Hub Promiscuity in Protein-Protein Interaction Networks

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
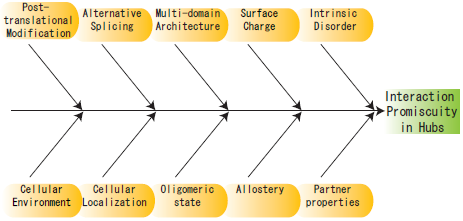
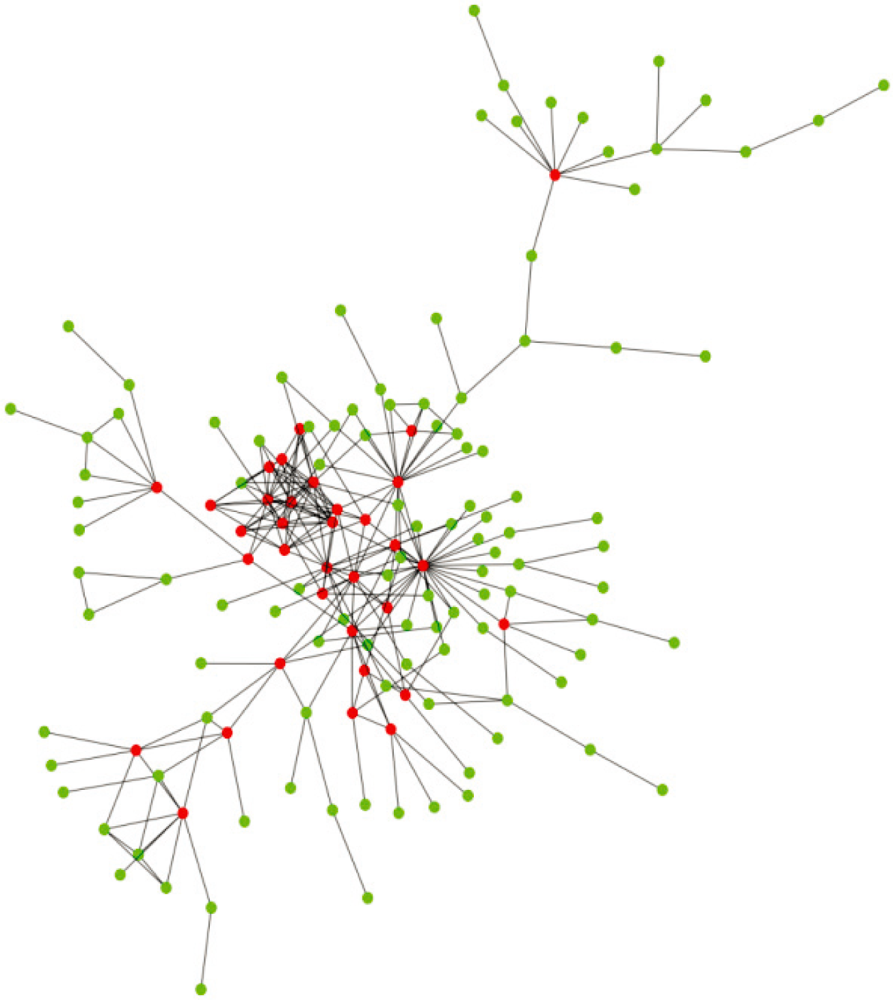
2. Structural Characteristics of Hub Proteins
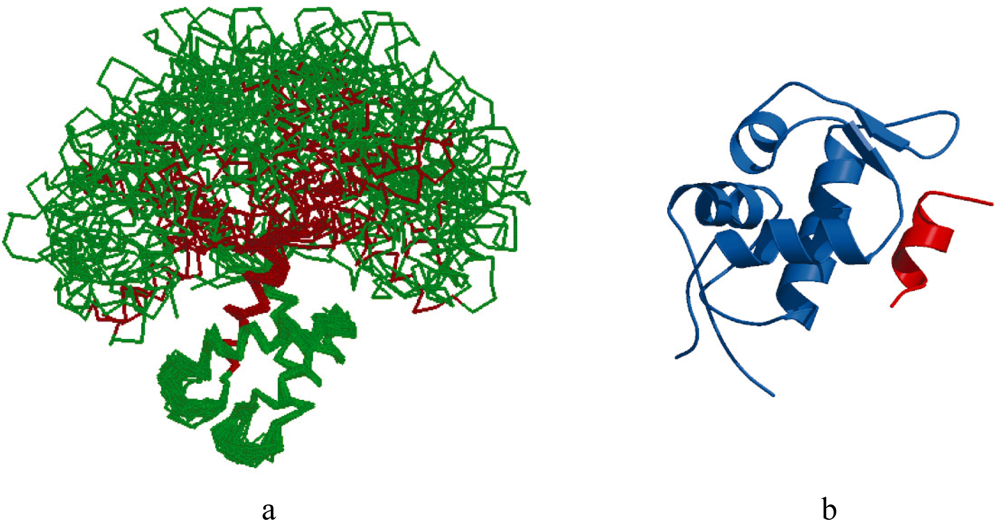
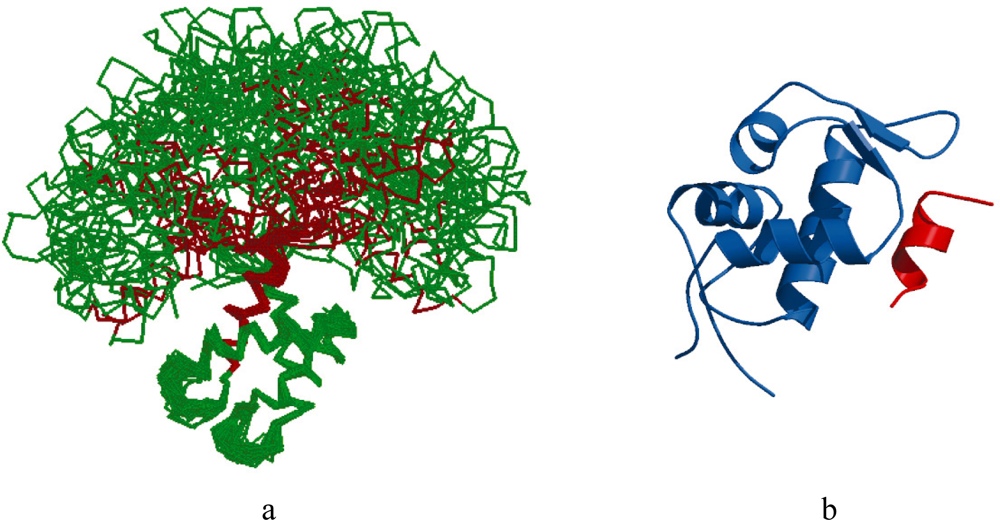
2.1. Intrinsic Disorder
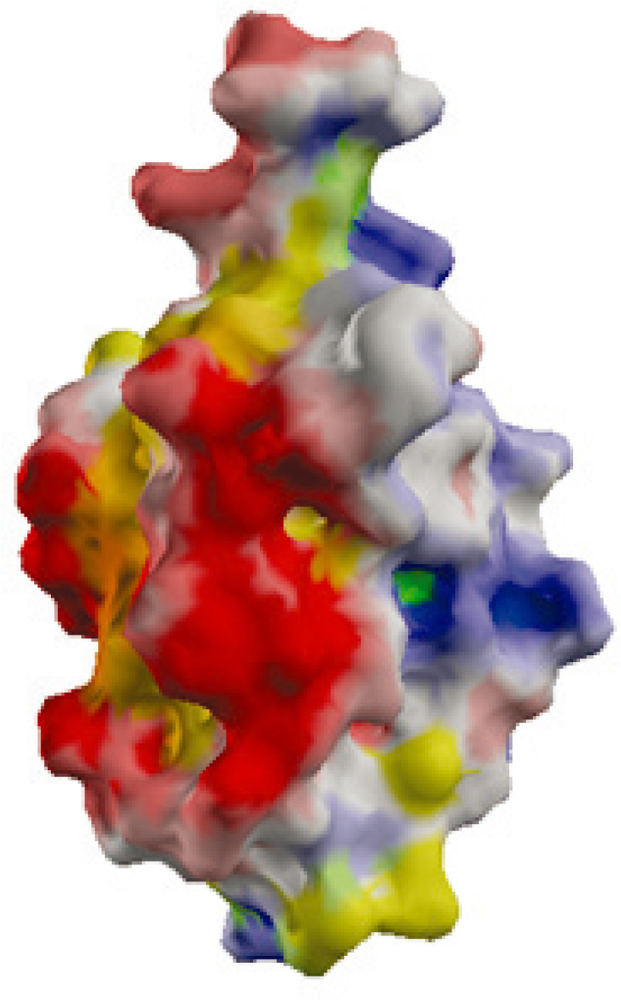
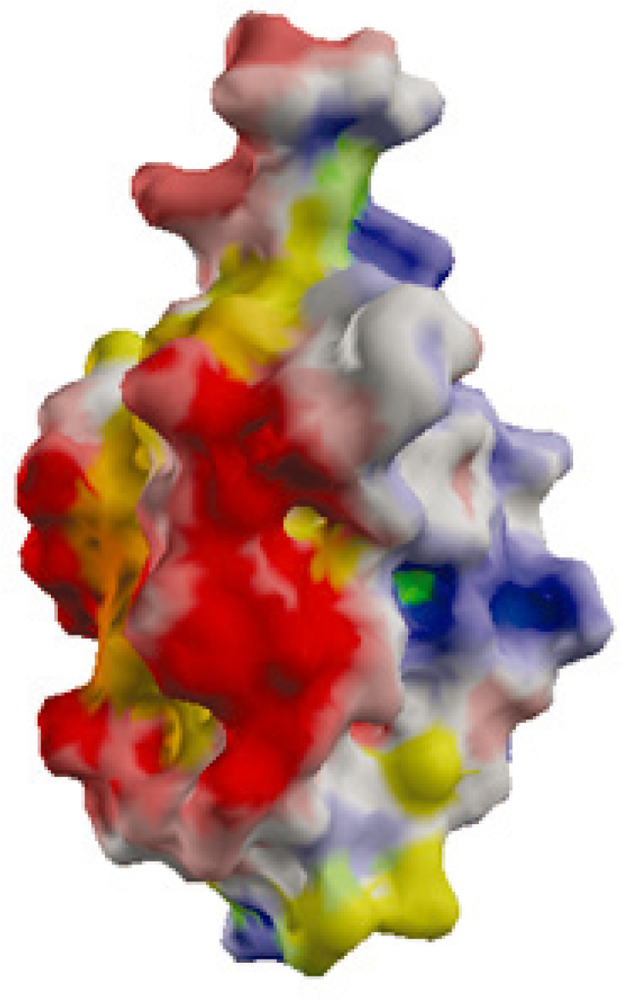
2.2. Surface Charge
2.3. Domain Distribution and Enrichment
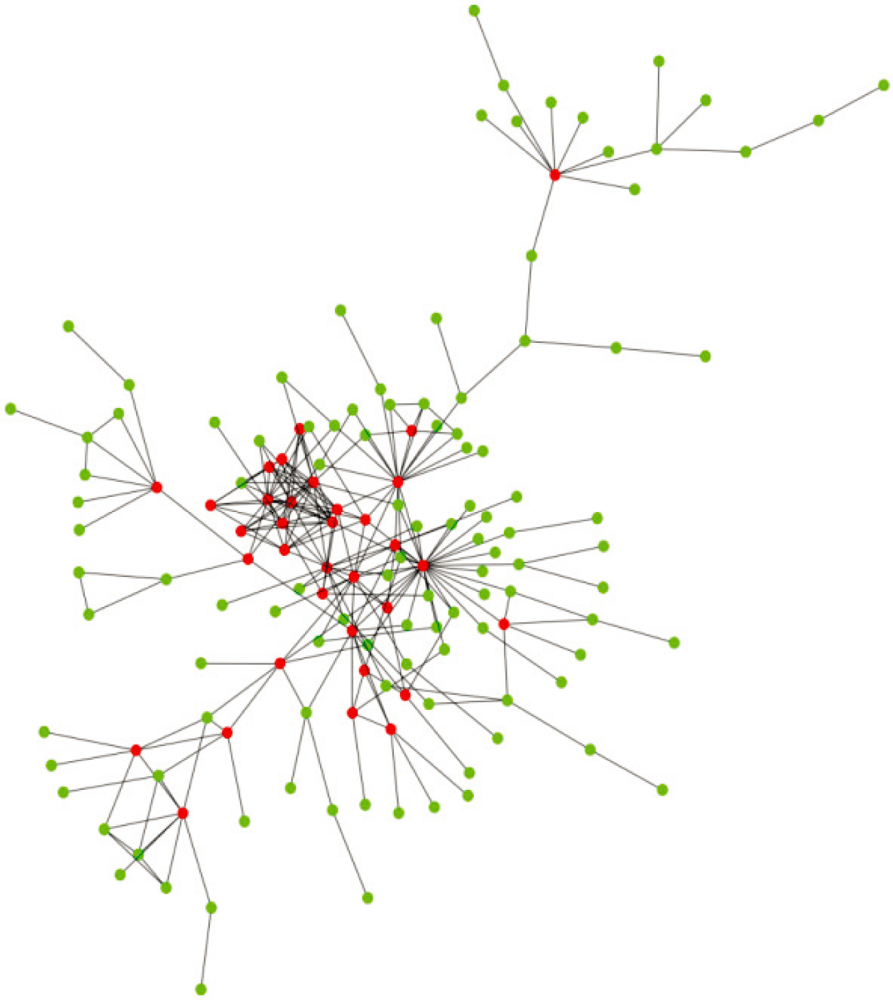
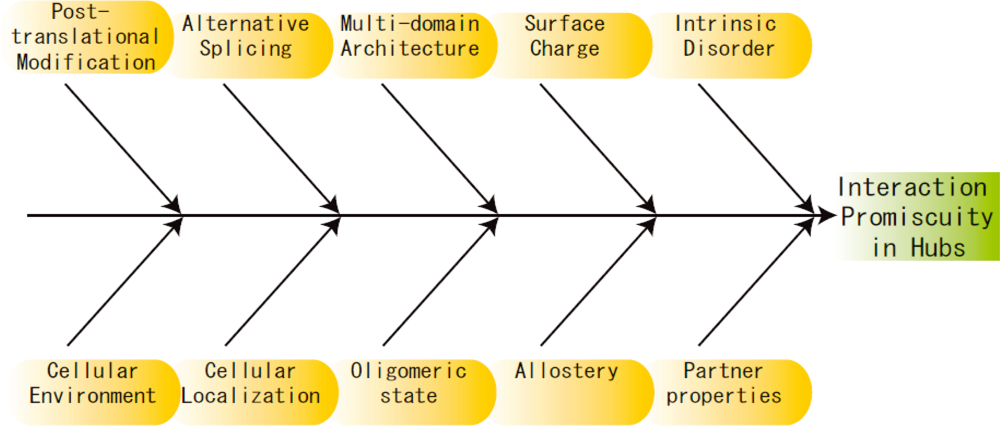
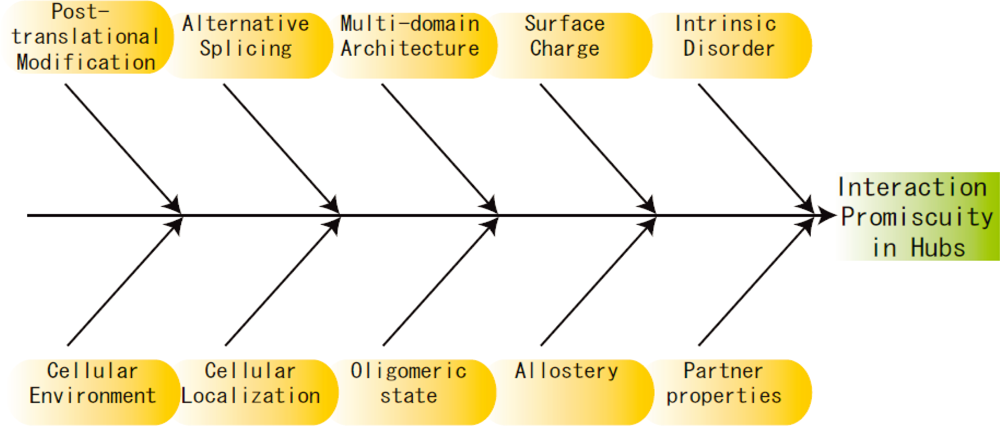
3. Other Perspectives
4. Future Directions and Challenges
Acknowledgments
References and Notes
- Uetz, P; Giot, L; Cagney, G; Mansfield, TA; Judson, RS; Knight, JR; Lockshon, D; Narayan, V; Srinivasan, M; Pochart, P; Qureshi-Emili, A; Li, Y; Godwin, B; Conover, D; Kalbfleisch, T; Vijayadamodar, G; Yang, M; Johnston, M; Fields, S; Rothberg, JM. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 2000, 403, 623–627. [Google Scholar]
- Ito, T; Chiba, T; Ozawa, R; Yoshida, M; Hattori, M; Sakaki, Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 2001, 98, 4569–4574. [Google Scholar]
- Gavin, A-C; Bosche, M; Krause, R; Grandi, P; Marzioch, M; Bauer, A; Schultz, J; Rick, JM; Michon, A-M; Cruciat, C-M; Remor, M; Hofert, C; Schelder, M; Brajenovic, M; Ruffner, H; Merino, A; Klein, K; Hudak, M; Dickson, D; Rudi, T; Gnau, V; Bauch, A; Bastuck, S; Huhse, B; Leutwein, C; Heurtier, M-A; Copley, RR; Edelmann, A; Querfurth, E; Rybin, V; Drewes, G; Raida, M; Bouwmeester, T; Bork, P; Seraphin, B; Kuster, B; Neubauer, G; Superti-Furga, G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 415, 141–147. [Google Scholar]
- Ho, Y; Gruhler, A; Heilbut, A; Bader, GD; Moore, L; Adams, S-L; Millar, A; Taylor, P; Bennett, K; Boutilier, K; Yang, L; Wolting, C; Donaldson, I; Schandorff, S; Shewnarane, J; Vo, M; Taggart, J; Goudreault, M; Muskat, B; Alfarano, C; Dewar, D; Lin, Z; Michalickova, K; Willems, AR; Sassi, H; Nielsen, PA; Rasmussen, KJ; Andersen, JR; Johansen, LE; Hansen, LH; Jespersen, H; Podtelejnikov, A; Nielsen, E; Crawford, J; Poulsen, V; Sorensen, BD; Matthiesen, J; Hendrickson, RC; Gleeson, F; Pawson, T; Moran, MF; Durocher, D; Mann, M; Hogue, CWV; Figeys, D; Tyers, M. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 2002, 415, 180–183. [Google Scholar]
- Giot, L; Bader, JS; Brouwer, C; Chaudhuri, A; Kuang, B; Li, Y; Hao, YL; Ooi, CE; Godwin, B; Vitols, E; Vijayadamodar, G; Pochart, P; Machineni, H; Welsh, M; Kong, Y; Zerhusen, B; Malcolm, R; Varrone, Z; Collis, A; Minto, M; Burgess, S; McDaniel, L; Stimpson, E; Spriggs, F; Williams, J; Neurath, K; Ioime, N; Agee, M; Voss, E; Furtak, K; Renzulli, R; Aanensen, N; Carrolla, S; Bickelhaupt, E; Lazovatsky, Y; DaSilva, A; Zhong, J; Stanyon, CA; Finley, RL, Jr; White, KP; Braverman, M; Jarvie, T; Gold, S; Leach, M; Knight, J; Shimkets, RA; McKenna, MP; Chant, J; Rothberg, JM. A protein interaction map of Drosophila melanogaster. Science 2003, 302, 1727–1736. [Google Scholar]
- Li, S; Armstrong, CM; Bertin, N; Ge, H; Milstein, S; Boxem, M; Vidalain, P-O; Han, J-DJ; Chesneau, A; Hao, T; Goldberg, DS; Li, N; Martinez, M; Rual, J-F; Lamesch, P; Xu, L; Tewari, M; Wong, SL; Zhang, LV; Berriz, GF; Jacotot, L; Vaglio, P; Reboul, J; Hirozane-Kishikawa, T; Li, Q; Gabel, HW; Elewa, A; Baumgartner, B; Rose, DJ; Yu, H; Bosak, S; Sequerra, R; Fraser, A; Mango, SE; Saxton, WM; Strome, S; van den Heuvel, S; Piano, F; Vandenhaute, J; Sardet, C; Gerstein, M; Doucette-Stamm, L; Gunsalus, KC; Harper, JW; Cusick, ME; Roth, FP; Hill, DE; Vidal, M. A Map of the interactome network of the metazoan C. elegans. Science 2004, 303, 540–543. [Google Scholar]
- Rual, JF; Venkatesan, K; Hao, T; Hirozane-Kishikawa, T; Dricot, A; Li, N; Berriz, GF; Gibbons, FD; Dreze, M; Ayivi-Guedehoussou, N; Klitgord, N; Simon, C; Boxem, M; Milstein, S; Rosenberg, J; Goldberg, DS; Zhang, LV; Wong, SL; Franklin, G; Li, S; Albala, JS; Lim, J; Fraughton, C; Llamosas, E; Cevik, S; Bex, C; Lamesch, P; Sikorski, RS; Vandenhaute, J; Zoghbi, HY; Smolyar, A; Bosak, S; Sequerra, R; Doucette-Stamm, L; Cusick, ME; Hill, DE; Roth, FP; Vidal, M. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar]
- Stelzl, U; Worm, U; Lalowski, M; Haenig, C; Brembeck, FH; Goehler, H; Stroedicke, M; Zenkner, M; Schoenherr, A; Koeppen, S; Timm, J; Mintzlaff, S; Abraham, C; Bock, N; Kietzmann, S; Goedde, A; Toksoz, E; Droege, A; Krobitsch, S; Korn, B; Birchmeier, W; Lehrach, H; Wanker, EE. A human protein-protein interaction network: A resource for annotating the proteome. Cell 2005, 122, 957–968. [Google Scholar]
- Gavin, A-C; Aloy, P; Grandi, P; Krause, R; Boesche, M; Marzioch, M; Rau, C; Jensen, LJ; Bastuck, S; Dumpelfeld, B; Edelmann, A; Heurtier, M-A; Hoffman, V; Hoefert, C; Klein, K; Hudak, M; Michon, A-M; Schelder, M; Schirle, M; Remor, M; Rudi, T; Hooper, S; Bauer, A; Bouwmeester, T; Casari, G; Drewes, G; Neubauer, G; Rick, JM; Kuster, B; Bork, P; Russell, RB; Superti-Furga, G. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006, 440, 631–636. [Google Scholar]
- Barabasi, AL; Oltvai, ZN. Network biology: Understanding the cell's functional organization. Nat. Rev. Genet 2004, 5, 101–113. [Google Scholar]
- Barabasi, AL; Albert, R. Emergence of scaling in random networks. Science 1999, 286, 509–512. [Google Scholar]
- Jeong, H; Mason, SP; Barabasi, AL; Oltvai, ZN. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar]
- Han, J-DJ; Bertin, N; Hao, T; Goldberg, DS; Berriz, GF; Zhang, LV; Dupuy, D; Walhout, AJM; Cusick, ME; Roth, FP; Vidal, M. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 2004, 430, 88–93. [Google Scholar]
- Batada, NN; Reguly, T; Breitkreutz, A; Boucher, L; Breitkreutz, BJ; Hurst, LD; Tyers, M. Stratus not altocumulus: A new view of the yeast protein interaction network. PLoS Biol 2006, 4, e317. [Google Scholar]
- Bertin, N; Simonis, N; Dupuy, D; Cusick, ME; Han, JD; Fraser, HB; Roth, FP; Vidal, M. Confirmation of organized modularity in the yeast interactome. PLoS Biol 2007, 5, e153. [Google Scholar]
- Batada, NN; Reguly, T; Breitkreutz, A; Boucher, L; Breitkreutz, BJ; Hurst, LD; Tyers, M. Still stratus not altocumulus: Further evidence against the date/party hub distinction. PLoS Biol 2007, 5, e154. [Google Scholar]
- Higurashi, M; Ishida, T; Kinoshita, K. Identification of transient hub proteins and the possible structural basis for their multiple interactions. Protein Sci 2008, 17, 72–78. [Google Scholar]
- Kim, PM; Lu, LJ; Xia, Y; Gerstein, MB. Relating three-dimensional structures to protein networks provides evolutionary insights. Science 2006, 314, 1938–1941. [Google Scholar]
- Patil, A; Nakamura, H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett 2006, 580, 2041–2045. [Google Scholar]
- Haynes, C; Oldfield, CJ; Ji, F; Klitgord, N; Cusick, ME; Radivojac, P; Uversky, VN; Vidal, M; Iakoucheva, LM. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol 2006, 2, e100. [Google Scholar]
- Dosztanyi, Z; Chen, J; Dunker, AK; Simon, I; Tompa, P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J. Proteome Res 2006, 5, 2985–2995. [Google Scholar]
- Vallabhajosyula, RR; Chakravarti, D; Lutfeali, S; Ray, A; Raval, A. Identifying Hubs in Protein Interaction Networks. PLoS ONE 2009, 4, e5344. [Google Scholar]
- Wright, PE; Dyson, HJ. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol 1999, 293, 321–331. [Google Scholar]
- Dyson, HJ; Wright, PE. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol 2005, 6, 197–208. [Google Scholar]
- Ward, JJ; Sodhi, JS; McGuffin, LJ; Buxton, BF; Jones, DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol 2004, 337, 635–645. [Google Scholar]
- Iakoucheva, LM; Brown, CJ; Lawson, JD; Obradovic, Z; Dunker, AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol 2002, 323, 573–584. [Google Scholar]
- Dunker, AK; Cortese, MS; Romero, P; Iakoucheva, LM; Uversky, VN. Flexible nets. FEBS J 2005, 272, 5129–5148. [Google Scholar]
- Dosztanyi, Z; Chen, J; Dunker, AK; Simon, I; Tompa, P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J. Proteome Res 2006, 5, 2985–2995. [Google Scholar]
- Ekman, D; Light, S; Bjorklund, A; Elofsson, A. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol 2006, 7, R45. [Google Scholar]
- Merkley, N; Shaw, GS. Solution structure of the flexible class II ubiquitin-conjugating enzyme Ubc1 provides insights for polyubiquitin chain assembly. J. Biol. Chem 2004, 279, 47139–47147. [Google Scholar]
- Wilson, MA; Brunger, AT. The 1.0 A crystal structure of Ca2+-bound calmodulin: An analysis of disorder and implications for functionally relevant plasticity. J. Mol. Biol 2000, 301, 1237–1256. [Google Scholar]
- Kussie, PH; Gorina, S; Marechal, V; Elenbaas, B; Moreau, J; Levine, AJ; Pavletich, NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar]
- Kriwacki, RW; Hengst, L; Tennant, L; Reed, SI; Wright, PE. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. USA 1996, 93, 11504–11509. [Google Scholar]
- Lacy, ER; Filippov, I; Lewis, WS; Otieno, S; Xiao, L; Weiss, S; Hengst, L; Kriwacki, RW. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat. Struct. Mol. Biol 2004, 11, 358–364. [Google Scholar]
- Mark, WY; Liao, JC; Lu, Y; Ayed, A; Laister, R; Szymczyna, B; Chakrabartty, A; Arrowsmith, CH. Characterization of segments from the central region of BRCA1: An intrinsically disordered scaffold for multiple protein-protein and protein-DNA interactions? J. Mol. Biol 2005, 345, 275–287. [Google Scholar]
- Wright, PE; Dyson, HJ. Linking folding and binding. Curr. Opin. Struct. Biol 2009, 19, 31–38. [Google Scholar]
- Shoemaker, BA; Portman, JJ; Wolynes, PG. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. USA 2000, 97, 8868–8873. [Google Scholar]
- Huang, Y; Liu, Z. Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: A critical assessment of the “fly-casting” mechanism. J. Mol. Biol 2009, 393, 1143–1159. [Google Scholar]
- Yura, K; Hayward, S. The interwinding nature of protein-protein interfaces and its implication for protein complex formation. Bioinformatics 2009, 25, 3108–3113. [Google Scholar]
- Liu, J; Faeder, JR; Camacho, CJ. Toward a quantitative theory of intrinsically disordered proteins and their function. Proc. Natl. Acad. Sci. USA 2009, 106, 19819–19823. [Google Scholar]
- Gsponer, J; Futschik, ME; Teichmann, SA; Babu, MM. Tight regulation of unstructured proteins: From transcript synthesis to protein degradation. Science 2008, 322, 1365–1368. [Google Scholar]
- Kim, PM; Sboner, A; Xia, Y; Gerstein, M. The role of disorder in interaction networks: A structural analysis. Mol Syst Biol 2008, 4. [Google Scholar]
- Shimizu, K; Toh, H. Interaction between intrinsically disordered proteins frequently occurs in a human protein-protein interaction network. J. Mol. Biol 2009, 392, 1253–1265. [Google Scholar]
- Dunker, AK; Silman, I; Uversky, VN; Sussman, JL. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol 2008, 18, 756–764. [Google Scholar]
- Singh, GP; Ganapathi, M; Dash, D. Role of intrinsic disorder in transient interactions of hub proteins. Proteins 2007, 66, 761–765. [Google Scholar]
- Patil, A; Nakamura, H. The role of charged surface residues in the binding ability of small hubs in protein-protein interaction networks. Biophysics 2007, 3, 27–35. [Google Scholar]
- Joughin, BA; Green, DF; Tidor, B. Action-at-a-distance interactions enhance protein binding affinity. Protein Sci 2005, 14, 1363–1369. [Google Scholar]
- Sheinerman, FB; Norel, R; Honig, B. Electrostatic aspects of protein-protein interactions. Curr. Opin. Struct. Biol 2000, 10, 153–159. [Google Scholar]
- Friedler, A; Veprintsev, DB; Rutherford, T; von Glos, KI; Fersht, AR. Binding of Rad51 and other peptide sequences to a promiscuous, highly electrostatic binding site in p53. J. Biol. Chem 2005, 280, 8051–8059. [Google Scholar]
- Ubbink, M. The courtship of proteins: Understanding the encounter complex. FEBS Lett 2009, 583, 1060–1066. [Google Scholar]
- Bogan, AA; Thorn, KS. Anatomy of hot spots in protein interfaces. J. Mol. Biol 1998, 280, 1–9. [Google Scholar]
- Hu, Z; Ma, B; Wolfson, H; Nussinov, R. Conservation of polar residues as hot spots at protein interfaces. Proteins 2000, 39, 331–342. [Google Scholar]
- Rajamani, D; Thiel, S; Vajda, S; Camacho, CJ. Anchor residues in protein-protein interactions. Proc. Natl. Acad. Sci. USA 2004, 101, 11287–11292. [Google Scholar]
- Kinoshita, K; Nakamura, H. eF-site and PDBjViewer: Database and viewer for protein functional sites. Bioinformatics 2004, 20, 1329–1330. [Google Scholar]
- Humphris, EL; Kortemme, T. Design of Multi-Specificity in Protein Interfaces. PLoS Comput. Biol 2007, 3, e164. [Google Scholar]
- Fromer, M; Yanover, C; Linial, M. Design of multispecific protein sequences using probabilistic graphical modeling. Proteins 2009, 78, 530–547. [Google Scholar]
- Patil, A; Kinoshita, K; Nakamura, H. Domain distribution and intrinsic disorder in the human protein-protein interaction network. Submitted for publication.
- Finn, RD; Tate, J; Mistry, J; Coggill, PC; Sammut, SJ; Hotz, H-R; Ceric, G; Forslund, K; Eddy, SR; Sonnhammer, ELL; Bateman, A. The Pfam protein families database. Nucl. Acids Res 2008, 36, D281–288. [Google Scholar]
- Tsai, CJ; Ma, B; Nussinov, R. Protein-protein interaction networks: How can a hub protein bind so many different partners? Trends Biochem. Sci 2009, 34, 594–600. [Google Scholar]
- Yura, K; Shionyu, M; Hagino, K; Hijikata, A; Hirashima, Y; Nakahara, T; Eguchi, T; Shinoda, K; Yamaguchi, A; Takahashi, K-i; Itoh, T; Imanishi, T; Gojobori, T; Go, M. Alternative splicing in human transcriptome: Functional and structural influence on proteins. Gene 2006, 380, 63–71. [Google Scholar]
- Romero, PR; Zaidi, S; Fang, YY; Uversky, VN; Radivojac, P; Oldfield, CJ; Cortese, MS; Sickmeier, M; LeGall, T; Obradovic, Z; Dunker, AK. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc. Natl. Acad. Sci. USA 2006, 103, 8390–8395. [Google Scholar]
- Iakoucheva, LM; Radivojac, P; Brown, CJ; O'Connor, TR; Sikes, JG; Obradovic, Z; Dunker, AK. The importance of intrinsic disorder for protein phosphorylation. Nucl. Acids Res 2004, 32, 1037–1049. [Google Scholar]
- Radivojac, P; Vacic, V; Haynes, C; Cocklin, RR; Mohan, A; Heyen, JW; Goebl, MG; Iakoucheva, LM. Identification, analysis, and prediction of protein ubiquitination sites. Proteins: Struct. Funct. Bioinformat 2010, 78, 365–380. [Google Scholar]
- Nobeli, I; Favia, AD; Thornton, JM. Protein promiscuity and its implications for biotechnology. Nat. Biotechnol 2009, 27, 157–167. [Google Scholar]
- Kitahara, R; Yokoyama, S; Akasaka, K. NMR snapshots of a fluctuating protein structure: ubiquitin at 30 bar-3 kbar. J. Mol. Biol 2005, 347, 277–285. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Patil, A.; Kinoshita, K.; Nakamura, H. Hub Promiscuity in Protein-Protein Interaction Networks. Int. J. Mol. Sci. 2010, 11, 1930-1943. https://doi.org/10.3390/ijms11041930
Patil A, Kinoshita K, Nakamura H. Hub Promiscuity in Protein-Protein Interaction Networks. International Journal of Molecular Sciences. 2010; 11(4):1930-1943. https://doi.org/10.3390/ijms11041930
Chicago/Turabian StylePatil, Ashwini, Kengo Kinoshita, and Haruki Nakamura. 2010. "Hub Promiscuity in Protein-Protein Interaction Networks" International Journal of Molecular Sciences 11, no. 4: 1930-1943. https://doi.org/10.3390/ijms11041930