Complexes in the Photocatalytic Reaction of CO2 and H2O: Theoretical Studies

Abstract
:1. Introduction
2. Computational Details
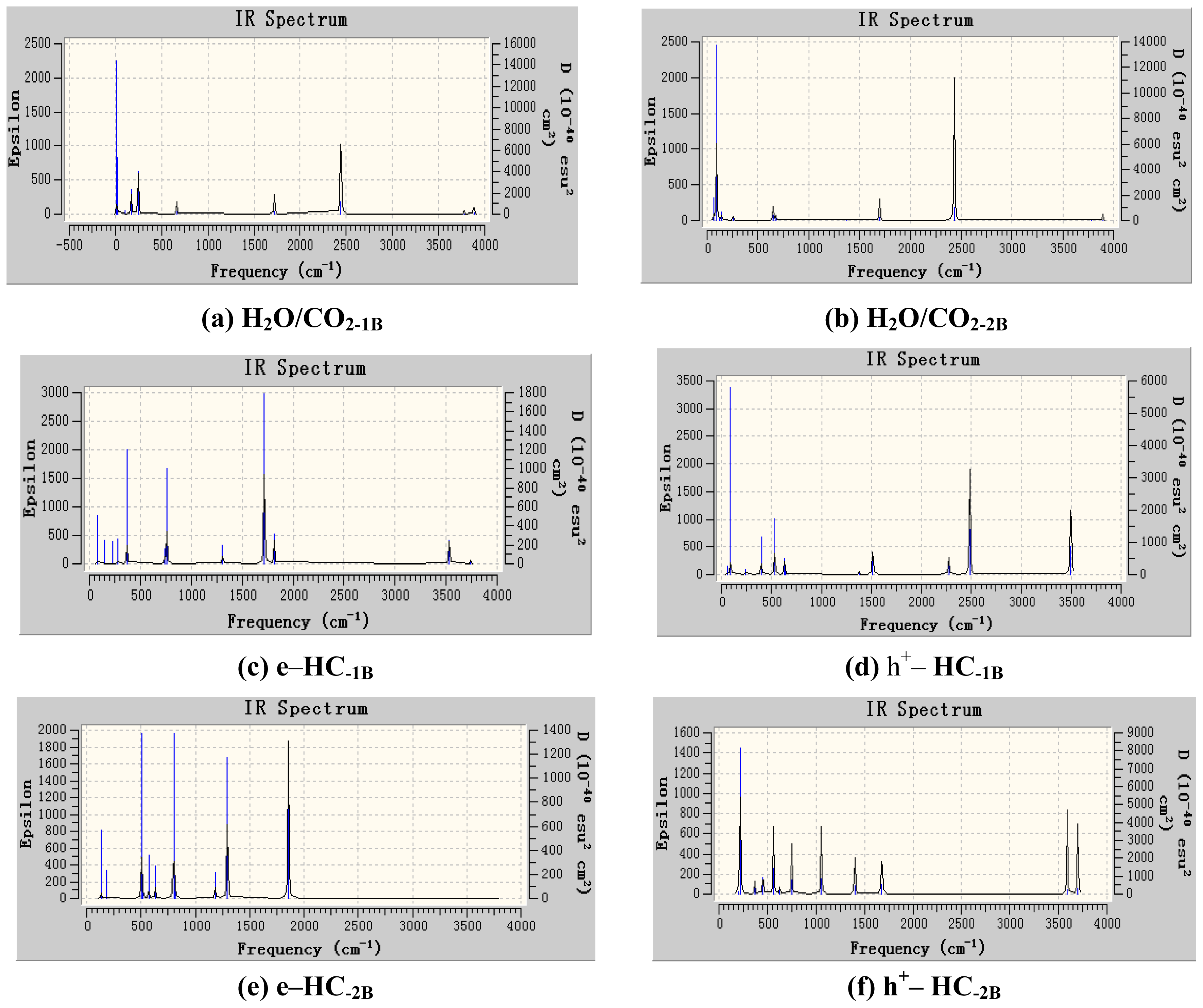
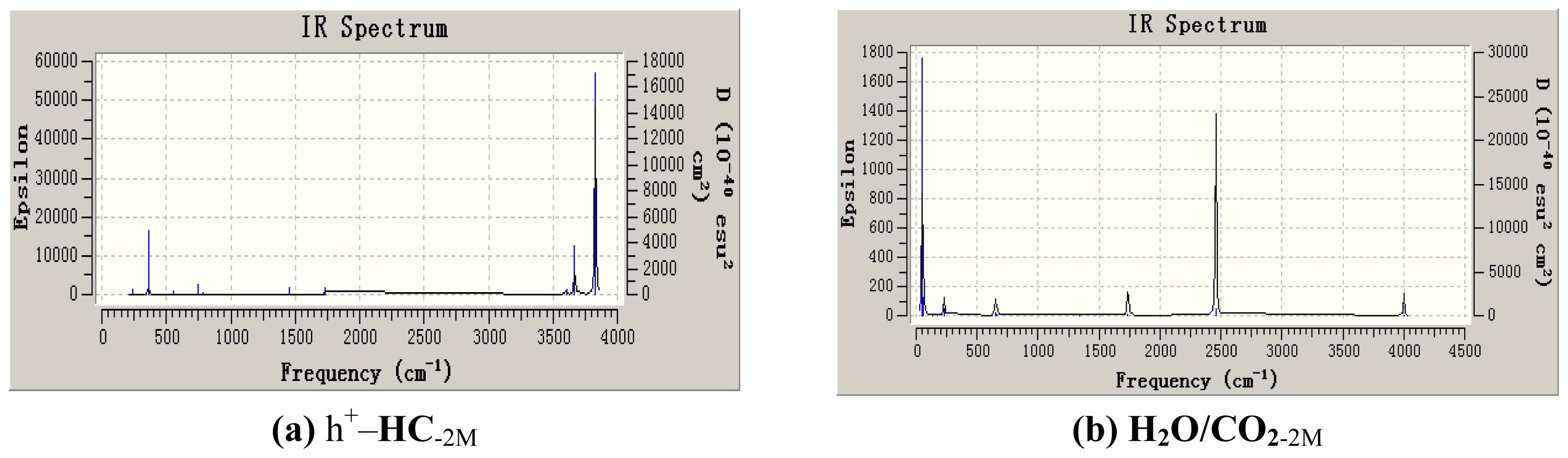
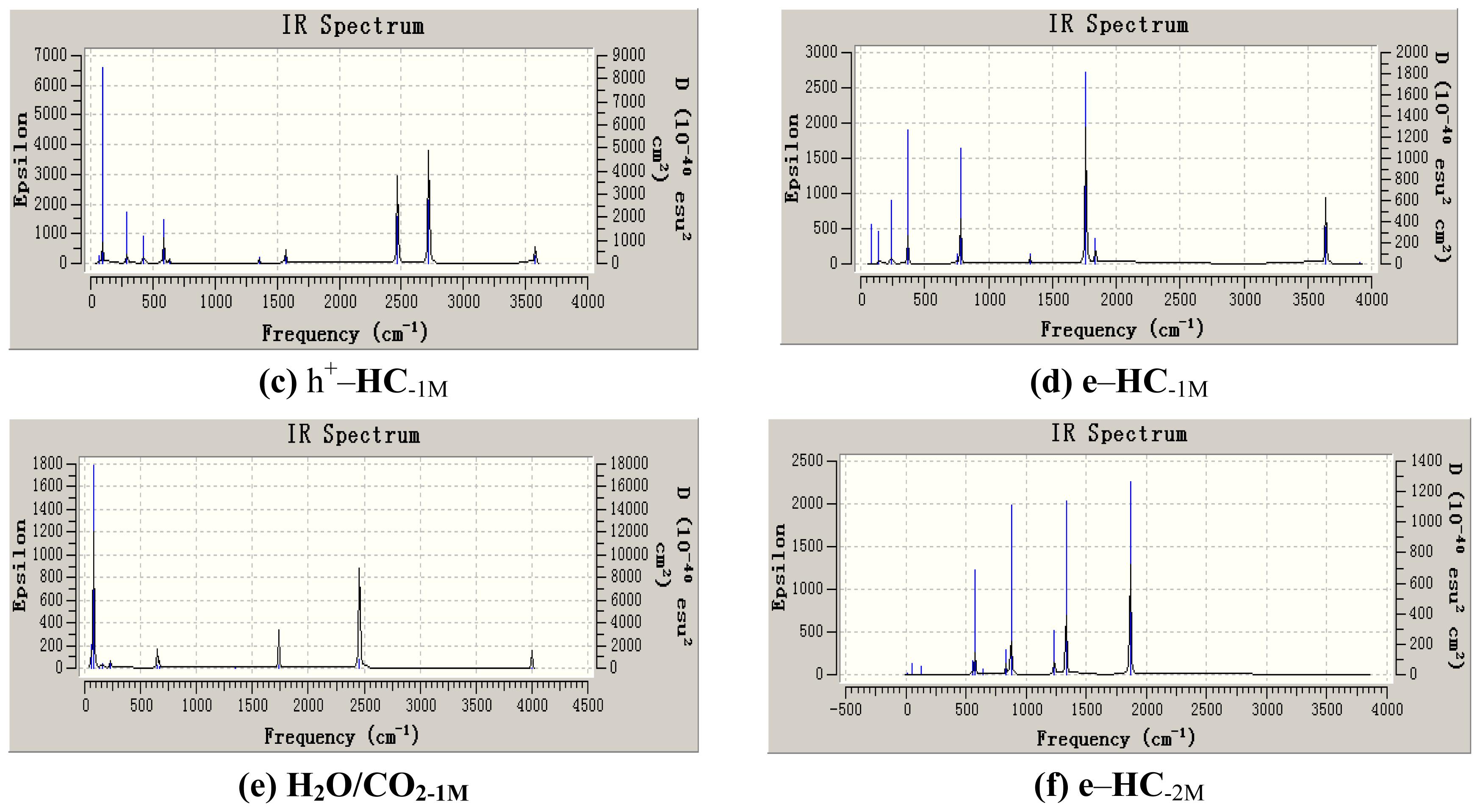
3. Results and Discussion
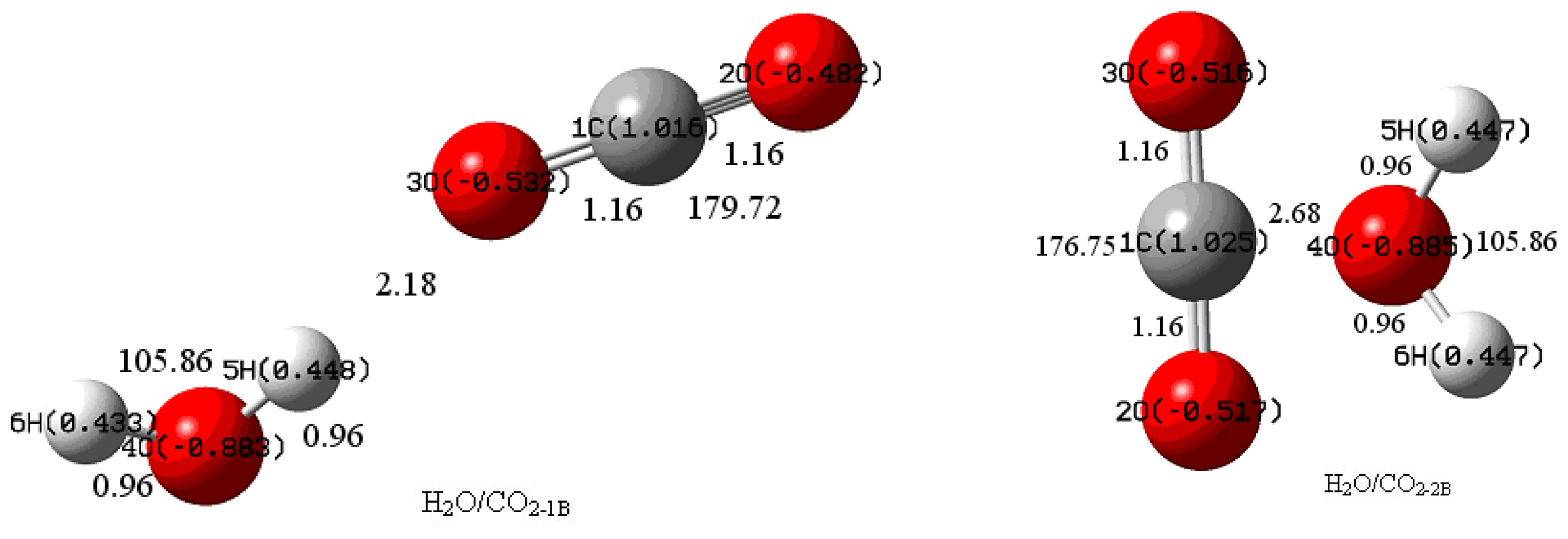
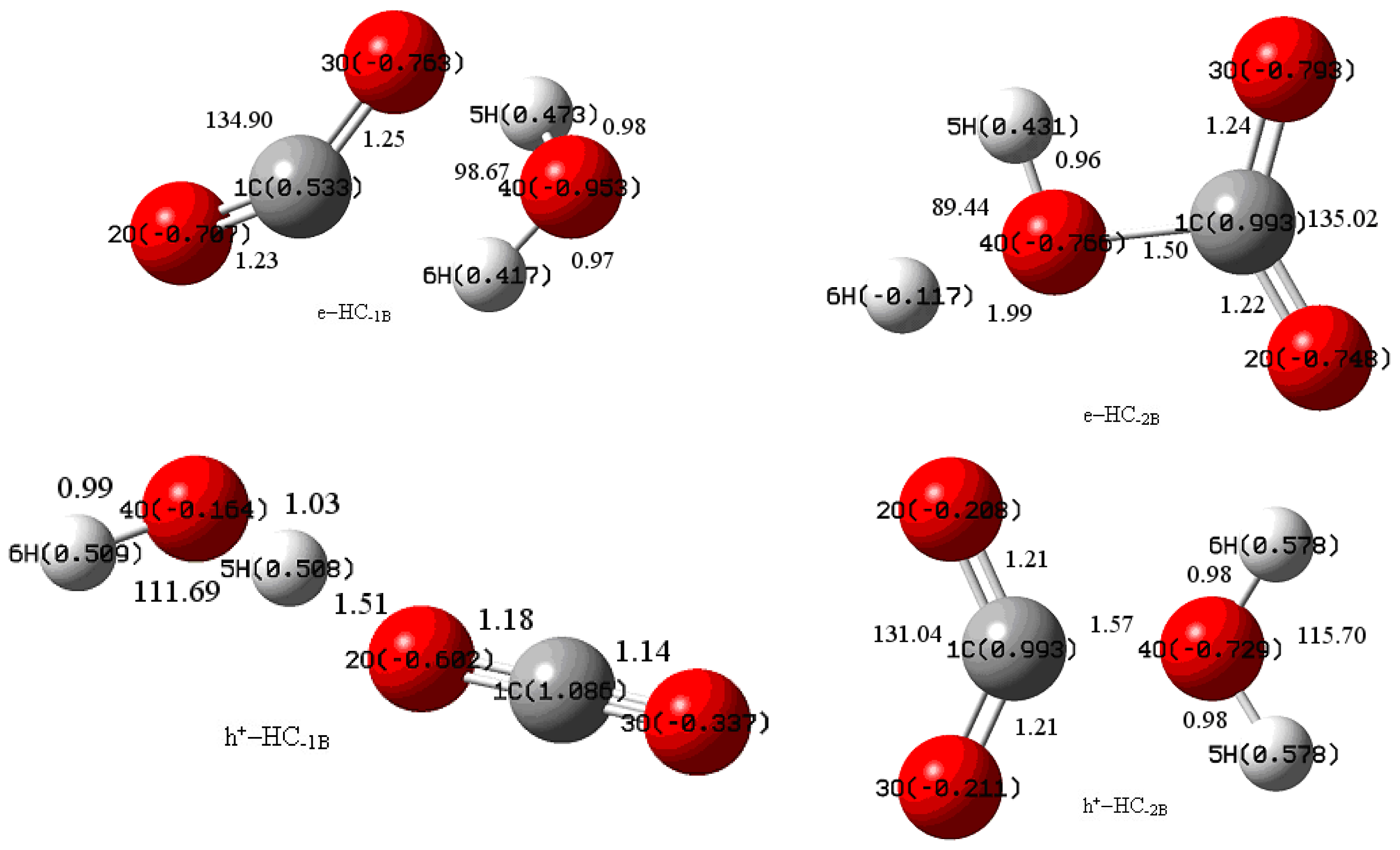
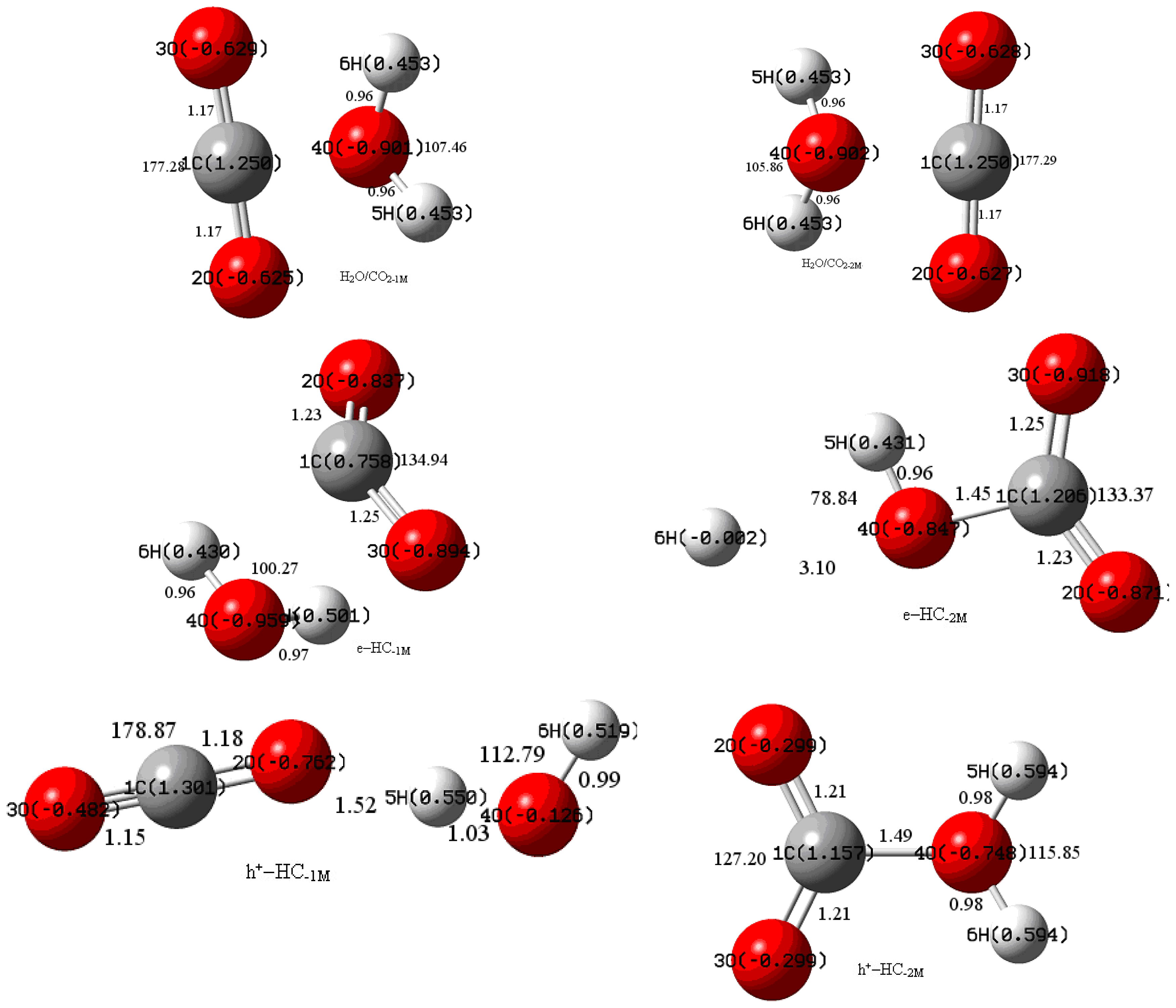
3.1. Geometric Parameters Analysis
3.2. NBO Atomic Charge Analysis
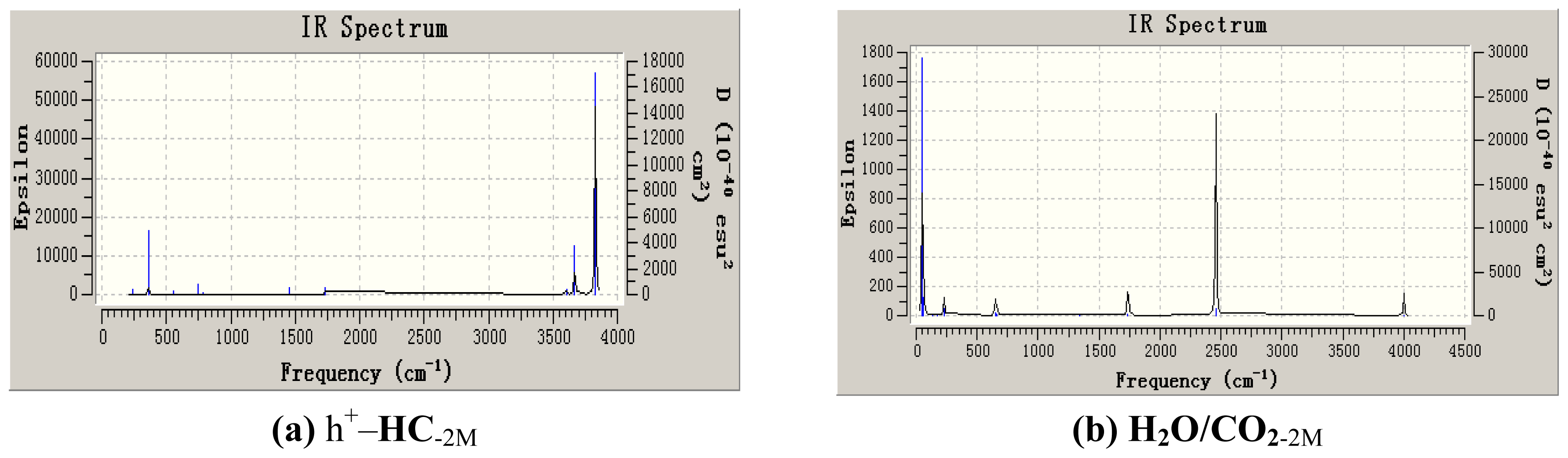
3.3. Thermochemistry Parameters Analysis
4. Conclusion
Acknowledgement
References
- Subrahmanyam, M; Kaneco, S; Alonso-Vante, N. A Screening for the Photo Reduction of Carbon Dioxide Supported on Metal Oxide Catalysts for C1–C3 Selectivity. Appl. Catal. B 1999, 23, 169–174. [Google Scholar]
- Tseng, IH; Chang, WC; Wu, JCS. Photoreduction of CO2 Using Sol-Gel Derived Titania and Titania-Supported Copper Catalysts. Appl. Catal. B 2002, 37, 37–48. [Google Scholar]
- Xie, TF; Wang, DJ; Zhu, LJ; Li, TJ; Xu, YJ. Application of Surface Photovoltage Technique in Photocatalysis Studies on Modified TiO2 Photo-catalysts for Photo-reduction of CO2. Mater. Chem. Phys 2001, 70, 103–106. [Google Scholar]
- Linsebigler, AL; Lu, GJ; Yates, JT. Photocatalysis on TiO2 Surfaces: Principles, Mechanisms, and Selected Results. Chem. Rev 1995, 95, 735–758. [Google Scholar]
- Sasirekha, N; Basha, S; Shanthi, K. Photocatalytic Performance of Ru Doped Anatase Mounted on Silica for Reduction of Carbon Dioxide. Appl. Catal. B Environ 2006, 62, 169–180. [Google Scholar]
- Yamashita, H; Fujii, Y; Ichihashi, Y; Zhang, SG; Ikeue, K; Park, DR; Koyano, K; Tatsumi, T; Anpo, M. Selective Formation of CH3OH in the Photocatalytic Reduction of CO2 with H2O on Titanium Oxides highly Dispersed within Zeolites and Mesoporous Molecular Sieves. Catal. Today 1998, 45, 221–227. [Google Scholar]
- Zhang, SG; Fujii, Y; Yamashita, K; Koyano, K; Tatsumi, T; Anpo, M. Photocatalytic Reduction of CO2 with H2O on Ti-MCM-41 and Ti-MCM-48 Mesoporous Zeolites at 328 K. Chem. Lett 1997, 7, 659–660. [Google Scholar]
- Rasko, J; Solymosi, F. Infrared Spectroscopic Study of the Photoinduced Activation of CO2 on TiO2 and Rh/TiO2 Catalysts. J. Phys. Chem 1994, 98, 7147–7152. [Google Scholar]
- Lin, WY; Han, HX; Frei, H. CO2 Splitting by H2O to CO and O2 under UV Light in TiMCM-41 Silicate Sieve. J. Phys. Chem. B 2004, 108, 18269–18273. [Google Scholar]
- Anpo, M; Yamashita, H; Ikeue, K; Fujii, Y; Zhang, SG; Ichihashi, Y; Park, DR; Suzuki, Y; Koyano, K; Tatsumi, T. Photocatalytic Reduction of CO2 with H2O on Ti-MCM-41 and Ti-MCM-48 Mesoporous Zeolite Catalysts. Catal. Today 1998, 44, 327–332. [Google Scholar]
- Anpo, M; Yamashita, H; Ichihashi, Y; Fujii, Y; Honda, M. Photocatalytic Reduction of CO2 with H2O on Titanium Oxides Anchored within Micropores of Zeolites: Effects of the Structure of the Active Sites and the Addition of Pt. J. Phys. Chem. B 1997, 101, 2632–2636. [Google Scholar]
- Anpo, M; Yamashita, H; Ichihashi, Y; Ehara, S. Photocatalytic Reduction of CO2 with H2O on Various Titanium Oxide Catalysts of Special Interest. J. Electroanal. Chem 1995, 396, 21–26. [Google Scholar]
- Xu, YJ; Chen, FM; Jiang, L; Zhou, LD. Photoreduction of CO2 in the Suspension System of Semiconductor Catalyst TiO2 Modified by Palladium. Photogr. Sci. Photochem 1999, 17, 61–65. [Google Scholar]
- Banfield, JF; Navrotsky, A. Reviews in Mineralogy and Geochemistry. Mineral. Soc. Am 2001, 44, 293–349. [Google Scholar]
- Huang, D; Xiao, ZD; Gu, JH; Huang, NP; Yuan, CW. TiO2 Thin-Films Formation on Industrial Glass through Self-Assembly Processing. Thin Solid Films 1997, 305, 110–115. [Google Scholar]
- Li, W; Shah, SI; Hunag, CP; Jung, O; Ni, C. Metallorganic Chemical Vapor Deposition and Characterization of TiO2 Nanoparticles. Mater. Sci. Eng. B 2002, 96, 247–253. [Google Scholar]
- Luo, DM; Chen, C; Zhang, N; Hong, SG; Wu, HW; Liu, ZH. Characterization and DFT Research of Nd/TiO2: Photocatalyst for Synthesis of Methanol from CO2 and H2O. Z. Phys. Chem 2009, 223, 1470–1476. [Google Scholar]
- Hemminger, JC; Carr, R; Somorjai, GA. The Photoassisted Reaction of Gaseous Water and Carbon Dioxide Adsorbed on the SrTiO3 (111) Crystal Face to form Methane. Chem. Phys. Lett 1978, 57, 100–104. [Google Scholar]
- Ikeue, K; Nozaki, S; Ogawa, M; Anpo, M. Characterization of Self-standing Ti-containing Porous Silica Thin Films and Their Reactivity for the Photocatalytic Reduction of CO2 with H2O. Catal. Today 2002, 74, 241–248. [Google Scholar]
- Inokuchi, Y; Kobayashi, Y; Muraoka, A; Nagata, T; Ebata, T. Structures of water-CO2 and methanol-CO2 cluster ions: [H2O•(CO2)n]+ and [CH3OH•(CO2)n]+ (n = 1–7). J. Chem. Phys 2009, 130, 1–12. [Google Scholar]
- Hohenberg, P; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. B 1964, 136, B864–B871. [Google Scholar]
- Kohn, W; Sham, LJ. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev 1965, 140, A1133–A1138. [Google Scholar]
- Gerratt, J; Mills, IM. Force Constants and Dipole-Moment Derivatives of Molecules from Perturbed Hartree–Fock Calculations. J. Chem. Phys 1968, 49, 1719–1729. [Google Scholar]
- Pulay, P. Second and Third Derivatives of Variational Energy Expressions-application to Multi-configurational Self-consistent Field Wave-functions. J. Chem. Phys 1983, 78, 5043–5051. [Google Scholar]
- Vosko, SH; Wilk, L; Nusair, M. Accurate Spin-dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys 1980, 58, 1200–1211. [Google Scholar]
- Lee, CT; Yang, WT; Parr, RG. Development of the Colle-Salvetti Correlation-energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Becke, AD. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- Becke, AD. Density-functional Exchange-Energy Approximation with Correct Asymptotic Behaviour. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar]
- McLean, AD; Chandler, GS. Contracted Gaussian-basis Sets for Molecular Calculations. 1. 2nd Row Atoms, Z = 11–18. J. Chem. Phys 1980, 72, 5639–5648. [Google Scholar]
- M⊘ller, C; Plesset, MS. Note on an Approximation Treatment for Many-electron Systems. Phys. Rev 1934, 46, 618–622. [Google Scholar]
- Krishnan, R; Binkley, JS; Seeger, R; Pople, JA. Self-Consistent Molecular Orbital Methods. 20. Basis set for Correlated Wave-functions. J. Chem. Phys 1980, 72, 650–654. [Google Scholar]
- Frisch, MJ. Gaussian 94; Revision E.2; Gaussian: Pittsburgh, PA, USA, 1995. [Google Scholar]
- Frisch, MJ; Trucks, GW; Schlegel, HB; Scuseria, GE; Robb, MA; Cheeseman, JR; Montgomery, JA, Jr; Vreven, T; Kudin, KN; Burant, JC; et al. Gaussian 03; Revision E.01; Gaussian Inc: Wallingford, CT, USA, 2004. [Google Scholar]
- Li, Z; Wang, L; Ran, H; Xie, D; Blinov, N; Roy, PN; Guo, H. Path integral Monte Carlo study of CO2 solvation in 4He clusters. J. Chem. Phys 2008, 128, 224513:1–224513:7. [Google Scholar]
- DelBene, JE. Ab initio Molecular Orbital Study of the Structures and Energies of Neutral and Charged Bimolecular Complexes of Water with the Hydrides AHn (A = nitrogen, oxygen, fluorine, phosphorus, sulfur, and chlorine). J. Phys. Chem 1988, 92, 2874–2880. [Google Scholar]
- Binkley, JS; Pople, JA. M⊘ller-Plesset Theory for Atomic Ground State Energies. Int. J. Quantum Chem 1975, 9, 229–236. [Google Scholar]
- Feller, D. Application of Systematic Sequences of Wave-functions to the Water Dimer. J. Chem. Phys 1992, 96, 6104–6114. [Google Scholar]
- Kim, KS; Mhin, BJ; Choi, US; Lee, K. Ab-initio Studies of the Water Dimer using large Basis set: The Structure and Thermodynamics Energies. J. Chem. Phys 1992, 97, 6649–6662. [Google Scholar]
- Szalewicz, K; Cole, SJ; Kolos, W; Bartlett, RJ. A Theoretical Study of the Water. Dimer Interaction. J. Chem. Phys 1988, 89, 3662–3673. [Google Scholar]
- Frisch, MJ; DelBene, JE; Binkley, JS; Schaefer, HF, III. Retical Studies of the Hydrogen-bonded Complexes (H2O)2, (H2O)2H+, (HF)2, (HF)2H+, F2H−, and (NH3)2. J. Chem. Phys 1986, 48, 2279–2289. [Google Scholar]

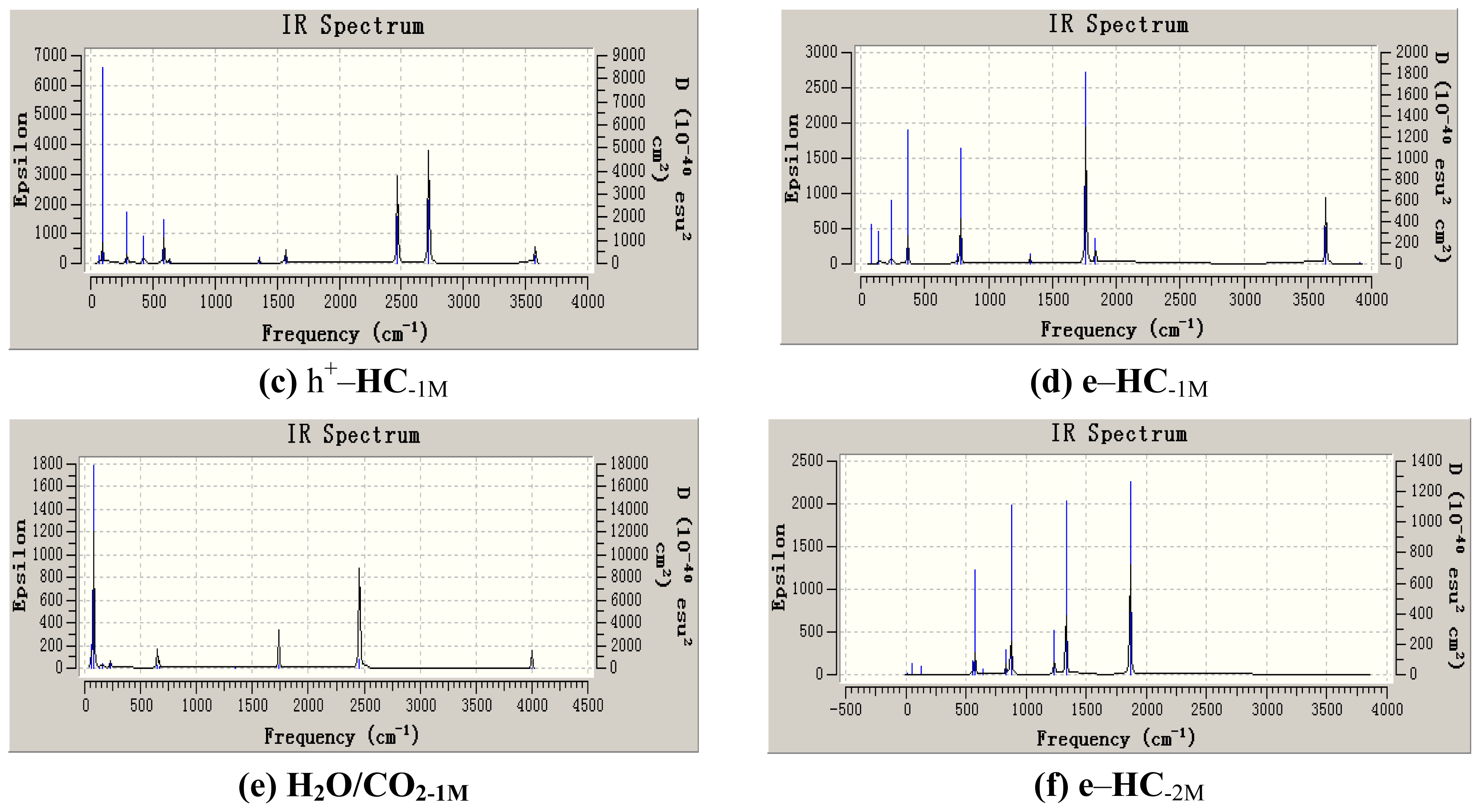

{kind=link}
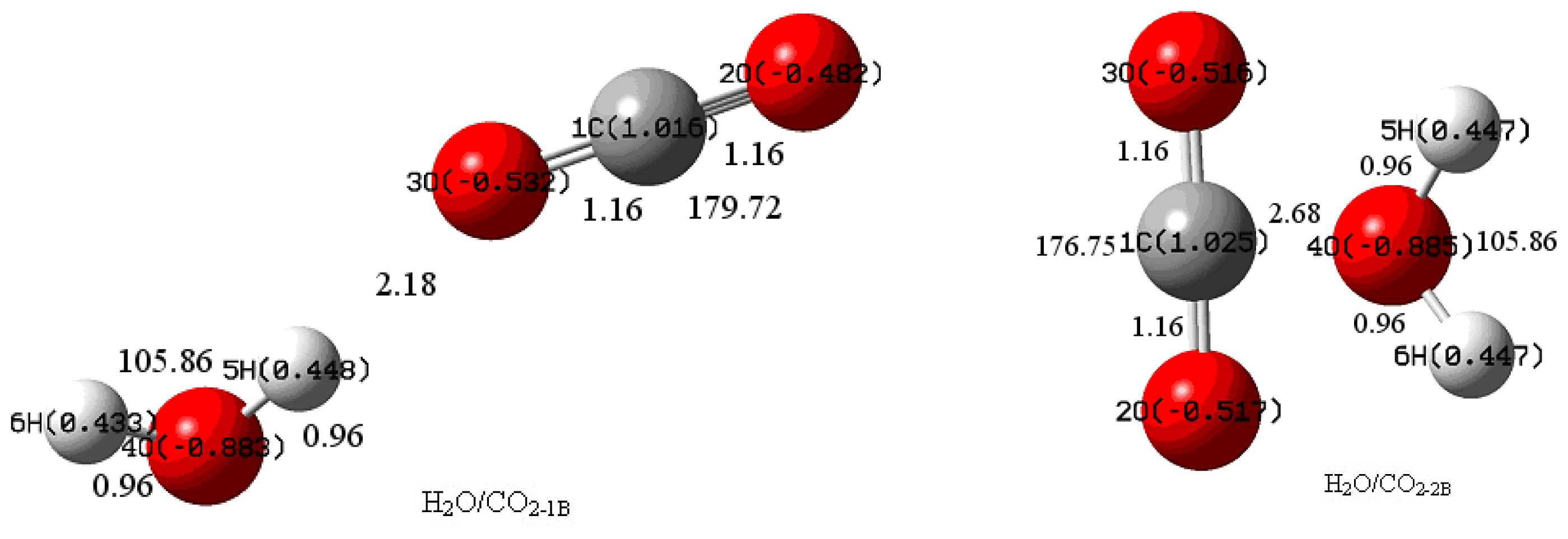
{kind=link}
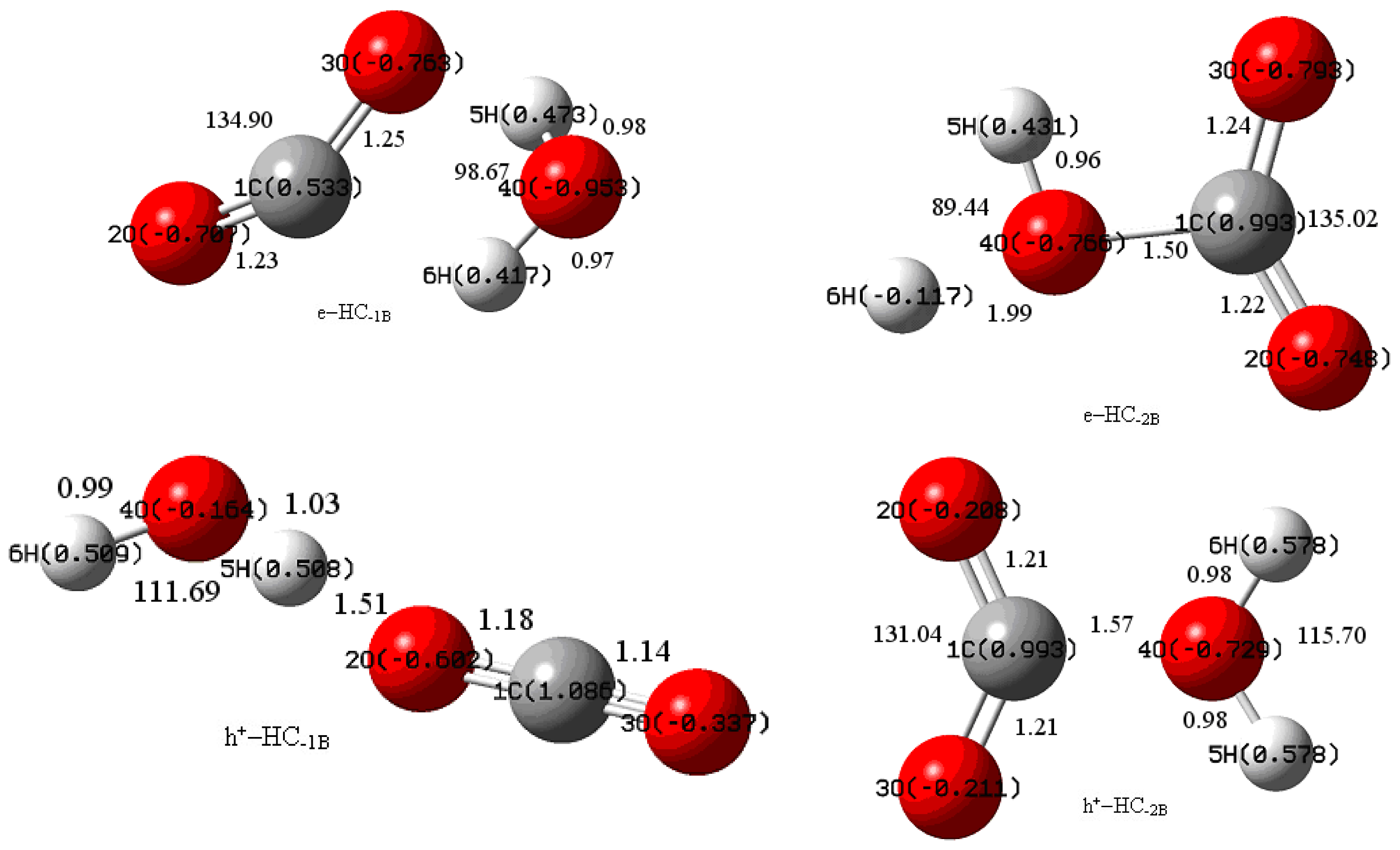
{kind=link}
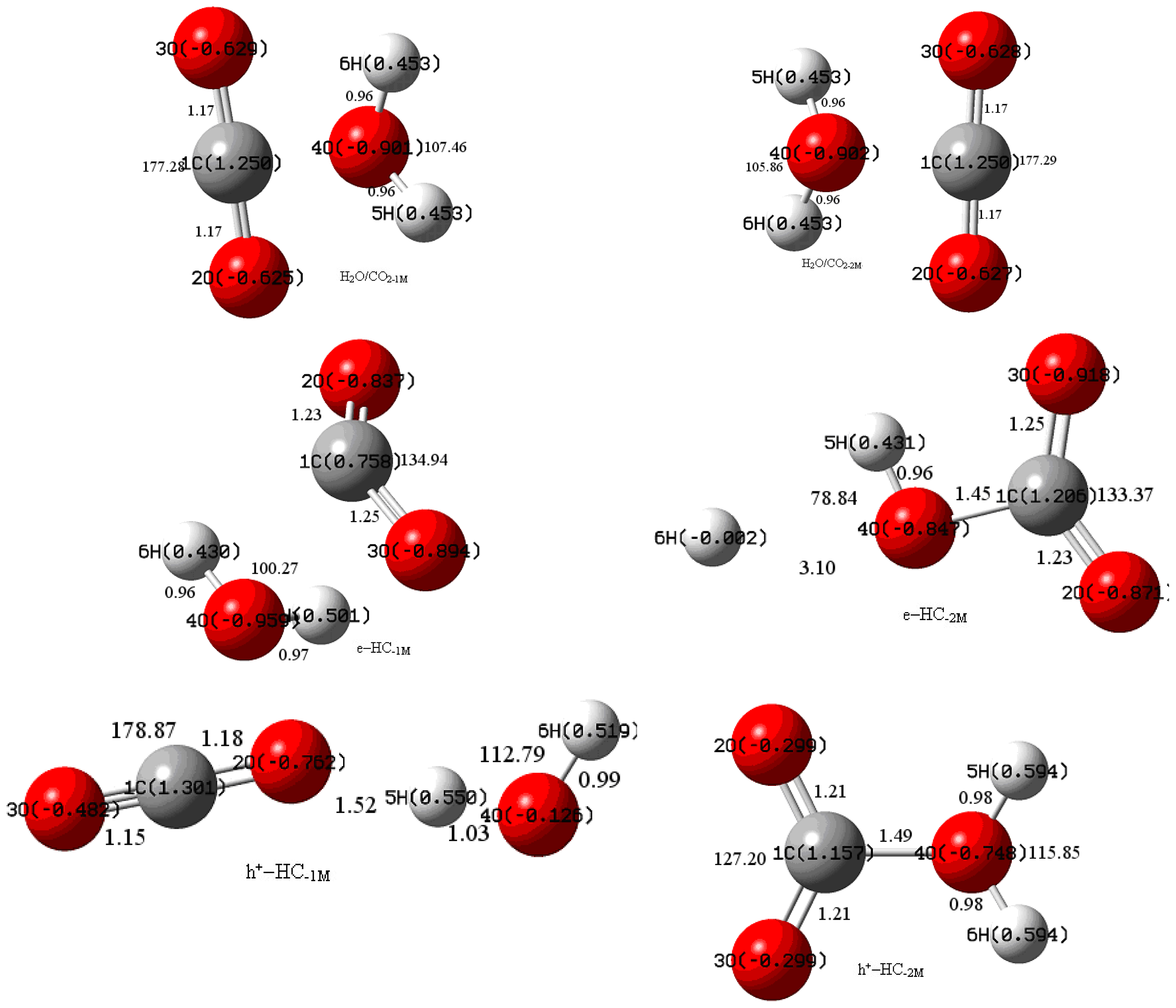
{kind=link}
{kind=link}
{kind=link}
| M | E | h+–HC−1 | h+–HC−2 | e-HC−1 | e-HC−2 | H2O/CO2-1 | H2O/CO2-2 |
|---|---|---|---|---|---|---|---|
| B | O | 2s(1.69) g | 2s(1.75) | 2s(1.74) | 2s(1.72) | 2s(1.72) | 2s(1.72) |
| 3 | 2p(4.90) h | 2p(4.45) | 2p(4.96) | 2p(5.02) | 2p(4.75) | 2p(4.78) | |
| L | 3d(0.01) i | 3d(0.01) | 3d(0.01) | 3d(0.01) | 3d( 0.01) | 3d( 0.01) | |
| Y | C | 2s(0.62) | 2s(0.68) | 2s(0.96) | 2s(0.67) | 2s(0.64) | 2s(0.64) |
| P | 2p(2.25) | 2p(2.27) | 2p(2.40) | 2p(2.27) | 2p(2.30) | 2p(2.29) | |
| 3p(0.02) | 3p(0.03) | 3s(0.06) | 3p(0.05) | 3p(0.03) | 3p(0.03) | ||
| 3d(0.02) | 3d(0.02) | 3p(0.04) | 3d(0.02) | 3d(0.02) | 3d(0.02) | ||
| B | 3d(0.01) | ||||||
| 3 | O | 2s(1.72) | 2s(1.75) | 2s(1.75) | 2s(1.73) | 2s(1.72) | 2s(1.72) |
| L | 2p(4.60) | 2p(4.45) | 2p(5.01) | 2p(5.05) | 2p(4.80) | 2p(4.78) | |
| Y | 3d(0.01) | 3d(0.01) | 3d( 0.01) | 3d(0.01) | 3d(0.01) | 3d(0.01) | |
| P | H | 1s( 0.49) | 1s(0.42) | 1s(0.52) | 1s(0.56) | 1s(0.55) | 1s(0.55) |
| 2s(0.01) | 2s(0.01) | ||||||
| B | O | 2s(1.79) | 2s(1.65) | 2s(1.75) | 2s(1.75) | 2s(1.75) | 2s(1.75) |
| 3 | 2p(4.37) | 2p(5.07) | 2p(5.19) | 2p(5.00) | 2p(5.13) | 2p(5.13) | |
| L | 3d( 0.01) | 3d(0.01) | 3d( 0.01) | 3d(0.01) | |||
| Y | H | 1s( 0.49) | 1s(0.42) | 1s(0.52) | 1s(1.12) | 1s( 0.56) | 1s(0.55) |
| P | 2s(0.01) | ||||||
| M | O | 2s(1.70) | 2s(1.75) | 2s(1.74) | 2s(1.73) | 2s(1.72) | 2s(1.72) |
| P | 2p(5.05) | 2p(4.54) | 2p(5.08) | 2p(5.13) | 2p(4.89) | 2p(4.89) | |
| 2 | 3p(0.01) | ||||||
| 3d(0.01) | 3d(0.01) | 3d(0.01) | 3d(0.01) | 3d( 0.01) | 3d( 0.01) | ||
| M | C | 2s(0.63) | 2s(0.66) | 2s(0.93) | 2s(0.66) | 2s(0.65) | 2s(0.65) |
| P | 2p(2.02) | 2p(2.11) | 2p(2.20) | 2p(2.06) | 2p(2.05) | 2p(2.05) | |
| 2 | 3p(0.02) | 3p(0.03) | 3s(0.05) | 3p(0.04) | 3p(0.03) | 3p(0.03) | |
| 3d(0.03) | 3d(0.03) | 3p(0.04) | 3d(0.03) | 3d(0.03) | 3d(0.03) | ||
| M | 3d(0.02) | ||||||
| P | O | 2s(1.72) | 2s(1.75) | 2s(1.75) | 2s(1.74) | 2s(1.72) | 2s(1.72) |
| 2 | 2p(4.75) | 2p(4.54) | 2p(5.13) | 2p(5.17) | 2p(4.89) | 2p(4.89) | |
| 3d(0.01) | 3d(0.01) | 3d( 0.01) | 3d(0.01) | d(0.01) | 33d(0.01) | ||
| M | H | 1s( 0.45) | 1s(0.40) | 1s(0.49) | 1s(0.56) | 1s(0.54) | 1s(0.54) |
| P | |||||||
| 2 | O | 2s(1.78) | 2s(1.64) | 2s(1.75) | 2s(1.73) | 2s(1.74) | 2s(1.74) |
| 2p(4.34) | 2p(5.11) | 2p(5.20) | 2p(5.10) | 2p(5.16) | 2p(5.16) | ||
| M | 3d( 0.01) | 3d(0.01) | 3d(0.01) | 3d( 0.01) | 3d(0.01) | ||
| P | H | 1s( 0.48) | 1s(0.40) | 1s(0.57) | 1s(1.00) | 1s( 0.54) | 1s(0.54) |
| 2 | 2s(0.01) | ||||||
| Method parameter | B3LYP | MP2 | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔEtot kJ/mol | ΔH kJ/mol | ΔG kJ/mol | ΔS J/mol·K | ΔEtot kJ/mol | ΔH kJ/mol | ΔG kJ/mol | ΔS J/mol·K | |
| HC−1 | 4.68 | 0.28 | 53.78 | −160.66 | −26.26 | −26.26 | 52.52 | −264.32 |
| HC−2 | −2.48 | −7.44 | 35.45 | −128.81 | −26.26 | −26.26 | 52.52 | −264.32 |
| e-HC−1 | −69.04 | −74.00 | −4.80 | −207.81 | −73.51 | −68.26 | 2.62 | −237.73 |
| e-HC−2 | 14.54 | 9.58 | 90.75 | −243.75 | −28.88 | −23.63 | 39.38 | −211.34 |
| h+–HC−1 | −84.70 | −93.49 | −37.77 | −167.33 | −77.75 | −58.17 | −8.13 | −167.83 |
| h+–HC−2 | −25.74 | −30.68 | 36.74 | −202.46 | 42.47 | 47.24 | 108.29 | −204.76 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Luo, D.; Zhang, N.; Hong, S.; Wu, H.; Liu, Z. Complexes in the Photocatalytic Reaction of CO2 and H2O: Theoretical Studies. Int. J. Mol. Sci. 2010, 11, 2792-2804. https://doi.org/10.3390/ijms11082792
Luo D, Zhang N, Hong S, Wu H, Liu Z. Complexes in the Photocatalytic Reaction of CO2 and H2O: Theoretical Studies. International Journal of Molecular Sciences. 2010; 11(8):2792-2804. https://doi.org/10.3390/ijms11082792
Chicago/Turabian StyleLuo, Dongmei, Ning Zhang, Sanguo Hong, Huanwen Wu, and Zhihua Liu. 2010. "Complexes in the Photocatalytic Reaction of CO2 and H2O: Theoretical Studies" International Journal of Molecular Sciences 11, no. 8: 2792-2804. https://doi.org/10.3390/ijms11082792