Dimerization of Protegrin-1 in Different Environments

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Microscopic Models for PG1 Dimers in the Water Phase, on the Surface of a POPG:POPE Membrane and inside a POPG:POPE Membrane
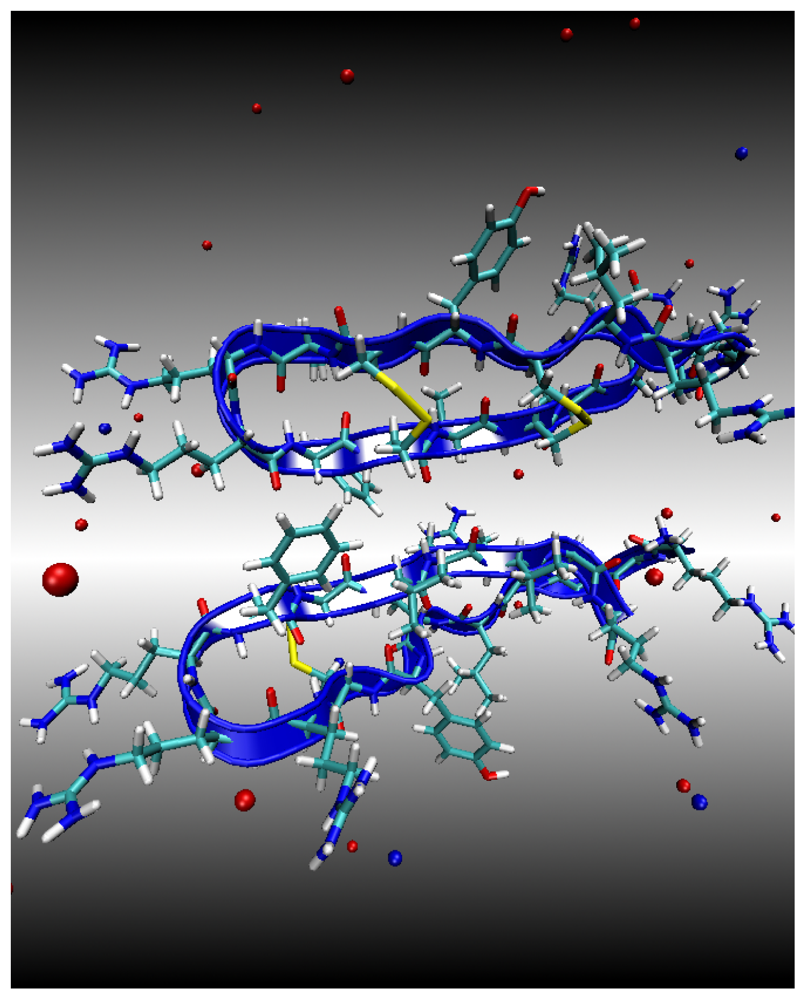
Environment 1: Water Subphase
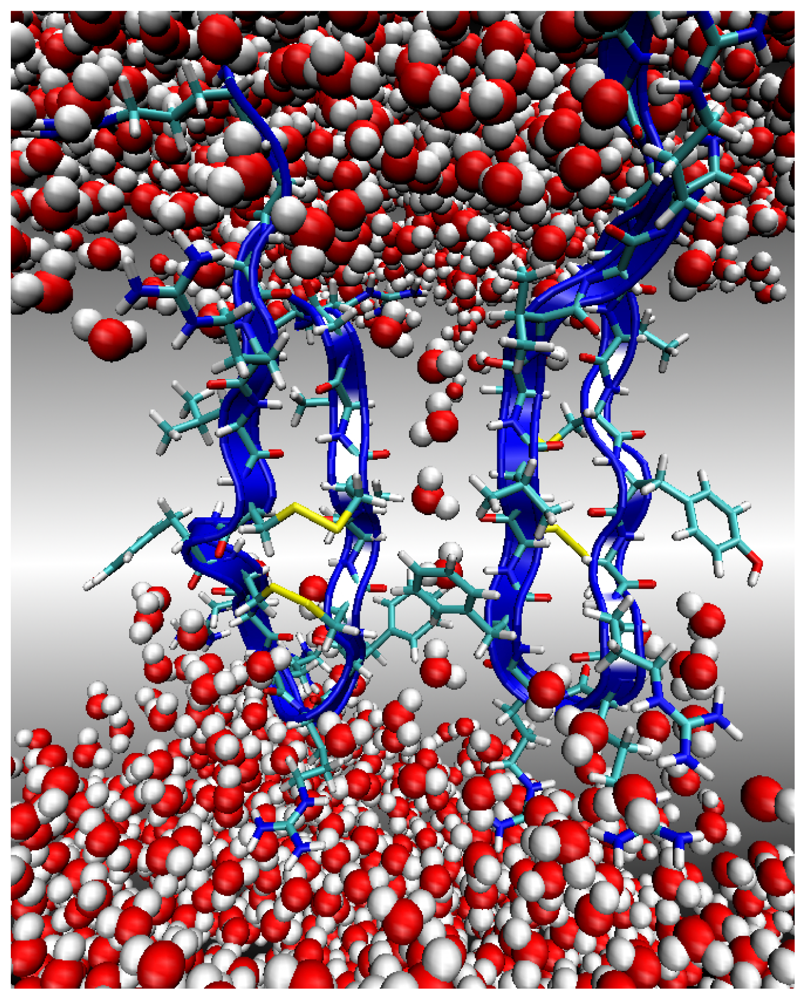
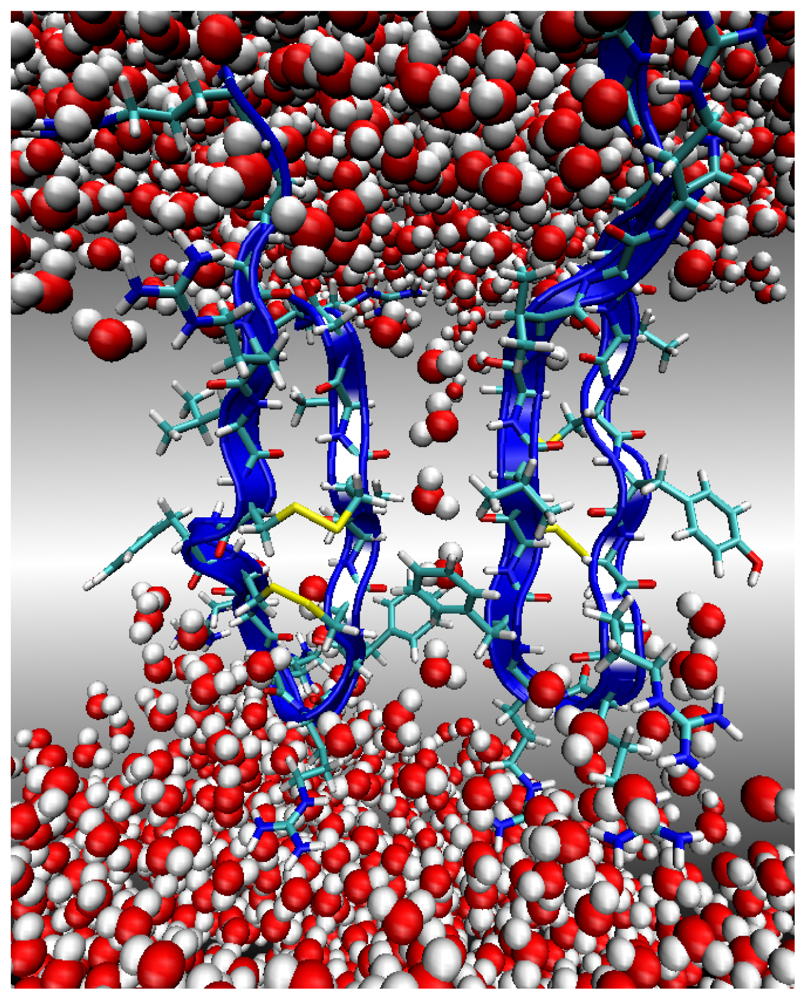
Environment 2: Lipid Bilayer Surface
Environment 3: Lipid Bilayer Core
2.2. Molecular Dynamics Protocol
2.3. Construction of Potential of Mean Force (PMF) for the Formation of PG1pd and PG1ad
- The two peptides are positioned in either the parallel or antiparallel orientation, at a distance D. The peptide separation, D, ranges from 9 Å to 25 Å in increments of 2 Å. There are thus 54 systems constructed (two orientations, three environments, nine separation distances). We should note that for PG1pd in Environment 3, we added another separation distance of D = 27 Å, in order to ascertain that the PMF attains a plateau at long distances, as discussed in more detail in Section 3. Each of the 55 constructed system is then equilibrated over 4 ns in the NPT ensemble. During this equilibration the PG1 peptides are restrained using harmonic springs with a a force constant 20 (kcal/mol)/Å2 applied to all peptide backbone atoms.
- A 4 ns production run is then conducted for each of the 55 initial equilibrated systems. In production runs, and in order to restrain the peptides and their orientations, we use harmonic springs coupled to the three carbon CB backbone atoms of Arg1, Arg10 and Cys15. All spring constants were 20 (kcal/mol)/Å2. In addition, and in order to better ascertain convergence of the PMF calculation, we extended the simulation of the parallel configuration, PG1pd, inside the membrane by an additional 4 ns, to 8 ns, for all examined distances.
- The instantaneous restraint forces are computed for each of the 55 system configurations for PG1 dimers with a sampling interval of 0.2 ps, and averaged to obtain the mean force F̄ (D) = −F̄res (D) for each position, where F̄res (D) is the force exerted on the harmonic restraint springs. We concentrate our efforts on reducing the statistical errors. A difficulty is that, on short time scales, the results are highly correlated, and thus unsuitable for statistical analysis. We find that the correlation time for estimating the error due to solvent force fluctuations is about 0.1 ns, and membrane fluctuations and systematic error due to the harmonic restraints require data for no less than 0.5 ns to compute reliable average forces. Using the block-averaging method [27] we find the statistical errors in F̄res (D) to be within 0.4 (kcal/mol)/Å in all cases. The total sampling time must therefore be long enough to ensure a collection of uncorrelated configurations.
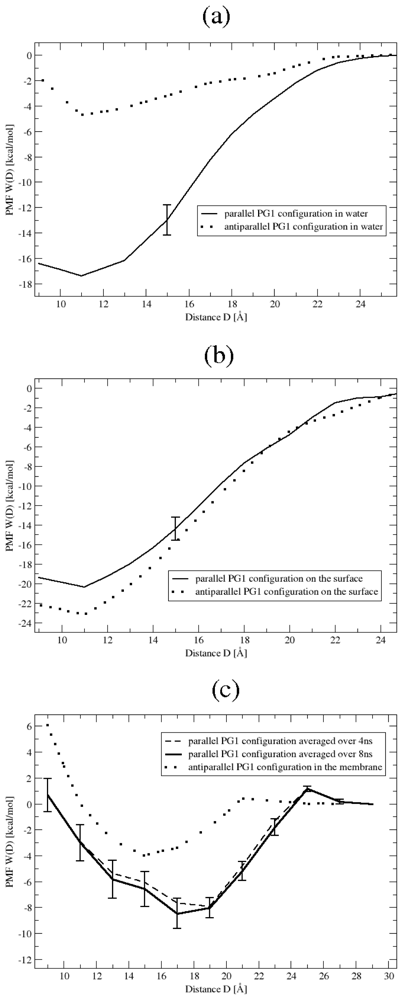
- The PMF can be evaluated by applying the mean force integration method which was developed for the PMF calculation of a peptide in the vicinity of a neutral POPC membrane [28]. This method is a variant of constrained MD and thermodynamic integration [29–34]. In particular, the PMF, W(D), is calculated using (1), where the integration over the D coordinate is performed using the trapezoidal rule:
3. Results and Discussion
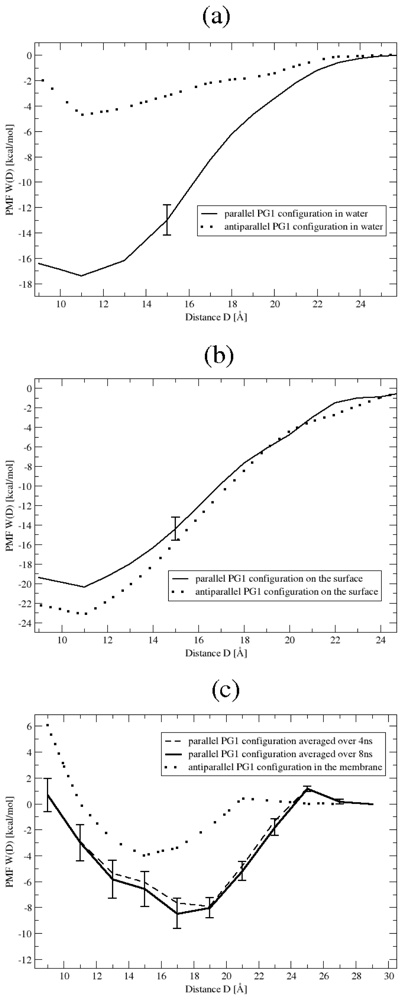
3.1. Binding Affinity of PG1 Peptides in the Parallel and Antiparallel β-Sheet Arrangements
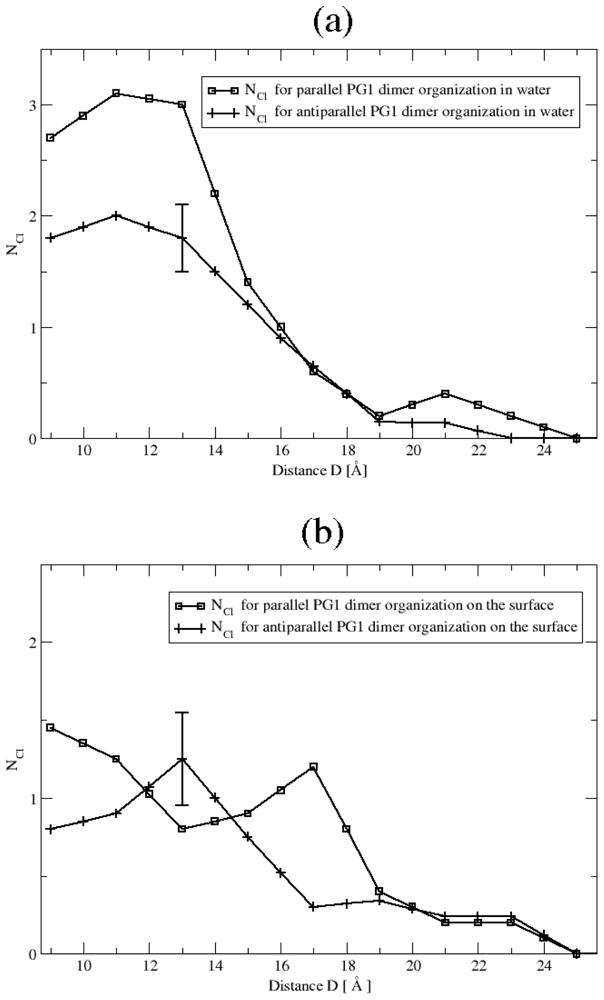
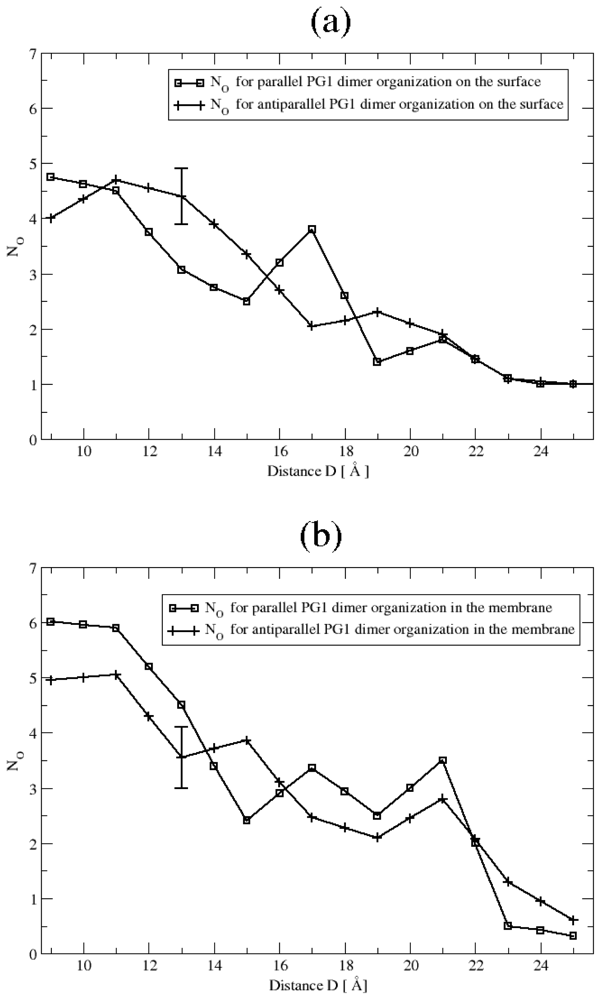
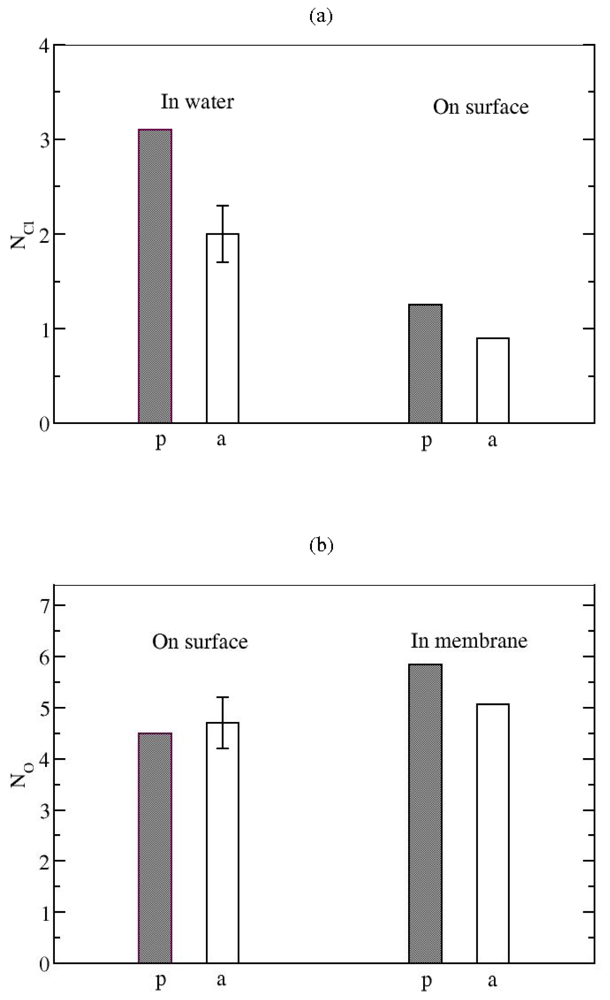
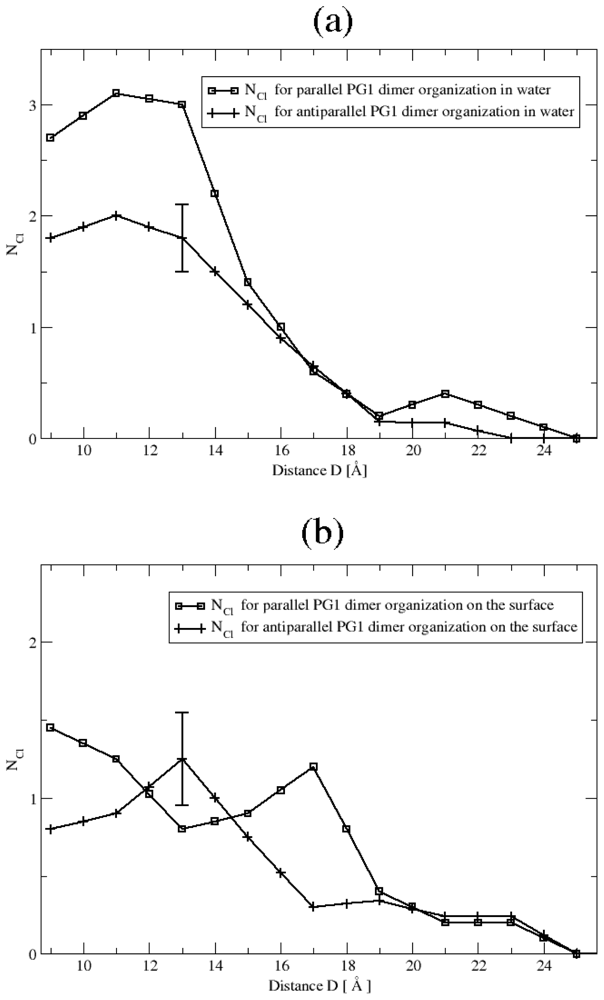
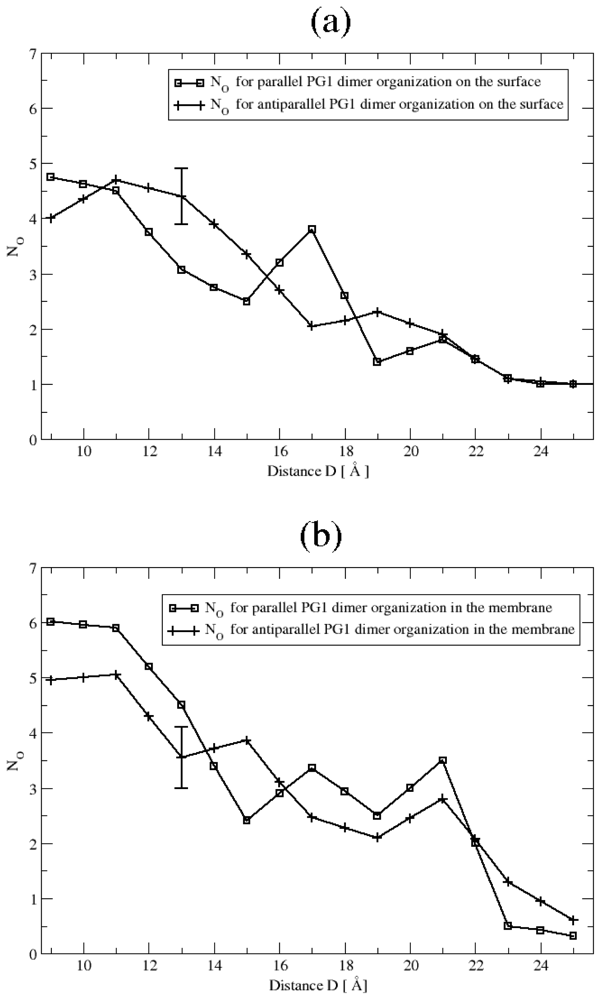
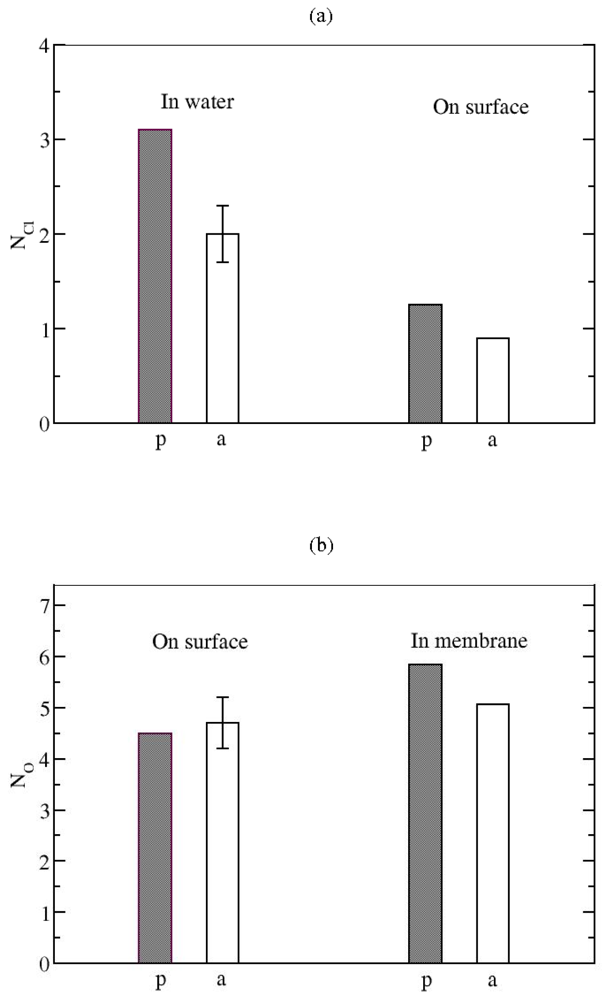
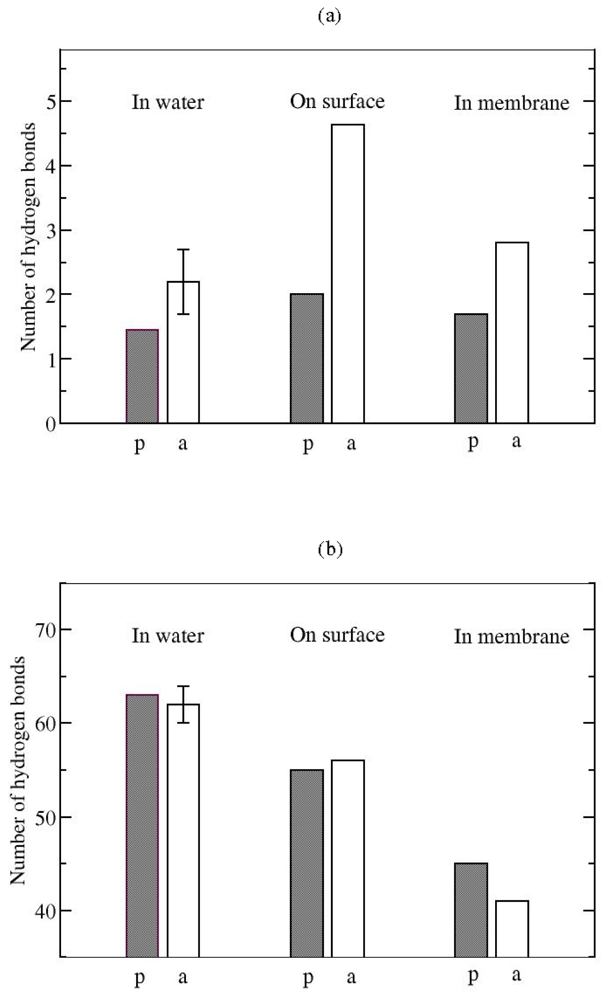
3.2. The Role of Ionic Bridge and Hydrogen Bond Networks in Dimerization Stability
Ionic Bridges
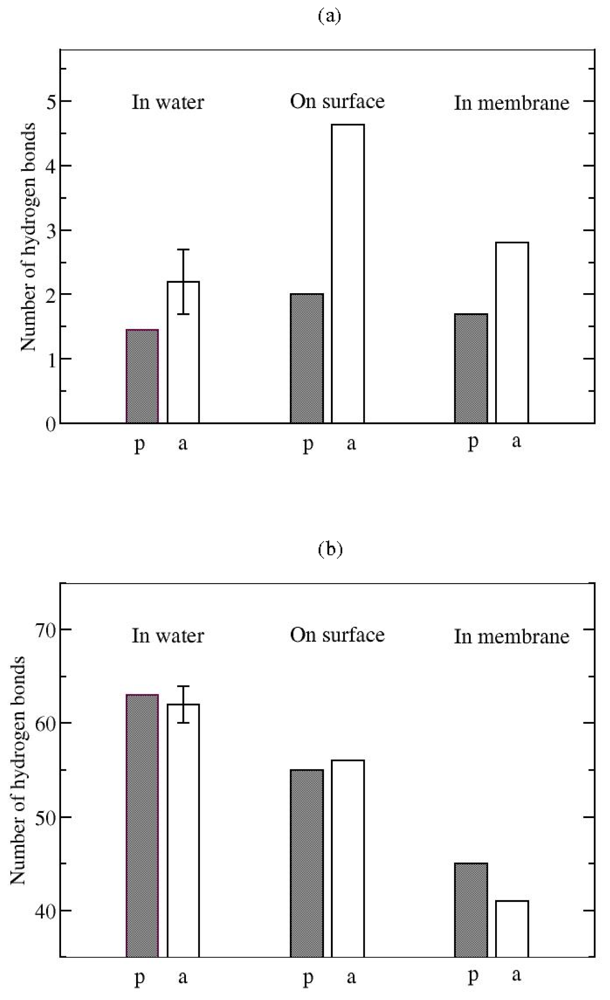
Hydrogen bonds
4. Conclusions
Acknowledgements
- Classification: PACS 87.15.-v
References
- Fahrner, RL; Dieckmann, T; Harwig, SS; Lehrer, RI; Eisenberg, D; Feigon, J. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol 1996, 3, 543–550. [Google Scholar]
- Jenssen, H; Hammil, P; Hancock, RE. Peptide antimicrobial agents. Clin. Microbiol. Rev 2006, 19, 491–511. [Google Scholar]
- Mani, R; Tang, M; Wu, X; Buffy, IJ; Waring, AJ; Sherman, MA; Hong, M. Membrane-bound dimer structure of a beta-hairpin antimicrobial peptide from rotational-echo double-resonance solid state NMR. Biochemistry 2006, 45, 8341–8349. [Google Scholar]
- Langham, AA; Sayyed-Ahmad, A; Kaznessis, YN. On the nature of antimicrobial activity: A model for protegrin-1 pores. J. Am. Chem. Soc 2008, 130, 4338–4346. [Google Scholar]
- Bolintineanu, D; Sayyed-Ahmad, A; Davis, HT; Kaznessis, YN. Poisson-Nernst-Planck models of nonequilibrium ion electrodiffusion through a protegrin transmembrane pore. PLoS Comput. Biol 2009, 5, e1000277. [Google Scholar]
- Bolintineanu, D; Hazrati, E; Davis, HT; Lahrer, RI; Kaznessis, YN. Antimicrobial mechanism of pore-forming protegrin peptides: 100 pores to kill E. coli. Peptides 2009, 31, 1–8. [Google Scholar]
- Roumestand, C; Louis, V; Aumelas, A; Grasi, G; Calas, B; Chavanieu, A. Oligomerization of protegrin-1 in the presence of DPC micells. A proton high-resolution NMR study. FEBS Lett 1998, 421, 263–267. [Google Scholar]
- Ostberg, N; Khandelia, H; Kaznessis, YN. Protegrin structure activity relationships: Using homology models of synthetic sequences to determine structural characteristics important for activity. Peptides 2005, 26, 297–306. [Google Scholar]
- Langham, AA; Khandelia, H; Kaznessis, YN. How can protegrin-1 be both a potent antimicrobial and harmfully toxic? Molecular dynamics simulations of a beta-sheet antimicrobial peptide in micelles. Biopolymers: Peptide Sci 2006, 84, 219–231. [Google Scholar]
- Langham, AA; Waring, AJ; Kaznessis, YN. Comparison of interactions between beta-hairpin decapeptides and SDS/DPC micelles from experimental and simulation data. BMC Biochem 2007, 8, 11. [Google Scholar]
- Khandelia, H; Langham, AA; Kaznessis, YN. Driving engineering of novel antimicrobial peptides from simulations of peptide-micelle interactions. BBA 2006, 1758, 1224–1234. [Google Scholar]
- Khandelia, H; Kaznessis, YN. Structure of the antimicrobial beta-hairpin peptide protegrin-1 in a DLPC lipid bilayer investigated by molecular dynamics simulation. BBA Biomembr 2007, 1768, 509–520. [Google Scholar]
- Jang, H; Ma, B; Lal, R; Nussinov, R. Models of toxic beta-sheet channels of protegrin-1 suggest a common subunit organization motif shared with toxic alzheimer beta-amyloid ion channels. Biophys. J 2008, 95, 4631–4642. [Google Scholar]
- Lee, J; Im, W. Beta-hairpin restraint potentials for calculations of potentials of mean force as a function of beta-hairpin tilt, rotation, and distance. J. Comput. Chem 2009, 30, 1334–1343. [Google Scholar]
- Vivcharuk, V; Kaznessis, Y. Free energy profile of the interaction between a monomer or a dimer of protegrin-1 in a specific binding orientation and a model lipid bilayer. J. Phys. Chem. B 2010, 114, 2790–2797. [Google Scholar]
- Sayyed-Ahmad, A; Kaznessis, YN. Determining the orientation of protegrin-1 in DLPC bilayers using an implicit solvent-membrane model. PLoS One 2009, 4, e4799. [Google Scholar]
- Jo, S; Kim, T; Iyer, VG; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem 2008, 29, 1859–1865. [Google Scholar]
- Brooks, BR; Brucolleri, RE; Olafsoan, BD; States, DJ; Swaminathan, S; Karplus, M. A program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem 1983, 4, 187–217. [Google Scholar]
- MacKerell, AD; Bashford, D; Bellot, M; Dunbrack, R.L; Evanseek, J; Field, MJ; Fischer, S; Gao, J; Guo, H; Ha, S; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar]
- MacKerell, AD; Feig, M; Brooks, CL. Improved treatment of the protein backbone in empirical force fields. J. Chem. Soc 2004, 126, 698–699. [Google Scholar]
- Phillips, JC; Braun, R; Wang, W; Gumbart, J; Tajkhorshid, E; Villa, E; Chipot, C; Skeel, RD; Kalle, L; Schulten, K. Scalable molecular dynamics with NAMD. J. Comp. Chem 2005, 26, 1781–1802. [Google Scholar]
- Martyna, GJ; Tobias, DJ; Klein, ML. Constant pressure molecular dynamics algorithms. J. Chem. Phys 1994, 101, 4177–4189. [Google Scholar]
- Feller, SE; Zhang, Y; Pastor, RW; Brooks, BR. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys 1995, 103, 4613–4621. [Google Scholar]
- Tang, M; Waring, J; Hong, M. Arginine dynamics in a membrane-bound cationic beta-hairpin peptide from solid-state NMR. ChemBioChem 2008, 9, 1487–1482. [Google Scholar]
- Jorgensen, WL; Chandrasekhar, J; Madura, JD; Impey, RW; Klein, ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- Darden, T; Perera, L; Li, L; Pedersen, L. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure 1999, 7, R55–R60. [Google Scholar]
- Flyvbjerg, H; Petersen, HG. Error-estimates on averages of correlated data. J. Chem. Phys 1989, 91, 461–466. [Google Scholar]
- Vivcharuk, V; Tomberli, B; Tolokh, IS; Gray, CG. Prediction of binding free energy for adsorption of antimicrobial peptide lactoferricin B on a POPC membrane. Phys. Rev. E 2008, 77, 031913. [Google Scholar]
- Chipot, C; Pohorille, A. Free Energy Calculations; Springer: Berlin, Germany, 2007. [Google Scholar]
- Tobias, DJ; Brooks, CL, III. Calculation of free energy surfaces using the methods of thermodynamic perturbation theory. Chem. Phys. Lett 1987, 142, 472–476. [Google Scholar]
- Roux, B; Karplus, M. Ion transport in a gramicidin-like channel: Structure and thermodynamics. Biophys. J 1991, 59, 961–981. [Google Scholar]
- Ciccotti, G; Ferrario, M; Hynes, JT; Kapral, R. Constrained molecular dynamics and the mean potential for an ion pair in a polar solvent. Chem. Phys 1989, 129, 241–251. [Google Scholar]
- Shinto, H; Morisada, S; Miyahara, M; Higashitani, K. A reexamination of mean force potentials for the methane pair and the constituent ion pairs of NaCl in water. J. Chem. Eng. Jap 2003, 36, 57–65. [Google Scholar]
- Darve, E. Chipot, C, Pohorille, A, Eds.; Free Energy Calculations; Springer: Berlin, Germany, 2007; p. 119. [Google Scholar]
- DeLoof, H; Nilsson, L; Rigler, R. Molecular dynamics simulation of galanin in aqueous and nonaqueous solution. J. Am. Soc 1992, 114, 4028–4035. [Google Scholar]
- Jang, H; Ma, B; Woolf, TB; Nussinov, R. Interaction of protegrin-1 with lipid bilayers: membrane thinning effect. Biophys. J 2006, 91, 2848–2859. [Google Scholar]
- Evans, DF; Wennerstrom, H. Colloidal Domain, 2nd ed; Wiley VCH: New York, NY, USA, 1999; p. 409. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vivcharuk, V.; Kaznessis, Y.N. Dimerization of Protegrin-1 in Different Environments. Int. J. Mol. Sci. 2010, 11, 3177-3194. https://doi.org/10.3390/ijms11093177
Vivcharuk V, Kaznessis YN. Dimerization of Protegrin-1 in Different Environments. International Journal of Molecular Sciences. 2010; 11(9):3177-3194. https://doi.org/10.3390/ijms11093177
Chicago/Turabian StyleVivcharuk, Victor, and Yiannis N. Kaznessis. 2010. "Dimerization of Protegrin-1 in Different Environments" International Journal of Molecular Sciences 11, no. 9: 3177-3194. https://doi.org/10.3390/ijms11093177