M.; Knight, R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res 2009, 19, 1141–1152. [Google Scholar]
- Delwart, E.L. Viral metagenomics. Rev. Med. Virol 2007, 17, 115–131. [Google Scholar]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar]
- Djikeng, A.; Kuzmickas, R.; Anderson, N.G.; Spiro, D.J. Metagenomic analysis of RNA viruses in a fresh water lake. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Lopez-Bueno, A.; Tamames, J.; Velazquez, D.; Moya, A.; Quesada, A.; Alcami, A. High diversity of the viral community from an Antarctic lake. Science 2009, 326, 858–861. [Google Scholar]
- Angly, F.E.; Felts, B.; Breitbart, M.; Salamon, P.; Edwards, R.A.; Carlson, C.; Chan, A.M.; Haynes, M.; Kelley, S.; Liu, H.; et al. The marine viromes of four oceanic regions. PLoS Biol 2006, 4. [Google Scholar] [CrossRef]
- Rosario, K.; Nilsson, C.; Lim, Y.W.; Ruan, Y.; Breitbart, M. Metagenomic analysis of viruses in reclaimed water. Environ. Microbiol 2009, 11, 2806–2820. [Google Scholar]
- Kristensen, D.M.; Mushegian, A.R.; Dolja, V.V.; Koonin, E.V. New dimensions of the virus world discovered through metagenomics. Trends Microbiol 2010, 18, 11–19. [Google Scholar]
- Bibby, K.; Viau, E.; Peccia, J. Viral metagenome analysis to guide human pathogen monitoring in environmental samples. Lett. Appl. Microbiol 2011, 52, 386–392. [Google Scholar]
- Finkbeiner, S.R.; Allred, A.F.; Tarr, P.I.; Klein, E.J.; Kirkwood, C.D.; Wang, D. Metagenomic analysis of human diarrhea: viral detection and discovery. PLoS Pathog 2008, 4. [Google Scholar] [CrossRef]
- Svraka, S.; Rosario, K.; Duizer, E.; van der Avoort, H.; Breitbart, M.; Koopmans, M. Metagenomic sequencing for virus identification in a public-health setting. J. Gen. Virol 2010, 91, 2846–2856. [Google Scholar]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar]
- Greninger, A.L.; Runckel, C.; Chiu, C.Y.; Haggerty, T.; Parsonnet, J.; Ganem, D.; DeRisi, J.L. The complete genome of klassevirus - a novel picornavirus in pediatric stool. Virol. J 2009, 6. [Google Scholar] [CrossRef]
- Legendre, M.; Santini, S.; Rico, A.; Abergel, C.; Claverie, J.M. Breaking the 1000-gene barrier for Mimivirus using ultra-deep genome and transcriptome sequencing. Virol. J 2011, 8, 99. [Google Scholar]
- Huang, Y.; Huang, X.; Liu, H.; Gong, J.; Ouyang, Z.; Cui, H.; Cao, J.; Zhao, Y.; Wang, X.; Jiang, Y.; Qin, Q. Complete sequence determination of a novel reptile iridovirus isolated from softshelled turtle and evolutionary analysis of Iridoviridae. BMC Genomics 2009, 10. [Google Scholar] [CrossRef]
- Hoper, D.; Hoffmann, B.; Beer, M. A comprehensive deep sequencing strategy for full-length genomes of influenza A. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Willerth, S.M.; Pedro, H.A.; Pachter, L.; Humeau, L.M.; Arkin, A.P.; Schaffer, D.V. Development of a low bias method for characterizing viral populations using next generation sequencing technology. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Monger, W.A.; Adams, I.P.; Glover, R.H.; Barrett, B. The complete genome sequence of Canna yellow streak virus. Arch. Virol 2010, 155, 1515–1518. [Google Scholar]
- Potgieter, A.C.; Page, N.A.; Liebenberg, J.; Wright, I.M.; Landt, O.; van Dijk, A.A. Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J. Gen. Virol 2009, 90, 1423–1432. [Google Scholar]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog 2010, 6. [Google Scholar] [CrossRef]
- Novella, I.S.; Domingo, E.; Holland, J.J. Rapid viral quasispecies evolution: implications for vaccine and drug strategies. Mol. Med. Today 1995, 1, 248–253. [Google Scholar]
- Ruiz-Jarabo, C.M.; Arias, A.; Baranowski, E.; Escarmis, C.; Domingo, E. Memory in viral quasispecies. J. Virol 2000, 74, 3543–3547. [Google Scholar]
- Eriksson, N.; Pachter, L.; Mitsuya, Y.; Rhee, S.Y.; Wang, C.; Gharizadeh, B.; Ronaghi, M.; Shafer, R.W.; Beerenwinkel, N. Viral population estimation using pyrosequencing. PLoS Comput. Biol 2008, 4, e1000074. [Google Scholar]
- Zagordi, O.; Geyrhofer, L.; Roth, V.; Beerenwinkel, N. Deep sequencing of a genetically heterogeneous sample: local haplotype reconstruction and read error correction. J. Comput. Biol 2010, 17, 417–428. [Google Scholar]
- Prosperi, M.C.; Prosperi, L.; Bruselles, A.; Abbate, I.; Rozera, G.; Vincenti, D.; Solmone, M.C.; Capobianchi, M.R.; Ulivi, G. Combinatorial analysis and algorithms for quasispecies reconstruction using next-generation sequencing. BMC Bioinform 2011, 12, 5. [Google Scholar]
- Beerenwinkel, N.; Zagordi, O. Ultra-deep sequencing for the analysis of viral populations. Curr. Opin. Virol 2011, 1, 1–6. [Google Scholar]
- Vrancken, B.; Lequime, S.; Theys, K.; Lemey, P. Covering all bases in HIV research: Unveiling a hidden world of viral evolution. AIDS Rev 2010, 12, 89–102. [Google Scholar]
- Tebit, D.M.; Arts, E.J. Tracking a century of global expansion and evolution of HIV to drive understanding and to combat disease. Lancet Infect. Dis 2011, 11, 45–56. [Google Scholar]
- Bruselles, A.; Rozera, G.; Bartolini, B.; Prosperi, M.; Del Nonno, F.; Narciso, P.; Capobianchi, M.R.; Abbate, I. Use of massive parallel pyrosequencing for near full-length characterization of a unique HIV Type 1 BF recombinant associated with a fatal primary infection. AIDS Res. Hum. Retrovir 2009, 25, 937–942. [Google Scholar]
- Wu, X.; Zhou, T.; Zhu, J.; Zhang, B.; Georgiev, I.; Wang, C.; Chen, X.; Longo, N.S.; Louder, M.; McKee, K.; et al. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science 2011, 333, 1593–1602. [Google Scholar]
- Tapparel, C.; Cordey, S.; Junier, T.; Farinelli, L.; Van Belle, S.; Soccal, P.M.; Aubert, J.D.; Zdobnov, E.; Kaiser, L. Rhinovirus genome variation during chronic upper and lower respiratory tract infections. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Gorzer, I.; Guelly, C.; Trajanoski, S.; Puchhammer-Stockl, E. Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. J. Virol 2010, 84, 7195–7203. [Google Scholar]
- Renzette, N.; Bhattacharjee, B.; Jensen, J.D.; Gibson, L.; Kowalik, T.F. Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog 2011, 7. [Google Scholar] [CrossRef]
- Szpara, M.L.; Parsons, L.; Enquist, L.W. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J. Virol 2010, 84, 5303–5313. [Google Scholar]
- Zemanick, M.C.; Strick, P.L.; Dix, R.D. Direction of transneuronal transport of herpes simplex virus 1 in the primate motor system is strain-dependent. Proc. Natl. Acad. Sci. USA 1991, 88, 8048–8051. [Google Scholar]
- Palmer, S.; Kearney, M.; Maldarelli, F.; Halvas, E.K.; Bixby, C.J.; Bazmi, H.; Rock, D.; Falloon, J.; Davey, R.T., Jr; Dewar, R.L.; et al. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol 2005, 43, 406–413. [Google Scholar]
- Jourdain, G.; Ngo-Giang-Huong, N.; Le Coeur, S.; Bowonwatanuwong, C.; Kantipong, P.; Leechanachai, P.; Ariyadej, S.; Leenasirimakul, P.; Hammer, S.; Lallemant, M. Intrapartum exposure to nevirapine and subsequent maternal responses to nevirapine-based antiretroviral therapy. N. Engl. J. Med 2004, 351, 229–240. [Google Scholar]
- Lecossier, D.; Shulman, N.S.; Morand-Joubert, L.; Shafer, R.W.; Joly, V.; Zolopa, A.R.; Clavel, F.; Hance, A.J. Detection of minority populations of HIV-1 expressing the K103N resistance mutation in patients failing nevirapine. J. Acquir. Immune Defic. Syndr 2005, 38, 37–42. [Google Scholar]
- Palmer, S.; Boltz, V.; Martinson, N.; Maldarelli, F.; Gray, G.; McIntyre, J.; Mellors, J.; Morris, L.; Coffin, J. Persistence of nevirapine-resistant HIV-1 in women after single-dose nevirapine therapy for prevention of maternal-to-fetal HIV-1 transmission. Proc. Natl. Acad. Sci. USA 2006, 103, 7094–7099. [Google Scholar]
- Wang, C.; Mitsuya, Y.; Gharizadeh, B.; Ronaghi, M.; Shafer, R.W. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res 2007, 17, 1195–1201. [Google Scholar]
- Mild, M.; Hedskog, C.; Jernberg, J.; Albert, J. Performance of Ultra-Deep Pyrosequencing in Analysis of HIV-1 pol Gene Variation. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Hoffmann, C.; Minkah, N.; Leipzig, J.; Wang, G.; Arens, M.Q.; Tebas, P.; Bushman, F.D. DNA bar coding and pyrosequencing to identify rare HIV drug resistance mutations. Nucleic Acids Res 2007, 35. [Google Scholar] [CrossRef]
- Simen, B.B.; Simons, J.F.; Hullsiek, K.H.; Novak, R.M.; MacArthur, R.D.; Baxter, J.D.; Huang, C.L.; Lubeski, C.; Turenchalk, G.S.; Braverman, M.S.; et al. Low-abundance drugresistant viral variants in chronically HIV-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. J. Infect. Dis 2009, 199, 693–701. [Google Scholar]
- Metzner, K.J.; Giulieri, S.G.; Knoepfel, S.A.; Rauch, P.; Burgisser, P.; Yerly, S.; Gunthard, H.F.; Cavassini, M. Minority quasispecies of drug-resistant HIV-1 that lead to early therapy failure in treatment-naive and -adherent patients. Clin. Infect. Dis 2009, 48, 239–247. [Google Scholar]
- Johnson, J.A.; Li, J.F.; Wei, X.; Lipscomb, J.; Irlbeck, D.; Craig, C.; Smith, A.; Bennett, D.E.; Monsour, M.; Sandstrom, P.; et al. Minority HIV-1 drug resistance mutations are present in antiretroviral treatment-naive populations and associate with reduced treatment efficacy. PLoS Med 2008, 5. [Google Scholar] [CrossRef]
- Le, T.; Chiarella, J.; Simen, B.B.; Hanczaruk, B.; Egholm, M.; Landry, M.L.; Dieckhaus, K.; Rosen, M.I.; Kozal, M.J. Low-abundance HIV drug-resistant viral variants in treatment-experienced persons correlate with historical antiretroviral use. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Codoner, F.M.; Pou, C.; Thielen, A.; Garcia, F.; Delgado, R.; Dalmau, D.; Alvarez-Tejado, M.; Ruiz, L.; Clotet, B.; Paredes, R. Added value of deep sequencing relative to population sequencing in heavily pre-treated HIV-1-infected subjects. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Peuchant, O.; Thiebaut, R.; Capdepont, S.; Lavignolle-Aurillac, V.; Neau, D.; Morlat, P.; Dabis, F.; Fleury, H.; Masquelier, B. Transmission of HIV-1 minority-resistant variants and response to first-line antiretroviral therapy. AIDS 2008, 22, 1417–1423. [Google Scholar]
- Jakobsen, M.R.; Tolstrup, M.; Sogaard, O.S.; Jorgensen, L.B.; Gorry, P.R.; Laursen, A.; Ostergaard, L. Transmission of HIV-1 drug-resistant variants: prevalence and effect on treatment outcome. Clin. Infect. Dis 2010, 50, 566–573. [Google Scholar]
- Mukherjee, R.; Jensen, S.T.; Male, F.; Bittinger, K.; Hodinka, R.L.; Miller, M.D.; Bushman, F.D. Switching between raltegravir resistance pathways analyzed by deep sequencing. AIDS 2011, 25, 1951–1959. [Google Scholar]
- Codoner, F.M.; Pou, C.; Thielen, A.; Garcia, F.; Delgado, R.; Dalmau, D.; Santos, J.R.; Buzon, M.J.; Martinez-Picado, J.; Alvarez-Tejado, M.; et al. Dynamic escape of pre-existing raltegravir-resistant HIV-1 from raltegravir selection pressure. Antiviral Res 2010, 88, 281–286. [Google Scholar]
- Gulick, R.M.; Lalezari, J.; Goodrich, J.; Clumeck, N.; DeJesus, E.; Horban, A.; Nadler, J.; Clotet, B.; Karlsson, A.; Wohlfeiler, M.; et al. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med 2008, 359, 1429–1441. [Google Scholar]
- Ogert, R.A.; Hou, Y.; Ba, L.; Wojcik, L.; Qiu, P.; Murgolo, N.; Duca, J.; Dunkle, L.M.; Ralston, R.; Howe, J.A. Clinical resistance to vicriviroc through adaptive V3 loop mutations in HIV-1 subtype D gp120 that alter interactions with the N-terminus and ECL2 of CCR5. Virology 2010, 400, 145–155. [Google Scholar]
- Tilton, J.C.; Amrine-Madsen, H.; Miamidian, J.L.; Kitrinos, K.M.; Pfaff, J.; Demarest, J.F.; Ray, N.; Jeffrey, J.L.; Labranche, C.C.; Doms, R.W. HIV type 1 from a patient with baseline resistance to CCR5 antagonists uses drug-bound receptor for entry. AIDS Res. Hum. Retrovir 2010, 26, 13–24. [Google Scholar]
- Tsibris, A.M.; Sagar, M.; Gulick, R.M.; Su, Z.; Hughes, M.; Greaves, W.; Subramanian, M.; Flexner, C.; Giguel, F.; Leopold, K.E.; et al. In vivo emergence of vicriviroc resistance in a human immunodeficiency virus type 1 subtype C-infected subject. J. Virol 2008, 82, 8210–8214. [Google Scholar]
- Tilton, J.C.; Wilen, C.B.; Didigu, C.A.; Sinha, R.; Harrison, J.E.; Agrawal-Gamse, C.; Henning, E.A.; Bushman, F.D.; Martin, J.N.; Deeks, S.G.; et al. A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J. Virol 2010, 84, 10863–10876. [Google Scholar]
- Hwang, S.S.; Boyle, T.J.; Lyerly, H.K.; Cullen, B.R. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science 1991, 253, 71–74. [Google Scholar]
- Poveda, E.; Alcami, J.; Paredes, R.; Cordoba, J.; Gutierrez, F.; Llibre, J.M.; Delgado, R.; Pulido, F.; Iribarren, J.A.; Garcia Deltoro, M.; et al. Genotypic determination of HIV tropism - clinical and methodological recommendations to guide the therapeutic use of CCR5 antagonists. AIDS Rev 2010, 12, 135–148. [Google Scholar]
- Swenson, L.C.; Mo, T.; Dong, W.W.Y.; Zhong, X.Y.; Woods, C.K.; Jensen, M.A.; Thielen, A.; Chapman, D.; Lewis, M.; James, I.; et al. Deep sequencing to infer HIV-1 co-receptor usage: Application to three clinical trials of Maraviroc in treatment-experienced patients. J. Infect. Dis 2011, 203, 237–245. [Google Scholar]
- Bunnik, E.M.; Swenson, L.C.; Edo-Matas, D.; Huang, W.; Dong, W.; Frantzell, A.; Petropoulos, C.J.; Coakley, E.; Schuitemaker, H.; Harrigan, P.R.; et al. Detection of inferred CCR5- and CXCR4-using HIV-1 variants and evolutionary intermediates using ultra-deep pyrosequencing. PLoS Pathog 2011, 7. [Google Scholar] [CrossRef]
- Abbate, I.; Vlassi, C.; Rozera, G.; Bruselles, A.; Bartolini, B.; Giombini, E.; Corpolongo, A.; D’Offizi, G.; Narciso, P.; Desideri, A.; et al. Detection of quasispecies variants predicted to use CXCR4 by ultra-deep pyrosequencing during early HIV infection. AIDS 2011, 25, 611–617. [Google Scholar]
- Archer, J.; Braverman, M.S.; Taillon, B.E.; Desany, B.; James, I.; Harrigan, P.R.; Lewis, M.; Robertson, D.L. Detection of low-frequency pretherapy chemokine (CXC motif) receptor 4 (CXCR4)-using HIV-1 with ultra-deep pyrosequencing. AIDS 2009, 23, 1209–1218. [Google Scholar]
- Tsibris, A.M.; Korber, B.; Arnaout, R.; Russ, C.; Lo, C.C.; Leitner, T.; Gaschen, B.; Theiler, J.; Paredes, R.; Su, Z.; et al. Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Margeridon-Thermet, S.; Shulman, N.S.; Ahmed, A.; Shahriar, R.; Liu, T.; Wang, C.; Holmes, S.P.; Babrzadeh, F.; Gharizadeh, B.; Hanczaruk, B.; et al. Ultra-deep pyrosequencing of hepatitis B virus quasispecies from nucleoside and nucleotide reverse-transcriptase inhibitor (NRTI)-treated patients and NRTI-naive patients. J. Infect. Dis 2009, 199, 1275–1285. [Google Scholar]
- Solmone, M.; Vincenti, D.; Prosperi, M.C.F.; Bruselles, A.; Ippolito, G.; Capobianchi, M.R. Use of massively parallel ultradeep pyrosequencing to characterize the genetic diversity of Hepatitis B Virus in drug-resistant and drug-naive patients and to detect minor variants in reverse transcriptase and Hepatitis B S antigen. J. Virol 2009, 83, 1718–1726. [Google Scholar]
- Verbinnen, T.; Van Marck, H.; Vandenbroucke, I.; Vijgen, L.; Claes, M.; Lin, T.I.; Simmen, K.; Neyts, J.; Fanning, G.; Lenz, O. Tracking the evolution of multiple in vitro hepatitis C virus replicon variants under protease inhibitor selection pressure by 454 deep sequencing. J. Virol 2010, 84, 11124–11133. [Google Scholar]
- Delang, L.; Vliegen, I.; Froeyen, M.; Neyts, J. Comparative study of the genetic barriers and pathways towards resistance of selective inhibitors of Hepatitis C Virus replication. Antimicrob. Agents Chemother 2011, 55, 4103–4113. [Google Scholar]
- Redd, A.D.; Collinson-Streng, A.; Martens, C.; Ricklefs, S.; Mullis, C.E.; Manucci, J.; Tobian, A.A.; Selig, E.J.; Laeyendecker, O.; Sewankambo, N.; et al. Identification of HIV superinfection in seroconcordant couples in Rakai, Uganda, by use of next-generation deep sequencing. J. Clin. Microbiol 2011, 49, 2859–2867. [Google Scholar]
- Campbell, M.S.; Mullins, J.I.; Hughes, J.P.; Celum, C.; Wong, K.G.; Raugi, D.N.; Sorensen, S.; Stoddard, J.N.; Zhao, H.; Deng, W.J.; et al. Viral linkage in HIV-1 seroconverters and their partners in an HIV-1 prevention clinical trial. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Fischer, W.; Ganusov, V.V.; Giorgi, E.E.; Hraber, P.T.; Keele, B.F.; Leitner, T.; Han, C.S.; Gleasner, C.D.; Green, L.; Lo, C.C.; et al. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Bull, R.A.; Luciani, F.; McElroy, K.; Gaudieri, S.; Pham, S.T.; Chopra, A.; Cameron, B.; Maher, L.; Dore, G.J.; White, P.A.; et al. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathog 2011, 7. [Google Scholar] [CrossRef]
- Neverov, A.; Chumakov, K. Massively parallel sequencing for monitoring genetic consistency and quality control of live viral vaccines. Proc. Natl. Acad. Sci. USA 2010, 107, 20063–20068. [Google Scholar]
- Victoria, J.G.; Wang, C.; Jones, M.S.; Jaing, C.; McLoughlin, K.; Gardner, S.; Delwart, E.L. Viral nucleic acids in live-attenuated vaccines: detection of minority variants and an adventitious virus. J. Virol 2010, 84, 6033–6040. [Google Scholar]
- Harismendy, O.; Ng, P.C.; Strausberg, R.L.; Wang, X.; Stockwell, T.B.; Beeson, K.Y.; Schork, N.J.; Murray, S.S.; Topol, E.J.; Levy, S.; et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol 2009, 10, R32. [Google Scholar]
- Zagordi, O.; Klein, R.; Daumer, M.; Beerenwinkel, N. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic Acids Res 2010, 38, 7400–7409. [Google Scholar]
- Quince, C.; Lanzen, A.; Davenport, R.J.; Turnbaugh, P.J. Removing noise from pyrosequenced amplicons. BMC Bioinform 2011, 12, 38. [Google Scholar]
- Finotello, F.; Lavezzo, E.; Fontana, P.; Peruzzo, D.; Albiero, A.; Barzon, L.; Di Camillo, B.; Toppo, S. Comparative analysis of algorithms for whole genome assembly of pyrosequencing data. Brief. Bioinform 2011. [Google Scholar] [CrossRef]

{kind=link}
| Maximum Throughput Mb/run | Mean Length (nucleotide) | Error rate * | Applications | Main source of errors | |
|---|---|---|---|---|---|
| 454 FLX | 700 | ~800 (for shotgun experiments) ~400 (for amplicon experiments) | 10−3–10−4 | De novo genome sequencing and resequencing, target resequencing, genotyping, metagenomics | Intensity cutoff, homopolymers, signal cross-talk interference among neighbors, amplification, mixed beads |
| Illumina | 6,000 | ~100 | 10−2–10−3 | Genome resequencing, quantitative transcriptomics, genotyping, metagenomics | Signal interference among neighboring clusters, homopolymers, phasing, nucleotide labeling, amplification, low coverage of AT rich regions |
| SOLiD | 20,000 | ~50 | 10−2–10−3 | Genome resequencing, quantitative transcriptomics, genotyping | Signal interference among neighbours, phasing, nucleotide labeling, signal degradation, mixed beads, low coverage of AT rich regions |
| Helicos | 21,000–35,000 | ~35 | 10−2 | Non amplifiable samples, PCR free and unbiased quantitative analyses | Polymerase employed, molecule loss, low intensities |
| Ion Torrent PGM | 1,000 | ~200 | 3 × 10−2 | De novo genome sequencing and resequencing, target resequencing, genotyping, RNA-seq on low-complexity transcriptome, metagenomics | Homopolymers, amplification |
| GS Junior | ~35 | ~400 | 10−3–10−4 | Target resequencing (amplicons), genotyping | Intensity cutoff, homopolymers, signal cross-talk interference among neighbors, amplification, mixed beads |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Barzon, L.; Lavezzo, E.; Militello, V.; Toppo, S.; Palù, G. Applications of Next-Generation Sequencing Technologies to Diagnostic Virology. Int. J. Mol. Sci. 2011, 12, 7861-7884. https://doi.org/10.3390/ijms12117861
Barzon L, Lavezzo E, Militello V, Toppo S, Palù G. Applications of Next-Generation Sequencing Technologies to Diagnostic Virology. International Journal of Molecular Sciences. 2011; 12(11):7861-7884. https://doi.org/10.3390/ijms12117861
Chicago/Turabian StyleBarzon, Luisa, Enrico Lavezzo, Valentina Militello, Stefano Toppo, and Giorgio Palù. 2011. "Applications of Next-Generation Sequencing Technologies to Diagnostic Virology" International Journal of Molecular Sciences 12, no. 11: 7861-7884. https://doi.org/10.3390/ijms12117861
APA StyleBarzon, L., Lavezzo, E., Militello, V., Toppo, S., & Palù, G. (2011). Applications of Next-Generation Sequencing Technologies to Diagnostic Virology. International Journal of Molecular Sciences, 12(11), 7861-7884. https://doi.org/10.3390/ijms12117861