Bacterial Expression of Mouse Argonaute 2 for Functional and Mutational Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
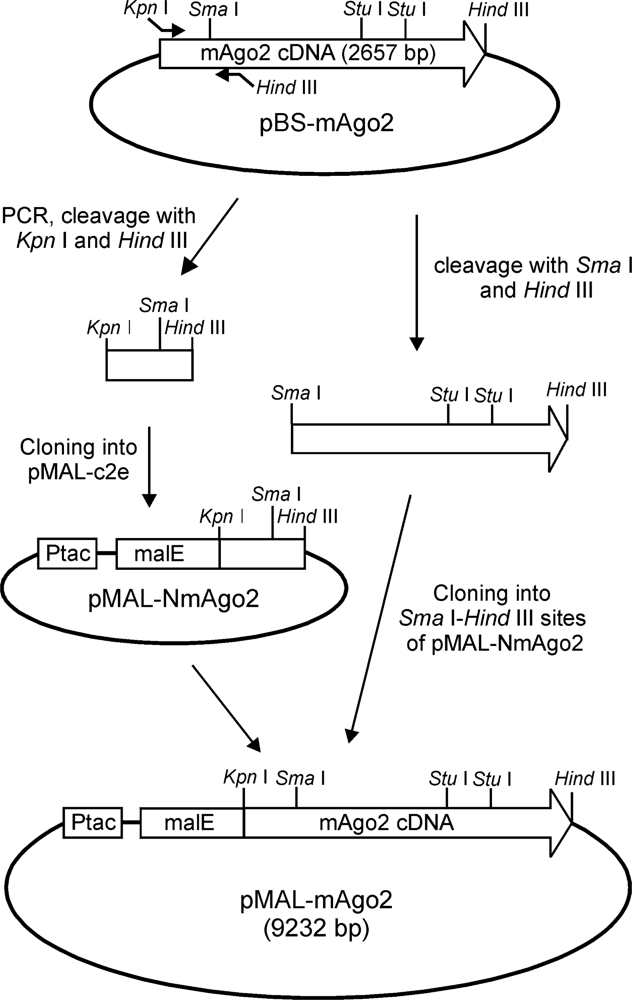
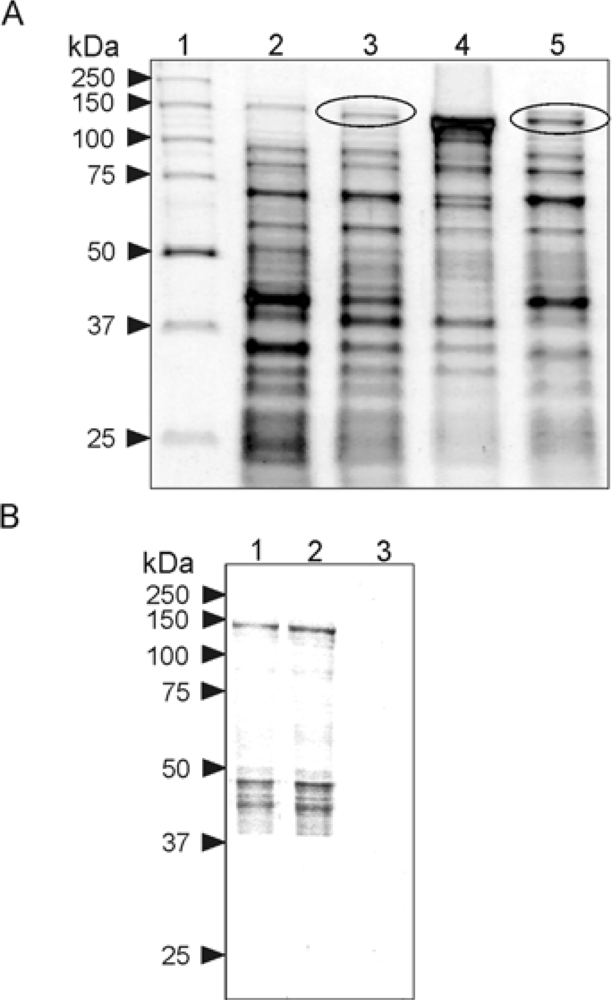
2.1. Expression in E. Coli and Purification of Mouse Argonaute 2
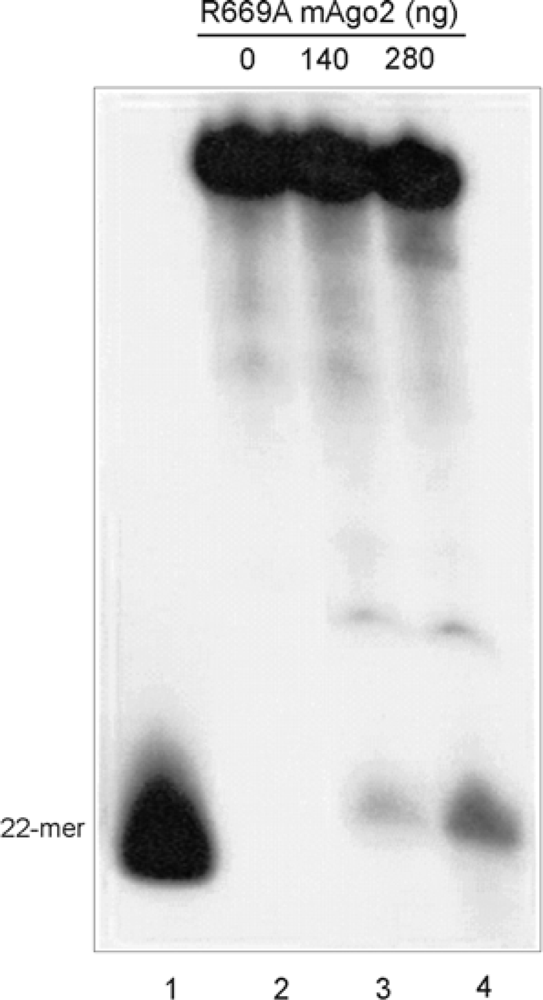
2.2. Functional Characterization of Mouse Argonaute 2
2.3. Mutational Analysis of mAgo2
3. Experimental Section
3.1. Materials and General Procedures
3.2. DNA Cloning and Site-Directed Mutagenesis
3.3. Protein Expression and Purification
3.4. Structural and Functional Analyses
4. Conclusions
Acknowledgments
References and Notes
- Fire, A; Xu, S; Montgomery, MK; Kostas, SA; Driver, SE; Mello, CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar]
- Meister, G; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar]
- Zhang, H; Kolb, FA; Brondani, V; Billy, E; Filipowicz, W. Human Dicer preferentially cleaves dsRNAs at their termini without a requirement for ATP. EMBO J 2002, 21, 5875–5885. [Google Scholar]
- Zhang, H; Kolb, FA; Jaskiewicz, L; Westhof, E; Filipowicz, W. Single processing center models for human Dicer and bacterial RNase III. Cell 2004, 118, 57–68. [Google Scholar]
- Martinez, J; Tuschl, T. RISC is a 5′ phosphomonoester producing RNA endonuclease. Genes Dev 2004, 18, 975–980. [Google Scholar]
- Hammond, SM; Bernstein, E; Beach, D; Hannon, GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar]
- Schwarz, DS; Tomari, Y; Zamore, PD. The RNA induced silencing complex is a Mg2+-dependent endonuclease. Curr. Biol 2004, 14, 787–791. [Google Scholar]
- Rand, TA; Ginalski, K; Grishin, NV; Wang, X. Biochemical identification of Argonaute 2 as the sole protein required for RNA-induced silencing complex activity. Proc. Natl. Acad. Sci. USA 2004, 101, 14385–14389. [Google Scholar]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar]
- Bartel, DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar]
- Parker, JS; Roe, SM; Barford, D. Crystal structure of a PIWI protein suggests mechanisms for siRNA recognition and slicer activity. EMBO J 2004, 23, 4727–4737. [Google Scholar]
- Liu, J; Carmell, MA; Rivas, FV; Marsden, CG; Thomson, JM; Song, JJ; Hammond, SM; Joshua-Tor, L; Hannon, GJ. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar]
- Song, JJ; Smith, SK; Hannon, GJ; Joshua-Tor, L. Crystal structure of Argonaute and its implications for RISC slicer activity. Science 2004, 305, 1434–1437. [Google Scholar]
- Rivas, FV; Tolia, NH; Song, JJ; Aragon, JP; Liu, J; Hannon, GJ; Joshua-Tor, L. Purified Argonaute 2 and an siRNA form recombinant human RISC. Nat. Struct. Mol. Biol 2005, 12, 340–349. [Google Scholar]
- Nowotny, M; Gaidamakov, SA; Crouch, RJ; Yang, W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell 2005, 121, 1005–1016. [Google Scholar]
- Doi, N; Zenno, S; Ueda, R; Ohki-Hamazaki, H; Ui-Tei, K; Saigo, K. Short-interfering-RNA-mediated gene silencing in mammalian cells requires Dicer and eIF2C translation initiation factors. Curr. Biol 2003, 13, 41–46. [Google Scholar]
- Laemmli, JK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar]

© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Salvatore, V.; Potenza, N.; Papa, U.; Nobile, V.; Russo, A. Bacterial Expression of Mouse Argonaute 2 for Functional and Mutational Studies. Int. J. Mol. Sci. 2010, 11, 745-753. https://doi.org/10.3390/ijms11020745
Salvatore V, Potenza N, Papa U, Nobile V, Russo A. Bacterial Expression of Mouse Argonaute 2 for Functional and Mutational Studies. International Journal of Molecular Sciences. 2010; 11(2):745-753. https://doi.org/10.3390/ijms11020745
Chicago/Turabian StyleSalvatore, Vincenzo, Nicoletta Potenza, Umberto Papa, Valentina Nobile, and Aniello Russo. 2010. "Bacterial Expression of Mouse Argonaute 2 for Functional and Mutational Studies" International Journal of Molecular Sciences 11, no. 2: 745-753. https://doi.org/10.3390/ijms11020745
APA StyleSalvatore, V., Potenza, N., Papa, U., Nobile, V., & Russo, A. (2010). Bacterial Expression of Mouse Argonaute 2 for Functional and Mutational Studies. International Journal of Molecular Sciences, 11(2), 745-753. https://doi.org/10.3390/ijms11020745