Molecular Photovoltaics in Nanoscale Dimension


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Transport in Organic Semiconductors: Achievements vs. Problems
1.2. Analytical Tools to Study Molecular Photovoltaic Device
1.3. Self-Assembly (SAM) and SAM Molecular Photovoltaic Devices
1.4. Charge Mobility in Naphthalene-Derivative (NTCDI) Thick Film and Self-Assembled (SAM) Devices
1.5. Transport in Naphthalene-Derivative (NTCDI) Self-Assembled (SAM) Devices
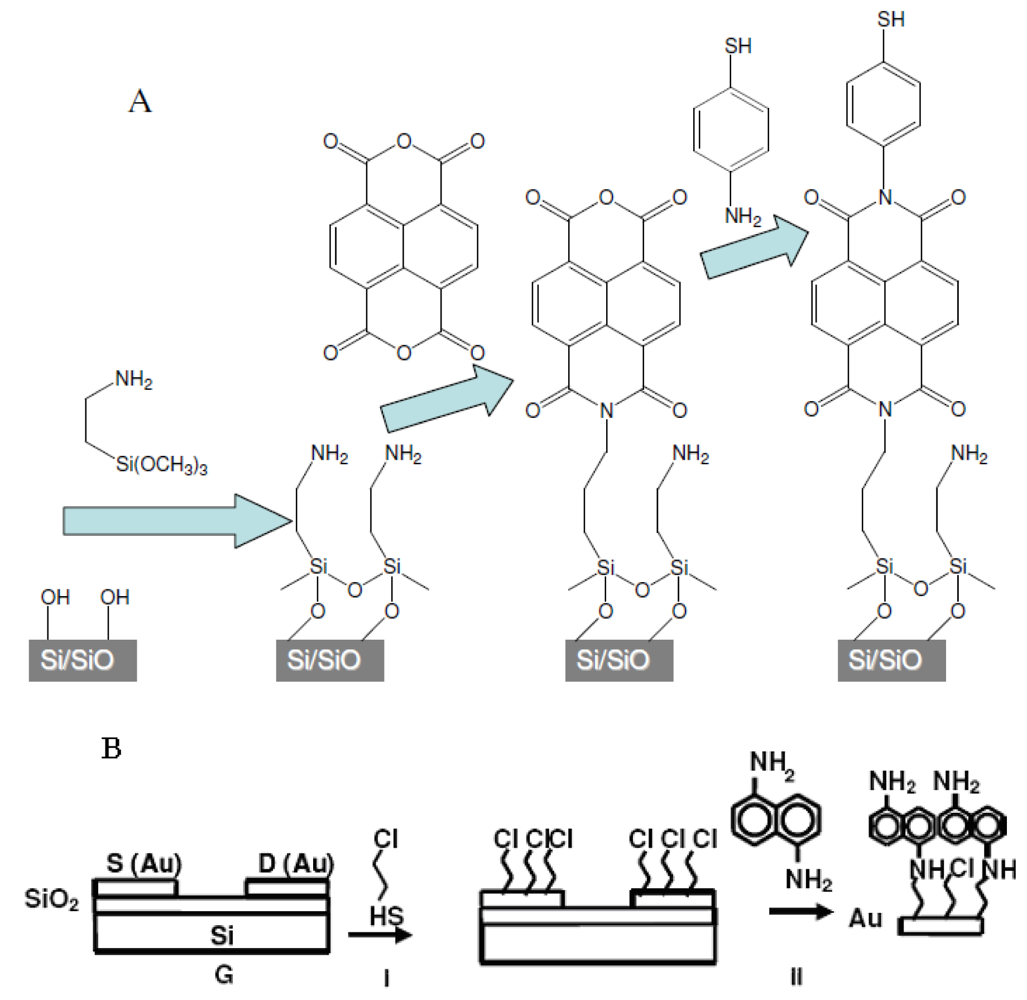
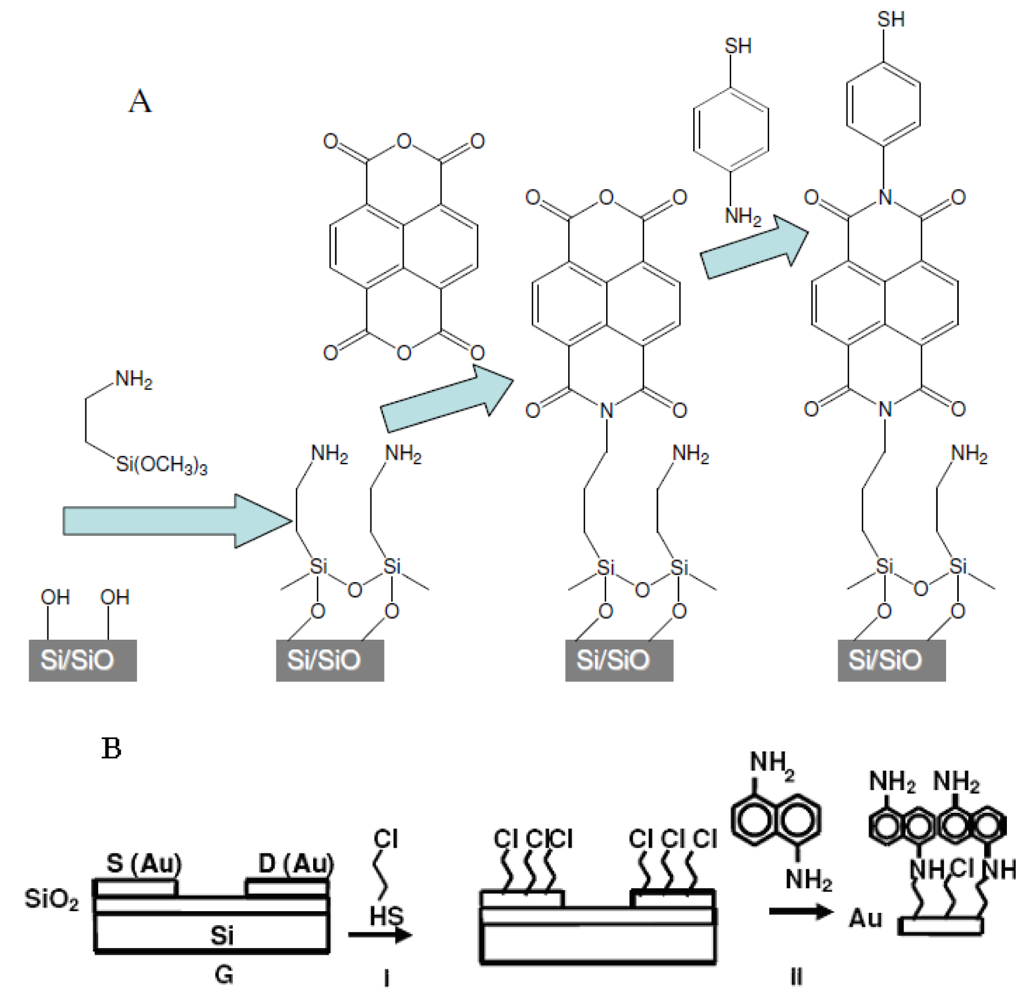
1.6. Background of SAM Fabrication Technology
1.7. The Scope of This Review
2. Materials and Methods
2.1. Preparation of Organic Films
2.2. Characterization of the NTCDI Monolayer
2.3. SAM-OFET Electrode Configuration
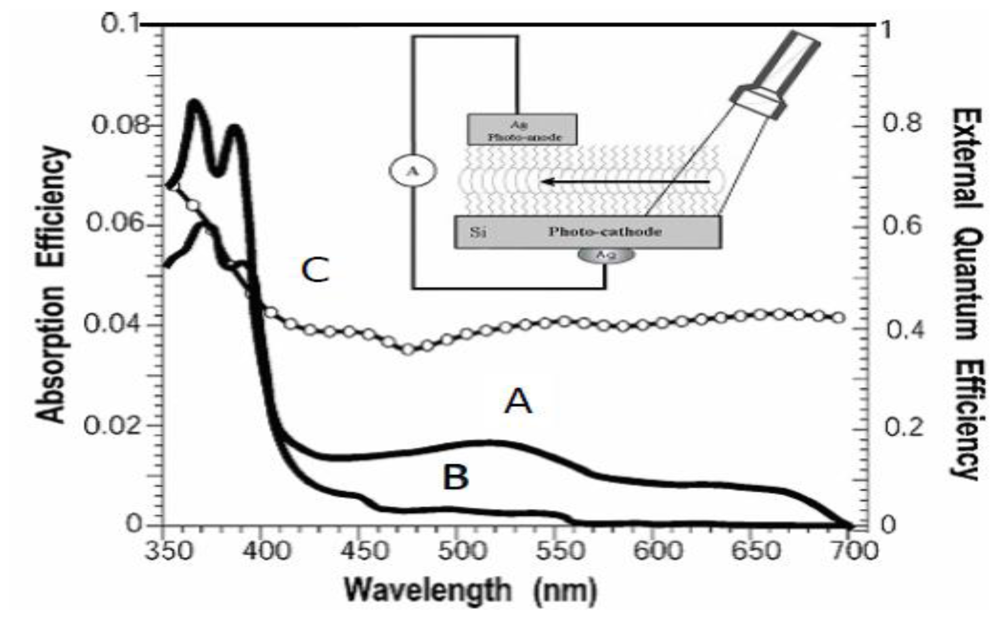
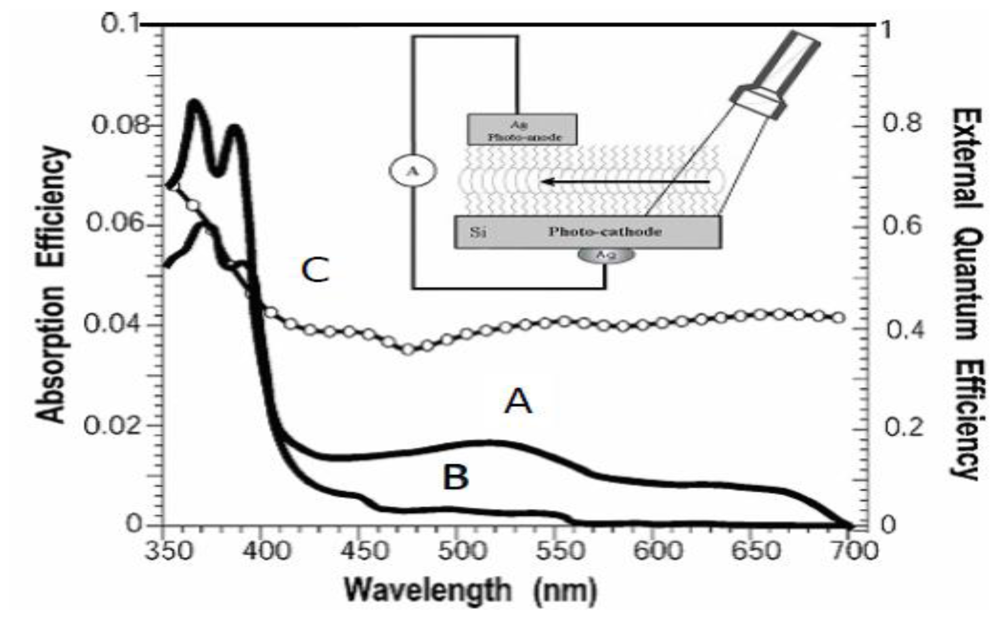
2.4. Determination of the Absorption and External Quantum Efficiencies in Closed-Circuit SAM-PVCs
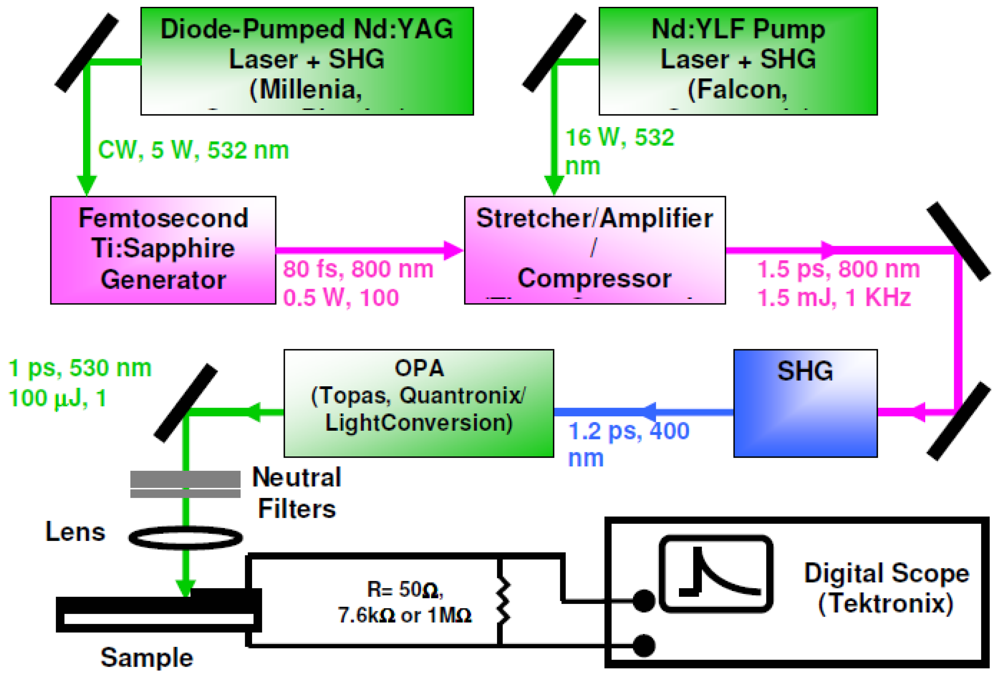
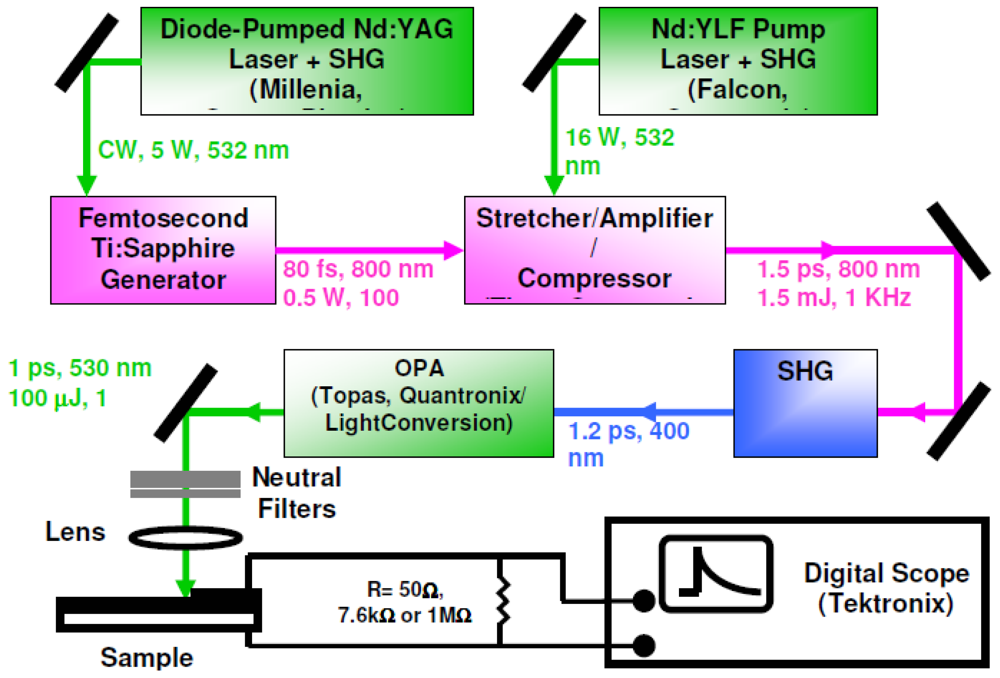
2.5. Time-Resolved Photovoltaic Measurements in Closed-Circuit SAM-PVCs
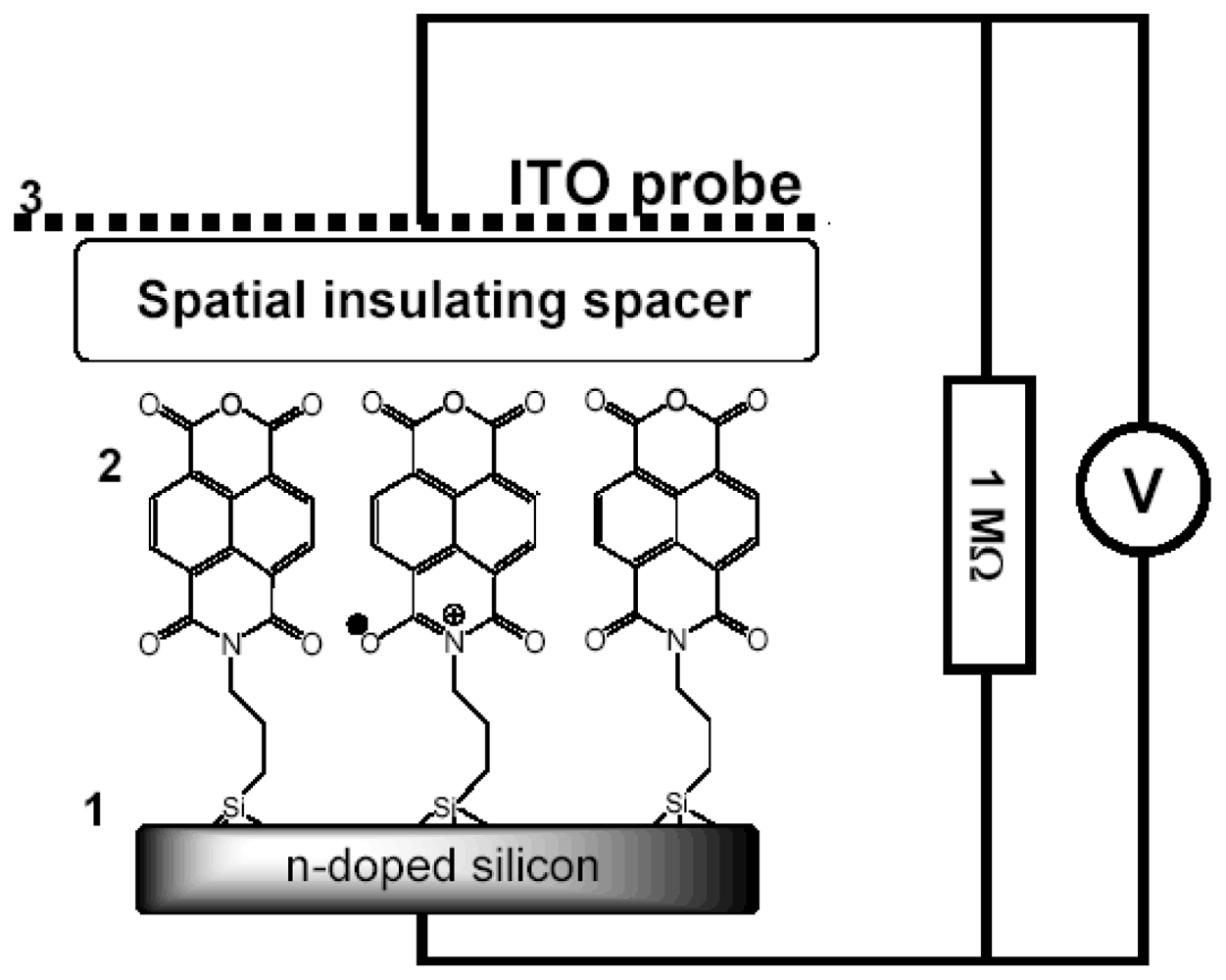
2.6. Capacitance Coupling (Open-Circuit) Measurements in SAM-PVC
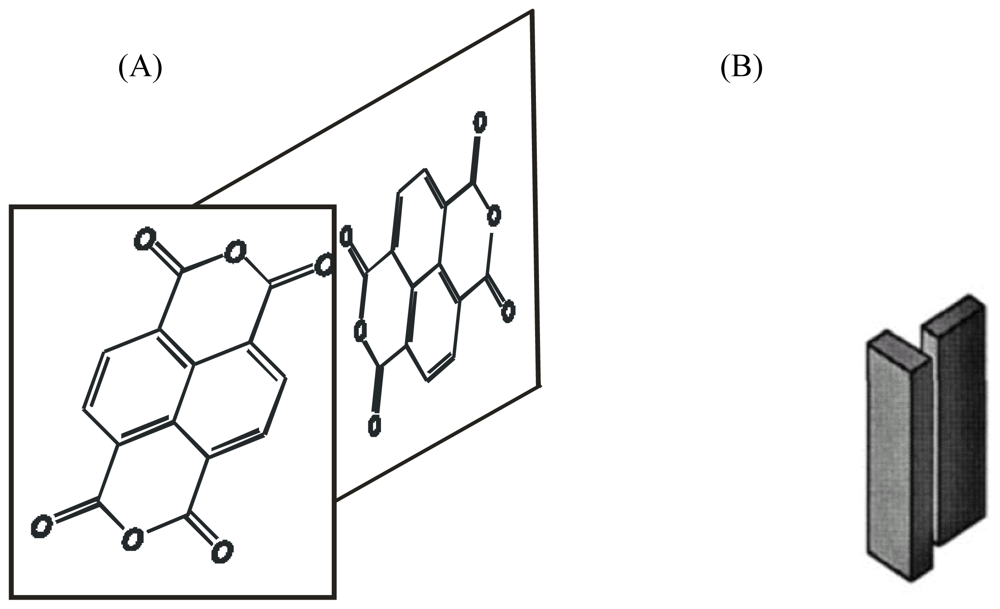
2.7. Synthesis of Naphthalene-Diimide Model Compounds
3. Results
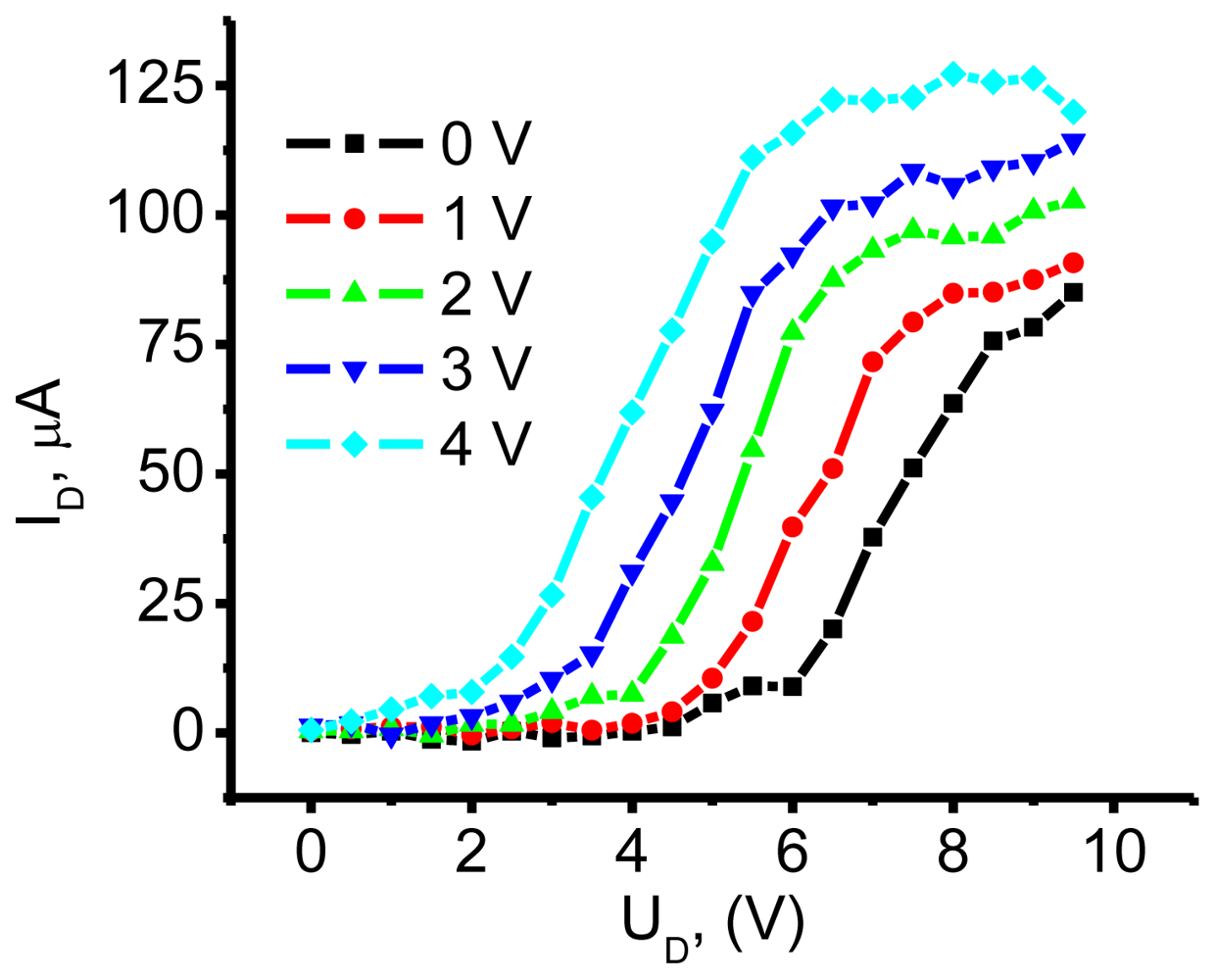
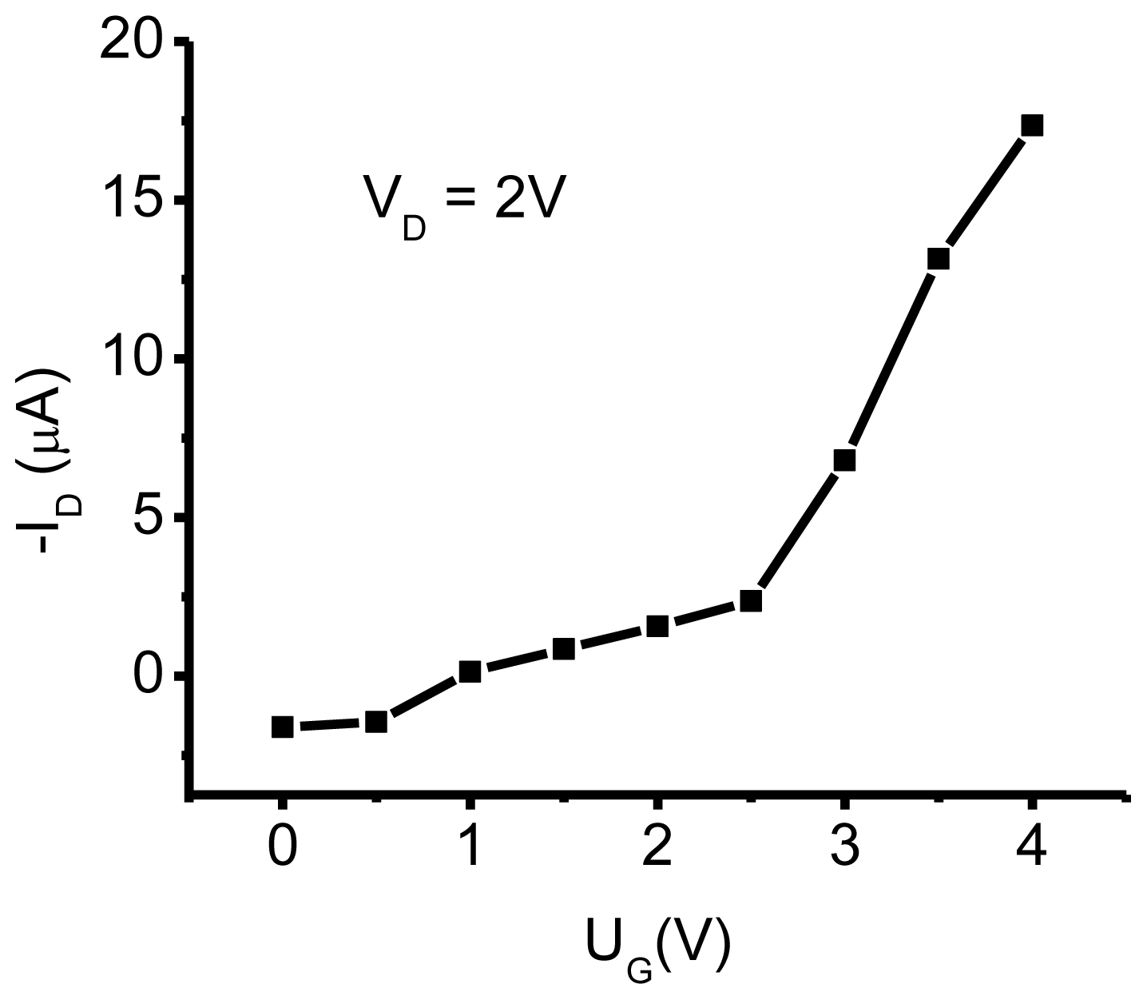
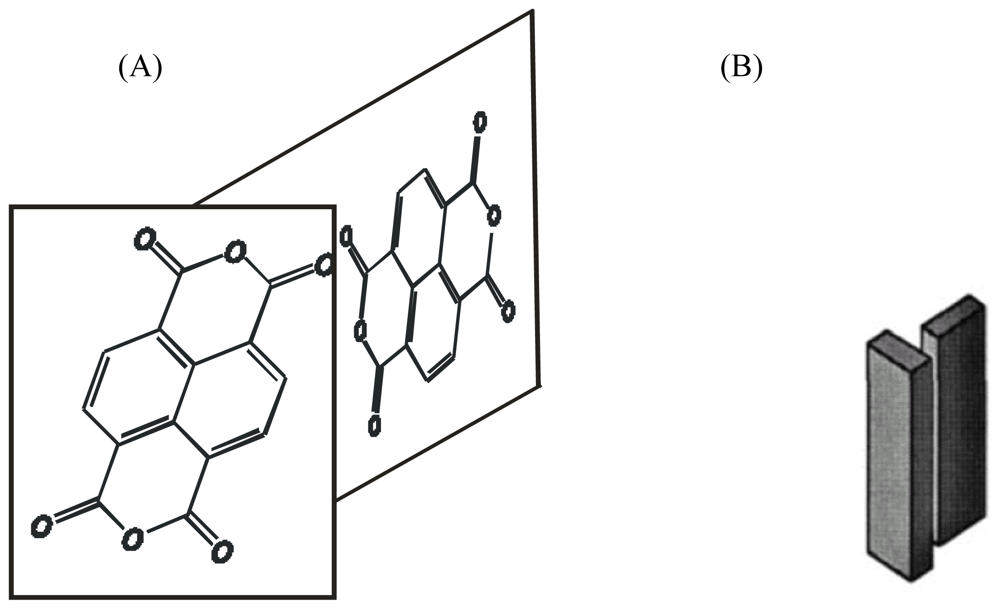
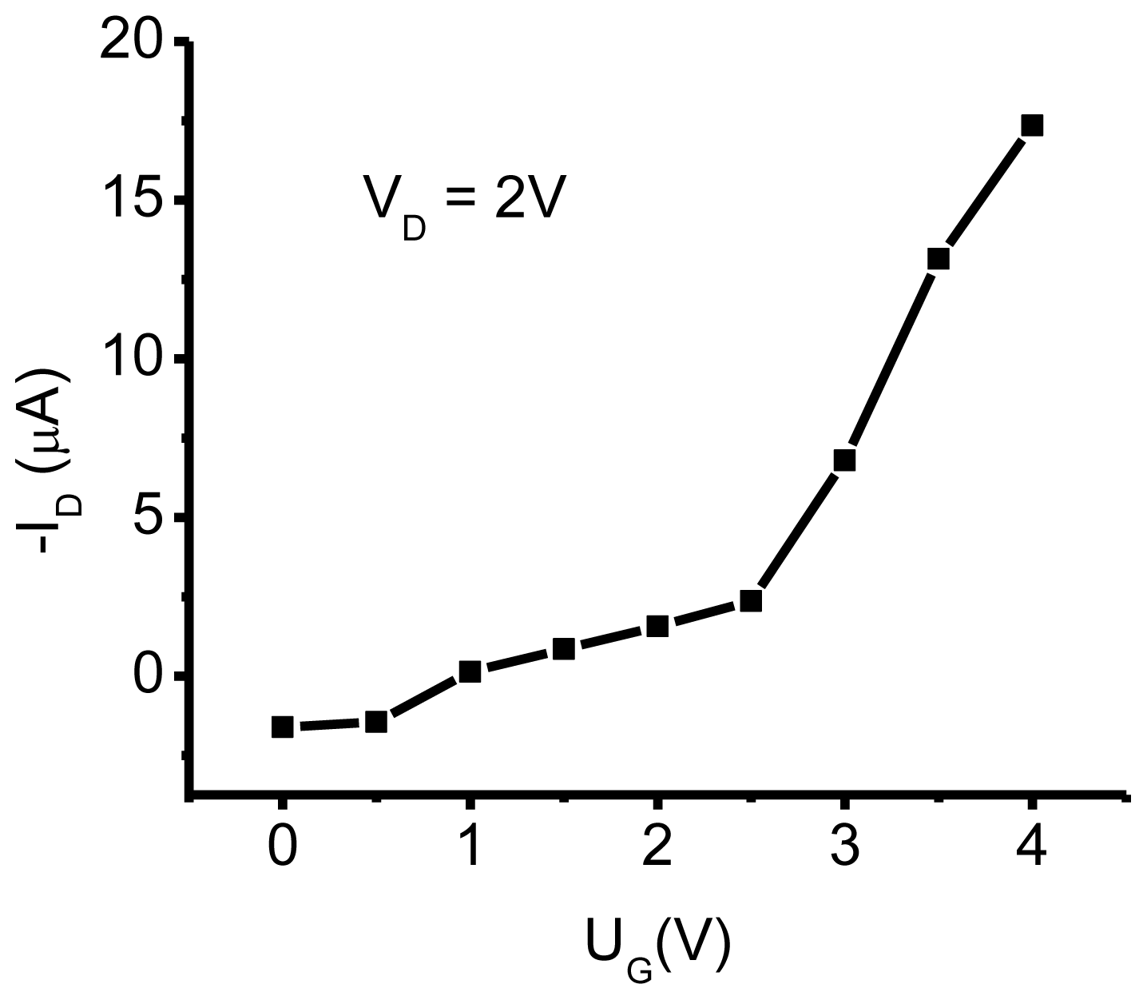
3.1. NTCDI SAM OFET Devices
3.2. Naphtalene Tetracarboxyldiimide (NTCDI) SAM PVC Devices
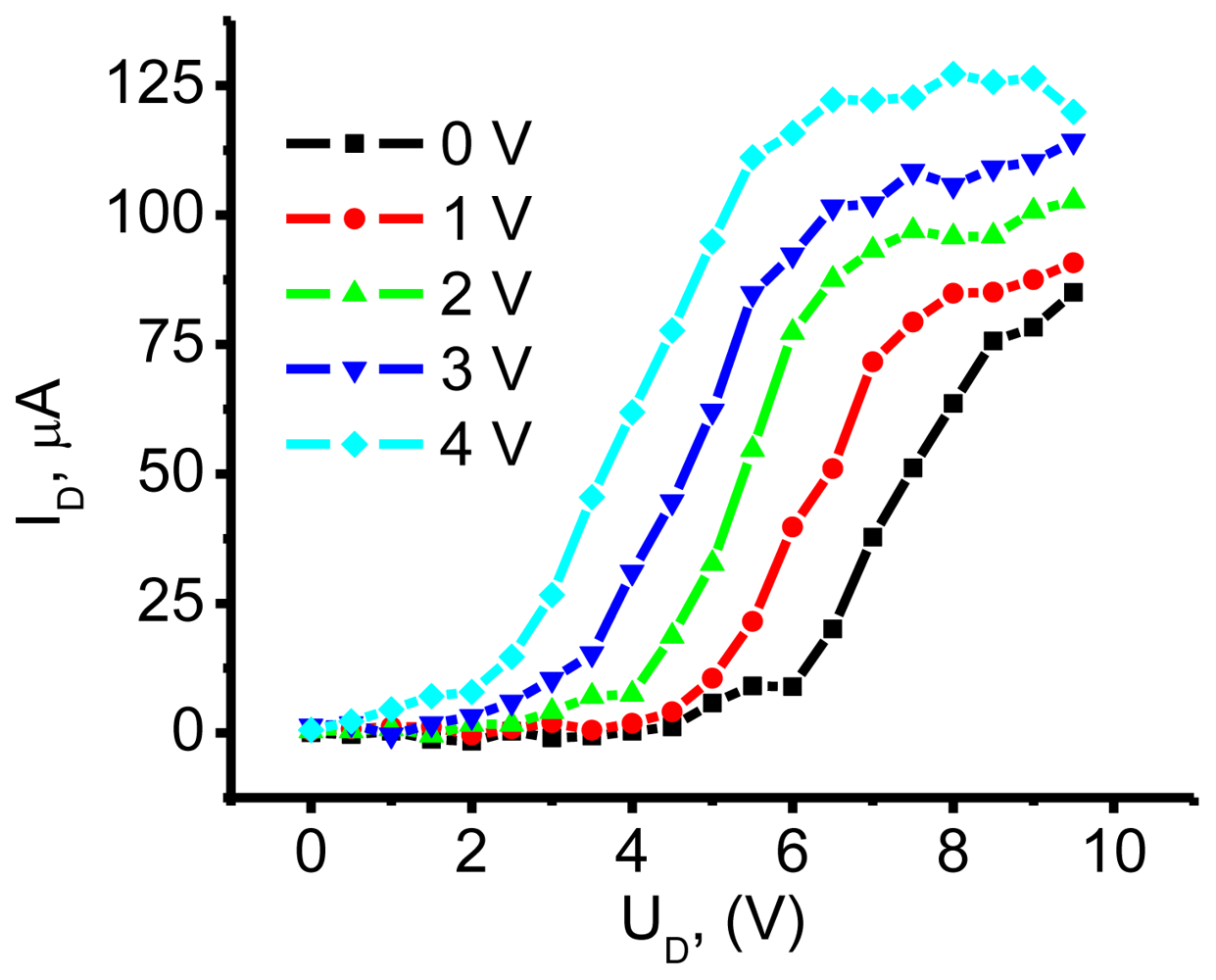
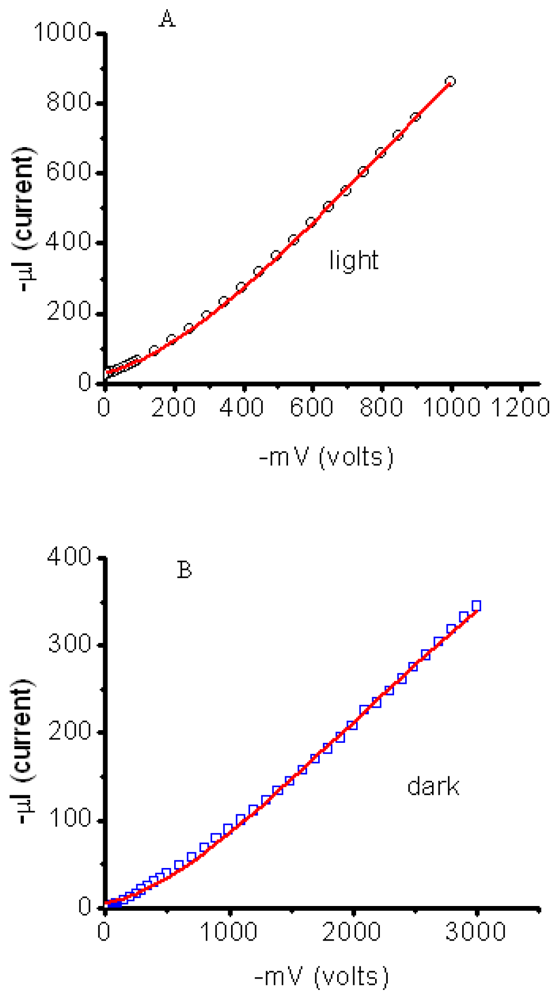
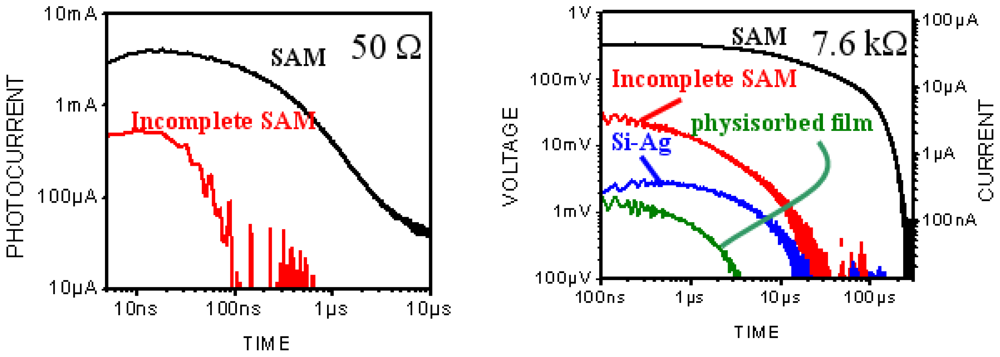
3.2.1. Measurements of In-Plane Currents in SAM-PVC: Closed-Circuit Electrical Measurements
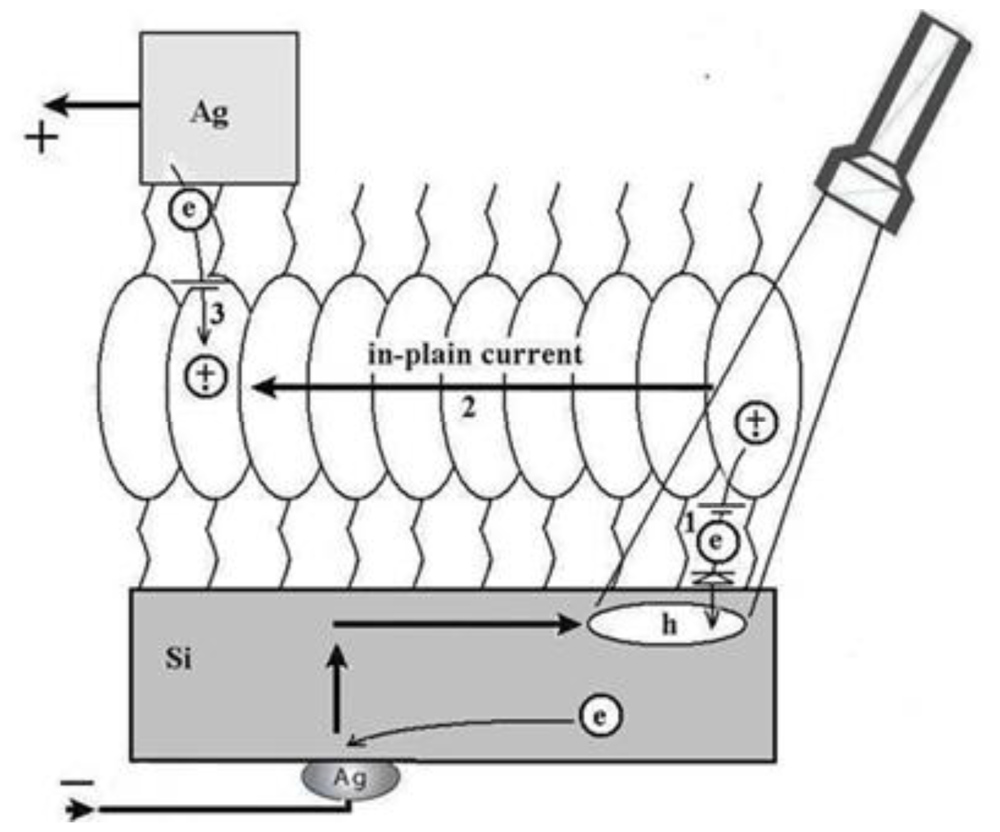
3.2.2. Measurement of Out-of-Plane Charge Recombination in SAM-PVC: Open-Circuit Electrical Measurements
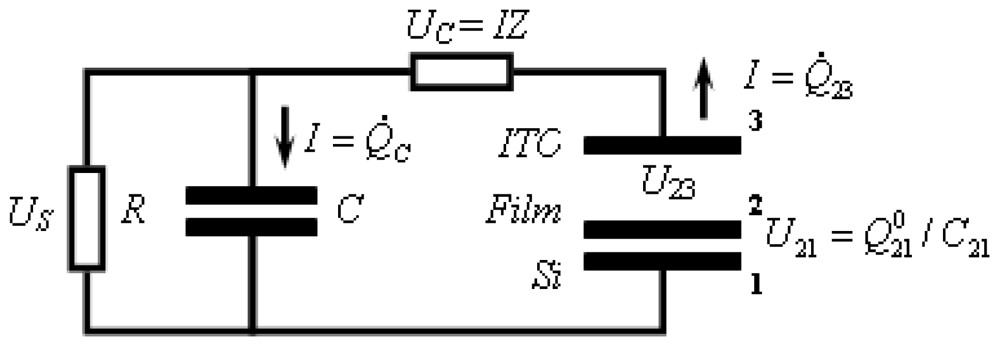
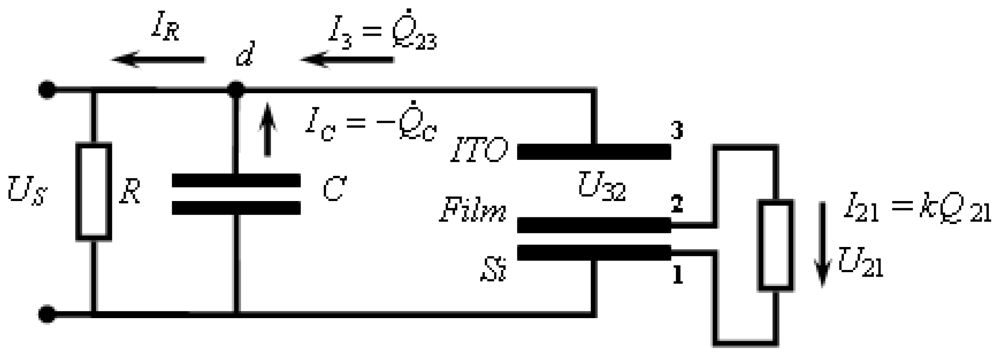
3.2.2.1. Theoretical Background for the Capacitive Probe Measurements
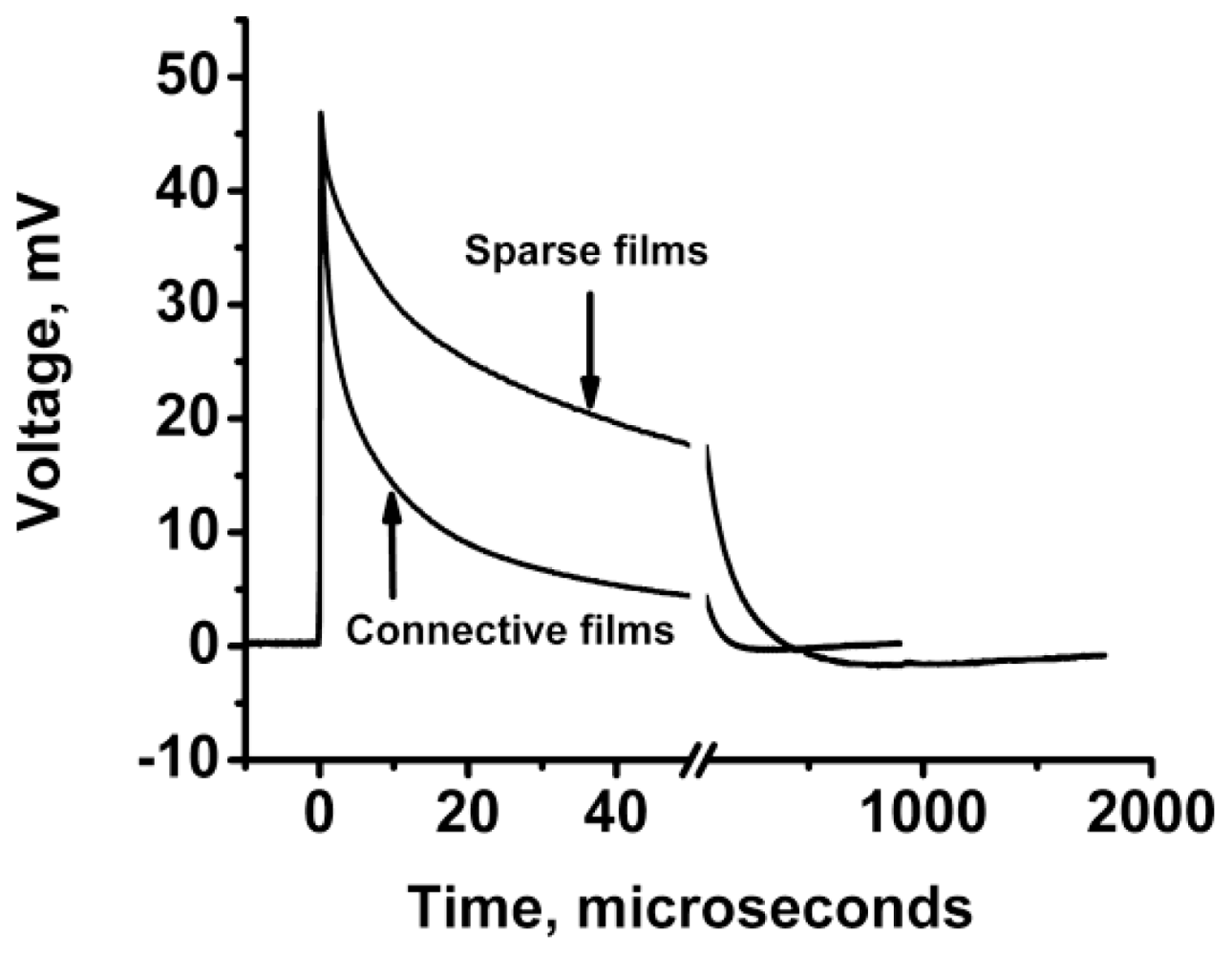
3.2.2.2. PVC Response in Out-of-Plane Direction
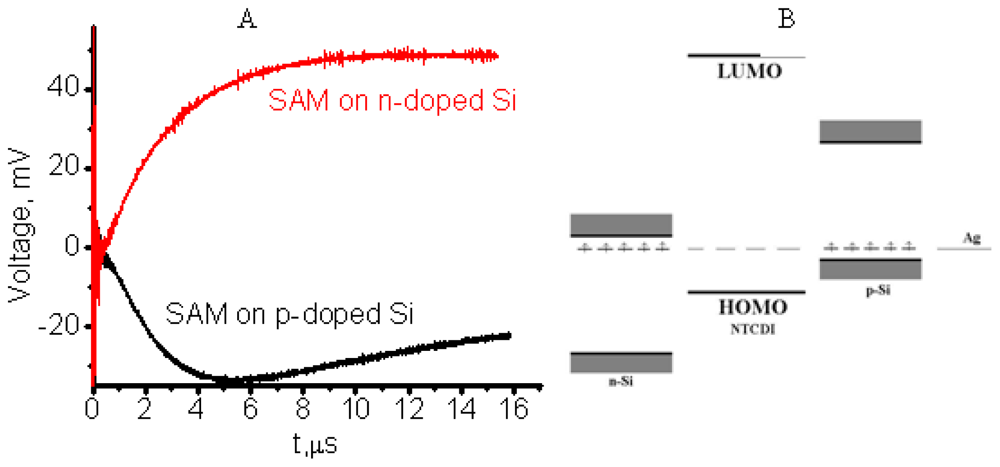
3.2.3. Reversing Conductivity Channel in SAM-PVC (Closed-Circuit Measurements)
4. Discussion
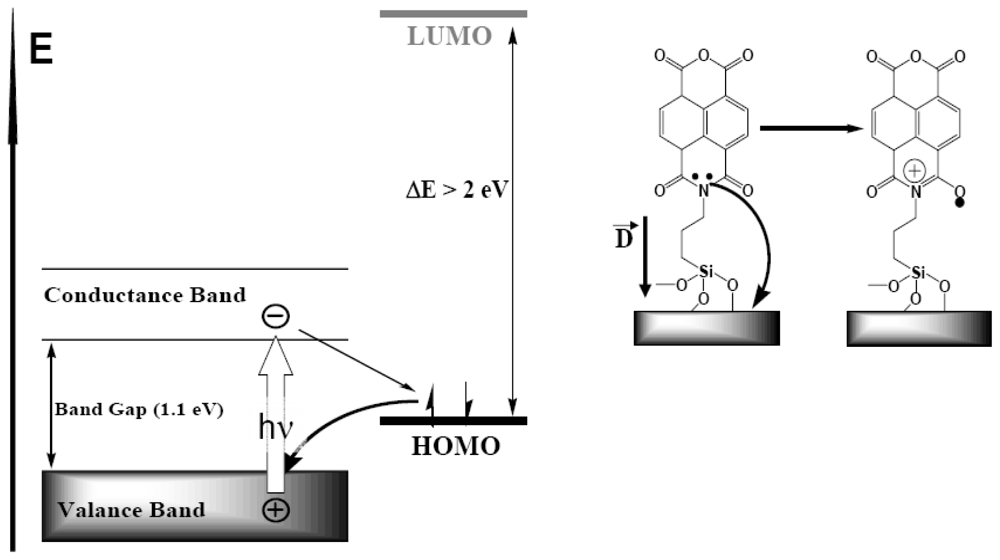
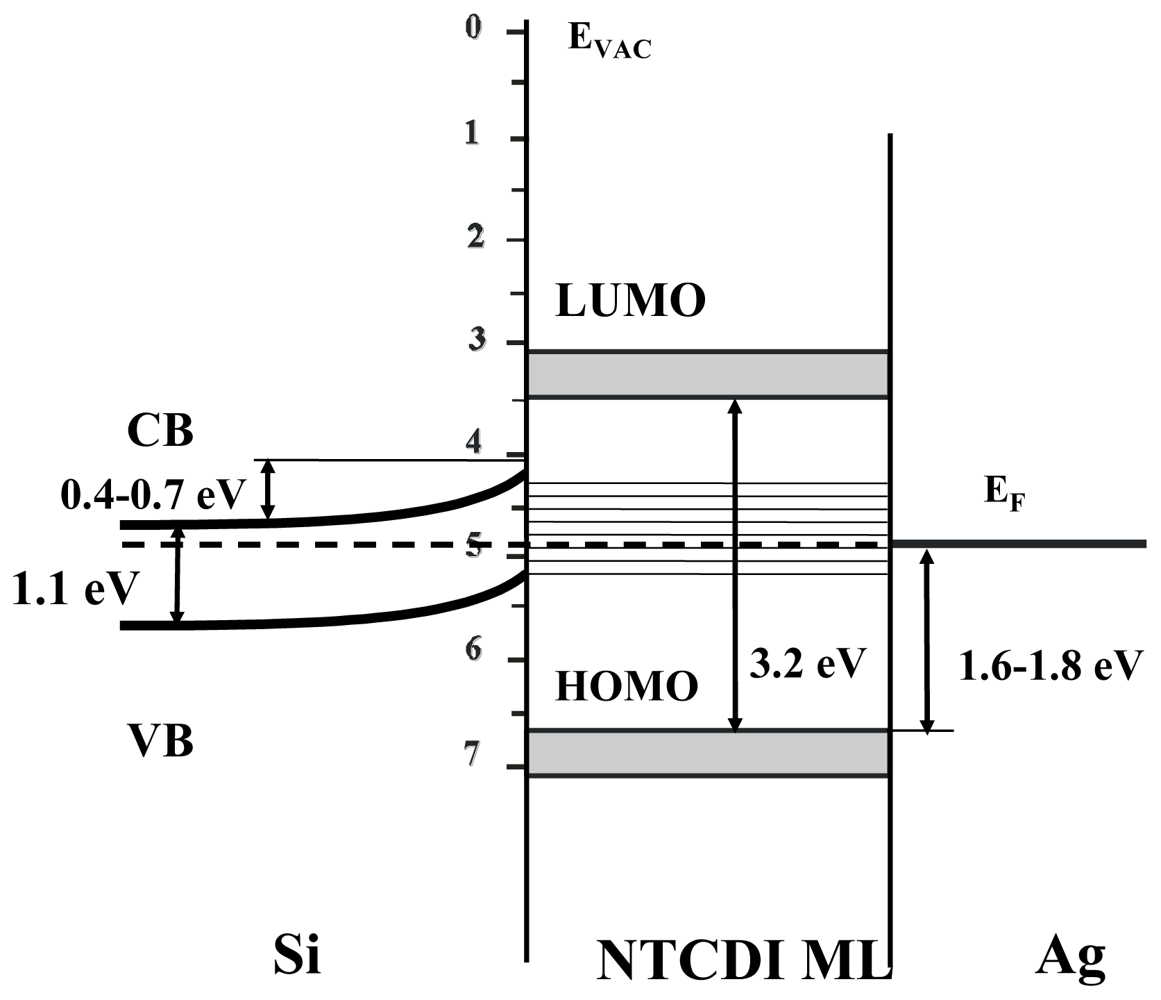
4.1. Polaron Transport Mechanism in SAM PVC Devices
4.2. Model Compounds
4.2.1. Structure of Model Compounds and Liquid Crystals (LCs)
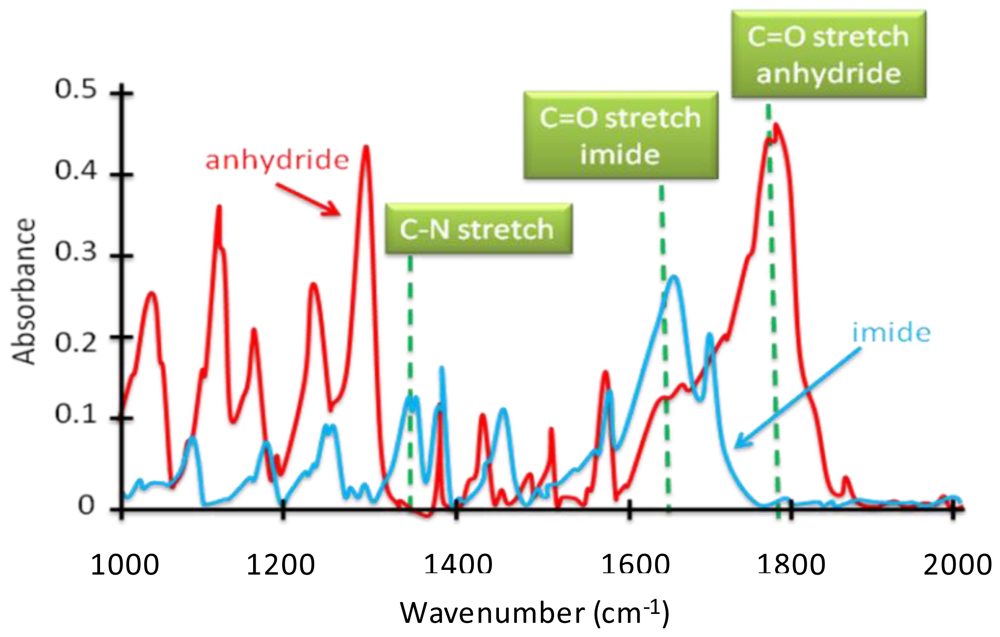
4.2.2. Tracing Imide Bonds in Model Compounds
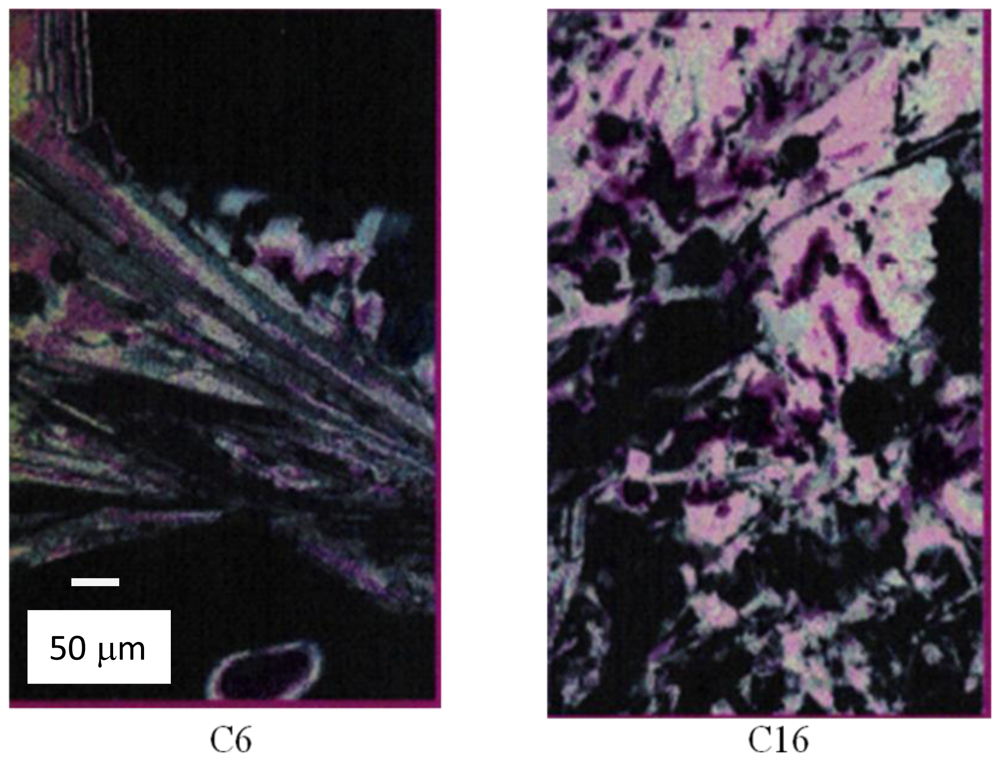
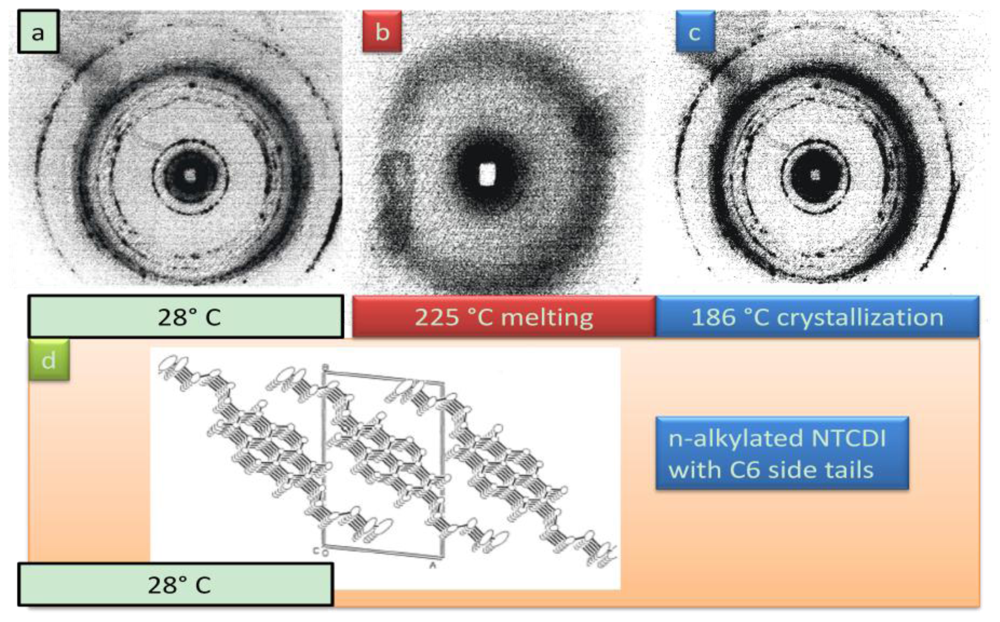
4.2.3. Phase Properties of Model Compounds
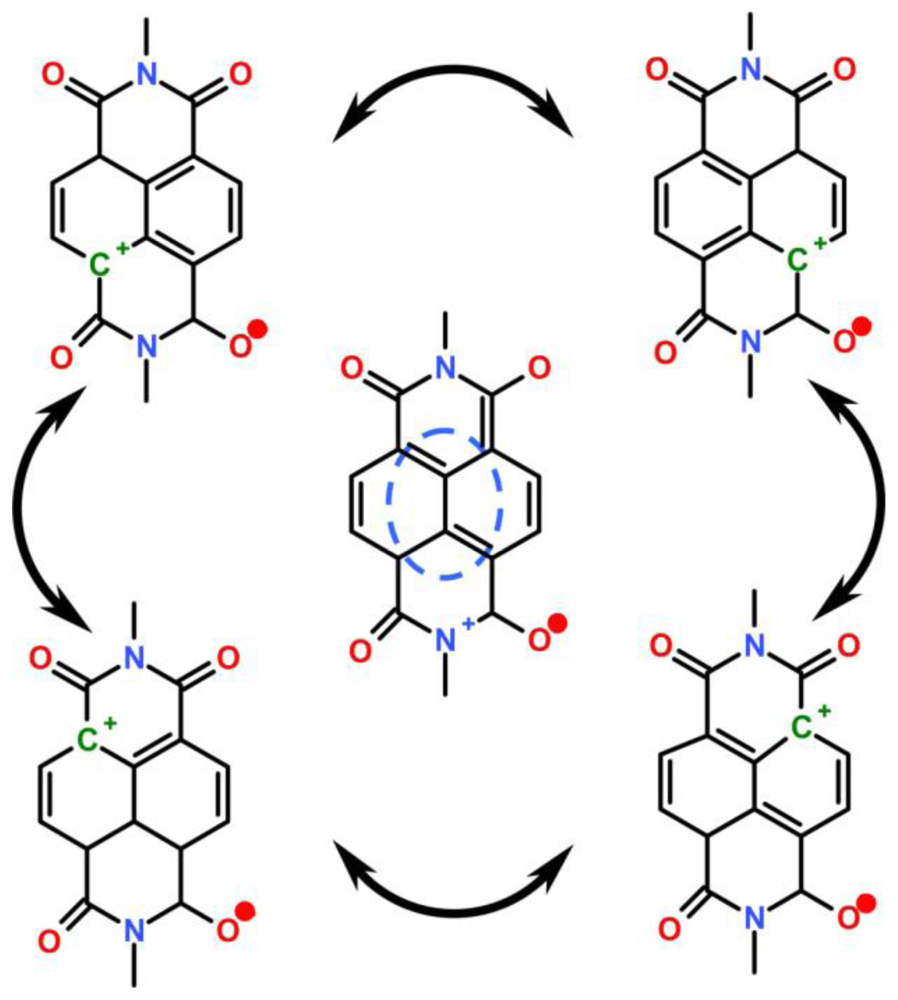
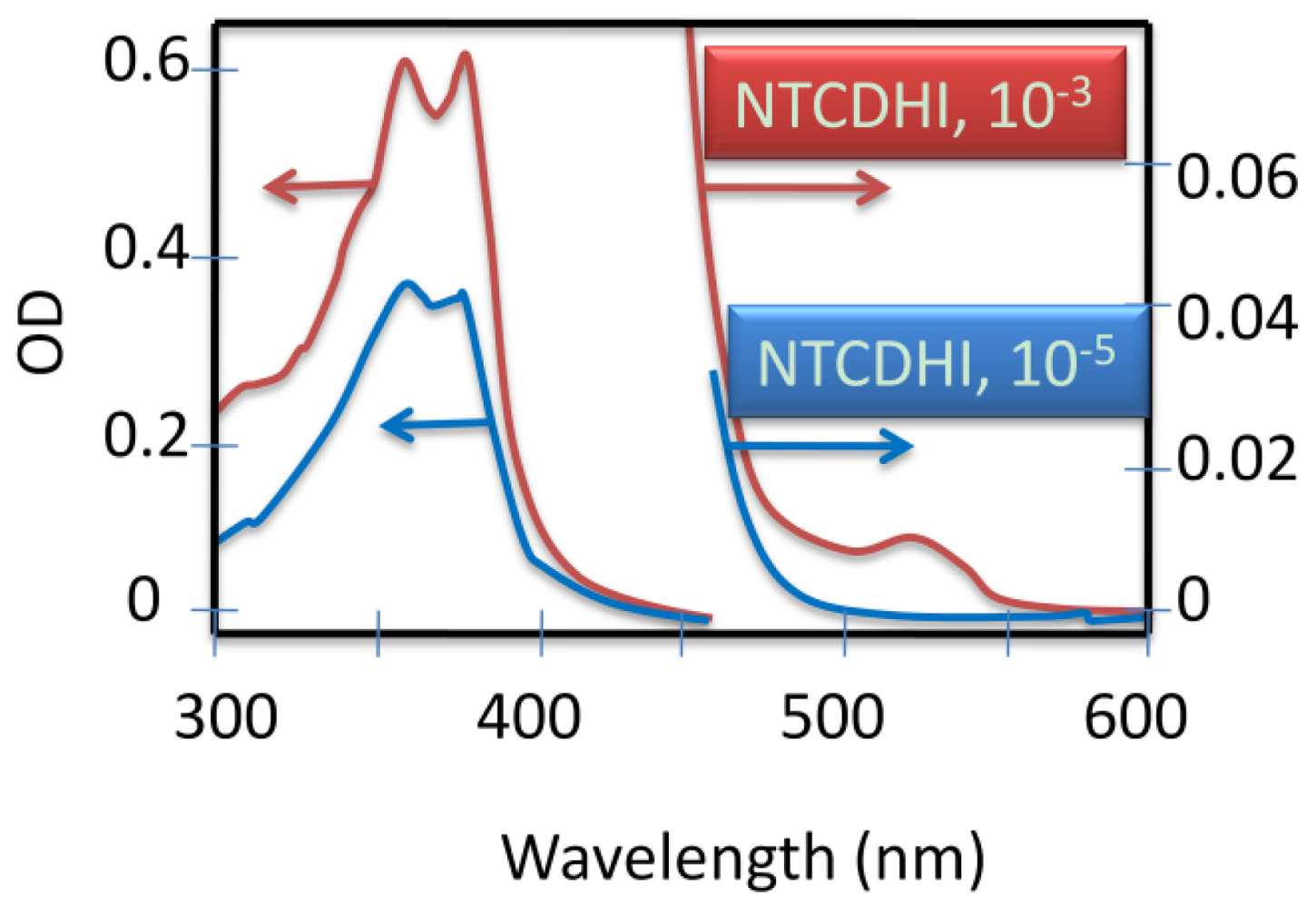
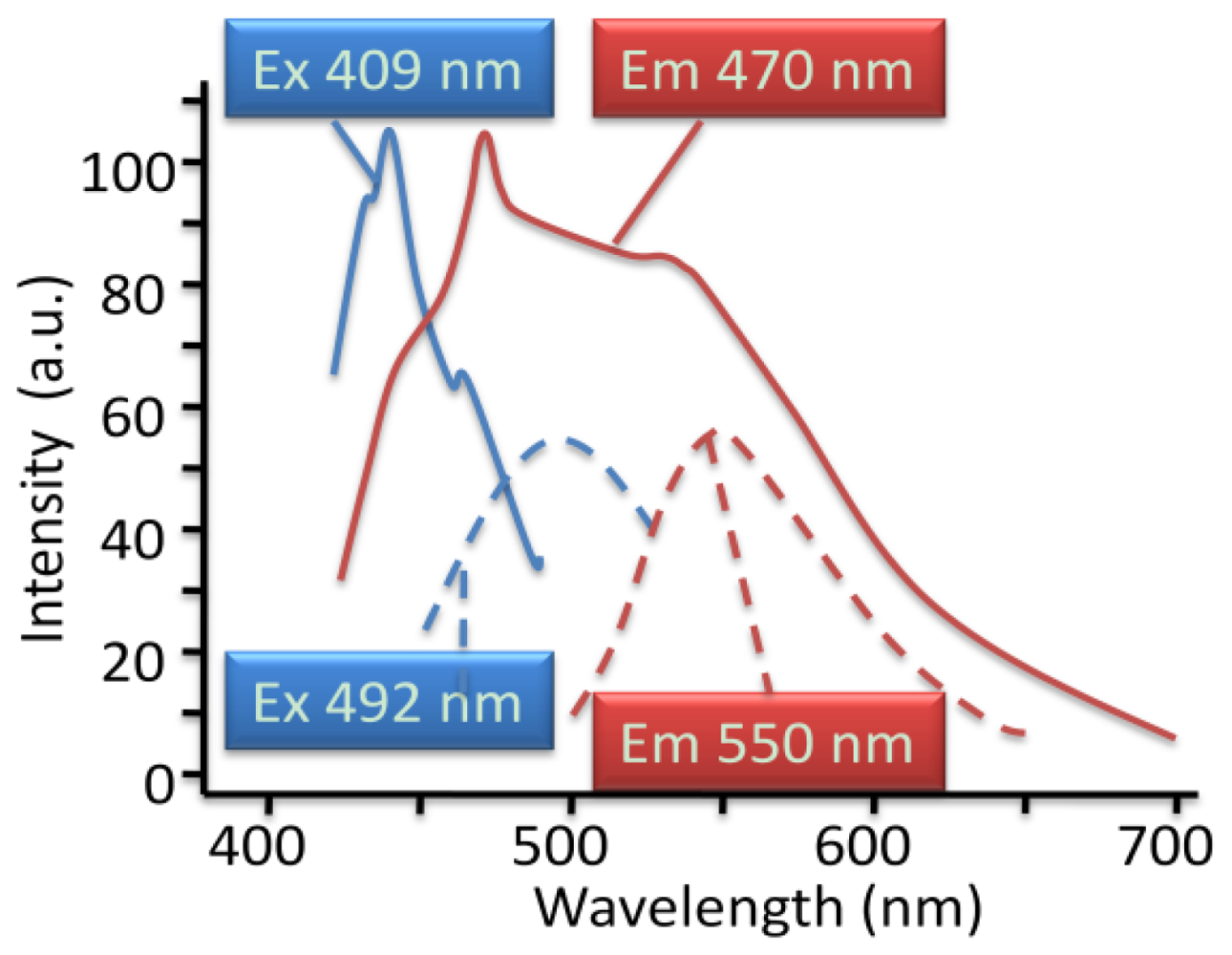
4.2.4. Analogy of π-Aggregation in Model Compounds and VP-SAM Structures
4.2.5. Weak Delocalization in SAMs and Model Compounds
5. Conclusion
Acknowledgments
References
- Aviram, A; Ratner, MA. Molecular rectifiers. Chem. Phys. Lett 1974, 29, 277–283. [Google Scholar]
- Katz, HE; Bao, Z. The Physical Chemistry of Organic Field-Effect Transistors. J. Phys. Chem B 2000, 104, 671–678. [Google Scholar]
- Horowitz, G. Organic Thin Film Transistors: From Theory to Real Devices. J. Mater. Res 2004, 19, 1946–1962. [Google Scholar]
- Newman, CR; Frisbie, CD; Da Silva Filho, DA; Bredas, JL; Ewbank, PC; Mann, KR. Introduction to Organic Thin Film Transistors and Design of n-Channel Organic Semiconductors. Chem. Mater 2004, 16, 4436–4451. [Google Scholar]
- Da Silva Pinheiro, C; Silva, GME. Dynamics of polarons and bipolarons with interchain coupling in conjugated polymers. Int. J. Quantum Chem 2005, 95, 153–158. [Google Scholar]
- Zelichenok, A; Burtman, V; Zenou, N; Yitzchaik, S; Di Bella, S; Meshulam, G; Kotler, Z. Quinolinium Derived Acentric Crystals for Second-Order NLO Applications with Transparency in the Blue. J. Phys. Chem B 1999, 103, 8702–8701. [Google Scholar]
- Moukouri, S; Allen, S; Lemay, F; Kyung, B; Poulin, D; Vilk, YM; Tremblay, MS. Many-body theory versus simulations for the pseudogap in the Hubbard model. Phys. Rev B 2000, 61, 7887–7892. [Google Scholar]
- Kobayashi, S; Takenobu, T; Mori, S; Fujiwara, A; Iwasa, Y. Fabrication and characterization of C60 thin-film transistors with high field-effect mobility. Appl. Phys. Lett 2003, 82, 4581. [Google Scholar]
- Chikamatsu, M; Nagamatsu, S; Yoshida, Y; Saito, K; Yase, K; Kikuchi, K. Solution-processed n-type organic thin-film transistors with high field-effect mobility. Appl. Phys. Lett 2005, 87, 203504. [Google Scholar]
- Letizia, JA; Facchetti, AC; Stem, L; Ratner, MA; Marks, TJ. High Electron Mobility in Solution-Cast and Vapor-Deposited Phenacyl-Quaterthiophene Based Field-Effect Transistors: Toward N-Type Polythiophenes. J. Am. Chem. Soc 2005, 127, 13476–13477. [Google Scholar]
- Jones, BA; Ahrens, MJ; Yoon, MH; Facchetti, A; Marks, TJ; Wasielewski, MR. High-Mobility Air-Stable n-Type Semiconductors with Processing Versatility: Dicyano Perylene-3,4:9,10-bis(dicarboximides). Angew. Chem. Int. Ed 2004, 43, 6363. [Google Scholar]
- Chen, HZ; Ling, MM; Mo, X; Shi, MM; Wang, M; Bao, Z. Air Stable n-Channel Organic Semiconductors for Thin Film Transistors Based on Fluorinated Derivatives of Perylene Diimides. Chem. Mater 2007, 19, 816–824. [Google Scholar]
- Ling, M-M; Erk, P; Gomez, M; Koenemann, M; Locklin, J; Bao, Z. Air-stable n-channel Organic Semiconductors Base on Perylene Diimide Derivatives without Strong Electron Withdrawing Groups. Adv. Mater 2007, 19, 1123–1127. [Google Scholar]
- Chesterfield, RJ; McKeen, JC; Newman, CR; Ewbank, PC; Da Silva Filho, DA; Brédas, JL; Miller, LL; Mann, KR; Frisbie, CD. Organic Thin Film Transistors Based on N-Alkyl Perylene Diimides: Charge Transport Kinetics as a Function of Gate Voltage and Temperature. J. Phys. Chem B 2004, 108, 19281–19292. [Google Scholar]
- Sirringhaus, H. Device Physics of Solution-Processed Organic Field-Effect Transistors. Adv. Mater 2005, 17, 2411–2425. [Google Scholar]
- Newman, CR; Frisbie, CD; Da Silva Filho, DA; Brédas, JL; Ewbank, PC; Mann, KR. Introduction to Organic Thin Film Transistors and Design of n-Channel Organic Semiconductors. Chem. Mater 2004, 16, 4436–4451. [Google Scholar]
- Yoo, B; Jung, T; Basu, D; Dodabalapur, A; Jones, BA; Facchetti, A; Wasielewski, MR; Marks, TJ. High Mobility Bottom-Contact n-Channel Organic Transistors and their Use in Complementary Ring Oscillators. Appl. Phys. Lett 2006, 88, 082104. [Google Scholar]
- Jones, BA; Facchetti, A; Marks, TJ; Wasielewski, MR. Cyanonaphthalene Diimide Semiconductors for Air-Stable, Flexible, and Optically Transparent n-Channel Field-Effect Transistors. Chem. Mater 2007, 19, 2703–2705. [Google Scholar]
- Ando, S; Murakami, R; Nishida, JI; Tuda, H; Inoue, Y; Tokito, S; Yamashita, Y. n-Type Organic Field-Effect Transistors with Very High Electron Mobility Based on Thiazole Oligomers with Trifluoromethylphenyl Groups. J. Am. Chem. Soc 2005, 127, 14996–14997. [Google Scholar]
- Salomon, A; Boecking, T; Chan, CK; Amy, F; Girshevitz, O; Cahen, D; Kahn, A. How Do Electronic Carriers Cross Si-Bound Alkyl Monolayers? Phys. Rev. Lett 2005, 95, 266807. [Google Scholar]
- Salomon, A; Cahen, D; Lindsay, S; Tomfohr, J; Engelkes, VB; Frisbie, CD. Comparison of Electronic Transport Measurements on Organic Molecules. Adv. Mater 2003, 15, 1881–1890. [Google Scholar]
- Zahid, F; Ghosh, AW; Paulsson, M; Polizzi, E; Datta, S. Charging induced asymmetry in molecular conductors. Phys. Rev B 2004, 70, 245317. [Google Scholar]
- Cahen, D; Kahn, A; Umbach, E. Energetics of molecular interfaces. Mater Today 2005, 8, 32–41. [Google Scholar]
- Burtman, V; Hukic, G; Ndobe, AS; Drori, T; Vardeny, ZV. Electrical and Optical Studies of Gap States in Self-Assembled Molecular Aggregates. J. Appl. Phys 2007, 101, 054502. [Google Scholar]
- Burtman, V; Ndobe, AS; Vardeny, ZV. Transport Studies of Isolated Molecular Wires in Self-Assembled Monolayer Devices. J. Appl. Phys 2005, 98, 034314. [Google Scholar]
- Ulman, A. Formation and Structure of Self-Assembled Monolayers. Chem. Rev 1996, 96, 1533–1554. [Google Scholar]
- Ulman, A. An Introduction to Ultrathin Organic Films; Academic Press: Boston, MA, USA, 1991. [Google Scholar]
- Lehn, JM. Toward Self-Organization and Complex Matter. Science 2002, 295, 2400–2403. [Google Scholar]
- Katz, HE; Lovinger, AJ; Johnson, J; Kloc, C; Seigrist, T; Li, W; Lin, YY; Dodabalapur, A. A soluble and air-stable organic semiconductor with high electron mobility. Nature 2000, 404, 478–481. [Google Scholar]
- Katz, HE; Johnson, J; Lovinger, AJ; Li, W. Naphthalenetetracarboxylic Diimide-Based n-Channel Transistor Semiconductors: Structural Variation and Thiol-Enhanced Gold Contacts. J. Am. Chem. Soc 2000, 122, 7787–7792. [Google Scholar]
- Singh, TB; Erten, S; Günes, S; Zafer, C; Turkmen, G; Kuhan, B; Teoman, Y; Sariciftci, NS; Icli, S. Soluble derivatives of perylene and naphthalene diimide for n-channel organic field-effect transistors. Org. Electron 2006, 7, 480–489. [Google Scholar]
- Kao, CC; Lin, P; Lee, CC; Wang, YK; Ho, JC; Shen, YY. High-performance bottom-contact devices based on an air-stable n-type organic semiconductor N,N′-bis(4-trifluoromethoxybenzyl)-1,4,5,8-naphthalene-tetracarboxylic di-imide. Appl. Phys. Lett 2007, 90, 212101. [Google Scholar]
- Letizia, JA; Facchetti, A; Stern, CL; Ratner, MA; Marks, TJ. High Electron Mobility in Solution-Cast and Vapor-Deposited Phenacyl-Quaterthiophene-Based Field-Effect Transistors: Toward N-Type Polythiophenes. J. Am. Chem. Soc 2005, 127, 13476–13477. [Google Scholar]
- Lee, Y-L; Hsu, H-L; Chen, S-Y; Yew, T-R. Solution-Processed Naphthalene Diimide Derivatives as n-Type Semiconductor Materials. J. Phys. Chem C 2008, 112, 1694–1699. [Google Scholar]
- Janssen, D. Long-term performance versus structure of polymer materials. Ph.D. Thesis, Katholieke University of Leuven, Belgium, 2006. [Google Scholar]
- Burtman, V; Zelichenok, A; Yitzchaik, S. Organic Superlattices via Chemical Vapor Molecular Layer Deposition (CV-MLD) Process. Polym. Prepr 1998, 39, 167–168. [Google Scholar]
- Burtman, V; Zelichenok, A; Yakimov, A; Yitzchaik, S. Yitzchaik, Vapor Phase Molecular Epitaxy via Self-Assembly Reactions. In Semiconductive Polymers: Applications, Properties, and Synthesis; Hsieh, BR, Ed.; American Chemical Society: Washington, DC, USA, 1999; pp. 399–419. [Google Scholar]
- Ofir, Y; Shwartzglass, O; Shappir, J; Yitzchaik, S. Organic Field Effect Transistors Based on Multilayer Films via Molecular Layer Epitaxy. In Organic Thin-Film Electronics; Arias, N, Tessler, L, Burgi, JA, Eds.; Emerson; Warrendale, PA, USA, 2005; pp. I3222–3226. [Google Scholar]
- Ofir, Y; Burtman, V; Yitzchaik, S. Levy, M, Ed.; Organic Field Effect Transistor. Proceeding of The 66th Annual Meeting of the Israel Chemical Society, David-Intercontinental Hotel, Tel-Aviv, Israel, February, 2001, Israel Chemical Society: Tel-Aviv, Israel, 2001; p. 24. [Google Scholar]
- Yitzchaik, S. 2D Conducting and Semiconducting Organic Heterostructures. Proceeding of The 66th Annual Meeting of the Israel Chemical Society, David-Intercontinental Hotel, Tel-Aviv, Israel, February, 2001.
- Holstein, T. Studies of polaron motion: Part II. The “small” polaron. Ann. Phys 1959, 8, 343–389. [Google Scholar]
- Emin, D. Small polarons. Phys Today 1982, 6, 34–35. [Google Scholar]
- Silinsh, EA; Cápek, VC. Organic Molecular Crystals: Interaction, Localization, and Transport Phenomena; American Institute of Physics: New York, NY, USA, 1994. [Google Scholar]
- Pope, M; Swenberg, CE. Electronic Processes in Organic Crystals and Polymers, 2nd ed; Oxford University Press: New York NY, USA, 1999. [Google Scholar]
- Gershenson, ME; Podzorov, V; Morpurgo, AF. Electronic transport in single-crystal organic transistors. Rev. Mod. Phys 2006, 78, 973–989. [Google Scholar]
- Troisi, A; Orlandi, G; Anthony, JE. Electronic Interactions and Thermal Disorder in Molecular Crystals Containing Cofacial Pentacene Units. Chem. Mater 2005, 17, 5024. [Google Scholar]
- Kenkre, VM; Andersen, JD; Dunlap, DH; Duke, CB. Unified theory of the mobilities of photoinjected electrons in naphthalene. Phys. Rev. Lett 1989, 62, 1165. [Google Scholar]
- Wu, M-W; Conwell, EM. Transport in α-sexithiophene films. Chem. Phys. Lett 1997, 266, 363–367. [Google Scholar]
- Fratini, S; Ciuchi, S. Dynamical Mean-Field Theory of Transport of Small Polarons. Phys. Rev. Lett 2003, 91, 256403. [Google Scholar]
- Deng, WQ; Goddard, WA. Predictions of Hole Mobilities in Oligoacene Organic Semiconductors from Quantum Mechanical Calculations. J. Phys. Chem B 2004, 108, 8614–8621. [Google Scholar]
- Hannewald, K; Bobbert, PA. Ab-Initio theory of charge-carrier conduction in ultrapure organic crystals. Appl. Phys. Lett 2004, 85, 1535. [Google Scholar]
- Hannewald, K; Bobbert, PA. Menendez, J, van de Walle, CG, Eds.; Ab-Initio Theory of Charge Transport in Organic Crystals. In Physics of Semiconductors; American Institute of Physics: Melville, NY, USA, 2005; p. 1101. [Google Scholar]
- Da Silva Filho, DA; Kim, EG; Bredas, JL. Transport Properties in the Rubrene Crystal: Electronic Coupling and Vibrational Reorganization Energy. Adv. Mater 2005, 17, 1072–1076. [Google Scholar]
- Karl, N. Conjugated Polymers and Low-Molecular Weight Organic Solids. In Organic Electronic Materials; Farchioni, R, Grosso, G, Eds.; Springer-Verlag: Berlin, Germany, 2001; pp. 283–326. [Google Scholar]
- Dodabalapur, A; Torsi, L; Katz, HE. Organic Transistors: Two-Dimensional Transport and Improved Electrical Characteristics. Science 1995, 268, 270–271. [Google Scholar]
- Chua, L-L; Zaumseil, J; Chang, J-F; Ou, EC-W; Ho, PKH; Sirringhaus, H; Friend, R. General observation of n-type field-effect behaviour in organic semiconductors. Nature 2005, 434, 194–199. [Google Scholar]
- Kiguchi, M; Nakayama, M; Shimada, T; Saiki, K. Electric-field-induced charge injection or exhaustion in organic thin film transistor. Phys. Rev B 2005, 71, 035332. [Google Scholar]
- Panzer, MJ; Frisbie, CD. Polymer Electrolyte Gate Dielectric Reveals Finite Windows of High Conductivity in Organic Thin Film Transistors at High Charge Carrier Densities. J. Am. Chem. Soc 2005, 127, 6960–6961. [Google Scholar]
- Houili, H; Picon, JD; Bussac, MN; Zuppiroli, L. Polarization effects in the channel of an organic field-effect transistor. J. Appl. Phys 2006, 100, 023702. [Google Scholar]
- Horowitz, G. Organic thin film transistors: From theory to real devices. J. Mater. Res 2004, 19, 1946–1962. [Google Scholar]
- Burtman, V; Zelichenok, A; Yitzchaik, S. Organic Quantum-Confined Structures through Molecular Layer Epitaxy. Angew. Chem. Int. Ed 1999, 38, 2041–2045. [Google Scholar]
- Burtman, V; Offir, Y; Yitzhaik, S. In Situ Spectroscopic Ellipsometry Monitoring of Multilayer Growth Dynamics via Molecular Layer Epitaxy. Langmuir 2001, 17, 2137–2142. [Google Scholar]
- Whitesides, GM; Laibinis, PE. Wet chemical approaches to the characterization of organic surfaces: self-assembled monolayers, wetting, and the physical-organic chemistry of the solid-liquid interface. Langmuir 1990, 6, 87–96. [Google Scholar]
- Ada, ET; Hanley, L; Etchin, S; Melngailis, J; Dressick, WJ; Chen, MS; Calvert, JM. Ion beam modification and patterning of organosilane self-assembled monolayers. J. Vac. Sci. Technol B 1995, 13, 2189–2196. [Google Scholar]
- Briggs, D; Seah, MP (Eds.) Practical Surface Analysis; Wiley: New York, NY, USA, 1990; p. 444.
- Kallury, KMR; Macdonald, PM; Thompson, M. Effect of Surface Water and Base Catalysis on the Silanization of Silica by (Aminopropyl)alkoxysilanes Studied by X-ray Photoelectron Spectroscopy and 13C Cross-Polarization/Magic Angle Spinning Nuclear Magnetic Resonance. Langmuir 1994, 10, 492–499. [Google Scholar]
- Forrest, SR. Ultrathin Organic Films Grown by Organic Molecular Beam Deposition and Related Techniques. Chem. Rev 1997, 97, 1793–1896. [Google Scholar]
- Dodabalapur, A; Katz, HE; Tordi, L; Haddon, RC. Complementary circuits with organic transistors. Appl. Phys. Lett 1996, 69, 4227. [Google Scholar]
- Werner, A; Li, F; Harada, K; Pfeiffer, M; Fritz, T; Leo, K; Machill, S. n-Type Doping of Organic Thin Films Using Cationic Dyes. Adv. Funct. Mater 2004, 14, 255–260. [Google Scholar]
- Burtman, V. Organic tunneling devices and field effect transistors using polycrystalline naphthalene diimine frameworks. Thin Solid Films 2008, 516, 3436–3440. [Google Scholar]
- Sze, S. Semiconductor Devices Physics and Technology; Wiley: New York, NY, USA, 1985. [Google Scholar]
- Hagfeldt, A; Gratzel, M. Molecular Photovoltaics. Acc. Chem. Res 2000, 33, 269–277. [Google Scholar]
- Grätzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar]
- Huynh, WU; Dittmer, JJ; Alivisatos, AP. Hybrid Nanorod-Polymer Solar Cells. Science 2002, 295, 2425–2427. [Google Scholar]
- Glazer, AN. Comparative Biochemistry of Photosynthetic Light-Harvesting Systems. Annu. Rev. Biochem 1983, 52, 125–157. [Google Scholar]
- di Bilio, AJ; Crane, BR; Wehbi, WA; Kiser, CN; Abu-Omar, MM; Carlos, RM; Richards, JH; Winkler, JR; Gray, HB. Properties of Photogenerated Tryptophan and Tyrosyl Radicals in Structurally Characterized Proteins Containing Rhenium(I) Tricarbonyl Diimines. J. Am. Chem. Soc 2001, 123, 3181–3182. [Google Scholar]
- Barry, BA; El-Deeb, MK; Sandusky, PO; Babcock, GT. Tyrosine radicals in photosystem II and related model compounds. Characterization by isotopic labeling and EPR spectroscopy. J. Biol. Chem 1990, 265, 20139–20143. [Google Scholar]
- Collet, J; Lenfant, S; Vuillaume, D; Bouloussa, O; Rondelez, F; Gay, JM; Kham, K. Chevrot, High anisotropic conductivity in organic insulator/semiconductor monolayer heterostructure. Appl. Phys. Lett 2000, 76, 1339–1341. [Google Scholar]
- Bryce, MR; Petty, MC. Electrically conductive Langmuir-Blodgett films of charge-transfer materials. Nature 1995, 374, 771–776. [Google Scholar]
- Bjornholm, T; Hassenkam, T; Reitzel, N. Supramolecular organization of highly conducting organic thin films by the Langmuir-Blodgett technique. J. Mater. Chem 1999, 9, 1975–1990. [Google Scholar]
- Advances in Synthetic Metals Twenty Years of Progress in Science and Technology; Bernier, P; Lefrant, S; Bidan, G (Eds.) Elsevier Science: Amsterdam, The Netherlands, 1999.
- Novàk, P; Muller, K; Santhanam, KSV; Haas, O. Electrochemically Active Polymers for Rechargeable Batteries. Chem. Rev 1997, 97, 207–281. [Google Scholar]
- Heath, JR; Ratner, MA. Molecular Electronics. Phys Today 2003, 56, 43–49. [Google Scholar]
- Rehm, JM; McLendon, GL; Nagasawa, Y; Yoshihara, K; Moser, J; Grattzel, M. Femtosecond Electron-Transfer Dynamics at a Sensitizing Dye-Semiconductor (TiO2) Interface. J. Phys. Chem 1996, 100, 9577–9578. [Google Scholar]
- Schroder, DK. Surface voltage and surface photovoltage: history, theory and applications. Meas. Sci. Technol 2001, 12, R16–R31. [Google Scholar]
- Bergmann, L. Uber einen lichtelektrischen Effekt in Halbleitern. Phys. Zeits 1932, 33, 209. [Google Scholar]
- Munakata, C; Matsubara, S. The photovoltaic observation of semiconductor surfaces. J. Phys. D: Appl. Phys 1982, 16, 1093–1098. [Google Scholar]
- Munakata, C; Yagi, K; Warabisako, T; Nanba, M; Matsubara, S. Observation of p-n Junctions with a Flying-Spot Scanner Using a Chopped Photon Beam. Jpn. J. Appl. Phys 1982, 21, 624–632. [Google Scholar]
- Munakata, C; Honma, N; Itoh, H. A Non-Destructive Method for Measuring Lifetimes for Minority Carriers in Semiconductor Wafers Using Frequency-Dependent ac Photovoltages. Jpn. J. Appl. Phys 1983, 22, L103–L105. [Google Scholar]
- Honma, N; Munakata, C; Shimizu, H. Comparison of Minority Carrier Lifetimes Measured by Photoconductive Decay and ac Photovoltaic Method. Jpn. J. Appl. Phys 1988, 27, 1498–1503. [Google Scholar]
- Witt, HT; Zickler, A. Electrical evidence for the field indicating absorption change in bioenergetic membranes. FEBS Lett 1973, 37, 307–310. [Google Scholar]
- Trissl, HW. Spatial correlation between primary redox components in reaction centers of Rhodopseudomonas sphaeroides measured by two electrical methods in the nanosecond range. Proc. Natl. Acad. Sci USA 1983, 80, 7173–7177. [Google Scholar]
- Cohen, R; Zenou, N; Cahen, D; Yitzchaik, S. Molecular electronic tuning of Si surfaces. Chem. Phys. Lett 1997, 279, 270–274. [Google Scholar]
- Pakoulev, A; Zaslavsky, D; Burtman, V. McIntire, R, Donnell, P, Eds.; Photocurrent Study of the Transport Mechanism in Molecular Self-Assembly Transistors. In Integrated Circuits, Photodiodes and Organic Field Effect Transistors; Nova Publishers: Hauppauge, NY, USA, 2009. [Google Scholar]
- Holman, MW; Liu, R; Adams, DM. Single-Molecule Spectroscopy of Interfacial Electron Transfer. J. Am. Chem. Soc 2003, 125, 12649–12654. [Google Scholar]
- Raistrick, ID. Electrochemical Capacitors. In Electrochemistry of Semiconductors and Electronics—Processes and Devices; McHardy, J, Ludwig, F, Eds.; William Andrew Publishing/Noyes: Norwich, NY, USA, 1992; p. 297. [Google Scholar]
- Wenger, B; Gratzel, M; Moser, JE. Rationale for Kinetic Heterogeneity of Ultrafast Light-Induced Electron Transfer from Ru(II) Complex Sensitizers to Nanocrystalline TiO2. J. Am. Chem. Soc 2005, 127, 12150–12151. [Google Scholar]
- Grätzel, M. Photoelectrochemical cells. Nature 2001, 414, 338. [Google Scholar]
- Hagfeldt, A; Grätzel, M. Light-Induced Redox Reactions in Nanocrystalline Systems. Chem. Rev 1995, 95, 49–68. [Google Scholar]
- Karl, N; Kraft, KH; Marktanner, J; Munch, M; Schatz, F; Stehle, R; Uhde, HM. Fast electronic transport in organic molecular solids? J. Vac. Sci. Technol A 1999, 174, 2318–2328. [Google Scholar]
- Meyer, JR; Arnold, DJ; Hoffman, CA; Bartoli, FJ. Free-carrier transport in superlattices: Smooth transition between the quasi-two-dimensional and uniform three-dimensional limits. Phys. Rev B 1992, 45, 1295–1304. [Google Scholar]
- Horovitz, B; Pazy, E. Photocurrent in conjugated polymers. Europhys. Lett 2004, 65, 386–392. [Google Scholar]
- Khondaker, SI; Shlimak, IS; Nicholls, JT; Pepper, M; Ritchie, DA. Two-dimensional hopping conductivity in a δ-doped GaAs/AlxGa1-xAs heterostructure. Phys. Rev B 1999, 59, 4580–4583. [Google Scholar]
- Laquindanum, JG; Katz, HE; Dodabalapur, A; Lovinger, AJ. n-Channel Organic Transistor Materials Based on Naphthalene Frameworks. J. Am. Chem. Soc 1996, 118, 11331–11332. [Google Scholar]
- Facchetti, A. Semiconductors for organic transistors. Mater Today 2007, 10, 28–37. [Google Scholar]
- Hiramoto, M; Imahigashi, T; Yokoyama, M. Photocurrent multiplication in organic pigment films. Appl. Phys. Lett 1994, 64, 187–189. [Google Scholar]
- Hiramoto, M; Sato, I; Yokoyama, M. Metal Nanostructure of Metal/Organic Interface Causing Photocurrent Multiplication Phenomenon. IEICE Trans Electron 2002, E85-C, 1253–1255. [Google Scholar]
- Burtman, V; Vardeny, ZV. Design and Characterization of Novel Systems for Molecular Nanoscale Self-Assembly. Jpn. J. Appl. Phys 2008, 47, 1165–1172. [Google Scholar]
- Reed, MA; Zhou, C; Deshpande, MR; Muller, CJ; Burgin, TP; Jones, L, II; Tour, JM. The Electrical Measurement of Molecular Junctions. Ann. N.Y. Acad. Sci 1998, 852, 133–144. [Google Scholar]
- Holmlin, RE; Haag, R; Chabinyc, ML; Ismagilov, RF; Cohen, AE; Terfort, A; Rampi, MA; Whitesides, GM. Electron Transport through Thin Organic Films in Metal-Insulator- Metal Junctions Based on Self-Assembled Monolayers. J. Am. Chem. Soc 2001, 123, 5075–5085. [Google Scholar]
- Petta, JR; Slater, SK; Ralph, DC. Spin-Dependent Transport in Molecular Tunnel Junctions. Phys. Rev. Lett 2004, 93, 136601. [Google Scholar]
- Curtis, MD; Cao, J; Kampf, JW. Solid-State Packing of Conjugated Oligomers: From π-Stacks to the Herringbone Structure. J. Am. Chem. Soc 2004, 126, 4318–4328. [Google Scholar]
- Desiraju, GR. Crystal engineering. From molecules to materials. J. Mol. Struct 2003, 656, 5–15. [Google Scholar]
- Gregg, BA; Fox, MA; Bard, AJ. 2,3,7,8,12,13,17,18-Octakis(beta.-hydroxyethyl)porphyrin (octaethanolporphyrin) and its liquid crystalline derivatives: synthesis and characterization. J. Am. Chem. Soc 1989, 111, 3024–3029. [Google Scholar]
- Cormier, RA; Gregg, BA. Synthesis and Characterization of Liquid Crystalline Perylene Diimides. Chem. Mater 1998, 10, 1309–1319. [Google Scholar]
- Sugiama, S; Yoshimura, D; Miyamae, T; Miyazaki, T; Ishii, H; Ouchi, Y; Seki, K. Electronic structures of organic molecular materials for organic electroluminescent devices studied by ultraviolet photoemission spectroscopy. J. Appl. Phys 1988, 83, 4928–4938. [Google Scholar]
- Nakahara, H; Seki, K; Asada, S; Fukuda, K; Inikuchi, H. UV Photoelectron Spectroscopic Study of the Photopolymerization of Long-Chain Diacetylene Monocarboxylic Acid in Langmuir-Blodgett Films. Chem. Phys 1987, 118, 123–131. [Google Scholar]
- Yin, S; Liu, X; Li, X; Huang, W; Li, W; He, B. Electroluminescent properties of naphthalimide derivative thin film devices. Thin Solid Films 1998, 325, 268–270. [Google Scholar]
- Adachi, C; Tsutsui, T; Saito, S. Blue light-emitting organic electroluminescent devices. Appl. Phys. Lett 1990, 56, 799–801. [Google Scholar]
- Angadi, MA; Gosztola, D; Waseilewski, MR. Characterization of photovoltaic cells using poly(phenylenevinylene) doped with perylenediimide electron acceptors. J. Appl. Phys 1998, 83, 6187–6189. [Google Scholar]
- Wiederrecht, GP; Yoon, BA; Wasielewski, MR. Photorefractive liquid crystals. Adv. Mater 1996, 8, 535–539. [Google Scholar]
- Mizuguchi, J. Synthesis and Characterization of Liquid Crystalline Perylene Diimides. J. Appl. Phys 1998, 84, 4479–4486. [Google Scholar]
- Pressner, D; Goeltner, C; Spiess, HW; Muellen, K; Ber, B-G. Liquid-crystalline perylene oligomers: Synthesis and phase behavior. Phys. Chem 1993, 97, 1362–1365. [Google Scholar]
- Aveline, BM; Matsugo, S; Redmond, RW. Photochemical Mechanisms Responsible for the Versatile Application of Naphthalimides and Naphthaldiimides in Biological Systems. J. Am. Chem. Soc 1997, 119, 11785–11795. [Google Scholar]
- Burtman, V; Zelichenok, A; Yitzchaik, S. Hsieh, BR, Ed.; Vapor Phase Molecular Layer Epitaxy via Self-Assembly Reactions. In Semiconductive Polymers; ACS Publications: Washington, DC, USA, 1998. [Google Scholar]
- Ofir, Y; Zelichenok, A; Yitzchaik, S. 1,4,5,8-naphthalene-tetracarboxylic Diimide Derivatives as Model Compounds for Molecular Layer Epitaxy. J. Mater. Chem 2006, 16, 2142–2149. [Google Scholar]
- Firestone, MA; Shank, ML; Sligar, SG; Bohn, PW. Film Architecture in Biomolecular Assemblies. Effect of Linker on the Orientation of Genetically Engineered Surface-Bound Proteins. J. Am. Chem. Soc 1996, 118, 9033–9041. [Google Scholar]
- Di Ventra, M. Electrical Transport in Nanoscale Systems; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Galperin, M; Ratner, MA; Nitzan, A. Molecular transport junctions: vibrational effects. J. Phys.: Condens. Mater 2007, 19, 103201. [Google Scholar]
- Dahnovsky, Y. Ab-Initio electron propagators in molecules with strong electron-phonon interaction: II. Electron Green’s function. J. Chem. Phys 2007, 127, 014104. [Google Scholar]







© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Burtman, V.; Zelichonok, A.; Pakoulev, A.V. Molecular Photovoltaics in Nanoscale Dimension. Int. J. Mol. Sci. 2011, 12, 173-225. https://doi.org/10.3390/ijms12010173
Burtman V, Zelichonok A, Pakoulev AV. Molecular Photovoltaics in Nanoscale Dimension. International Journal of Molecular Sciences. 2011; 12(1):173-225. https://doi.org/10.3390/ijms12010173
Chicago/Turabian StyleBurtman, Vladimir, Alexander Zelichonok, and Andrei V. Pakoulev. 2011. "Molecular Photovoltaics in Nanoscale Dimension" International Journal of Molecular Sciences 12, no. 1: 173-225. https://doi.org/10.3390/ijms12010173