Effect of Gadolinium Chloride on Liver Regeneration Following Thioacetamide-Induced Necrosis in Rats

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals and Treatment
2.3. Processing of the Samples
2.4. Determination of Parameters of Injury and TNFα in Serum
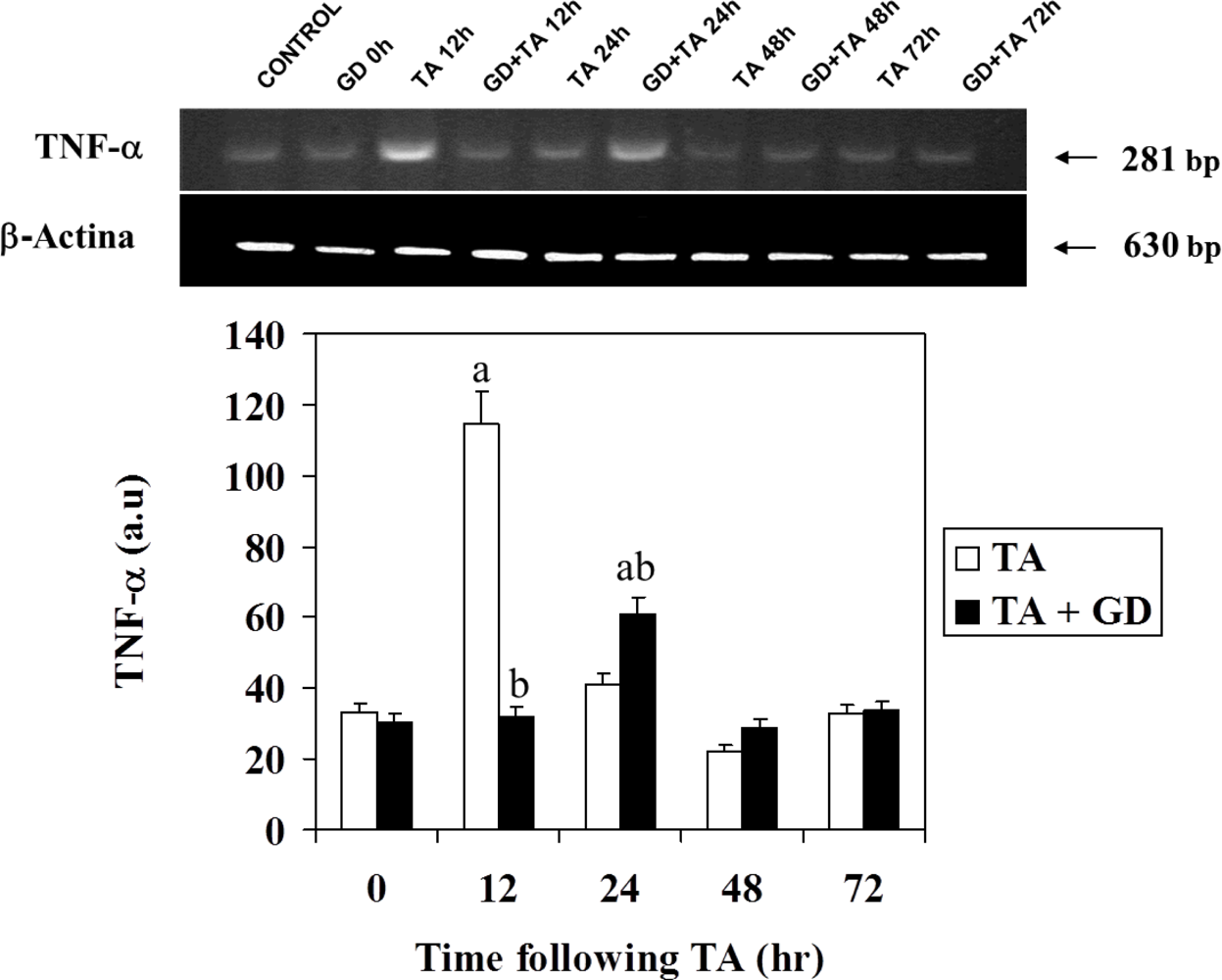
2.5. RT-PCR Analysis of TNFα
2.6. Flow Cytometry Analysis of DNA Content
2.7. Statistical Analysis
3. Results
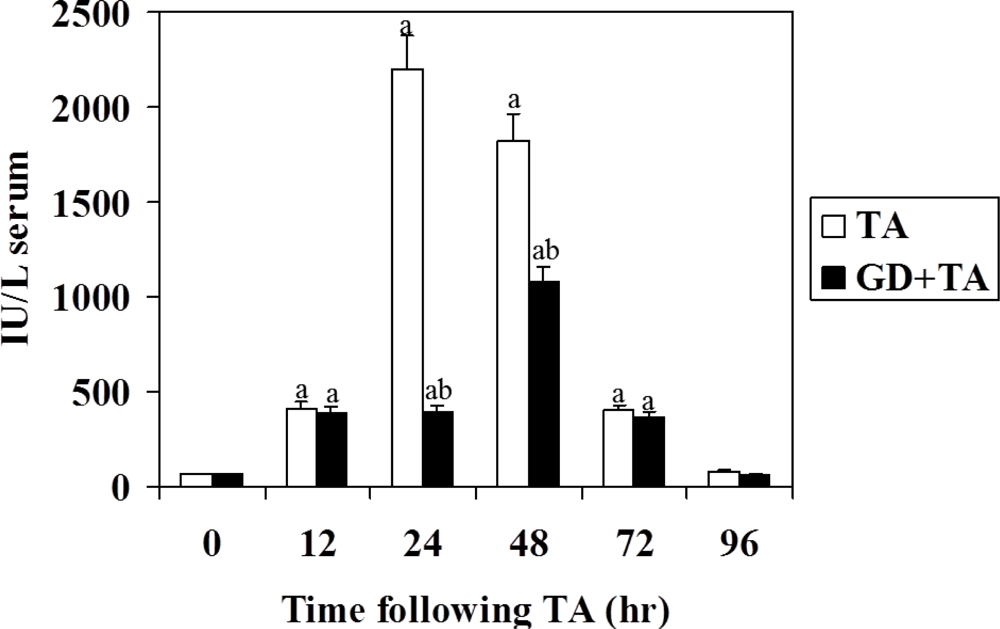
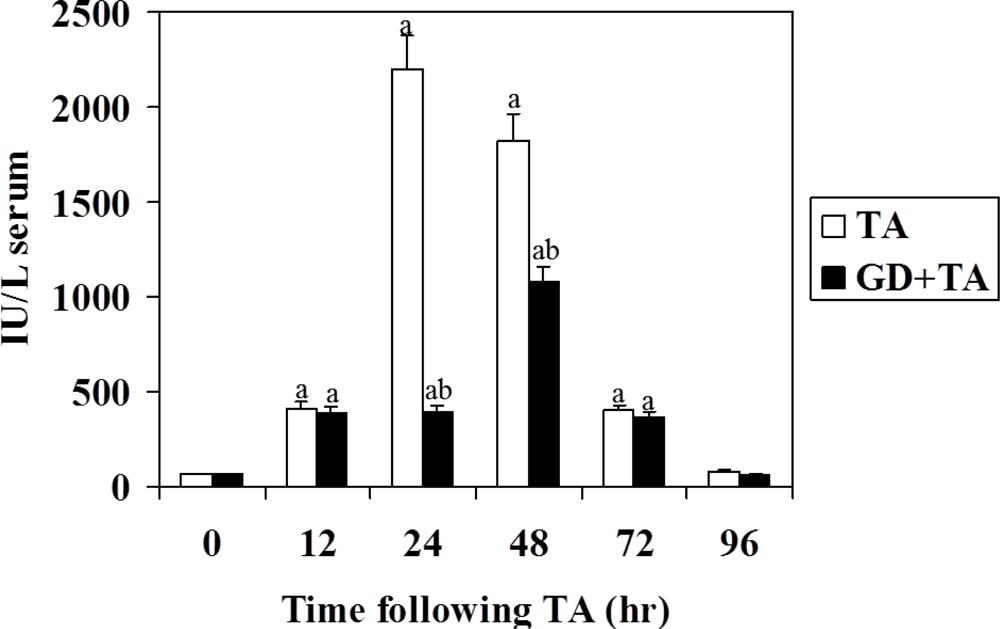
3.1. Effect of GD on Parameters of Liver Necrosis
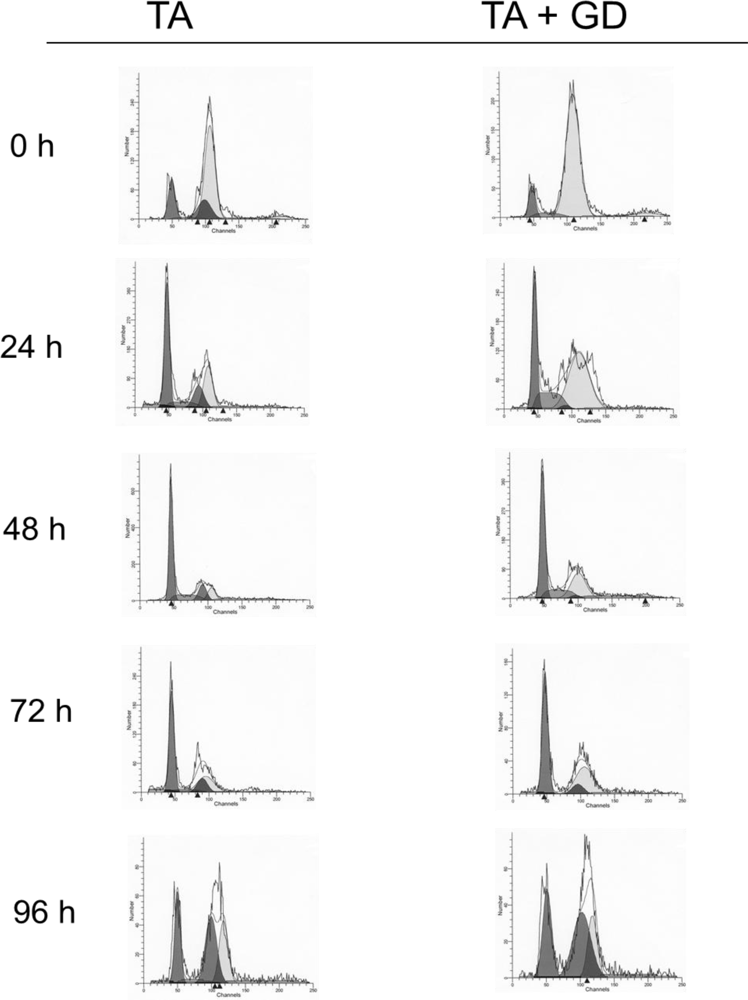
3.2. Effect of GD Pretreatment on the Time Course of Genomic DNA Ploidy and Distribution in Hepatocytes Isolated from TA-Treated Rats
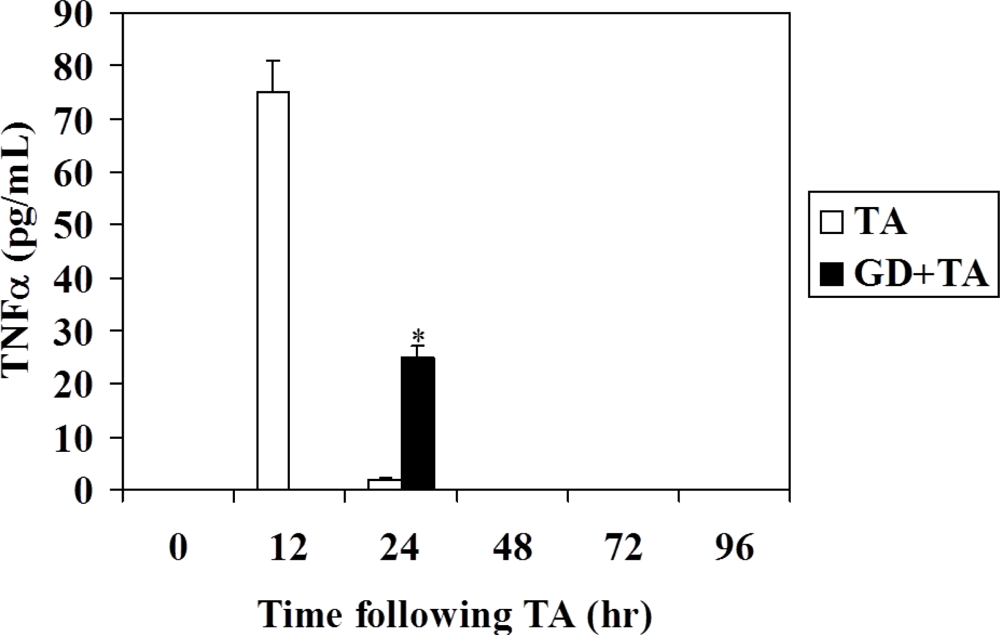
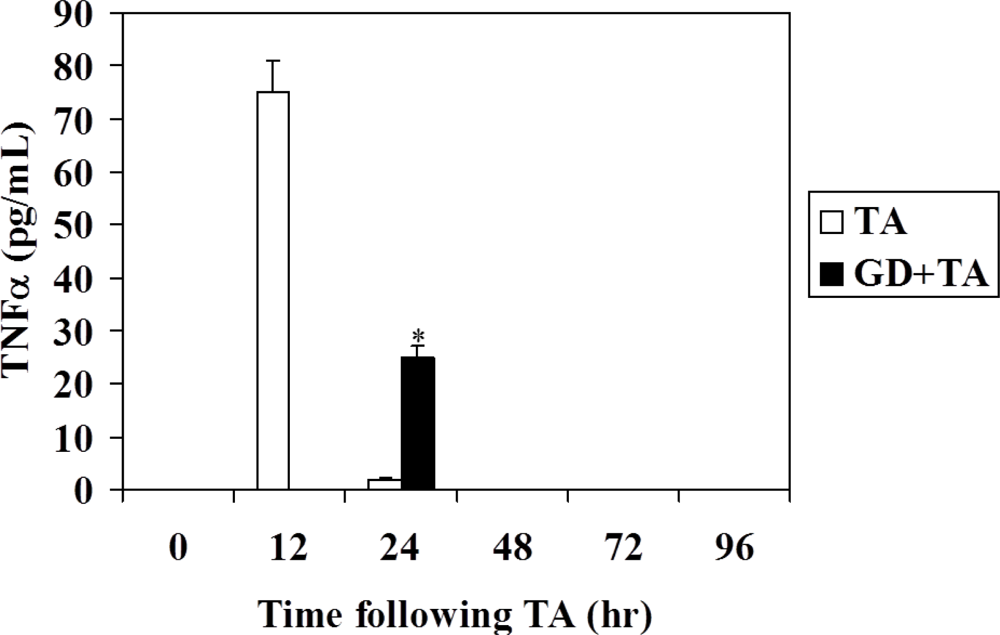
3.3. Effect of GD Pretreatment on Serum TNFα Level and TNFα Expression in Rat Liver Following Intoxication with TA
4. Discussion
- That pretreatment with GD increases the cellular dedifferentiation induced by thioacetamide,
- That the analogy with the fetal pattern in hepatocellular injury is more severe in the group pretreated with GD.(A) The liver of mammals contains polyploid hepatocytes, whose number depends on the species and age of the animal [32]. Fetal rat hepatocytes are mostly diploid with an allocation of 85.3% of cells involved in this phase (diploid), 7.3% in DNA synthesis phase and 7.4% polyploid (tetraploid + octoploid) [9]. Fetal liver cells of animals have a greater number of cells in S (S1 and S2) and diploid phases and fewer diploid and polyploid (tetraploid + octoploid). In adult animals, the polyploidization increases and the synthesis is reduced to values of approximately 1%.
5. Conclusion
Acknowledgments
References
- Rose, ML; Bradford, BU; Germolec, DR; Lin, M; Tsukamoto, H; Thurman, RG. Gadolinium chloride-induced hepatocytes proliferation is prevented by antibodies to Tumor Necrosis Factor α. Toxicol. Appl. Pharmacol 2001, 170, 39–45. [Google Scholar]
- Laskin, DL. Nonparenchymal cells and hepatotoxicity. Semin. Liver Dis 1990, 10, 293–304. [Google Scholar]
- Bautista, AP; Skrepnic, N; Niesman, MR; Bagby, GJ. Elimination of macrophages by liposome-encapsulated dichloromethylene diphosphonate suppresses the endotoxin-induced priming of Kupffer cells. J. Leukoc. Biol 1994, 55, 321–327. [Google Scholar]
- Ding, H; Peng, R; Reed, E; Li, QQ. Effects of Kupffer cell inhibition on liver function and hepatocellular activity in mice. Int. J. Mol. Med 2003, 12, 549–557. [Google Scholar]
- Zhong, Z; Connor, HD; Mason, RP; Qu, W; Lemasters, JJ; Thurman, RG. Role of Kupffer cells in reperfusion injury in fat-loaded livers from ethanol-treated rats. J. Pharmacol. Exp. Ther 1995, 27, 1512–1517. [Google Scholar]
- Andres, D; Sanchez-Reus, I; Bautista, M; Cascales, M. Depletion of Kupffer cell function by gadolinium chloride attenuates thioacetamide-induced hepatotoxicity. Expression of metallothionein and HSP70. Biochem. Pharmacol 2003, 66, 917–926. [Google Scholar]
- Landon, EJ; Naukam, RJ; Rama Sastry, BV. Effects of calcium channel blocking agents on calcium and centrilobular necrosis in the liver of rats treated with hepatotoxic agents. Biochem. Pharmacol 1986, 35, 697–705. [Google Scholar]
- Cascales, M; Martín-Sanz, P; Alvarez, A; Sanchez-Pérez, M; Díez-Fernández, C; Boscá, L. Isoenzymes of carbohydrate metabolism in primary cultures of hepatocytes from thioacetamide-induced rat liver necrosis: Responses to growth factors. Hepatology 1992, 16, 232–240. [Google Scholar]
- Diez-Fernandez, C; Boscá, L; Fernandez-Simón, L; Alvarez, A; Cascales, M. Relationship between genomic DNA parameters of liver damage during necrosis and regeneration induced by thiacetamide. Hepatology 1993, 18, 912–918. [Google Scholar]
- Sanz, N; Diez-Fernández, C; Andrés, D; Cascales, M. Hepatotoxicity and aging: endogenous antioxidant systems in hepatocytes from 2-, 6-, 12-, 18- and 30-month-old rats following a necrogenic dose of thioacetamide. Biochim. Biophys. Acta 2002, 1587, 12–20. [Google Scholar]
- Decker, K. Biologically active products of stimulated liver macrophages (Kupffer cells). Eur. J. Biochem 1990, 192, 245–261. [Google Scholar]
- Olynyk, JK; Matuschak, GM; Lechner, AJ; Britton, RS; Tredway, TL; O'Neill, R; Bacon, BR. Differential production of TNF by Kupffer cells after phagocytosis of E. coli and C. albicans. Am. J. Physiol 1994, 267, G213–G219. [Google Scholar]
- Fausto, N. Liver Regeneration. J. Hepatol 2000, 32, 19–31. [Google Scholar]
- Taub, R. Liver regeneration: From myth to mechanism. Nat. Rev. Mol. Cell. Biol 2004, 5, 836–847. [Google Scholar]
- Fujita, J; Marino, MW; Wada, H; Jungbluth, AA; Mackrell, PJ; Rivadeneira, DE; Stapleton, PP; Daly, JM. Effect of TNF gene depletion on liver regeneration after partial hepatectomy in mice. Surgery 2001, 129, 48–54. [Google Scholar]
- Iimuro, Y; Yamamoto, M; Kohno, H; Itakura, J; Fujii, H; Matsumoto, Y. Blockade of liver macrophages by gadolinium chloride reduces lethality in endotoxemic rats. Analysis of mechanisms lethality in endotoxemia. J. Leukoc. Biol 1994, 55, 723–728. [Google Scholar]
- Lazar, G, Jr; Lazar, G; Kaszaki, J; Olah, J; Kiss, I; Husztik, E. Inhibition of anaphylactic shock by gadolinium chloride-induced Kupffer cell blockade. Agents Actions, 1994; 41 Spec No:C97-8. [Google Scholar]
- Fujita, S; Arii, S; Monden, K; Adachi, Y; Funaki, N; Higashitsuji, H; Furutani, M; Mise, M; Ishiguro, S; Kitao, T; et al. Participation of hepatic macrophages and plasma factors in endotoxin-induced liver injury. J. Surg. Res 1995, 59, 263–270. [Google Scholar]
- Kono, Y; Fridovich, I. Superoxide radical inhibits catalase. J. Biol. Chem 1982, 257, 5751–5754. [Google Scholar]
- Rai, RM; Yang, SQ; McClain, C; Karp, CL; Klein, AS; Diehl, AM. Kupffer cell depletion by gadolinium chloride enhances liver regeneration after partial hepatectomy in rats. Am. J. Physiol 1996a, 270, G909–G918. [Google Scholar]
- Rai, RM; Zhang, JX; Clemens, MG; Diehl, AM. Gadolinium chloride alters the acinar distribution of phagocytosis and balance between pro- and anti-inflammatory cytokines. Shock 1996b, 6, 243–247. [Google Scholar]
- Rai, RM; Loffreda, S; Karp, CL; Yang, SQ; Lin, HZ; Diehl, AM. Kupffer cell depletion abolishes induction of interleukin-10 and permits sustained overexpression of tumor necrosis factor alpha messenger RNA in the regenerating rat liver. Hepatology 1997, 25, 889–895. [Google Scholar]
- Sanz, N; Díez-Fernández, C; Alvarez, AM; Fernández-Simón, L; Cascales, M. Age-related changes on parameters of experimentally-induced liver injury and regeneration. Toxicol. Appl. Pharmacol 2000, 154, 40–49. [Google Scholar]
- Zaragoza, A; Andrés, D; Sarrión, D; Cascales, M. Potentiation of thioacetamide hepatotoxicity by phenobarbital pretreatment in rats. Inducibility of FAD monooxygenase system and age effect. Chem. Biol. Interact 2000, 124, 87–101. [Google Scholar]
- Seglen, PO. Isolation of hepatocytes by collagenase perfusion. In Methods in Toxicology In vitro Biological Systems; Tyson, CA, Frazier, JM, Eds.; Academia Press: New York, NY, USA, 1993; pp. 231–243. [Google Scholar]
- Sanz, N; Díez-Fernández, C; Fernández-Simón, L; Alvarez, A; Cascales, M. Necrogenic and regenerative responses of liver of newly weaned rats. Biochem. Biophys. Acta 1998, 1384, 66–78. [Google Scholar]
- Diez-Fernandez, C; Sanz, N; Cascales, M. Changes in glucose-6-phosphate dehydrogenase and malic enzyme gene expression in acute hepatic injury induced by thioacetamide. Biochem. Pharmacol 1996, 51, 1159–1163. [Google Scholar]
- Ramírez-Farías, C; Madrigal-Santillán, E; Gutiérrez-Salinas, J; Rodríguez-Sánchez, N; Martínez-Cruz, M; Valle-Jones, I; Gramlich-Martínez, I; Hernández-Ceruelos, A; Morales-González, JA. Protective effect of some vitamins against the toxic action of ethanol on liver regeneration induced by partial hepatectomy in rats. World J. Gastroenterol 2008, 14, 899–907. [Google Scholar]
- Vindelov, LL; Christensen, IJ; Nissen, NI. A detergent trypsin method for the preparation of nuclei for flow cytometric. Cytometry 1983, 3, 323–327. [Google Scholar]
- Cascales, M; Martin-Sanz, P; Craciunescu, DC; Mayo, I; Aguilar, A; Robles-Chillida, EM; Cascales, C. Alterations in hepatic peroxidation mechanisms in thioacetamide-induced tumors in rats. Effect of a rhodium complex. Carcinogenesis 1991, 12, 233–240. [Google Scholar]
- Mehendale, HM; Roth, RA; Gandolfo, AJ; Klauning, JE; Lemasters, JJ; Curtis, RL. Novel mechanisms in chemical-induced hepatotoxicity. FASEB J 1994, 8, 1285–1295. [Google Scholar]
- Saeter, G; Schwarze, PE; Nesland, JM; Juul, N; Pettersen, EO; Seglen, PO. The polyploidizing growth pattern of normal rat liver is replaced by divisional, diploid growth in hepatocellular nodules and carcinomas. Carcinogenesis 1988, 9, 939–945. [Google Scholar]
- Sandgren, EP; Palmiter, RD; Heckel, JL; Daugherty, CC; Brinster, RL; Degen, JL. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene. Cell 1991, 66, 245–256. [Google Scholar]
- Fausto, N. Liver regeneration and repair: Hepatocytes, progenitor cells, and stem cells. Hepatology 2004, 39, 1477–1487. [Google Scholar]
- Michalopoulos, GK; DeFrances, M. Liver regeneration. Adv. Biochem. Eng. Biotechnol 2005, 93, 101–134. [Google Scholar]
- Jungermann, K; Katz, N. Functional hepatocellular heterogeneity. Hepatology 1982, 2, 385–395. [Google Scholar]
- Baker, SJ; Reddy, EP. Transducers of life and death: TNF receptor superfamily and associated proteins. Oncogene 1996, 12, 1–9. [Google Scholar]
- Akerman, PA; Cote, PM; Yang, SQ; McClain, C; Nelson, S; Bagby, G; Diehl, AM.
- Long-term ethanol consumption alters the hepatic response to the regenerative effects of tumor necrosis factor-alpha. Hepatology 1993, 17, 1066–1073.
- Xing, Z; Richards, CD; Braciak, T; Thibault, V; Gauldie, J. Citokine regulation of hepatic acute phase protein espression. In Cytokines and the Liver; Gerok, W, Decker, K, Andus, T, Gross, V, Eds.; Kluwer Academic Publishers: Dordrecht, Germany, 1995; pp. 164–171. [Google Scholar]
- Lee, CM; Yeoh, GC; Olynyk, JK. Differential effects of gadolinium chloride on Kupffer cells in vivo and in vitro. Int. J. Biochem. Cell. Biol 2004, 36, 481–488. [Google Scholar]
- Bautista, AP; Deaciuc, IV; Jaeschke, H. Hepatic responses to bacterial endotoxin (LPS). In Pathophysiology of Shock, Sepsis and Organ Failure; Schlag, G, Redl, H, Eds.; Springer-Verlag: Berlin Heidelberg, Germany, 1993; pp. 915–934. [Google Scholar]
- Theocharis, SE; Kanelli, H; Margeli, AP; Spiliopoulou, CA; Koutselinis, AS. Metallothionein and heat shock protein expression during acute liver injury and regeneration in rats. Clin. Chem. Lab. Med 2000, 38, 1137–1140. [Google Scholar]
- Oliver, JR; Mara, TW; Cherian, MG. Impaired hepatic regeneration in metallothionein-I/II knockout mice after partial hepatectomy. Exp. Biol. Med 2005, 230, 61–67. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hypodiploid (<2C) | Diploid (2C) | S1 Phase (2C → 4C) | Tetraploid (4C) | S2 Phase (4C → 8C) | Octoploid (8N) | |
|---|---|---|---|---|---|---|
| Control | 0.54 | 13.0 | 1.1 | 78.35 | 2.62 | 4.35 |
| Control Gd | 0.70 | 17.86 | 0.9 | 72.17 | 4.60 | 3.72 |
| TA 24 | 2.80 | 42.76a | 10.01a | 40.2ª | 6.72ª | 0.30 |
| TA-Gd 24 | 1.36 | 26.15ab | 17.17ab | 50.96ab | 4.32 | 0.32 |
| TA 48 | 1.66 | 50.86a | 14.45a | 23.66a | 9.18a | 0 |
| TA-Gd 48 | 2.35 | 42.77a | 14.92a | 28.0a | 10.25ª | 1.70 |
| TA 72 | 4.08ª | 46.11a | 6.89a | 36.32ª | 4.49ª | 2.12 |
| TA-Gd 72 | 2.86ab | 47.98a | 1.74b | 40.61ª | 6.26 | 0.53 |
| TA 96 | 1.17 | 23.94a | 4.98a | 60.56 | 9.06ª | 0.29 |
| TA-Gd 96 | 1.15 | 28.56a | 0 | 63.65 | 6.62 | 0.02 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bautista, M.; Andres, D.; Cascales, M.; Morales-González, J.A.; Sánchez-Reus, M.I. Effect of Gadolinium Chloride on Liver Regeneration Following Thioacetamide-Induced Necrosis in Rats. Int. J. Mol. Sci. 2010, 11, 4426-4440. https://doi.org/10.3390/ijms11114426
Bautista M, Andres D, Cascales M, Morales-González JA, Sánchez-Reus MI. Effect of Gadolinium Chloride on Liver Regeneration Following Thioacetamide-Induced Necrosis in Rats. International Journal of Molecular Sciences. 2010; 11(11):4426-4440. https://doi.org/10.3390/ijms11114426
Chicago/Turabian StyleBautista, Mirandeli, David Andres, María Cascales, José A. Morales-González, and María Isabel Sánchez-Reus. 2010. "Effect of Gadolinium Chloride on Liver Regeneration Following Thioacetamide-Induced Necrosis in Rats" International Journal of Molecular Sciences 11, no. 11: 4426-4440. https://doi.org/10.3390/ijms11114426