Ionocovalency and Applications 1. Ionocovalency Model and Orbital Hybrid Scales

Abstract
:1. Introduction
2. Methodology
2.1. IC Model
2.2. IC Electronegativity XIC
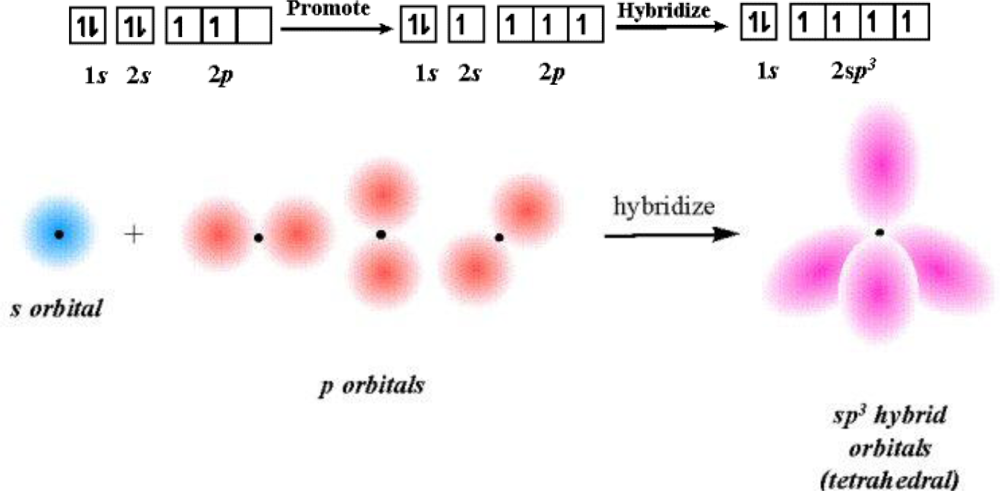
2.3. IC Orbital Hybrid Bonding Procedures

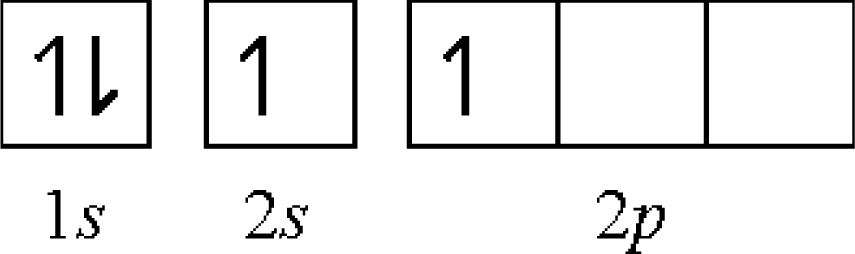
- first: Be[He](2s)2 → Be+[He] (2s)1 − 9.32 eV
- second: Be+[He](2s)1 → Be2+[He] − 18.20 eV
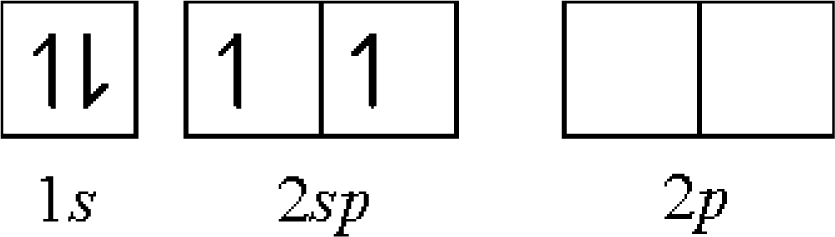
- first: C[He](2s)2(2p)2 → C+[He](2s)2(2p)1 − 11.26 eV
- second: C+[He](2s)2(2p)1 → C2+[He](2s)2 − 24.40 eV
- third: C2+[He](2s)2 → C3+[He](2s)1 − 47.90 eV
- fourth: C3+[He](2s)1 → C4+[He] − 64.50 eV
3. Results
3.1. General Trend of Periodic Table
3.2. Hydrogen
3.3. Diagonal Relationship (Top Periods)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H | He | ||||||||||||||||
| (1)2.297 | (2)5.173 | ||||||||||||||||
| Li | Be | B | C | N | O | F | Ne | ||||||||||
| (1)1.023 | (2)2.064 | (3)3.291 | (4)4.302 | (5)5.329 | (6)6.273 | (7)7.370 | (4)6.949 | ||||||||||
| (2)2.998 | (3)4.030 | (2)3.648 | (2)5.203 | ||||||||||||||
| Na | Mg | Al | Si | P | S | Cl | Ar | ||||||||||
| 1.130 | (2)1.933 | (3)2.730 | (4)3.371 | (5)4.355 | (6)5.165 | (7)6.048 | (4)4.845 | ||||||||||
| (1)1.585 | (2)2.322 | (4)3.872 | (4)4.093 | (5)4.977 | (2)3.762 | ||||||||||||
| (3)3.286 | (2)2.121 | (1)2.851 | |||||||||||||||
| K | Ca | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | Ga | Ge | As | Se | Br | Kr |
| (1)0.999 | (2)1.617 | (3)2.494 | (4)3.374 | (5)4.359 | (6)5.287 | (7)6.012 | (6)5.520 | (4)4.261 | (4)4.537 | (3)3.918 | (2)2.772 | (3)3.281 | (4)3.895 | (5)4.502 | (6)5.209 | (7)5.941 | (6)5.422 |
| (3)2.824 | (4)3.772 | (5)4.691 | (6)5.426 | (5)4.879 | (3)3.591 | (3)3.802 | (2)3.155 | (1)2.331 | (1)1.841 | (2)2.639 | (3)3.364 | (4)4.090 | (5)4.869 | (4)4.562 | |||
| (2)2.258 | (3)3.143 | (4)4.058 | (5)4.810 | (4)4.203 | (2)2.874 | (2)3.029 | (1)2.343 | (1)2.422 | (2)3.146 | (1)2.815 | (2)3.538 | ||||||
| (2)2.502 | (3)3.395 | (4)4.161 | (3)3.431 | (1)2.113 | (1)2.330 | ||||||||||||
| (1)2.077 | (2)2.719 | (1)2.253 | |||||||||||||||
| (1)2.185 | |||||||||||||||||
| Rb | Sr | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd | In | Sn | Sb | Te | I | Xe |
| (1)0.988 | (2)1.576 | (3)2.332 | (4)3.716 | (5)4.043 | (6)4.964 | (7)5.593 | (8)6.394 | (6)5.450 | (5)4.842 | (3)3.592 | (2)2.658 | (3)2.923 | (4)3.601 | (5)4.057 | (6)4.641 | (7)5.339 | (8)5.553 |
| (3)2.267 | (4)3.575 | (5)4.547 | (6)5.090 | (7)5.890 | (4)4.233 | (4)4.277 | (2)2.973 | (1)2.174 | (1)1.678 | (2)2.472 | (3)3.036 | (4)3.675 | (5)3.817 | (6)4.667 | |||
| (2)1.884 | (3)3.052 | (4)3.980 | (5)4.563 | (6)5.355 | (3)3.638 | (3)3.672 | (1)2.147 | (2)2.832 | (1)2.530 | (4)3.900 | |||||||
| (2)2.532 | (3)3.318 | (4)4.008 | (5)4.791 | (2)2.993 | (2)3.042 | (2)3.044 | |||||||||||
| (1)2.040 | (2)2.760 | (3)3.425 | (4)4.189 | (1)2.287 | |||||||||||||
| (1)2.155 | (2)2.762 | (3)3.525 | |||||||||||||||
| Δ | (1)2.218 | (2)2.924 | |||||||||||||||
| Cs | Ba | La | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg | Tl | Pb | Bi | Po | At | Rn |
| (1)0.992 | (2)1.646 | (3)2.185 | (4)3.624 | (5)4.393 | (6)5.178 | (7)5.874 | (8)6.616 | (6)5.683 | (6)5.640 | (5)5.033 | (2)3.118 | (3)3.307 | (4)3.780 | (5)4.273 | (6)4.707 | (7)5.430 | (8)5.567 |
| (3)3.170 | (4)3.924 | (5)4.697 | (6)5.383 | (7)6.122 | (5)5.107 | (5)5.056 | (3)3.965 | (1)2.653 | (1)1.887 | (2)2.574 | (3)3.162 | (4)3.700 | (5)4.419 | (6)5.046 | |||
| (2)2.691 | (3)3.462 | (4)4.200 | (5)4.881 | (6)5.608 | (4)4.478 | (4)4.518 | (2)3.412 | (3)3.285 | (3)3.522 | (4)4.183 | |||||||
| (2)3.056 | (3)3.707 | (4)4.344 | (5)5.066 | (3)3.910 | (3)3.963 | (1)2.689 | (2)2.811 | (1)2.473 | |||||||||
| (1)2.473 | (2)3.261 | (3)3.795 | (4)4.483 | (2)3.324 | (2)3.405 | ||||||||||||
| (2)3.237 | (3)3.869 | (1)2.831 | (1)2.749 | ||||||||||||||
| (1)2.597 | (2)3.370 | ||||||||||||||||
| ΔΔ | (1)2.779 | ||||||||||||||||
| Fr | Ra | Ac | Δ | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu |
| (1)0.959 | (2)1.400 | (3)2.093 | (4)3.074 | (4)3.037 | (3)2.572 | (3)2.606 | (3)2.604 | (3)2.385 | (3)2.637 | (4)3.301 | (3)2.730 | (3)2.754 | (3)2.780 | (3)2.829 | (3)2.649 | (3)2.592 | |
| (3)2.506 | (3)2.368 | (2)2.050 | (2)1.858 | (2)2.213 | (3)2.686 | (2)2.286 | (2)2.112 | ||||||||||
| ΔΔ | Th | Pa | U | Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | Lw | |||
| (4)2.789 | (3)2.279 | (3)2.316 | (4)2.341 | (4)2.354 | (4)2.349 | (3)2.165 | (3)2.215 | (3)2.229 | (3)2.262 | (3)2.278 | (3)2.309 | (3)2.323 | (3)3.900 | ||||
| (3)2.431 |
| Be2+ (2.252) | B3+ (3.291) | C4+ (4.320) | N5+ (5.554) | O6+ (6.939) |
| Mg2+ (1.933) | Al3+ (2.730) | Si4+ (3.371) | P5+ (4.355) | S6+ (5.165) |
| Be2+ (1.315) | B3+ (1.743) | C4+ (2.167) | N5+ (2.675) | O6+ (3.246) |
| Mg2+ (1.184) | Al3+ (1.418) | Si4+ (1.776) | P5+ (2.181) | S6+ (2.515) |
| Be2+ (13.76) | B3+ (23.800) | C4+ (37.015) | N5+ (53.406) | O6+ (72.020) |
| Mg2+ (11.325) | Al3+ (17.763) | Si4+ (25.763) | P5+ (35.358) | S6+ (46.077) |
| Be2+ (2.238) | B3+ (2.488) | C4+ (2.618) | N5+ (2.803) | O6+ (3.015) |
| Mg2+ (2.119) | Al3+ (2.189) | Si4+ (2.449) | P5+ (2.701) | S6+ (2.806) |
3.4. Carbon, Sulfur, P-elements and Hydrogen
3.5. 3d and 4f Electron Inefficient Screening (p-Block)
3.6. Standard Potential Redox E0 (Transition Elements)
3.7. Inert Pair Effect (6s2 Elements)
3.8. Color of Copper, Silver and Gold
4. Applications
4.1. Covalency Result Is Retrieved
4.2. Mössbauer Parameters δ and Δ
4.3. Effective Polarizing Power and Fajans Rules
4.4. Melting Points and Bond Properties
4.5. Lattice Energy
4.6. Lowis Acid Strengths
| Compound | Cation | Z* | Iav | rc−1 | n*rc−1 | XIC | IC | U(kJmol 1) [44] |
|---|---|---|---|---|---|---|---|---|
| LiH | Li+ | 1.253 | 5.39 | 0.816 | 2.238 | 0.808 | 1.023 | 905.4 |
| NaH | Na+ | 1.777 | 5.14 | 0.636 | 1.838 | 0.853 | 1.13 | 810.9 |
| KH | K+ | 1.949 | 4.34 | 0.513 | 1.769 | 0.799 | 0.999 | 714.2 |
| AgF | Ag+ | 2.874 | 7.58 | 0.747 | 2.875 | 1.271 | 2.147 | 954 |
| NaF | Na+ | 1.777 | 5.14 | 0.636 | 1.838 | 0.853 | 1.13 | 903.9 |
| KF | K+ | 1.949 | 4.34 | 0.513 | 1.769 | 0.799 | 0.999 | 801.2 |
| AgCl | Ag+ | 2.874 | 7.58 | 0.747 | 2.875 | 1.271 | 2.147 | 904 |
| TlCl | Tl+ | 2.922 | 6.11 | 0.646 | 2.815 | 1.164 | 1.887 | 732 |
| KCl | K+ | 1.949 | 4.34 | 0.513 | 1.769 | 0.799 | 0.999 | 697.9 |
| RbCl | Rb+ | 2.134 | 4.18 | 0.455 | 1.75 | 0.787 | 0.97 | 677.8 |
5. Dual Method
5.1. An Interesting Comparison
5.2. “Inverted” Sodium-Lithium Electronegativity
5.3. Predicting Raw Material for InN Nanocrystals
6. Conclusions
References
- Lewis, GN. The Atom and the Molecule. J. Am. Chem. Soc 1916, 38, 762–785. [Google Scholar]
- Pauling, L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, 2nd ed; Cornell University Press: Ithaca, NY; USA, 1940. [Google Scholar]
- Pauling, L. The Nature of the Chemical Bond IV. The energy of single bonds and the relative electronegativity atoms. J. Am. Chem. Soc 1932, 54, 3570–3582. [Google Scholar]
- Mulliken, RS. A New Electroaffinity Scale; Together with Data on Valence States and on Valence Ionization Potentials and Electron Affinities. J. Chem. Phys 1934, 2, 782. [Google Scholar]
- Gordy, WA. New Method of Determining Electronegativity from Other Atomic Properties. Phys. Rev 1946, 69, 604. [Google Scholar]
- Sanderson, RT. Electronegativities in inorganic chemistry. III. J. Chem. Educ 1955, 31, 238. [Google Scholar]
- Allred, EG; Rochow, AL. A scale of electronegativity based on electrostatic force. J. Inorg. Nucl. Chem 1958, 5, 264–268. [Google Scholar]
- Zhang, Y. Electronegativity from Ionization Potentials. J. Mol. Sci. (Chinese) 1981, 1, 125. [Google Scholar]
- Zhang, Y. Electronegativities of Elements in Valence States and Their Appplications.1. Electronegativities of Elements in Valence States. Inorg. Chem 1982, 21, 3886. [Google Scholar]
- Zhang, Y. Electronegativities of Elements in Valence States and Their Appplications.2. A Scale for Strengths of Lewis Acids. Inorg. Chem 1982, 21, 3889. [Google Scholar]
- Huheey, JM. Inorganic Chemistry: Principles of Structure and Reactivity, 3rd ed; Harper and Row: New York, NY, USA, 1983; pp. 140–160. [Google Scholar]
- Mullay, J. Structure & Bonding; Springer-Verlag, Berlin Heidelberg: New York, NY, USA, 1987; pp. 1–25. [Google Scholar]
- Datta, D. Ab initio calculation, electronegatiity equalization and group electronegativity. Proc. Indian Acad. Sci. (Chem. Sci.) 1988, 6, 549–557. [Google Scholar]
- Allen, LC. Electronegativity is the average one-electron energy of the valence-shell electrons in ground-state free atoms. J. Am. Chem. Soc 1989, 111, 9003. [Google Scholar]
- Luo, Y-R; Benson, SW. A new electronegativity scale. 12. Intrinsic Lewis acid strength for main-group elements. Inorg. Chem 1991, 30, 1676. [Google Scholar]
- Zhen, NW; Li, G-S. Electronegativity: Average Nuclear Potential of the Valence Electron. J. Phys. Chem 1994, 98, 3964. [Google Scholar]
- Bergmann, D; Hinze, J. Electronegativity and Molecular Properties. J. Angew. Chem. Int. Ed 1996, 35, 150–163. [Google Scholar]
- Cherkasov, AR; Galkin, VI; Zueva, EM; Cherkasov, RA. The concept of electronegativity and the current state of the problem. Russ. Chem. Rev 1998, 5, 375–392. [Google Scholar]
- Mackay, KM; Mackay, RA; Henderson, W. Introduction to Modern Inorganic Chemistry, 6th ed; Nelson, Thornes: Cheltenham, UK, 2002; pp. 53–55. [Google Scholar]
- Portier, J; Campet, G; Etoumeau, J; Shastry, MCR; Tanguy, B. A simple approach to materials design: Role played by an ionic-covalent parameter based on polarizing power and electronegativity. J. Alloys Compd 1994, 209, 59–64. [Google Scholar]
- Lenglet, M. Iono-covalent character of the metal-oxygen bonds in oxides: A comparison of experimental and theoretical data. Act. Passive Electron. Compon 2004, 27, 1–60. [Google Scholar]
- Li, Z-H; Dai, Y-M; Wen, S-N; Nie, C-M; Zhou, C-Y. Relationship between atom valence shell electron quantum topological indices and electronegativity of elements. Acta Chimica. Sinica 2005, 14, 1348. [Google Scholar]
- Villesuzanne, A; Elissalde, C; Pouchard, M; Ravez, J. New considerations on the role of covalency in ferroelectric niobates and tantalates. Eur. Phys. J. B 1998, 6, 307. [Google Scholar]
- Ravez, J; Pouchard, M; Hagenmuller, P. Chemical bonding and ferroelectric perovskites. Eur. J. Solid State Inorg. Chem 1991, 25, 1107. [Google Scholar]
- Lower, S. Why atoms don’t collapse. Chem1 Virtual Textbook. Available online: http://www.chem1.com/acad/webtext/virtualtextbook.html (accessed on 18 October 2010).
- Duffy, JA. General Inorganic Chemistry, 2nd ed; Longmans: London, UK, 1974; pp. 19–20. [Google Scholar]
- Blaber, M. General Chemistry 1 Virtual Textbook, Web Course Tutorial. Available online: www.mikeblaber.org/oldwine/chm1045/chm1045.htm (accessed on 18 October 2010).
- Mackay, KM; Mackay, RA; Henderson, W. Introduction to Modern Inorganic Chemistry, 6th ed; Nelson, Thornes: Cheltenham, UK, 2002; pp. 45–46. [Google Scholar]
- Pauling, L. Atomic Radii and Interatomic Distances in Metals. J. Am. Chem. Soc 1947, 69, 542. [Google Scholar]
- Batsanov, SS. Experimental determination of covalent radii of elements. Russ. Chem. Bull 1995, 12, 2245–2250. [Google Scholar]
- Cordero, B; Gomez, V; Platero-Prats, AE; Reves, M; Echeverris, J; Cremades, E; Barragan, F; Alvarez, S. Covalent Radius. Dalton Trans 2008, 2832–2838. [Google Scholar]
- Rappe, AK; Casewit, CJ; Colwell, KS; Goddard, WA, III; Skiff, WM. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc 1992, 114, 10024–10035. [Google Scholar]
- Mo, Y-R. Tow issues on the polarity of chemical bond. Univ. Chem 1993, 4, 15. [Google Scholar]
- Computational Chemistry Comparison and Benchmark Database. Available online: http://cccbdb.nist.gov/default.htm (accessed on 18 October 2010).
- Dalian Technology Institute. Inorg. Chem. (in Chinese), 3rd ed.; High Education Press: Beijing, China, 1990. [Google Scholar]
- Thomas, JM. Principles and Practice of Heterogeneous Catalysis; Wiley-VCH: Weinheim, Germany, 1996; p. 448. [Google Scholar]
- Xu, G-X. Material Structure (in Chinise); People’s Education Press: Beijing, China, 1961; p. 160. [Google Scholar]
- Housecroft, CE; Sharepe, AG. Inorganic Chemistry, 2nd ed; Pearson Education: Harlow, UK, 2005. [Google Scholar]
- Pyykkö, P. Relativistic Effects in Structural Chemistry. Chem. Rev 2002, 3, 563–594. [Google Scholar]
- Reguera, E; Bertran, JF; Miranda, J; Portilla, C. Study of the dependence of Mossbauer parameters on the outer cation in nitroprussides. J. Radioanal. Nucl. Chem. Lett 1992, 3, 191–201. [Google Scholar]
- Reguera, E; Rodriguez-Hernandez, J; Champi, A; Duque, JG; Granado, E; Rettori, C. Unique coordination of copper in hexacyanometallates. Zeitschrift für physikalische chemie 2006, 12, 1609–1619. [Google Scholar]
- Heslop, RB; Jones, K. Inorganic Chemistry; Elsevier Scientific Publishing: Amsterdam, The Netherland, 1976; p. 31. [Google Scholar]
- Mackay, KM; Mackay, RA; Henderson, W. Introduction to Modern Inorganic Chemistry, 6th ed; Nelson Thornes: Cheltenham, UK, 2002; p. 120. [Google Scholar]
- Huheey, JE. Inorganic Chemistry: Principles of Structure & Reactivity, 2nd ed; Harper and Row, Publishers: New York, NY, USA, 1978; p. 93. [Google Scholar]
- Lambert, C; Kaupp, M; von Rague Schleyer, P. “Inverted” sodium-lithium electronegativity: polarity and metalation energies of organic and inorganic alkali-metal compounds. Organometallics 1993, 12, 853–859. [Google Scholar]
- Wu, C; Li, T; Lei, L; Hu, S; Liu, Y; Xie, T. Indium nitride from indium iodide at low temperatures: Synthesis and their optical properties. New J Chem 2005, 29, 1610. [Google Scholar]
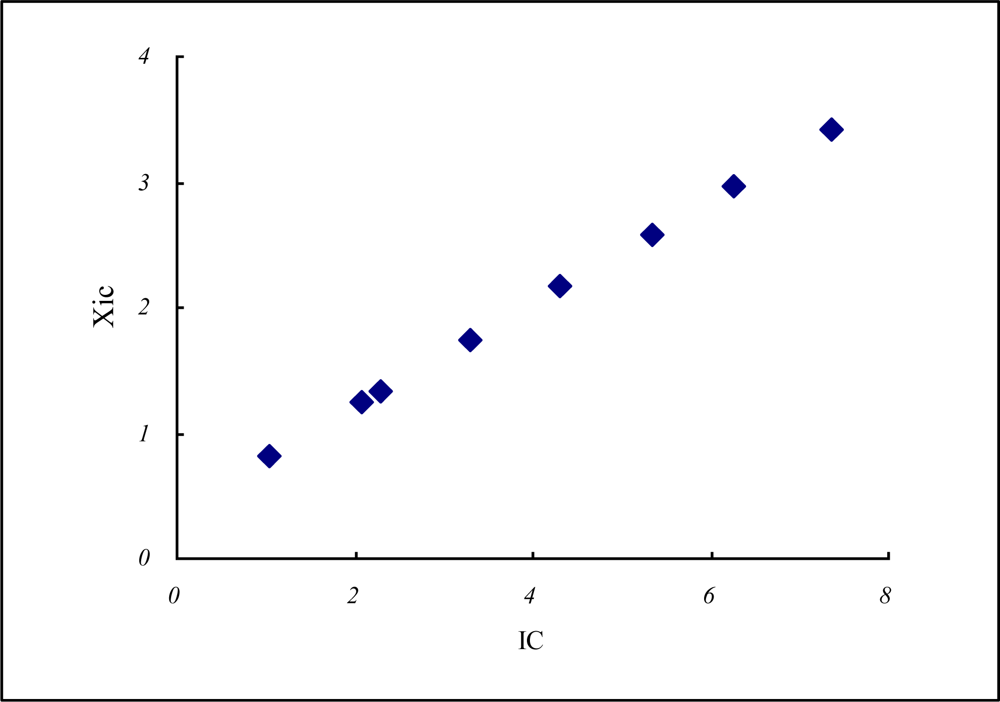
| Atm.No | Cations | n* | Iz | Iav | rc−1 | n*rc−1 | Z* | rc | Xz | XIC | IC | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | H+ | 0.85 | 13.600 | 13.600 | 2.703 | 2.297 | 0.850 | 0.370 | 2.271 | 1.333 | 2.297 | |
| 2 | He2+ | 0.85 | 54.400 | 39.500 | 3.571 | 3.036 | 1.449 | 0.280 | 6.001 | 2.518 | 5.173 | |
| 3 | Li+ | 1.99 | 5.3900 | 5.3900 | 0.816 | 1.624 | 1.253 | 1.225 | 0.976 | 0.808 | 1.023 | |
| 4 | Be2+ | 1.99 | 18.200 | 13.760 | 1.031 | 2.052 | 2.002 | 0.970 | 1.365 | 1.237 | 2.064 | |
| 5 | B3+ | 1.99 | 37.900 | 23.800 | 1.250 | 2.488 | 2.633 | 0.800 | 2.026 | 1.743 | 3.291 | |
| 6 | C4+ | 1.99 | 64.500 | 37.015 | 1.316 | 2.618 | 3.283 | 0.760 | 2.583 | 2.167 | 4.320 | |
| 6 | C3+ | 1.99 | 47.900 | 27.853 | 1.316 | 2.618 | 2.848 | 0.760 | 2.333 | 1.931 | 3.747 | |
| 6 | C2+ | 1.99 | 24.400 | 17.830 | 1.316 | 2.618 | 2.279 | 0.760 | 1.887 | 1.622 | 2.998 | |
| 7 | N5+ | 1.99 | 97.900 | 53.406 | 1.351 | 2.689 | 3.943 | 0.740 | 3.125 | 2.583 | 5.329 | |
| 7 | N4+ | 1.99 | 77.500 | 42.283 | 1.351 | 2.689 | 3.509 | 0.740 | 2.866 | 2.341 | 4.742 | |
| 7 | N3+ | 1.99 | 47.500 | 30.543 | 1.351 | 2.689 | 2.982 | 0.740 | 2.412 | 2.047 | 4.030 | |
| 7 | N2+ | 1.99 | 29.600 | 22.065 | 1.351 | 2.689 | 2.535 | 0.740 | 2.067 | 1.798 | 3.425 | |
| 7 | N+ | 1.99 | 14.530 | 14.530 | 1.351 | 2.689 | 2.057 | 0.740 | 1.680 | 1.532 | 2.780 | |
| 8 | O6+ | 1.99 | 138.00 | 72.020 | 1.370 | 2.726 | 4.579 | 0.730 | 3.642 | 2.972 | 6.273 | |
| 8 | O2+ | 1.99 | 35.100 | 24.360 | 1.370 | 2.726 | 2.663 | 0.730 | 2.221 | 1.890 | 3.648 | |
| 9 | F7+ | 1.99 | 185.00 | 94.030 | 1.408 | 2.803 | 5.233 | 0.710 | 4.284 | 3.423 | 7.370 | |
| 10 | Ne4+ | 1.99 | 97.100 | 55.785 | 1.724 | 3.431 | 4.030 | 0.580 | 4.584 | 3.250 | 6.949 | |
| 10 | Ne2+ | 1.99 | 41.000 | 31.270 | 1.724 | 3.431 | 3.018 | 0.580 | 3.250 | 2.530 | 5.203 | |
| 11 | Na+ | 2.89 | 5.1400 | 5.1400 | 0.636 | 1.838 | 1.777 | 1.572 | 0.948 | 0.853 | 1.130 | |
| 12 | Mg2+ | 2.89 | 15.000 | 11.325 | 0.733 | 2.119 | 2.637 | 1.364 | 1.168 | 1.184 | 1.933 | |
| 13 | Al3+ | 2.89 | 28.500 | 17.763 | 0.826 | 2.388 | 3.303 | 1.210 | 1.464 | 1.512 | 2.730 | |
| 13 | Al+ | 2.89 | 5.9900 | 5.9900 | 0.826 | 2.388 | 1.918 | 1.210 | 1.091 | 1.040 | 1.585 | |
| 14 | Si4+ | 2.89 | 45.100 | 25.763 | 0.847 | 2.449 | 3.978 | 1.180 | 1.686 | 1.776 | 3.371 | |
| 14 | Si2+ | 2.89 | 16.300 | 12.225 | 0.847 | 2.449 | 2.740 | 1.180 | 1.323 | 1.344 | 2.322 | |
| 15 | P5+ | 2.89 | 65.000 | 35.358 | 0.935 | 2.701 | 4.660 | 1.070 | 2.105 | 2.181 | 4.355 | |
| 15 | P4+ | 2.89 | 51.400 | 27.948 | 0.935 | 2.701 | 4.143 | 1.070 | 1.958 | 1.982 | 3.872 | |
| 15 | P3+ | 2.89 | 30.200 | 20.130 | 0.935 | 2.701 | 3.516 | 1.070 | 1.682 | 1.741 | 3.286 | |
| 15 | P+ | 2.89 | 10.490 | 10.490 | 0.935 | 2.701 | 2.538 | 1.070 | 1.309 | 1.364 | 2.372 | |
| 16 | S6+ | 2.89 | 88.000 | 46.077 | 0.971 | 2.806 | 5.319 | 1.030 | 2.445 | 2.515 | 5.165 | |
| 16 | S4+ | 2.89 | 47.300 | 28.940 | 0.971 | 2.806 | 4.216 | 1.030 | 1.999 | 2.073 | 4.093 | |
| 16 | S2+ | 2.89 | 23.300 | 16.830 | 0.971 | 2.806 | 3.215 | 1.030 | 1.634 | 1.673 | 3.121 | |
| 17 | Cl7+ | 2.89 | 114.00 | 58.381 | 1.010 | 2.919 | 5.988 | 0.990 | 2.832 | 2.879 | 6.048 | |
| 17 | Cl5+ | 2.89 | 67.800 | 39.534 | 1.010 | 2.919 | 4.927 | 0.990 | 2.362 | 2.438 | 4.977 | |
| 17 | Cl3+ | 2.89 | 39.600 | 25.457 | 1.010 | 2.919 | 3.954 | 0.990 | 1.988 | 2.032 | 3.994 | |
| 17 | Cl+ | 2.89 | 12.970 | 12.970 | 1.010 | 2.919 | 2.822 | 0.990 | 1.469 | 1.562 | 2.851 | |
| 18 | Ar4+ | 2.89 | 59.800 | 35.965 | 1.031 | 2.979 | 4.700 | 0.970 | 2.327 | 2.383 | 4.845 | |
| 18 | Ar2+ | 2.89 | 27.600 | 21.680 | 1.031 | 2.979 | 3.649 | 0.970 | 1.830 | 1.937 | 3.762 | |
| 19 | K+ | 3.45 | 4.3400 | 4.3400 | 0.513 | 1.769 | 1.949 | 1.950 | 0.899 | 0.799 | 0.999 | |
| 20 | Ca2+ | 3.45 | 11.900 | 9.0050 | 0.576 | 1.987 | 2.807 | 1.736 | 1.033 | 1.053 | 1.617 | |
| 21 | Sc3+ | 3.45 | 24.800 | 14.713 | 0.695 | 2.397 | 3.588 | 1.439 | 1.317 | 1.414 | 2.494 | |
| 22 | Ti4+ | 3.45 | 43.300 | 22.805 | 0.755 | 2.606 | 4.468 | 1.324 | 1.621 | 1.777 | 3.374 | |
| 22 | Ti3+ | 3.45 | 27.500 | 15.973 | 0.755 | 2.606 | 3.739 | 1.324 | 1.449 | 1.550 | 2.824 | |
| 22 | Ti2+ | 3.45 | 13.600 | 10.210 | 0.755 | 2.606 | 2.989 | 1.324 | 1.249 | 1.317 | 2.258 | |
| 23 | V5+ | 3.45 | 65.200 | 32.528 | 0.817 | 2.819 | 5.336 | 1.224 | 1.990 | 2.183 | 4.359 | |
| 23 | V4+ | 3.45 | 46.700 | 24.360 | 0.817 | 2.819 | 4.617 | 1.224 | 1.803 | 1.941 | 3.772 | |
| 23 | V3+ | 3.45 | 29.300 | 16.913 | 0.817 | 2.819 | 3.847 | 1.224 | 1.590 | 1.682 | 3.143 | |
| 23 | V2+ | 3.45 | 14.700 | 10.720 | 0.817 | 2.819 | 3.063 | 1.224 | 1.352 | 1.418 | 2.502 | |
| 23 | V+ | 3.45 | 6.7400 | 6.7400 | 0.817 | 2.819 | 2.429 | 1.224 | 1.166 | 1.205 | 1.984 | |
| 24 | Cr6+ | 3.45 | 90.600 | 43.878 | 0.853 | 2.944 | 6.197 | 1.172 | 2.337 | 2.565 | 5.287 | |
| 24 | Cr5+ | 3.45 | 69.300 | 34.534 | 0.853 | 2.944 | 5.498 | 1.172 | 2.141 | 2.320 | 4.691 | |
| 24 | Cr4+ | 3.45 | 49.100 | 25.843 | 0.853 | 2.944 | 4.756 | 1.172 | 1.925 | 2.059 | 4.058 | |
| 24 | Cr3+ | 3.45 | 31.000 | 18.090 | 0.853 | 2.944 | 3.979 | 1.172 | 1.689 | 1.786 | 3.395 | |
| 24 | Cr2+ | 3.45 | 16.500 | 11.635 | 0.853 | 2.944 | 3.191 | 1.172 | 1.442 | 1.509 | 2.723 | |
| 24 | Cr+ | 3.45 | 6.7700 | 6.7700 | 0.853 | 2.944 | 2.434 | 1.172 | 1.202 | 1.243 | 2.077 | |
| 25 | Mn7+ | 3.45 | 119.00 | 56.334 | 0.856 | 2.954 | 7.022 | 1.168 | 2.578 | 2.864 | 6.012 | |
| 25 | Mn6+ | 3.45 | 95.000 | 45.890 | 0.856 | 2.954 | 6.337 | 1.168 | 2.386 | 2.622 | 5.426 | |
| 25 | Mn5+ | 3.45 | 72.400 | 36.068 | 0.856 | 2.954 | 5.618 | 1.168 | 2.181 | 2.369 | 4.810 | |
| 25 | Mn4+ | 3.45 | 51.200 | 26.985 | 0.856 | 2.954 | 4.860 | 1.168 | 1.958 | 2.101 | 4.161 | |
| 25 | Mn3+ | 3.45 | 33.700 | 18.913 | 0.856 | 2.954 | 4.068 | 1.168 | 1.734 | 1.822 | 3.483 | |
| 25 | Mn2+ | 3.45 | 15.600 | 11.520 | 0.856 | 2.954 | 3.175 | 1.168 | 1.428 | 1.507 | 2.719 | |
| 25 | Mn+ | 3.45 | 7.4400 | 7.4400 | 0.856 | 2.954 | 2.552 | 1.168 | 1.226 | 1.287 | 2.185 | |
| 26 | Fe6+ | 3.45 | 99.000 | 47.262 | 0.858 | 2.961 | 6.431 | 1.165 | 2.428 | 2.661 | 5.520 | |
| 26 | Fe5+ | 3.45 | 75.000 | 36.914 | 0.858 | 2.961 | 5.684 | 1.165 | 2.214 | 2.397 | 4.879 | |
| 26 | Fe4+ | 3.45 | 54.800 | 27.393 | 0.858 | 2.961 | 4.896 | 1.165 | 2.005 | 2.119 | 4.203 | |
| 26 | Fe3+ | 3.45 | 30.700 | 18.257 | 0.858 | 2.961 | 3.997 | 1.165 | 1.695 | 1.801 | 3.431 | |
| 26 | Fe2+ | 3.45 | 16.200 | 12.035 | 0.858 | 2.961 | 3.245 | 1.165 | 1.444 | 1.535 | 2.786 | |
| 26 | Fe+ | 3.45 | 7.8700 | 7.8700 | 0.858 | 2.961 | 2.624 | 1.165 | 1.241 | 1.315 | 2.253 | |
| 27 | Co4+ | 3.45 | 51.300 | 27.440 | 0.870 | 3.000 | 4.901 | 1.150 | 1.996 | 2.143 | 4.261 | |
| 27 | Co3+ | 3.45 | 33.500 | 19.487 | 0.870 | 3.000 | 4.130 | 1.150 | 1.762 | 1.867 | 3.591 | |
| 27 | Co2+ | 3.45 | 17.100 | 12.480 | 0.870 | 3.000 | 3.305 | 1.150 | 1.480 | 1.571 | 2.874 | |
| 27 | Co+ | 3.45 | 7.8600 | 7.8600 | 0.870 | 3.000 | 2.623 | 1.150 | 1.253 | 1.327 | 2.281 | |
| 28 | Ni4+ | 3.45 | 54.900 | 28.985 | 0.901 | 3.108 | 5.037 | 1.110 | 2.131 | 2.256 | 4.537 | |
| 28 | Ni3+ | 3.45 | 35.200 | 20.347 | 0.901 | 3.108 | 4.220 | 1.110 | 1.861 | 1.953 | 3.802 | |
| 28 | Ni2+ | 3.45 | 18.200 | 12.920 | 0.901 | 3.108 | 3.363 | 1.110 | 1.556 | 1.635 | 3.029 | |
| 28 | Ni+ | 3.45 | 7.6400 | 7.6400 | 0.901 | 3.108 | 2.586 | 1.110 | 1.281 | 1.347 | 2.330 | |
| 29 | Cu3+ | 3.45 | 36.800 | 21.610 | 0.901 | 3.108 | 4.349 | 1.110 | 1.885 | 2.001 | 3.918 | |
| 29 | Cu2+ | 3.45 | 20.300 | 14.015 | 0.901 | 3.108 | 3.502 | 1.110 | 1.599 | 1.687 | 3.155 | |
| 29 | Cu+ | 3.45 | 7.7300 | 7.7300 | 0.901 | 3.108 | 2.601 | 1.110 | 1.284 | 1.352 | 2.343 | |
| 30 | Zn2+ | 3.45 | 18.000 | 13.695 | 0.801 | 2.762 | 3.462 | 1.249 | 1.388 | 1.529 | 2.772 | |
| 30 | Zn+ | 3.45 | 9.3900 | 9.3900 | 0.801 | 2.762 | 2.867 | 1.249 | 1.218 | 1.333 | 2.295 | |
| 31 | Ga3+ | 3.45 | 30.700 | 19.067 | 0.803 | 2.771 | 4.085 | 1.245 | 1.581 | 1.739 | 3.281 | |
| 31 | Ga+ | 3.45 | 6.0000 | 6.0000 | 0.803 | 2.771 | 2.292 | 1.245 | 1.131 | 1.145 | 1.841 | |
| 32 | Ge4+ | 3.45 | 45.700 | 25.925 | 0.818 | 2.821 | 4.763 | 1.223 | 1.794 | 1.992 | 3.895 | |
| 32 | Ge2+ | 3.45 | 15.900 | 11.900 | 0.818 | 2.821 | 3.227 | 1.223 | 1.376 | 1.474 | 2.639 | |
| 33 | As5+ | 3.45 | 62.600 | 33.902 | 0.826 | 2.851 | 5.447 | 1.210 | 1.993 | 2.242 | 4.502 | |
| 33 | As3+ | 3.45 | 28.400 | 18.937 | 0.826 | 2.851 | 4.071 | 1.210 | 1.596 | 1.773 | 3.364 | |
| 33 | As+ | 3.45 | 9.810 | 9.8100 | 0.826 | 2.851 | 2.930 | 1.210 | 1.257 | 1.385 | 2.422 | |
| 34 | Se6+ | 3.45 | 81.700 | 42.442 | 0.855 | 2.949 | 6.095 | 1.170 | 2.264 | 2.533 | 5.209 | |
| 34 | Se4+ | 3.45 | 42.900 | 26.163 | 0.855 | 2.949 | 4.785 | 1.170 | 1.854 | 2.072 | 4.090 | |
| 34 | Se2+ | 3.45 | 21.200 | 15.480 | 0.855 | 2.949 | 3.681 | 1.170 | 1.533 | 1.683 | 3.146 | |
| 35 | Br7+ | 3.45 | 103.00 | 52.601 | 0.876 | 3.021 | 6.785 | 1.142 | 2.529 | 2.835 | 5.941 | |
| 35 | Br5+ | 3.45 | 59.700 | 35.322 | 0.876 | 3.021 | 5.560 | 1.142 | 2.111 | 2.393 | 4.869 | |
| 35 | Br+ | 3.45 | 11.810 | 11.810 | 0.876 | 3.021 | 3.215 | 1.142 | 1.369 | 1.547 | 2.815 | |
| 36 | Kr6+ | 3.45 | 78.500 | 45.200 | 0.862 | 2.974 | 6.290 | 1.160 | 2.260 | 2.621 | 5.422 | |
| 36 | Kr4+ | 3.45 | 52.500 | 32.000 | 0.862 | 2.974 | 5.292 | 1.160 | 1.989 | 2.267 | 4.562 | |
| 36 | Kr2+ | 3.45 | 24.500 | 19.250 | 0.862 | 2.974 | 4.105 | 1.160 | 1.604 | 1.845 | 3.538 | |
| 37 | Rb+ | 3.85 | 4.1800 | 4.1800 | 0.463 | 1.782 | 2.134 | 2.160 | 0.885 | 0.794 | 0.988 | |
| 38 | Sr2+ | 3.85 | 11.000 | 8.3500 | 0.522 | 2.011 | 3.017 | 1.914 | 1.003 | 1.036 | 1.576 | |
| 39 | Y3+ | 3.85 | 20.500 | 13.027 | 0.619 | 2.382 | 3.768 | 1.616 | 1.211 | 1.348 | 2.332 | |
| 40 | Zr4+ | 3.85 | 34.300 | 19.310 | 0.688 | 2.648 | 4.588 | 1.454 | 1.472 | 1.687 | 3.155 | |
| 40 | Zr3+ | 3.85 | 23.000 | 14.313 | 0.688 | 2.648 | 3.950 | 1.454 | 1.346 | 1.506 | 2.716 | |
| 40 | Zr2+ | 3.85 | 13.100 | 9.9700 | 0.688 | 2.648 | 3.296 | 1.454 | 1.206 | 1.321 | 2.267 | |
| 41 | Nb5+ | 3.85 | 50.600 | 27.016 | 0.745 | 2.869 | 5.426 | 1.342 | 1.769 | 2.053 | 4.043 | |
| 41 | Nb4+ | 3.85 | 38.300 | 21.120 | 0.745 | 2.869 | 4.798 | 1.342 | 1.640 | 1.860 | 3.575 | |
| 41 | Nb3+ | 3.85 | 25.000 | 15.393 | 0.745 | 2.869 | 4.096 | 1.342 | 1.474 | 1.644 | 3.052 | |
| 41 | Nb2+ | 3.85 | 14.300 | 10.590 | 0.745 | 2.869 | 3.397 | 1.342 | 1.303 | 1.430 | 2.532 | |
| 41 | Nb+ | 3.85 | 6.8800 | 6.8800 | 0.745 | 2.869 | 2.738 | 1.342 | 1.141 | 1.228 | 2.040 | |
| 42 | Mo6+ | 3.85 | 68.000 | 37.683 | 0.775 | 2.982 | 6.409 | 1.291 | 2.020 | 2.432 | 4.964 | |
| 42 | Mo5+ | 3.85 | 61.200 | 31.620 | 0.775 | 2.982 | 5.870 | 1.291 | 1.956 | 2.260 | 4.547 | |
| 42 | Mo+4 | 3.85 | 46.400 | 24.225 | 0.775 | 2.982 | 5.138 | 1.291 | 1.803 | 2.027 | 3.980 | |
| 42 | Mo+3 | 3.85 | 27.200 | 16.833 | 0.775 | 2.982 | 4.283 | 1.291 | 1.562 | 1.754 | 3.318 | |
| 42 | Mo+2 | 3.85 | 16.200 | 11.650 | 0.775 | 2.982 | 3.563 | 1.291 | 1.383 | 1.524 | 2.760 | |
| 42 | Mo+ | 3.85 | 7.1000 | 7.1000 | 0.775 | 2.982 | 2.782 | 1.291 | 1.177 | 1.275 | 2.155 | |
| 43 | Tc7+ | 3.85 | 94.000 | 46.297 | 0.787 | 3.031 | 7.103 | 1.270 | 2.287 | 2.691 | 5.593 | |
| 43 | Tc6+ | 3.85 | 76.000 | 38.347 | 0.787 | 3.031 | 6.465 | 1.270 | 2.135 | 2.484 | 5.090 | |
| 43 | Tc5+ | 3.85 | 59.000 | 30.816 | 0.787 | 3.031 | 5.795 | 1.270 | 1.973 | 2.267 | 4.563 | |
| 43 | Tc4+ | 3.85 | 43.000 | 23.770 | 0.787 | 3.031 | 5.090 | 1.270 | 1.798 | 2.038 | 4.008 | |
| 43 | Tc3+ | 3.85 | 29.500 | 17.360 | 0.787 | 3.031 | 4.350 | 1.270 | 1.622 | 1.798 | 3.425 | |
| 43 | Tc2+ | 3.85 | 15.300 | 11.290 | 0.787 | 3.031 | 3.508 | 1.270 | 1.385 | 1.525 | 2.762 | |
| 43 | Tc+ | 3.85 | 7.2800 | 7.2800 | 0.787 | 3.031 | 2.817 | 1.270 | 1.196 | 1.301 | 2.218 | |
| 44 | Ru8+ | 3.85 | 119.00 | 57.771 | 0.806 | 3.102 | 7.935 | 1.241 | 2.557 | 3.021 | 6.394 | |
| 44 | Ru7+ | 3.85 | 100.00 | 49.024 | 0.806 | 3.102 | 7.310 | 1.241 | 2.409 | 2.814 | 5.890 | |
| 44 | Ru6+ | 3.85 | 81.000 | 40.528 | 0.806 | 3.102 | 6.646 | 1.241 | 2.245 | 2.593 | 5.355 | |
| 44 | Ru5+ | 3.85 | 63.000 | 32.434 | 0.806 | 3.102 | 5.946 | 1.241 | 2.072 | 2.361 | 4.791 | |
| 44 | Ru4+ | 3.85 | 46.500 | 24.793 | 0.806 | 3.102 | 5.198 | 1.241 | 1.889 | 2.113 | 4.189 | |
| 44 | Ru3+ | 3.85 | 28.500 | 17.557 | 0.806 | 3.102 | 4.374 | 1.241 | 1.647 | 1.839 | 3.525 | |
| 44 | Ru2+ | 3.85 | 16.800 | 12.085 | 0.806 | 3.102 | 3.629 | 1.241 | 1.445 | 1.592 | 2.924 | |
| 45 | Rh6+ | 3.85 | 85.000 | 42.377 | 0.802 | 3.087 | 6.796 | 1.247 | 2.267 | 2.632 | 5.450 | |
| 45 | Rh4+ | 3.85 | 45.600 | 25.565 | 0.802 | 3.087 | 5.279 | 1.247 | 1.868 | 2.131 | 4.233 | |
| 45 | Rh3+ | 3.85 | 31.100 | 18.887 | 0.802 | 3.087 | 4.537 | 1.247 | 1.677 | 1.886 | 3.638 | |
| 45 | Rh2+ | 3.85 | 18.100 | 12.780 | 0.802 | 3.087 | 3.732 | 1.247 | 1.463 | 1.620 | 2.993 | |
| 45 | Rh+ | 3.85 | 7.4600 | 7.4600 | 0.802 | 3.087 | 2.851 | 1.247 | 1.217 | 1.329 | 2.287 | |
| 46 | Pd6+ | 3.85 | 90.000 | 44.273 | 0.782 | 3.013 | 6.946 | 1.278 | 2.236 | 2.626 | 5.435 | |
| 46 | Pd5+ | 3.85 | 66.000 | 35.128 | 0.782 | 3.013 | 6.188 | 1.278 | 2.026 | 2.382 | 4.842 | |
| 46 | Pd4+ | 3.85 | 49.000 | 27.410 | 0.782 | 3.013 | 5.466 | 1.278 | 1.853 | 2.149 | 4.277 | |
| 46 | Pd3+ | 3.85 | 32.900 | 20.210 | 0.782 | 3.013 | 4.693 | 1.278 | 1.659 | 1.900 | 3.672 | |
| 46 | Pd2+ | 3.85 | 19.400 | 13.870 | 0.782 | 3.013 | 3.888 | 1.278 | 1.453 | 1.640 | 3.042 | |
| 47 | Ag3+ | 3.85 | 34.600 | 21.227 | 0.747 | 2.875 | 4.810 | 1.339 | 1.600 | 1.867 | 3.592 | |
| 47 | Ag2+ | 3.85 | 21.500 | 14.540 | 0.747 | 2.875 | 3.981 | 1.339 | 1.426 | 1.612 | 2.973 | |
| 47 | Ag+ | 3.85 | 7.5800 | 7.5800 | 0.747 | 2.875 | 2.874 | 1.339 | 1.161 | 1.271 | 2.147 | |
| 48 | Cd2+ | 3.85 | 16.900 | 12.945 | 0.708 | 2.725 | 3.756 | 1.413 | 1.293 | 1.482 | 2.658 | |
| 48 | Cd+ | 3.85 | 8.9900 | 8.9900 | 0.694 | 2.674 | 3.130 | 1.440 | 1.139 | 1.283 | 2.174 | |
| 49 | In3+ | 3.85 | 28.000 | 17.563 | 0.668 | 2.572 | 4.375 | 1.497 | 1.369 | 1.591 | 2.923 | |
| 49 | In+ | 3.85 | 5.7900 | 5.7900 | 0.668 | 2.572 | 2.512 | 1.497 | 1.045 | 1.078 | 1.678 | |
| 50 | Sn4+ | 3.85 | 40.700 | 23.285 | 0.715 | 2.752 | 5.038 | 1.399 | 1.595 | 1.871 | 3.601 | |
| 50 | Sn2+ | 3.85 | 14.600 | 10.970 | 0.715 | 2.752 | 3.458 | 1.399 | 1.266 | 1.405 | 2.472 | |
| 51 | Sb5+ | 3.85 | 55.500 | 30.028 | 0.709 | 2.730 | 5.721 | 1.410 | 1.718 | 2.059 | 4.057 | |
| 51 | Sb3+ | 3.85 | 25.300 | 16.813 | 0.709 | 2.730 | 4.281 | 1.410 | 1.412 | 1.638 | 3.036 | |
| 52 | Te6+ | 3.85 | 70.700 | 37.085 | 0.730 | 2.810 | 6.358 | 1.370 | 1.902 | 2.299 | 4.641 | |
| 52 | Te4+ | 3.85 | 37.400 | 23.253 | 0.730 | 2.810 | 5.034 | 1.370 | 1.595 | 1.901 | 3.675 | |
| 52 | Te2+ | 3.85 | 18.600 | 13.810 | 0.730 | 2.810 | 3.880 | 1.370 | 1.353 | 1.554 | 2.832 | |
| 53 | I7+ | 3.85 | 92.344 | 46.535 | 0.750 | 2.886 | 7.122 | 1.334 | 2.134 | 2.587 | 5.339 | |
| 53 | I5+ | 3.85 | 52.395 | 23.794 | 0.750 | 2.886 | 5.092 | 1.334 | 1.798 | 1.960 | 3.817 | |
| 53 | I+ | 3.85 | 10.450 | 10.450 | 0.750 | 2.886 | 3.375 | 1.334 | 1.232 | 1.429 | 2.530 | |
| 54 | Xe8+ | 3.85 | 112.91 | 55.46 | 0.714 | 2.750 | 7.775 | 1.400 | 2.139 | 2.675 | 5.553 | |
| 54 | Xe6+ | 3.85 | 68.718 | 39.18 | 0.714 | 2.750 | 6.534 | 1.400 | 1.839 | 2.310 | 4.667 | |
| 54 | Xe4+ | 3.85 | 43.956 | 27.35 | 0.714 | 2.750 | 5.459 | 1.400 | 1.626 | 1.994 | 3.900 | |
| 54 | Xe2+ | 3.85 | 21.200 | 16.670 | 0.714 | 2.750 | 4.262 | 1.400 | 1.366 | 1.641 | 3.044 | |
| 55 | Cs+ | 4.36 | 3.8900 | 3.8900 | 0.426 | 1.855 | 2.332 | 2.350 | 0.877 | 0.796 | 0.992 | |
| 56 | Ba2+ | 4.36 | 10.000 | 7.6050 | 0.505 | 2.201 | 3.260 | 1.981 | 1.005 | 1.065 | 1.646 | |
| 57 | La3+ | 4.36 | 19.200 | 11.960 | 0.534 | 2.330 | 4.089 | 1.871 | 1.132 | 1.287 | 2.185 | |
| 58 | Ce4+ | 4.36 | 36.720 | 18.311 | 0.608 | 2.649 | 5.059 | 1.646 | 1.412 | 1.653 | 3.074 | |
| 58 | Ce3+ | 4.36 | 20.199 | 12.174 | 0.608 | 2.649 | 4.125 | 1.646 | 1.248 | 1.420 | 2.506 | |
| 59 | Pr4+ | 4.36 | 38.979 | 17.918 | 0.607 | 2.646 | 5.005 | 1.648 | 1.430 | 1.638 | 3.037 | |
| 59 | Pr3+ | 4.36 | 21.619 | 10.898 | 0.607 | 2.646 | 3.903 | 1.648 | 1.263 | 1.363 | 2.368 | |
| 60 | Nd4+ | 4.36 | 40.420 | 19.676 | 0.609 | 2.655 | 5.244 | 1.642 | 1.447 | 1.703 | 3.194 | |
| 60 | Nd3+ | 4.36 | 22.075 | 12.762 | 0.609 | 2.655 | 4.223 | 1.642 | 1.272 | 1.447 | 2.572 | |
| 60 | Nd2+ | 4.36 | 10.716 | 8.105 | 0.609 | 2.655 | 3.366 | 1.642 | 1.121 | 1.232 | 2.050 | |
| 61 | Pm3+ | 4.36 | 22.283 | 12.914 | 0.613 | 2.675 | 4.249 | 1.630 | 1.281 | 1.461 | 2.606 | |
| 62 | Sm3+ | 4.36 | 23.423 | 13.373 | 0.602 | 2.627 | 4.323 | 1.660 | 1.275 | 1.460 | 2.604 | |
| 63 | Eu3+ | 4.36 | 24.874 | 13.929 | 0.541 | 2.357 | 4.412 | 1.850 | 1.190 | 1.370 | 2.385 | |
| 63 | Eu2+ | 4.36 | 11.245 | 8.4570 | 0.541 | 2.357 | 3.438 | 1.850 | 1.054 | 1.153 | 1.858 | |
| 64 | Gd3+ | 4.36 | 20.624 | 12.962 | 0.620 | 2.701 | 4.256 | 1.614 | 1.272 | 1.474 | 2.637 | |
| 64 | Gd2+ | 4.36 | 12.126 | 9.1310 | 0.620 | 2.701 | 3.572 | 1.614 | 1.156 | 1.299 | 2.213 | |
| 65 | Tb4+ | 4.36 | 39.798 | 19.759 | 0.628 | 2.739 | 5.255 | 1.592 | 1.484 | 1.747 | 3.301 | |
| 65 | Tb3+ | 4.36 | 21.868 | 13.079 | 0.628 | 2.739 | 4.276 | 1.592 | 1.301 | 1.494 | 2.686 | |
| 66 | Dy3+ | 4.36 | 22.801 | 13.466 | 0.629 | 2.744 | 4.339 | 1.589 | 1.314 | 1.512 | 2.730 | |
| 67 | Ho3+ | 4.36 | 22.801 | 13.542 | 0.633 | 2.759 | 4.351 | 1.580 | 1.320 | 1.521 | 2.754 | |
| 68 | Er3+ | 4.36 | 22.697 | 13.577 | 0.638 | 2.782 | 4.356 | 1.567 | 1.328 | 1.532 | 2.780 | |
| 69 | Tm3+ | 4.36 | 23.671 | 13.970 | 0.640 | 2.791 | 4.419 | 1.562 | 1.343 | 1.553 | 2.829 | |
| 69 | Tm2+ | 4.36 | 12.053 | 9.1190 | 0.640 | 2.791 | 3.570 | 1.562 | 1.180 | 1.329 | 2.286 | |
| 70 | Yb3+ | 4.36 | 25.029 | 14.487 | 0.589 | 2.566 | 4.500 | 1.699 | 1.269 | 1.478 | 2.649 | |
| 70 | Yb2+ | 4.36 | 12.178 | 9.2160 | 0.589 | 2.566 | 3.589 | 1.699 | 1.119 | 1.257 | 2.112 | |
| 71 | Lu3+ | 4.36 | 20.956 | 13.423 | 0.598 | 2.609 | 4.332 | 1.671 | 1.242 | 1.455 | 2.592 | |
| 72 | Hf4+ | 4.36 | 33.300 | 19.538 | 0.693 | 3.024 | 5.226 | 1.442 | 1.566 | 1.880 | 3.624 | |
| 72 | Hf3+ | 4.36 | 23.300 | 14.950 | 0.693 | 2.024 | 4.571 | 1.442 | 1.436 | 1.693 | 3.170 | |
| 72 | Hf2+ | 4.36 | 14.900 | 10.775 | 0.693 | 2.024 | 3.881 | 1.442 | 1.304 | 1.496 | 2.691 | |
| 73 | Ta5+ | 4.36 | 45.000 | 24.898 | 0.745 | 3.246 | 5.899 | 1.343 | 1.835 | 2.197 | 4.393 | |
| 73 | Ta4+ | 4.36 | 33.100 | 19.873 | 0.745 | 3.246 | 5.270 | 1.343 | 1.684 | 2.004 | 3.924 | |
| 73 | Ta3+ | 4.36 | 22.300 | 15.463 | 0.745 | 3.246 | 4.649 | 1.343 | 1.521 | 1.813 | 3.462 | |
| 73 | Ta2+ | 4.36 | 16.200 | 12.050 | 0.745 | 3.246 | 4.104 | 1.343 | 1.411 | 1.646 | 3.056 | |
| 73 | Ta+ | 4.36 | 7.8900 | 7.8900 | 0.745 | 3.246 | 3.321 | 1.343 | 1.219 | 1.406 | 2.473 | |
| 74 | W6+ | 4.36 | 61.000 | 32.363 | 0.770 | 3.356 | 6.726 | 1.299 | 2.094 | 2.520 | 5.178 | |
| 74 | W5+ | 4.36 | 48.000 | 26.636 | 0.770 | 3.356 | 6.102 | 1.299 | 1.945 | 2.322 | 4.697 | |
| 74 | W4+ | 4.36 | 35.400 | 21.295 | 0.770 | 3.356 | 5.456 | 1.299 | 1.780 | 2.117 | 4.200 | |
| 74 | W3+ | 4.36 | 24.100 | 16.593 | 0.770 | 3.356 | 4.816 | 1.299 | 1.604 | 1.914 | 3.707 | |
| 74 | W2+ | 4.36 | 17.700 | 12.840 | 0.770 | 3.356 | 4.236 | 1.299 | 1.485 | 1.731 | 3.261 | |
| 75 | Re7+ | 4.36 | 79.000 | 40.311 | 0.782 | 3.412 | 7.506 | 1.278 | 2.326 | 2.807 | 5.874 | |
| 75 | Re6+ | 4.36 | 64.000 | 33.863 | 0.782 | 3.412 | 6.880 | 1.278 | 2.171 | 2.605 | 5.383 | |
| 75 | Re5+ | 4.36 | 51.000 | 27.836 | 0.782 | 3.412 | 6.238 | 1.278 | 2.021 | 2.398 | 4.881 | |
| 75 | Re4+ | 4.36 | 37.700 | 22.045 | 0.782 | 3.412 | 5.551 | 1.278 | 1.846 | 2.177 | 4.344 | |
| 75 | Re3+ | 4.36 | 26.000 | 16.827 | 0.782 | 3.412 | 4.850 | 1.278 | 1.665 | 1.950 | 3.795 | |
| 75 | Re2+ | 4.36 | 16.600 | 12.240 | 0.782 | 3.412 | 4.136 | 1.278 | 1.486 | 1.720 | 3.237 | |
| 75 | Re+ | 4.36 | 7.8800 | 7.8800 | 0.782 | 3.412 | 3.319 | 1.278 | 1.265 | 1.457 | 2.597 | |
| 76 | Os8+ | 4.36 | 99.000 | 49.325 | 0.797 | 3.474 | 8.303 | 1.255 | 2.575 | 3.113 | 6.616 | |
| 76 | Os7+ | 4.36 | 83.000 | 42.230 | 0.797 | 3.474 | 7.683 | 1.255 | 2.423 | 2.909 | 6.122 | |
| 76 | Os6+ | 4.36 | 68.000 | 35.433 | 0.797 | 3.474 | 7.038 | 1.255 | 2.267 | 2.697 | 5.608 | |
| 76 | Os5+ | 4.36 | 54.000 | 28.920 | 0.797 | 3.474 | 6.358 | 1.255 | 2.104 | 2.474 | 5.066 | |
| 76 | Os4+ | 4.36 | 40.000 | 22.650 | 0.797 | 3.474 | 5.627 | 1.255 | 1.919 | 2.234 | 4.483 | |
| 76 | Os3+ | 4.36 | 25.000 | 16.867 | 0.797 | 3.474 | 4.855 | 1.255 | 1.680 | 1.981 | 3.869 | |
| 76 | Os2+ | 4.36 | 16.900 | 12.800 | 0.797 | 3.474 | 4.230 | 1.255 | 1.519 | 1.776 | 3.370 | |
| 76 | Os+ | 4.36 | 8.7000 | 8.7000 | 0.797 | 3.474 | 3.487 | 1.255 | 1.309 | 1.532 | 2.779 | |
| 77 | Ir6+ | 4.36 | 72.000 | 36.683 | 0.794 | 3.460 | 7.161 | 1.260 | 2.298 | 2.728 | 5.683 | |
| 77 | Ir5+ | 4.36 | 57.000 | 29.620 | 0.794 | 3.460 | 6.434 | 1.260 | 2.130 | 2.491 | 5.107 | |
| 77 | Ir4+ | 4.36 | 39.000 | 22.775 | 0.794 | 3.460 | 5.642 | 1.260 | 1.896 | 2.232 | 4.478 | |
| 77 | Ir3+ | 4.36 | 27.000 | 17.367 | 0.794 | 3.460 | 4.927 | 1.260 | 1.708 | 1.998 | 3.910 | |
| 77 | Ir2+ | 4.36 | 16.000 | 12.550 | 0.794 | 3.460 | 4.188 | 1.260 | 1.493 | 1.757 | 3.324 | |
| 77 | Ir+ | 4.36 | 9.1000 | 9.1000 | 0.794 | 3.460 | 3.566 | 1.260 | 1.316 | 1.553 | 2.831 | |
| 78 | Pt6+ | 4.36 | 75.000 | 37.867 | 0.775 | 3.380 | 7.275 | 1.290 | 2.258 | 2.711 | 5.640 | |
| 78 | Pt5+ | 4.36 | 55.000 | 30.440 | 0.775 | 3.380 | 6.523 | 1.290 | 2.045 | 2.470 | 5.056 | |
| 78 | Pt4+ | 4.36 | 41.100 | 24.300 | 0.775 | 3.380 | 5.828 | 1.290 | 1.873 | 2.248 | 4.518 | |
| 78 | Pt3+ | 4.36 | 28.500 | 18.700 | 0.775 | 3.380 | 5.113 | 1.290 | 1.689 | 2.020 | 3.963 | |
| 78 | Pt2+ | 4.36 | 18.600 | 13.800 | 0.775 | 3.380 | 4.392 | 1.290 | 1.513 | 1.790 | 3.405 | |
| 78 | Pt+ | 4.36 | 9.0000 | 9.0000 | 0.775 | 3.380 | 3.547 | 1.290 | 1.289 | 1.520 | 2.749 | |
| 79 | Au5+ | 4.36 | 58.000 | 32.346 | 0.749 | 3.263 | 6.724 | 1.336 | 1.991 | 2.461 | 5.033 | |
| 79 | Au3+ | 4.36 | 30.500 | 20.077 | 0.749 | 3.263 | 5.297 | 1.336 | 1.657 | 2.021 | 3.965 | |
| 79 | Au2+ | 4.36 | 20.500 | 14.865 | 0.749 | 3.263 | 4.558 | 1.336 | 1.498 | 1.793 | 3.412 | |
| 79 | Au+ | 4.36 | 9.2300 | 9.2300 | 0.749 | 3.263 | 3.592 | 1.336 | 1.260 | 1.495 | 2.689 | |
| 80 | Hg2+ | 4.33 | 18.800 | 14.620 | 0.694 | 3.008 | 4.490 | 1.440 | 1.367 | 1.672 | 3.118 | |
| 80 | Hg+ | 4.36 | 10.440 | 10.440 | 0.694 | 3.028 | 3.820 | 1.440 | 1.219 | 1.480 | 2.653 | |
| 81 | Tl+3 | 4.36 | 29.800 | 18.770 | 0.646 | 2.815 | 5.122 | 1.549 | 1.423 | 1.749 | 3.307 | |
| 81 | Tl+ | 4.36 | 6.1100 | 6.1100 | 0.646 | 2.815 | 2.922 | 1.549 | 1.069 | 1.164 | 1.887 | |
| 82 | Pb4+ | 4.36 | 42.300 | 24.180 | 0.650 | 2.835 | 5.814 | 1.538 | 1.558 | 1.944 | 3.780 | |
| 82 | Pb2+ | 4.36 | 15.000 | 11.210 | 0.650 | 2.835 | 3.958 | 1.538 | 1.242 | 1.447 | 2.574 | |
| 83 | Bi5+ | 4.36 | 56.000 | 30.178 | 0.658 | 2.868 | 6.495 | 1.520 | 1.698 | 2.147 | 4.273 | |
| 83 | Bi+3 | 4.36 | 25.600 | 16.530 | 0.658 | 2.868 | 4.807 | 1.520 | 1.399 | 1.690 | 3.162 | |
| 84 | Po6+ | 4.36 | 71.488 | 37.111 | 0.654 | 2.850 | 7.202 | 1.530 | 1.804 | 2.326 | 4.707 | |
| 84 | Po4+ | 4.36 | 37.448 | 22.922 | 0.654 | 2.850 | 5.660 | 1.530 | 0.775 | 1.911 | 3.700 | |
| 84 | Po3+ | 4.36 | 27.745 | 18.072 | 0.654 | 2.850 | 5.026 | 1.530 | 1.416 | 1.740 | 3.285 | |
| 84 | Po2+ | 4.36 | 18.051 | 13.235 | 0.654 | 2.850 | 4.301 | 1.530 | 1.292 | 1.545 | 2.811 | |
| 85 | At7+ | 4.36 | 88.291 | 44.345 | 0.690 | 3.007 | 7.873 | 1.450 | 2.048 | 2.624 | 5.430 | |
| 85 | At5+ | 4.36 | 88.291 | 29.370 | 0.690 | 3.007 | 6.407 | 1.450 | 2.048 | 2.208 | 4.419 | |
| 85 | At3+ | 4.36 | 29.262 | 18.660 | 0.690 | 3.007 | 5.107 | 1.450 | 1.508 | 1.838 | 3.522 | |
| 85 | At+ | 4.36 | 9.2000 | 9.2000 | 0.690 | 3.007 | 3.586 | 1.450 | 1.186 | 1.406 | 2.473 | |
| 86 | Rn8+ | 4.36 | 106.54 | 44.700 | 0.704 | 3.070 | 7.904 | 1.420 | 2.234 | 2.680 | 5.567 | |
| 86 | Rn6+ | 4.36 | 65.490 | 36.730 | 0.704 | 3.070 | 7.165 | 1.420 | 1.919 | 2.466 | 5.046 | |
| 86 | Rn4+ | 4.36 | 41.900 | 25.250 | 0.704 | 3.070 | 5.941 | 1.420 | 1.690 | 2.111 | 4.183 | |
| 87 | Fr+ | 4.36 | 3.9800 | 3.9800 | 0.407 | 1.772 | 2.359 | 2.460 | 0.869 | 0.782 | 0.959 | |
| 88 | Ra2+ | 4.36 | 10.200 | 7.740 | 0.426 | 1.855 | 3.289 | 2.350 | 0.940 | 0.964 | 1.400 | |
| 89 | Ac3+ | 4.36 | 19.692 | 12.321 | 0.504 | 2.199 | 4.150 | 1.983 | 1.097 | 1.249 | 2.093 | |
| 90 | Th4+ | 4.36 | 28.800 | 16.595 | 0.581 | 2.533 | 4.816 | 1.721 | 1.291 | 1.540 | 2.798 | |
| 90 | Th3+ | 4.36 | 20.000 | 12.527 | 0.581 | 2.533 | 4.184 | 1.721 | 1.205 | 1.389 | 2.431 | |
| 91 | Pa4+ | 4.36 | 0.584 | 2.548 | 3.900 | 1.711 | 1.096 | 1.326 | 2.279 | |||
| 92 | U4+ | 4.36 | 0.594 | 2.589 | 3.900 | 1.684 | 1.106 | 1.341 | 2.316 | |||
| 93 | Np4+ | 4.36 | 0.600 | 2.617 | 3.900 | 1.666 | 1.114 | 1.351 | 2.341 | |||
| 94 | Pu4+ | 4.36 | 0.604 | 2.631 | 3.900 | 1.657 | 1.117 | 1.357 | 2.354 | |||
| 95 | Am4+ | 4.36 | 0.602 | 2.627 | 3.900 | 1.660 | 1.116 | 1.355 | 2.349 | |||
| 96 | Cm3+ | 4.36 | 0.555 | 2.421 | 3.900 | 1.801 | 1.065 | 1.279 | 2.165 | |||
| 97 | Bk3+ | 4.36 | 0.568 | 2.476 | 3.900 | 1.761 | 1.078 | 1.299 | 2.215 | |||
| 98 | Cf3+ | 4.36 | 0.571 | 2.491 | 3.900 | 1.750 | 1.082 | 1.305 | 2.229 | |||
| 99 | Es3+ | 4.36 | 0.580 | 2.529 | 3.900 | 1.724 | 1.091 | 1.319 | 2.262 | |||
| 100 | Fm3+ | 4.36 | 0.584 | 2.547 | 3.900 | 1.712 | 1.096 | 1.326 | 2.278 | |||
| 101 | Md3+ | 4.36 | 0.592 | 2.581 | 3.900 | 1.689 | 1.104 | 1.338 | 2.309 | |||
| 102 | No3+ | 4.36 | 0.596 | 2.597 | 3.900 | 1.679 | 1.108 | 1.344 | 2.323 | |||
| 103 | Lw3+ | 4.36 | 1.000 | 4.360 | 3.900 | 1.000 | 1.715 | 1.994 | 3.900 | |||
| Elements | Li | Be | H | B | C | N | O | F |
|---|---|---|---|---|---|---|---|---|
| IC | 1.023 | 2.064 | 2.297 | 3.291 | 4.302 | 5.329 | 6.273 | 7.37 |
| XIC | 0.808 | 1.237 | 1.333 | 1.743 | 2.167 | 2.583 | 2.972 | 3.423 |
| Hydrides | LiH | BeH | BH | CH | NH | OH | FH |
|---|---|---|---|---|---|---|---|
| Bond Length (expr) | 1.595 | 1.343 | 1.233 | 1.12 | 1.038 | 0.971 | 0.917 |
| Dipole Moment (expr) | 5.88 | - | - | 1.46 | - | 1.66 | 1.82 |
| Dipole Moment (calc) | 5.999 | 0.281 | −1.689 | −1.647 | −1.743 | −1.864 | −2.02 |
| qA | 0.783 | 0.044 | −0.285 | −0.306 | −0.350 | −0.400 | −0.459 |
| Bond | H-Na | H-Mg | H-Al | H-Si | H-Se | H-P | H-S | H-Cl |
|---|---|---|---|---|---|---|---|---|
| Bond Length (expr) | 1.887 | 1.73 | 1.648 | 1.52 | 1.475 | 1.422 | 1.341 | 1.275 |
| Dipole Moment (cal) | 5.966 | 1.231 | −0.169 | −0.332 | −0.634 | −0.651 | −1.06 | −1.468 |
| dn | 0 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| E0/V | −2.87 | −1.63 | −1.18 | −0.971 | −1.19 | −0.44 | −0.28 | −0.25 | 0.34 | −0.76 |
| IC | 1.617 | 2.258 | 2.502 | 2.723 | 2.719 | 2.786 | 2.874 | 3.029 | 3.155 | 2.772 |
| Cations | Tl+ | Tl2+ | Tl3+ | Pb2+ | Pb3+ | Pb4+ | Bi3+ | Bi4+ | Bi5+ |
|---|---|---|---|---|---|---|---|---|---|
| Iz | 6.11 | 20.4 | 29.8 | 15 | 32 | 42.3 | 25.6 | 45.3 | 56 |
| Iav | 6.11 | 13.26 | 18.77 | 11.21 | 18.14 | 24.18 | 16.63 | 23.72 | 30.18 |
| XIC | 1.16 | 1.59 | 1.75 | 1.45 | 1.74 | 1.94 | 1.69 | 1.95 | 2.15 |
| IC | 1.89 | 2.92 | 3.31 | 2.58 | 3.44 | 3.78 | 3.16 | 3.81 | 4.27 |
| Cations | Cu3+ | Ag3+ | Au3+ |
|---|---|---|---|
| n* | 3.45 | 3.85 | 4.36 |
| Z* | 4.349 | 4.81 | 5.297 |
| rc | 1.11 | 1.339 | 1.336 |
| rc−1 | 0.901 | 0.747 | 0.749 |
| n*rc−1 | 3.108 | 2.875 | 3.263 |
| Iav | 21.610 | 21.227 | 18.77 |
| XIC | 1.885 | 1.867 | 2.021 |
| IC | 3.918 | 3.592 | 3.965 |
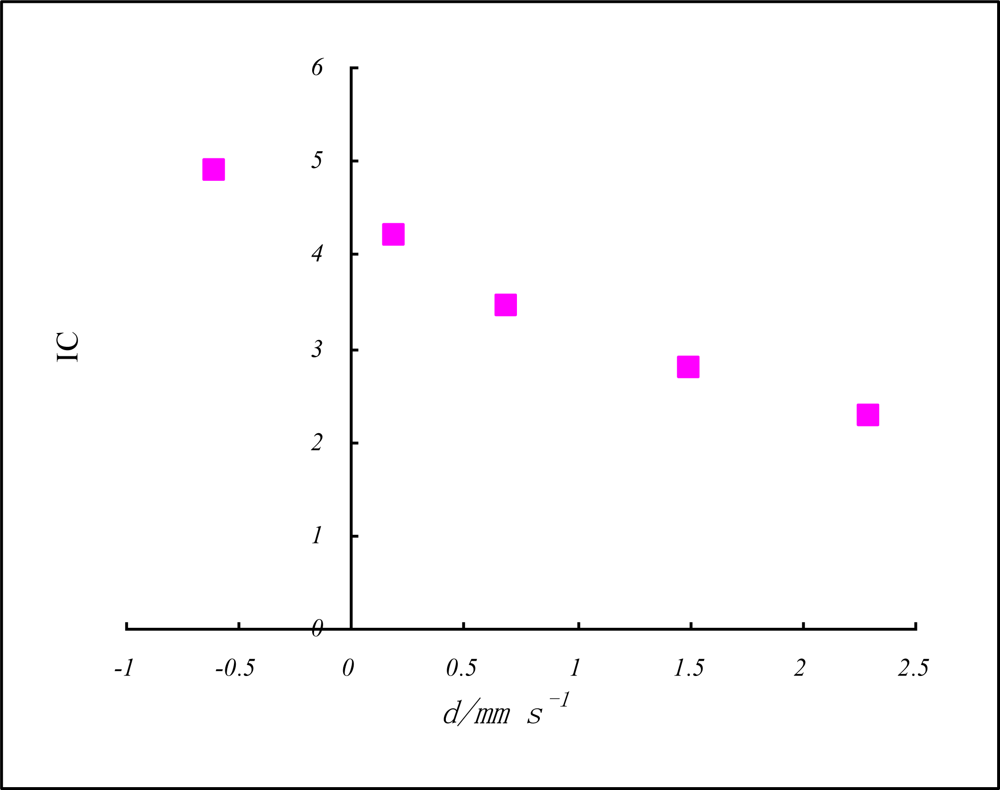
| Iron-57 | FeI | FeII | FeIII | FeIV | FeV |
|---|---|---|---|---|---|
| δ/mm s−1 | 2.3 | 1.5 | 0.7 | 0.2 | −0.6 |
| Z* = n*(Iav/R)½ | 2.624 | 3.245 | 3.997 | 4.896 | 5.684 |
| IC = n*(Iav/R)½rc−1 | 2.253 | 2.786 | 3.431 | 4.203 | 4.879 |
| Compound | Cation | Z* | Iav | rc−1 | n*rc−1 | XIC | IC | Melt.pt (°C) [43] |
|---|---|---|---|---|---|---|---|---|
| KF | K+ | 1.949 | 4.34 | 0.513 | 1.769 | 0.799 | 0.999 | 880⋄ |
| AgF | Ag+ | 2.874 | 7.58 | 0.747 | 2.875 | 1.271 | 2.147 | 435⋄ |
| CaCl2 | Ca+2 | 2.807 | 9.005 | 0.576 | 1.987 | 1.053 | 1.617 | 772⋄ |
| HgCl2 | Hg+2 | 4.49 | 14.82 | 0.694 | 3.008 | 1.672 | 3.118 | 276⋄ |
| CaCl2 | Ca+2 | 2.807 | 9.005 | 0.576 | 1.987 | 1.053 | 1.617 | 772⋄ |
| BeCl2 | Be+2 | 2.002 | 13.76 | 1.125 | 2.238 | 1.315 | 2.252 | 405⋄ |
| NaBr | Na+ | 1.777 | 5.14 | 0.636 | 1.838 | 0.853 | 1.13 | 755⋄ |
| MgBr2 | Mg+2 | 2.637 | 11.33 | 0.733 | 2.119 | 1.184 | 1.933 | 700⋄ |
| AlBr3 | Al+3 | 3.303 | 17.76 | 0.826 | 2.388 | 1.512 | 2.73 | 97.5⋄ |
| KBr | K+ | 1.949 | 4.34 | 0.513 | 1.769 | 0.799 | 0.999 | 730⋄ |
| CaBr2 | Ca+2 | 2.807 | 9.005 | 0.576 | 1.987 | 1.053 | 1.617 | 765⋄ |
| CsF | Cs+ | 2.332 | 3.89 | 0.41 | 1.787 | 0.781 | 0.956 | 684⋄ |
| BaF2 | Ba+2 | 3.26 | 7.605 | 0.505 | 2.201 | 1.065 | 1.646 | 1280⋄ |
| dn | 0 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| U | 2260 | 2500 | 2580 | 2580 | 2550 | 2650 | 2700 | 2790 | 2840 | 2760 |
| IC | 1.617 | 2.258 | 2.502 | 2.723 | 2.719 | 2.786 | 2.874 | 3.029 | 3.155 | 2.772 |
| Mn2+ | Fe2+ | Co2+ | Ni2+ | Cu2+ | Zn2+ | |
|---|---|---|---|---|---|---|
| IC | 2.719 | 2.786 | 2.874 | 3.029 | 3.155 | 2.772 |
| Xic | 1.507 | 1.535 | 1.571 | 1.635 | 1.687 | 1.529 |
| Log β for [M(en)3]2− | 5.7 | 9.5 | 13.8 | 18.6 | 18.7 | 12.1 |
| Log β for [M(EDTA)]2− | 13.8 | 14.3 | 16.3 | 18.6 | 18.7 | 16.1 |
| Compound | Cation | Z* | Iav | rc−1 | n*rc−1 | XIC | IC | Melt.pt (°C) [43] |
|---|---|---|---|---|---|---|---|---|
| KBr | K+ | 1.949 | 4.34 | 0.513 | 1.769 | 0.799 | 0.999 | 730 |
| CaBr2 | Ca+2 | 2.807 | 9.005 | 0.576 | 1.987 | 1.053 | 1.617 | 765 |
| CsF | Cs+ | 2.332 | 3.89 | 0.41 | 1.787 | 0.781 | 0.956 | 684 |
| BaF2 | Ba+2 | 3.26 | 7.605 | 0.505 | 2.201 | 1.065 | 1.646 | 1280 |
| Compound | Cation | Z* | Iav | rc−1 | n*rc−1 | XIC | IC | U (kJmol−1) [44] |
|---|---|---|---|---|---|---|---|---|
| LiH | Li+ | 1.253 | 5.39 | 0.816 | 2.238 | 0.808 | 1.023 | 905.4 |
| NaH | Na+ | 1.777 | 5.14 | 0.636 | 1.838 | 0.853 | 1.13 | 810.9 |
| KH | K+ | 1.949 | 4.34 | 0.513 | 1.769 | 0.799 | 0.999 | 714.2 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, Y. Ionocovalency and Applications 1. Ionocovalency Model and Orbital Hybrid Scales. Int. J. Mol. Sci. 2010, 11, 4381-4406. https://doi.org/10.3390/ijms11114381
Zhang Y. Ionocovalency and Applications 1. Ionocovalency Model and Orbital Hybrid Scales. International Journal of Molecular Sciences. 2010; 11(11):4381-4406. https://doi.org/10.3390/ijms11114381
Chicago/Turabian StyleZhang, Yonghe. 2010. "Ionocovalency and Applications 1. Ionocovalency Model and Orbital Hybrid Scales" International Journal of Molecular Sciences 11, no. 11: 4381-4406. https://doi.org/10.3390/ijms11114381
APA StyleZhang, Y. (2010). Ionocovalency and Applications 1. Ionocovalency Model and Orbital Hybrid Scales. International Journal of Molecular Sciences, 11(11), 4381-4406. https://doi.org/10.3390/ijms11114381