The Role of Reactive-Oxygen-Species in Microbial Persistence and Inflammation

Abstract
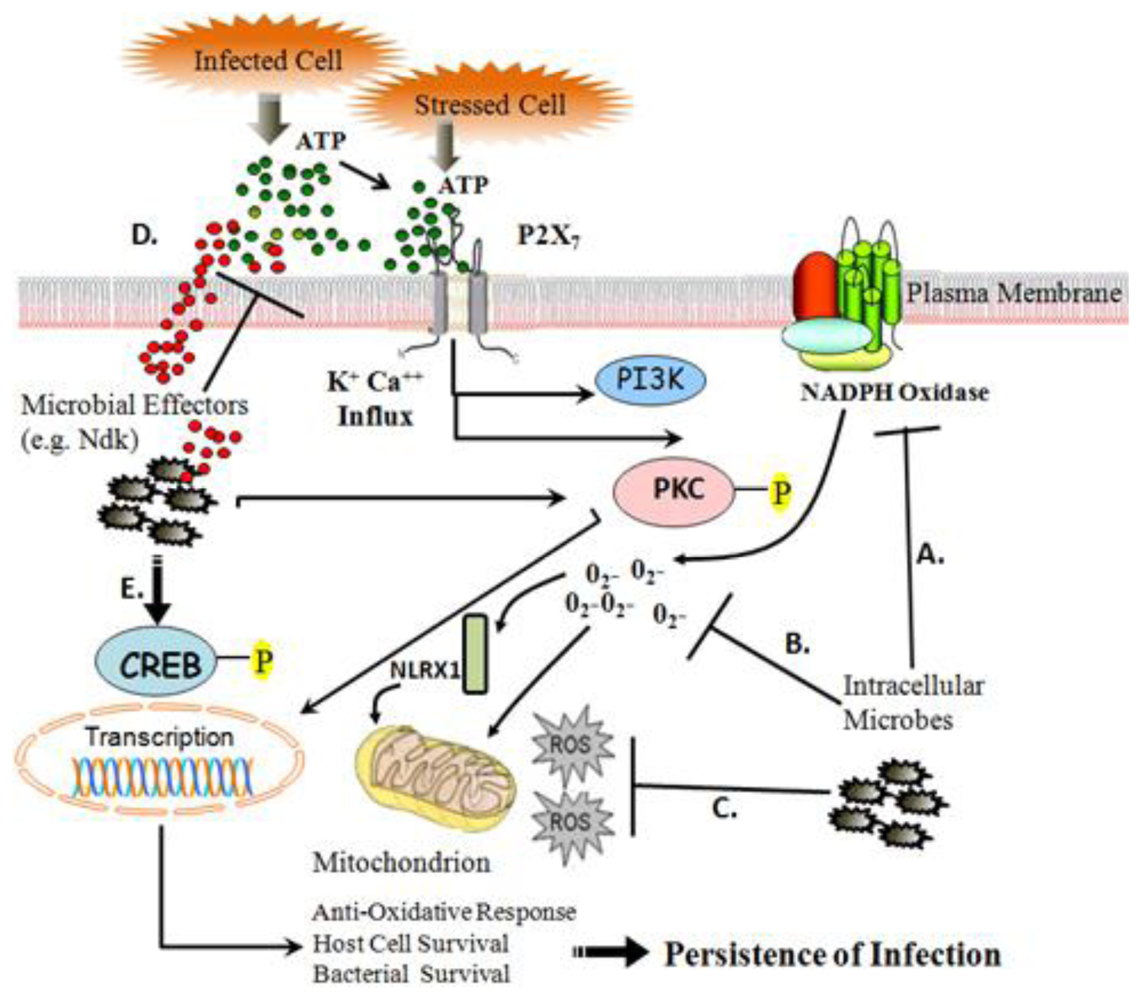
: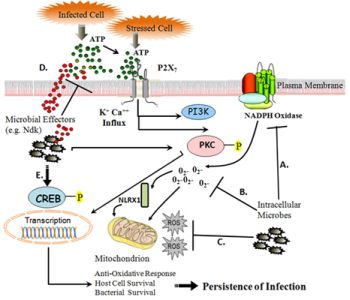
{kind=link}
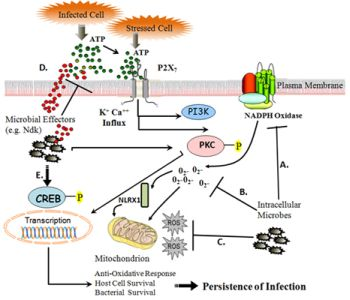{kind=link}
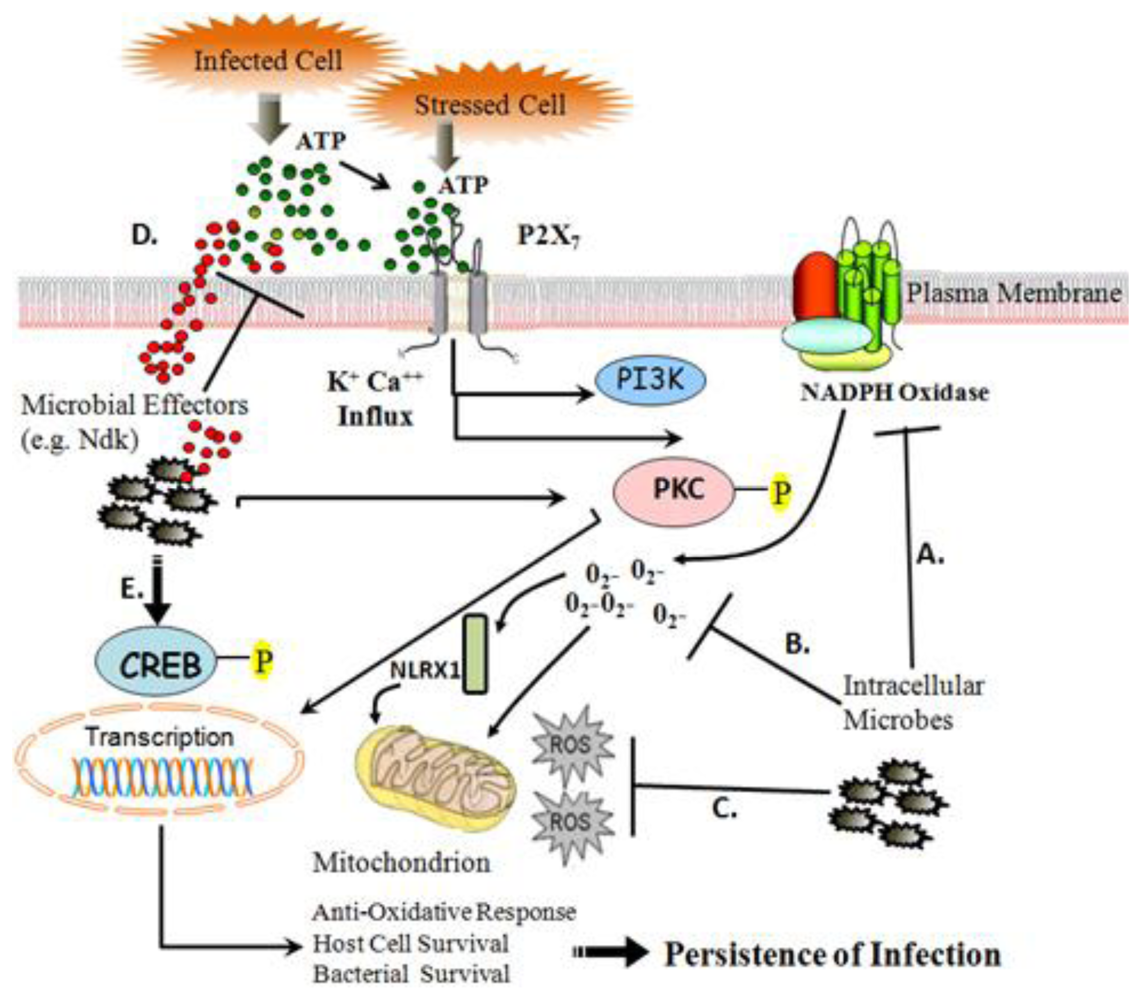{kind=link}
1. Introduction
2. Cellular ROS and Its Implications for Microbial Infections
2. 1. Sources of Intracellular ROS Production as an Early Response
2. 2. Modulation of Intracellular ROS by Infection
3. eATP-P2X7 Receptor Signaling in Controlling Infection and Inflammation via Cellular ROS
3. 1. eATP as a “Danger Signal”
3. 2. P2X7 Receptor Activation by eATP Elicits Signaling Cascades through ROS
3. 3. eATP-P2X7 Receptor Signaling Mediates Pathogen Killing
3. 4. Subversion of eATP-P2X7 Pathway by Persistent Microbes
4. Conclusions
Acknowledgement
References
- Circu, ML; Aw, TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 2010, 48(6), 749–762. [Google Scholar]
- Forman, HJ; Maiorino, M; Ursini, F. Signaling functions of reactive oxygen species. Biochemistry 2010, 49(5), 835–842. [Google Scholar]
- Kowaltowski, AJ; de Souza-Pinto, NC; Castilho, RF; Vercesi, AE. Mitochondria and reactive oxygen species. Free Radic Biol Med 2009, 47(4), 333–343. [Google Scholar]
- Guerra, AN; Gavala, ML; Chung, HS. Nucleotide receptor signalling and the generation of reactive oxygen species. Purinergic Signal 2007, 3 . [Google Scholar]
- Hewinson, J; Mackenzie, AB. P2X (7) receptor-mediated reactive oxygen and nitrogen species formation: from receptor to generators. Biochem Soc Trans 2007, 35 Pt 5, 1168–1170. [Google Scholar]
- Lau, AT; Wang, Y; Chiu, JF. Reactive oxygen species: current knowledge and applications in cancer research and therapeutic. J Cell Biochem 2008, 104(2), 657–667. [Google Scholar]
- Sareila, O; Kelkka, T; Pizzolla, A; Hultqvist, M; Holmdahl, R. NOX2 complex derived ROS as immune regulators. Antioxid Redox Signal 3635. [CrossRef]
- Koopman, WJ; Nitjmans, LG; Dieteren, CE; Roestenberg, P; Valsecchi, F; Smeitink, JA; Willems, PH. Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal 2010, 12(12), 1431–1470. [Google Scholar]
- Barja, G. Mitochondrial free radical production and aging in mammals and birds. Ann. N.Y. Acad. Sci 1998, 854, 224–238. [Google Scholar]
- Arnoult, D; Soares, F; Tattoli, I; Castanier, C; Philpott, DJ; Girardin, SE. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J Cell Sci 2009, 122 Pt 17, 3161–3168. [Google Scholar]
- Moore, CB; Bergstralh, DT; Duncan, JA; Lei, Y; Morrison, TE; Zimmerman, AG; Accavitti-Loper, MA; Madden, VJ; Sun, L; Ye, Z; Lich, JD; Heise, MT; Chen, Z; Ting, JP. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 2008, 451(7178), 573–577. [Google Scholar]
- Groeger, G; Quiney, C; Cotter, TG. Hydrogen peroxide as a cell-survival signaling molecule. Antioxid Redox Signal 2009, 11(11), 2655–2671. [Google Scholar]
- Dworakowski, R; Alom-Ruiz, SP; Shah, AM. NADPH oxidase-derived reactive oxygen species in the regulation of endothelial phenotype. Pharmacol Rep 2008, 60(1), 21–28. [Google Scholar]
- Liou, GY; Storz, P. Reactive oxygen species in cancer. Free Radic Res 2010, 44(5), 479–496. [Google Scholar]
- Arnoult, D; Carneiro, L; Tattoli, I; Girardin, SE. The role of mitochondria in cellular defense against microbial infection. Semin Immunol 2009, 21(4), 223–232. [Google Scholar]
- Tattoli, I; Carneiro, L; Jéhanno, M; Magalhaes, JG; Shu, Y; Philpott, DJ; Arnoult, D; Girardin, SE. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep 2008, 9(3), 293–300. [Google Scholar]
- Abdul-Sater, AA; Saïd-Sadier, N; Lam, VM; Singh, B; Pettengill, MA; Soares, F; Tattoli, I; Lipinski, S; Girardin, SE; Rosenstiel, P; Ojcius, DM. Enhancement of reactive oxygen species production and chlamydial infection by the mitochondrial Nod-like family member, NLRX 1. J Biol Chem 2010, 285(53), 41637–41645. [Google Scholar]
- Grandvaux, N; Soucy-Faulkner, A; Fink, K. Innate host defense: Nox and Duox on phox’s tail. Biochimie 2007, 89(9), 1113–1122. [Google Scholar]
- Leto, TL; Morand, S; Hurt, D; Ueyama, T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid Redox Signal 2009, 11(10), 2607–2619. [Google Scholar]
- Boots, AW; Hristova, M; Kasahara, DI; Haenan, GR; Bast, A; van der Vliet, A. ATP-mediated activation of the NADPH oxidase DUOX1 mediates airway epithelial responses to bacterial stimuli. J Biol Chem 2009, 284(26), 17858–17867. [Google Scholar]
- Mo, Y; Wan, R; Chien, S; Tollerud, DJ; Zhang, Q. Activation of endothelial cells after exposure to ambient ultrafine particles: the role of NADPH oxidase. Toxicol Appl Pharmacol 2009, 236(2), 183–193. [Google Scholar]
- Vignais, PV. The superoxide-generating NADPH oxidase: Structural aspects and activation mechanism. Cell Mol Life Sci 2002, 59(9), 1428–1459. [Google Scholar]
- Petry, A; Weitnauer, M; Gorlach, A. Receptor activation of NADPH oxidases. Antioxid Redox Signal 2010, 13(4), 467–487. [Google Scholar]
- Williams, LM; Lali, F; Willetts, K; Balague, C; Godessart, N; Brennan, F; Feldmann, M; Foxwell, BM. Rac mediates TNF-induced cytokine production via modulation of NF-kappaB. Mol Immunol 2008, 45(9), 2446–2454. [Google Scholar]
- Dovas, A; Couchman, JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J 2005, 390 Pt 1, 1–9. [Google Scholar]
- DerMardirossian, C; Bokoch, GM. GDIs: Central regulatory molecules in Rho GTPase activation. Trends Cell Biol 2005, 15(7), 356–363. [Google Scholar]
- Hawkins, PT; Davidson, K; Stephens, LR. The role of PI3Ks in the regulation of the neutrophil NADPH oxidase. Biochem Soc Symp 2007, (74), 59–67. [Google Scholar]
- Wu, W; Hsu, YM; Bi, L; Songyang, Z; Lin, X. CARD9 facilitates microbe-elicited production of reactive oxygen species by regulating the LyGDI-Rac1 complex. Nat Immunol 2009, 10(11), 1208–1214. [Google Scholar]
- Cirillo, SL; Subbian, S; Chen, B; Weisbrod, TR; Jacobs, WR, Jr; Cirillo, JD. Protection of Mycobacterium tuberculosis from reactive oxygen species conferred by the mel2 locus impacts persistence and dissemination. Infect Immun 2009, 77(6), 2557–2567. [Google Scholar]
- McCaffrey, RL; Allen, LA. Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J Leukoc Biol 2006, 80(6), 1224–1230. [Google Scholar]
- Carlyon, JA; Abdel-Latif, D; Pypaert, M; Lacy, P; Fikrig, E. Anaplasma phagocytophilum utilizes multiple host evasion mechanisms to thwart NADPH oxidase-mediated killing during neutrophil infection. Infect Immun 2004, 72(8), 4772–4783. [Google Scholar]
- Woldehiwet, Z. Immune evasion and immunosuppression by Anaplasma phagocytophilum, the causative agent of tick-borne fever of ruminants and human granulocytic anaplasmosis. Vet J 2008, 175(1), 37–44. [Google Scholar]
- Boncompain, G; Schneider, B; Delevoye, C; Kellermann, O; Dautry-Varsat, A; Subtil, A. Production of reactive oxygen species is turned on and rapidly shut down in epithelial cells infected with Chlamydia trachomatis. Infect Immun 2010, 78(1), 80–87. [Google Scholar]
- Siemsen, DW; Kirpotina, LN; Jutila, MA; Quinn, MT. Inhibition of the human neutrophil NADPH oxidase by Coxiella burnetii. Microbes Infect 2009, 11 . [Google Scholar]
- Harada, T; Miyake, M; Imai, Y. Evasion of Legionella pneumophila from the bactericidal system by reactive oxygen species (ROS) in macrophages. Microbiol Immunol 2007, 51(12), 1161–1170. [Google Scholar]
- Kim, JS; Bokoch, GM. Anthrax edema toxin inhibits Nox1-mediated formation of reactive oxygen species by colon epithelial cells. J Innate Immun 2009, 1(2), 145–152. [Google Scholar]
- Bylund, J; Burgess, LA; Cescutti, P; Ernst, RK; Speert, DP. Exopolysaccharides from Burkholderia cenocepacia inhibit neutrophil chemotaxis and scavenge reactive oxygen species. J Biol Chem 2006, 281(5), 2526–2532. [Google Scholar]
- Keith, KE; Hynes, DW; Sholdice, JE; Valvano, MA. Delayed association of the NADPH oxidase complex with macrophage vacuoles containing the opportunistic pathogen Burkholderia cenocepacia. Microbiology 2009, 155 Pt 4, 1004–1015. [Google Scholar]
- Bianchi, SM; Prince, LR; McPhillips, K; Allen, L; Marriott, HM; Taylor, GW; Hellewell, PG; Sabroe, I; Dockrell, DH; Henson, PW; Whyte, MK. Impairment of apoptotic cell engulfment by pyocyanin, a toxic metabolite of Pseudomonas aeruginosa. Am J Respir Crit Care Med 2008, 177(1), 35–43. [Google Scholar]
- Criss, AK; Seifert, HS. Neisseria gonorrhoeae suppresses the oxidative burst of human polymorphonuclear leukocytes. Cell Microbiol 2008, 10(11), 2257–2270. [Google Scholar]
- Wellington, M; Dolan, K; Krysan, DJ. Live Candida albicans suppresses production of reactive oxygen species in phagocytes. Infect Immun 2009, 77(1), 405–413. [Google Scholar]
- Orciuolo, E; Stanzani, M; Canestraro, M; Galimberti, S; Carulli, G; Lewis, R; Petrini, M; Komanduri, KV. Effects of Aspergillus fumigatus gliotoxin and methylprednisolone on human neutrophils: implications for the pathogenesis of invasive aspergillosis. J Leukoc Biol 2007, 82(4), 839–848. [Google Scholar]
- Saïd-Sadier, N; Padilla, E; Langsley, G; Ojcius, DM. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS One 2010, 5(4), e10008. [Google Scholar]
- Sim, S; Yong, TS; PArk, SJ; Im, KI; Kong, Y; Ryu, JS; Min, DY; Shin, MH. NADPH oxidase-derived reactive oxygen species-mediated activation of ERK1/2 is required for apoptosis of human neutrophils induced by Entamoeba histolytica. J Immunol 2005, 174(7), 4279–4288. [Google Scholar]
- Stanciu, M; Wang, Y; Kentor, R; Burke, N; Watkins, S; Kress, G; Reynolds, I; Klann, E; Angiolieri, MR; Johnson, JW; DeFranco, DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem 2000, 275(16), 12200–12206. [Google Scholar]
- Wang, X; Martindale, JL; Holbrook, NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem 2000, 275(50), 39435–39443. [Google Scholar]
- Singh, S; Upadhyay, AK; Ajay, AK; Bhat, MK. p53 regulates ERK activation in carboplatin induced apoptosis in cervical carcinoma: a novel target of p53 in apoptosis. FEBS Lett 2007, 581(2), 289–295. [Google Scholar]
- Yang, TC; Lai, CC; Shiu, SL; Chuang, PH; Tzou, BC; Lin, YY; Tsai, FJ; Lin, CW. Japanese encephalitis virus down-regulates thioredoxin and induces ROS-mediated ASK1-ERK/p38 MAPK activation in human promonocyte cells. Microbes Infect 2010, 12 . [Google Scholar]
- Gupta, S; Bhatia, V; Wen, JJ; Wu, Y; Huang, MH; Garg, NJ. Trypanosoma cruzi infection disturbs mitochondrial membrane potential and ROS production rate in cardiomyocytes. Free Radic Biol Med 2009, 47(10), 1414–1421. [Google Scholar]
- Zacks, MA; Wen, JJ; Vyatkina, G; Bhatia, V; Garg, N. An overview of chagasic cardiomyopathy: pathogenic importance of oxidative stress. An Acad Bras Cienc 2005, 77(4), 695–715. [Google Scholar]
- Paauw, A; Leverstein-van Hall, MA; Van Kessel, KP; Verhoef, J; Fluit, AC. Yersiniabactin reduces the respiratory oxidative stress response of innate immune cells. PLoS One 2009, 4(12), e8240. [Google Scholar]
- Songsungthong, W; Higgins, MC; Rolán, HC; Murphy, JL; Mecsas, J. ROS-inhibitory activity of YopE is required for full virulence of Yersinia in mice. Cell Microbiol 2010, 12(7), 988–1001. [Google Scholar]
- Spinner, JL; Seo, KS; O’Loughlin, JL; Cundiff, JA; Minnich, SA; Bohach, GA; Kobayashi, SD. Neutrophils are resistant to Yersinia YopJ/P-induced apoptosis and are protected from ROS-mediated cell death by the type III secretion system. PLoS One 2010, 5(2), e9279. [Google Scholar]
- O’Loughlin, JL; Spinner, JL; Minnich, SA; Kobayashi, SD. Yersinia pestis two-component gene regulatory systems promote survival in human neutrophils. Infect Immun 2010, 78(2), 773–782. [Google Scholar]
- Walsh, ER; Sahu, N; Kearley, J; Benjamin, E; Kang, BH; Humbles, A; August, A. Strain-specific requirement for eosinophils in the recruitment of T cells to the lung during the development of allergic asthma. J Exp Med 2008, 205(6), 1285–1292. [Google Scholar]
- Bours, MJ; Swennen, EL; Di Virgilio, F; Cronstein, BN; Dagnelie, PC. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther 2006, 112(2), 358–404. [Google Scholar]
- Trautmann, A. Extracellular ATP in the immune system: more than just a “danger signal”. Sci Signal 2009, 2(56), e6. [Google Scholar]
- Hanley, PJ; Musset, B; Reigunta, V; Limberg, SH; Dalpke, AH; Sus, R; Heeg, KM; Preisig-Müller, R; Daut, J. Extracellular ATP induces oscillations of intracellular Ca2+ and membrane potential and promotes transcription of IL-6 in macrophages. Proc Natl Acad Sci USA 2004, 101(25), 9479–9484. [Google Scholar]
- Mitani, H; Katayama, N; Araki, H; Ohishi, K; Kobayashi, K; Suzuki, H; Nishii, K; Masuya, M; Yasukawa, K; Minami, N; Shiku, H. Activity of interleukin 6 in the differentiation of monocytes to macrophages and dendritic cells. Br J Haematol 2000, 109(2), 288–295. [Google Scholar]
- Marteau, F; Gonzalez, NS; Communi, D; Goldman, M; Boeynaems, JM; Communi, D. Thrombospondin-1 and indoleamine 2,3-dioxygenase are major targets of extracellular ATP in human dendritic cells. Blood 2005, 106(12), 3860–3866. [Google Scholar]
- Idzko, M; Hammad, H; van Nimwegen, M; Kool, M; Willart, MA; Muskens, F; Hoogsteden, HC; Luttmann, W; Ferrari, D; Di Virgilio, F; Virchow, JC, Jr; Lambrecht, BN. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med 2007, 13(8), 913–919. [Google Scholar]
- Grahames, CB; Michel, AD; Chessell, IP; Humphrey, PP. Pharmacological characterization of ATP- and LPS-induced IL-1beta release in human monocytes. Br J Pharmacol 1999, 127(8), 1915–1921. [Google Scholar]
- Liu, JS; John, GR; Sikora, A; Lee, SC; Brosnan, CF. Modulation of interleukin-1beta and tumor necrosis factor alpha signaling by P2 purinergic receptors in human fetal astrocytes. J Neurosci 2000, 20(14), 5292–5299. [Google Scholar]
- John, GR; Simpson, GE; Woodroofe, MN; Lee, SC; Brosnan, CF. Extracellular nucleotides differentially regulate interleukin-1beta signaling in primary human astrocytes: implications for inflammatory gene expression. J Neurosci 2001, 21(12), 4134–4142. [Google Scholar]
- He, X; Mekasha, S; Mavrogiorgos, N; Fitzgerald, KA; Lien, E; Ingalls, RR. Inflammation and fibrosis during Chlamydia pneumoniae infection is regulated by IL-1 and the NLRP3/ASC inflammasome. J Immunol 2010, 184(10), 5743–5754. [Google Scholar]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur J Immunol 2010, 40(3), 616–619. [Google Scholar]
- Sarkar, A; Duncan, M; Hart, J; Hertlein, E; Guttridge, DC; Wewers, MD. ASC directs NF-kappaB activation by regulating receptor interacting protein-2 (RIP2) caspase-1 interactions. J Immunol 2006, 176(8), 4979–4986. [Google Scholar]
- Yilmaz, O; Sater, AA; Yao, L; Koutouzis, T; Pettengill, M; Ojcius, DM. ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell Microbiol 2010, 12(2), 188–198. [Google Scholar]
- Ghiringhelli, F; Apetoh, L; Tesniere, A; Aymeric, L; Ma, Y; Ortiz, C; Vermaelen, K; Panaretakis, T; Mignot, G; Ullrich, E; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 2009, 15(10), 1170–1178. [Google Scholar]
- Aymeric, L; Apetoh, L; Ghiringhelli, F; Tesniere, A; Martins, I; Kroemer, G; Smyth, MJ; Zitvogel, L. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res 2010, 70(3), 855–858. [Google Scholar]
- Qu, Y; Ramachandra, L; Mohr, S; Franchi, L; Harding, CV; Nunez, G; Dubyak, GR. P2X7 receptor-stimulated secretion of MHC class II-containing exosomes requires the ASC/NLRP3 inflammasome but is independent of caspase- 1. J Immunol 2009, 182(8), 5052–5062. [Google Scholar]
- Qu, Y; Franchi, L; Nunez, G; Dubyak, GR. Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol 2007, 179(3), 1913–1925. [Google Scholar]
- Ramachandra, L; Qu, Y; Wang, Y; Lewis, CJ; Cobb, BA; Takatsu, K; Boom, WH; Dubyak, GR; Harding, CV. Mycobacterium tuberculosis synergizes with ATP to induce release of microvesicles and exosomes containing MHC-II molecules capable of antigen presentation. Infect Immun 2010, 78(12), 5116–5125. [Google Scholar]
- Hewinson, J; Moore, SF; Glover, C; Watts, AG; MacKenzie, AB. A key role for redox signaling in rapid P2X7 receptor-induced IL-1 beta processing in human monocytes. J Immunol 2008, 180(12), 8410–8420. [Google Scholar]
- Noguchi, T; Ishii, K; Fukutomi, H; Naguro, I; Matsuzawa, A; Takeda, K; Ichijo, H. Requirement of reactive oxygen species-dependent activation of ASK1-p38 MAPK pathway for extracellular ATP-induced apoptosis in macrophage. J Biol Chem 2008, 283(12), 7657–7665. [Google Scholar]
- Parvathenani, LK; Tertyshnikova, S; Greco, CR; Roberts, SB; Robertson, B; Posmantur, R. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J Biol Chem 2003, 278(15), 13309–13317. [Google Scholar]
- Ferrari, D; Wesselborg, S; Bauer, MK; Schulze-Osthoff, K. Extracellular ATP activates transcription factor NF-kappaB through the P2Z purinoreceptor by selectively targeting NF-kappaB p 65. J Cell Biol 1997, 139(7), 1635–1643. [Google Scholar]
- Zou, J; Crews, F. CREB and NF-kappaB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cell Mol Neurobiol 2006, 26 . [Google Scholar]
- O’Driscoll, C; Wallace, D; Cotter, TG. bFGF promotes photoreceptor cell survival in vitro by PKA-mediated inactivation of glycogen synthase kinase 3beta and CREB-dependent Bcl-2 up-regulation. J Neurochem 2007, 103(3), 860–870. [Google Scholar]
- Bedogni, B; Pani, G; Colavitti, R; Riccio, A; Borrello, S; Murphy, M; Smith, R; Eboli, ML; Galeotti, T. Redox regulation of cAMP-responsive element-binding protein and induction of manganous superoxide dismutase in nerve growth factor-dependent cell survival. J Biol Chem 2003, 278(19), 16510–16519. [Google Scholar]
- Brautigam, VM; Frasier, C; Nikodemova, M; Watters, JJ. Purinergic receptor modulation of BV-2 microglial cell activity: potential involvement of p38 MAP kinase and CREB. J Neuroimmunol 2005, 166 . [Google Scholar]
- Corrêa, G; Marques da Silva, C; de Abreu Moreira-Souza, AC; Vommaro, RC; Coutinho-Silva, R. Activation of the P2X (7) receptor triggers the elimination of Toxoplasma gondii tachyzoites from infected macrophages. Microbes Infect 2010, 12(6), 497–504. [Google Scholar]
- Lees, MP; Fuller, SJ; McLeod, R; Boulter, NR; Miller, CM; Zakrzewski, AM; Mui, EJ; Witola, WH; Coyne, JJ; Hargrave, AC; Jamieson, SE; Blackwell, JM; Wiley, JS; Smith, NC. P2X7 receptor-mediated killing of an intracellular parasite, Toxoplasma gondii, by human and murine macrophages. J Immunol 2010, 184(12), 7040–7046. [Google Scholar]
- Lammas, DA; Stober, C; Harvey, CJ; Kendrick, N; Panchalingam, S; Kumararatne, DS. ATP-induced killing of mycobacteria by human macrophages is mediated by purinergic P2Z (P2X7) receptors. Immunity 1997, 7(3), 433–444. [Google Scholar]
- Kusner, DJ; Adams, J. ATP-induced killing of virulent Mycobacterium tuberculosis within human macrophages requires phospholipase D. J Immunol 2000, 164(1), 379–388. [Google Scholar]
- Biswas, D; Qureshi, OS; Lee, WY; Croudace, JE; Mura, M; Lammas, DA. ATP-induced autophagy is associated with rapid killing of intracellular mycobacteria within human monocytes/macrophages. BMC Immunol 2008, 9, 35. [Google Scholar]
- Fairbairn, IP; Stober, CB; Kumararatne, DS; Lammas, DA. ATP-mediated killing of intracellular mycobacteria by macrophages is a P2X (7)-dependent process inducing bacterial death by phagosome-lysosome fusion. J Immunol 2001, 167(6), 3300–3307. [Google Scholar]
- Britton, WJ; Fernando, SL; Saunders, BM; Sluyter, R; Wiley, JS. The genetic control of susceptibility to Mycobacterium tuberculosis. Novartis Found Symp 2007, 281, 79–89, discussion 89–92. [Google Scholar]
- Nino-Moreno, P; Portales-Pérez, D; Hernández-Castro, B; Portales-Cervantes, L; Flores-Meraz, V; Baranda, L; Gómez-Gómez, A; Acuna-Alonzo, V; Granados, J; González-Amaro, R. P2X7 and NRAMP1/SLC11 A1 gene polymorphisms in Mexican mestizo patients with pulmonary tuberculosis. Clin Exp Immunol 2007, 148(3), 469–477. [Google Scholar]
- Franco-Martinez, S; Nino-Moreno, P; Bernal-Silva, S; Baranda, L; Rocha-Meza, M; Portales-Cervantes, L; Lyseca-Espinosa, E; Gonzzalez-Amaro, R; Portales-Perez, D. Expression and function of the purinergic receptor P2X7 in patients with pulmonary tuberculosis. Clin Exp Immunol 2006, 146(2), 253–261. [Google Scholar]
- Fernando, SL; Saunders, BM; Sluyter, R; Skarratt, KK; Goldberg, H; Marks, GB; Wiley, JS; Britton, WJ. A polymorphism in the P2X7 gene increases susceptibility to extrapulmonary tuberculosis. Am J Respir Crit Care Med 2007, 175(4), 360–366. [Google Scholar]
- Sambasivan, V; Murthy, KJ; Reddy, R; Vijayalakshimi, V; Hasan, Q. P2X7 gene polymorphisms and risk assessment for pulmonary tuberculosis in Asian Indians. Dis Markers 2010, 28(1), 43–48. [Google Scholar]
- Darville, T; Welter-Stahl, L; Cruz, C; Sater, AA; Andrews, CW, Jr; Ojcius, DM. Effect of the purinergic receptor P2X7 on Chlamydia infection in cervical epithelial cells and vaginally infected mice. J Immunol 2007, 179(6), 3707–3714. [Google Scholar]
- Coutinho-Silva, R; Stahl, L; Raymond, MN; Jungas, T; Verbeke, P; Burnstock, G; Darville, T; Ojcius, DM. Inhibition of chlamydial infectious activity due to P2X7R-dependent phospholipase D activation. Immunity 2003, 19(3), 403–412. [Google Scholar]
- Perfettini, JL; Darville, T; Gachelin, G; Souque, P; Huerre, M; Dautry-Varsat, A; Ojcius, DM. Effect of Chlamydia trachomatis infection and subsequent tumor necrosis factor alpha secretion on apoptosis in the murine genital tract. Infect Immun 2000, 68(4), 2237–2244. [Google Scholar]
- Coutinho-Silva, R; Perfettini, JL; Persechini, PM; Dautry-Varsat, A; Ojcius, DM. Modulation of P2Z/P2X (7) receptor activity in macrophages infected with Chlamydia psittaci. Am J Physiol Cell Physiol 2001, 280(1), C81–89. [Google Scholar]
- Coutinho-Silva, R; Corrêa, G; Sater, AA; Ojcius, DM. The P2X (7) receptor and intracellular pathogens: a continuing struggle. Purinergic Signal 2009, 5(2), 197–204. [Google Scholar]
- Kolli, BK; Kostal, J; Zaborina, O; Chakrabarty, AM; Chang, KP. Leishmania-released nucleoside diphosphate kinase prevents ATP-mediated cytolysis of macrophages. Mol Biochem Parasitol 2008, 158(2), 163–175. [Google Scholar]
- Zaborina, O; Li, X; Cheng, G; Kapatral, V; Chakrabarty, AM. Secretion of ATP-utilizing enzymes, nucleoside diphosphate kinase and ATPase, by Mycobacterium bovis BCG: sequestration of ATP from macrophage P2Z receptors. Mol Microbiol 1999, 31(5), 1333–1343. [Google Scholar]
- Yilmaz, O. The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology 2008, 154 Pt 10, 2897–2903. [Google Scholar]
- Yilmaz, O; Verbeke, P; Lamont, RJ; Ojcius, DM. Intercellular spreading of Porphyromonas gingivalis infection in primary gingival epithelial cells. Infect Immun 2006, 74(1), 703–710. [Google Scholar]
- Avila, M; Ojcius, DM; Yilmaz, O. The oral microbiota: living with a permanent guest. DNA Cell Biol 2009, 28(8), 405–411. [Google Scholar]
- Yilmaz, O; Yao, L; Maeda, K; Rose, TM; Lewis, EL; Duman, M; Lamont, RJ; Ojcius, DM. ATP scavenging by the intracellular pathogen Porphyromonas gingivalis inhibits P2X7- mediated host-cell apoptosis. Cell Microbiol 2008, 10(4), 863–875. [Google Scholar]
- Choi, CH; DeGuzman, J; Yilmaz, O. Modulating of “danger signal” ATP induced reactiveoxygen- species production in primary gingival epithelial cells by Porphyromonas gingivalis; submitted for publication; 2011. [Google Scholar]
- Yilmaz, O; Jungas, T; Verbeke, P; Ojcius, DM. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun 2004, 72(7), 3743–3751. [Google Scholar]
- Yao, L; Jermanus, C; Barbetta, B; Choi, C; Verbeke, P; Ojcius, DM; Yilmaz, O. Porphyromonas gingivalis infection sequesters pro-apoptotic Bad through Akt in primary gingival epithelial cells. Mol Oral Microbiol 2010, 25(2), 89–101. [Google Scholar]
- Mao, S; Park, Y; Hasegawa, Y; Tribble, GD; James, CE; Handfield, M; Stavropoulos, MF; Yilmaz, O; Lamont, RJ. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol 2007, 9(8), 1997–2007. [Google Scholar]
- Hu, Y; Fisette, PL; Denlinger, LC; Guadarrama, AG; Sommer, JA; Proctor, RA; Bertics, PJ. Purinergic receptor modulation of lipopolysaccharide signaling and inducible nitric-oxide synthase expression in RAW 264.7 macrophages. J Biol Chem 1998, 273(42), 27170–27175. [Google Scholar]
- Auger, R; Motta, I; Benihoud, K; Ojcius, DM; Kanellopoulos, JM. A role for mitogen-activated protein kinase (Erk1/2) activation and non-selective pore formation in P2X7 receptor-mediated thymocyte death. J Biol Chem 2005, 280(30), 28142–28151. [Google Scholar]
- Saunders, JA; Rogers, LC; Klomsiri, C; Poole, LB; Daniel, LW. Reactive oxygen species mediate lysophosphatidic acid induced signaling in ovarian cancer cells. Free Radic Biol Med 2010, 49(12), 2058–2067. [Google Scholar]
- Abdul-Sater, AA; Koo, E; Häcker, G; Ojcius, DM. Inflammasome-dependent caspase-1 activation in cervical epithelial cells stimulates growth of the intracellular pathogen Chlamydia trachomatis. J Biol Chem 2009, 284(39), 26789–26796. [Google Scholar]
© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Spooner, R.; Yilmaz, Ö. The Role of Reactive-Oxygen-Species in Microbial Persistence and Inflammation. Int. J. Mol. Sci. 2011, 12, 334-352. https://doi.org/10.3390/ijms12010334
Spooner R, Yilmaz Ö. The Role of Reactive-Oxygen-Species in Microbial Persistence and Inflammation. International Journal of Molecular Sciences. 2011; 12(1):334-352. https://doi.org/10.3390/ijms12010334
Chicago/Turabian StyleSpooner, Ralee, and Özlem Yilmaz. 2011. "The Role of Reactive-Oxygen-Species in Microbial Persistence and Inflammation" International Journal of Molecular Sciences 12, no. 1: 334-352. https://doi.org/10.3390/ijms12010334