Lipid Based Therapy for Ulcerative Colitis—Modulation of Intestinal Mucus Membrane Phospholipids as a Tool to Influence Inflammation

{kind=link}
Abstract
:1. Introduction
2. Concept of Ulcerative Colitis
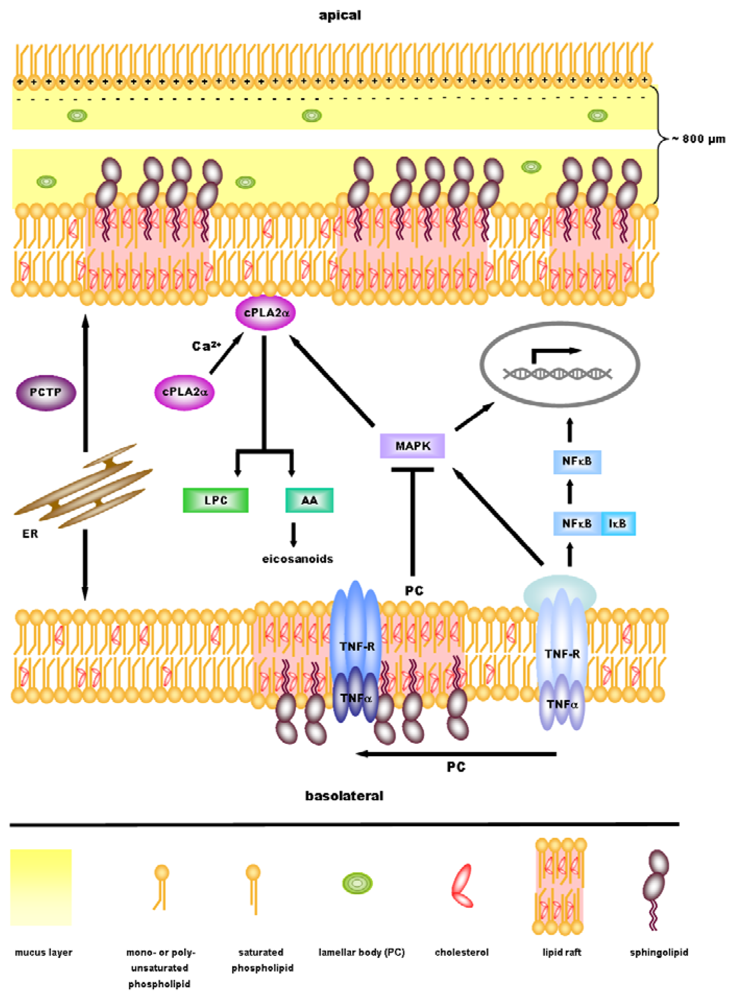
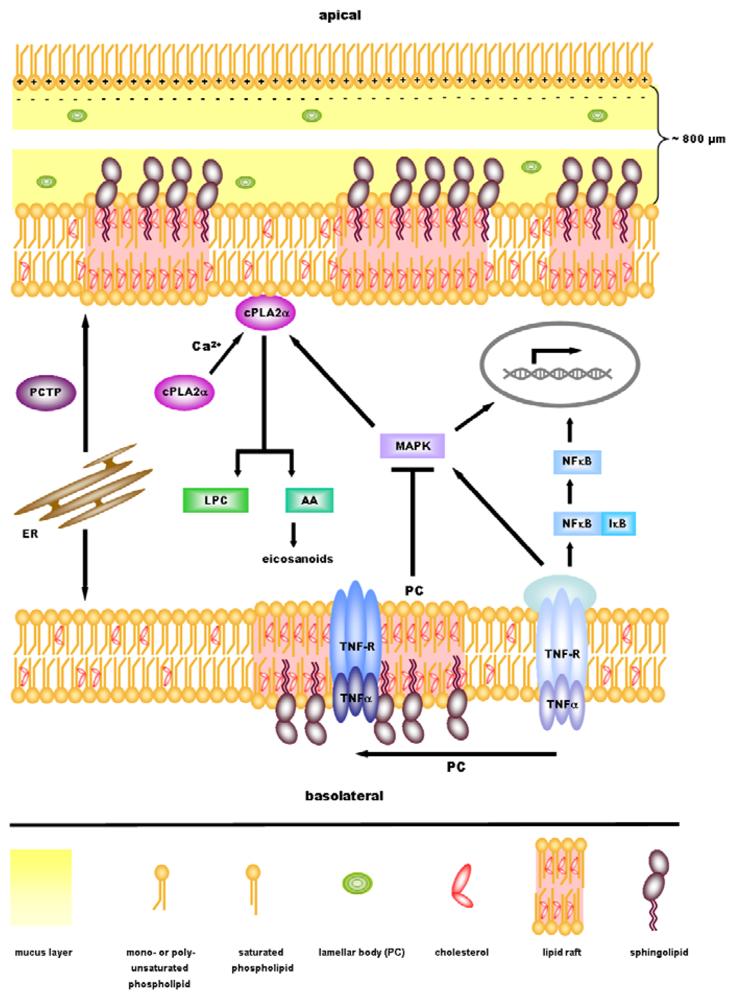
3. Role of PC in Ulcerative Colitis
4. Anti-Inflammatory Signaling by PC
5. cPLA2α—A Key Regulator in Inflammatory Signaling Downstream of PC?
6. Perspectives
Acknowledgements
References
- Best, CH; Hershey, JM; Huntsman, ME. The effect of lecithine on fat deposition in the liver of the normal rat. J. Physiol 1932, 75, 56–66. [Google Scholar]
- Eros, G; Ibrahim, S; Siebert, N; Boros, M; Vollmar, B. Oral phosphatidylcholine pretreatment alleviates the signs of experimental rheumatoid arthritis. Arthritis Res. Ther 2009, 11, R43. [Google Scholar]
- Eros, G; Varga, G; Varadi, R; Czobel, M; Kaszaki, J; Ghyczy, M; Boros, M. Anti-inflammatory action of a phosphatidylcholine, phosphatidylethanolamine and N-acylphosphatidylethanolamine-enriched diet in carrageenan-induced pleurisy. Eur. Surg. Res 2009, 42, 40–48. [Google Scholar]
- Lichtenberger, LM; Graziani, LA; Dial, EJ; Butler, BD; Hills, BA. Role of surface-active phospholipids in gastric cytoprotection. Science 1983, 219, 1327–1329. [Google Scholar]
- Lichtenberger, LM. The hydrophobic barrier properties of gastrointestinal mucus. Annu. Rev. Physiol 1995, 57, 565–583. [Google Scholar]
- Zhou, Y; Dial, EJ; Doyen, R; Lichtenberger, LM. Effect of indomethacin on bile acid-phospholipid interactions: Implication for small intestinal injury induced by nonsteroidal anti-inflammatory drugs. Am. J. Physiol. Gastrointest. Liver Physiol 2010, 298, G722–G731. [Google Scholar]
- Lugea, A; Antolin, M; Mourelle, M; Guarner, F; Malagelada, JR. Deranged hydrophobic barrier of the rat gastroduodenal mucosa after parenteral nonsteroidal anti-inflammatory drugs. Gastroenterology 1997, 112, 1931–1939. [Google Scholar]
- Lichtenberger, LM; Romero, JJ; Dial, EJ. Gastrointestinal safety and therapeutic efficacy of parenterally administered phosphatidylcholine-associated indomethacin in rodent model systems. Br. J. Pharmacol 2009, 157, 252–257. [Google Scholar]
- Lanza, FL; Marathi, UK; Anand, BS; Lichtenberger, LM. Clinical trial: Comparison of ibuprofen-phosphatidylcholine and ibuprofen on the gastrointestinal safety and analgesic efficacy in osteoarthritic patients. Aliment Pharmacol. Ther 2008, 28, 431–442. [Google Scholar]
- Cryer, B; Bhatt, DL; Lanza, FL; Dong, J-F; Lichtenberger, LM; Marathi, UK. Reduction of gastroduodenal ulceration with aspirin-phosphatidylcholine complex versus aspirin—potential importance of local mucosal injury. Gastroenterology 2010, 138, S497. [Google Scholar]
- Podolsky, DK. Inflammatory bowel disease. N. Engl. J. Med 2002, 347, 417–429. [Google Scholar]
- Sartor, RB. Therapeutic manipulation of the enteric microflora in inflammatory bowel diseases: Antibiotics, probiotics, and prebiotics. Gastroenterology 2004, 126, 1620–1633. [Google Scholar]
- Backhed, F; Ley, RE; Sonnenburg, JL; Peterson, DA; Gordon, JI. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar]
- Rutgeerts, P; Goboes, K; Peeters, M; Hiele, M; Penninckx, F; Aerts, R; Kerremans, R; Vantrappen, G. Effect of faecal stream diversion on recurrence of Crohn's disease in the neoterminal ileum. Lancet 1991, 338, 771–774. [Google Scholar]
- Duchmann, R; May, E; Heike, M; Knolle, P; Neurath, M; Meyer zum Buschenfelde, KH. T cell specificity and cross reactivity towards enterobacteria, bacteroides, bifidobacterium, and antigens from resident intestinal flora in humans. Gut 1999, 44, 812–818. [Google Scholar]
- Swidsinski, A; Ladhoff, A; Pernthaler, A; Swidsinski, S; Loening-Baucke, V; Ortner, M; Weber, J; Hoffmann, U; Schreiber, S; Dietel, M; Lochs, H. Mucosal flora in inflammatory bowel disease. Gastroenterology 2002, 122, 44–54. [Google Scholar]
- Wen, Z; Fiocchi, C. Inflammatory bowel disease: Autoimmune or immune-mediated pathogenesis? Clin. Dev. Immunol 2004, 11, 195–204. [Google Scholar]
- Pullan, RD; Thomas, GA; Rhodes, M; Newcombe, RG; Williams, GT; Allen, A; Rhodes, J. Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut 1994, 35, 353–359. [Google Scholar]
- Einerhand, AW; Renes, IB; Makkink, MK; van der Sluis, M; Buller, HA; Dekker, J. Role of mucins in inflammatory bowel disease: Important lessons from experimental models. Eur. J. Gastroenterol. Hepatol 2002, 14, 757–765. [Google Scholar]
- Ehehalt, R; Wagenblast, J; Erben, G; Lehmann, WD; Hinz, U; Merle, U; Stremmel, W. Phosphatidylcholine and lysophosphatidylcholine in intestinal mucus of ulcerative colitis patients. A quantitative approach by nanoElectrospray-tandem mass spectrometry. Scand. J. Gastroenterol 2004, 39, 737–742. [Google Scholar]
- Braun, A; Treede, I; Gotthardt, D; Tietje, A; Zahn, A; Ruhwald, R; Schoenfeld, U; Welsch, T; Kienle, P; Erben, G; Lehmann, WD; Fuellekrug, J; Stremmel, W; Ehehalt, R. Alterations of phospholipid concentration and species composition of the intestinal mucus barrier in ulcerative colitis: A clue to pathogenesis. Inflamm. Bowel. Dis 2009, 15, 1705–1720. [Google Scholar]
- Longman, RJ; Poulsom, R; Corfield, AP; Warren, BF; Wright, NA; Thomas, MG. Alterations in the composition of the supramucosal defense barrier in relation to disease severity of ulcerative colitis. J. Histochem. Cytochem 2006, 54, 1335–1348. [Google Scholar]
- Wehkamp, J; Schmid, M; Stange, EF. Defensins and other antimicrobial peptides in inflammatory bowel disease. Curr. Opin. Gastroenterol 2007, 23, 370–378. [Google Scholar]
- Schulzke, JD; Ploeger, S; Amasheh, M; Fromm, A; Zeissig, S; Troeger, H; Richter, J; Bojarski, C; Schumann, M; Fromm, M. Epithelial tight junctions in intestinal inflammation. Ann. N.Y. Acad. Sci 2009, 1165, 294–300. [Google Scholar]
- Langmann, T; Moehle, C; Mauerer, R; Scharl, M; Liebisch, G; Zahn, A; Stremmel, W; Schmitz, G. Loss of detoxification in inflammatory bowel disease: Dysregulation of pregnane X receptor target genes. Gastroenterology 2004, 127, 26–40. [Google Scholar]
- Macfarlane, GT; Blackett, KL; Nakayama, T; Steed, H; Macfarlane, S. The gut microbiota in inflammatory bowel disease. Curr. Pharm. Des 2009, 15, 1528–1536. [Google Scholar]
- Fabia, R; Ar'Rajab, A; Willen, R; Andersson, R; Ahren, B; Larsson, K; Bengmark, S. Effects of phosphatidylcholine and phosphatidylinositol on acetic-acid-induced colitis in the rat. Digestion 1992, 53, 35–44. [Google Scholar]
- Fabia, R; Ar'Rajab, A; Willen, R; Andersson, R; Bengmark, S. Effect of putative phospholipase A2 inhibitors on acetic acid-induced acute colitis in the rat. Br. J. Surg 1993, 80, 1199–1204. [Google Scholar]
- Tatsumi, Y; Lichtenberger, LM. Molecular association of trinitrobenzenesulfonic acid and surface phospholipids in the development of colitis in rats. Gastroenterology 1996, 110, 780–789. [Google Scholar]
- Mourelle, M; Guarner, F; Malagelada, JR. Polyunsaturated phosphatidylcholine prevents stricture formation in a rat model of colitis. Gastroenterology 1996, 110, 1093–1097. [Google Scholar]
- Barrios, JM; Lichtenberger, LM. Role of biliary phosphatidylcholine in bile acid protection and NSAID injury of the ileal mucosa in rats. Gastroenterology 2000, 118, 1179–1186. [Google Scholar]
- Lugea, A; Salas, A; Casalot, J; Guarner, F; Malagelada, JR. Surface hydrophobicity of the rat colonic mucosa is a defensive barrier against macromolecules and toxins. Gut 2000, 46, 515–521. [Google Scholar]
- Sturm, A; Zeeh, J; Sudermann, T; Rath, H; Gerken, G; Dignass, AU. Lisofylline and lysophospholipids ameliorate experimental colitis in rats. Digestion 2002, 66, 23–29. [Google Scholar]
- Treede, I; Braun, A; Sparla, R; Kuhnel, M; Giese, T; Turner, JR; Anes, E; Kulaksiz, H; Fullekrug, J; Stremmel, W; Griffiths, G; Ehehalt, R. Anti-inflammatory effects of phosphatidylcholine. J. Biol. Chem 2007, 282, 27155–27164. [Google Scholar]
- Anes, E; Kuhnel, MP; Bos, E; Moniz-Pereira, J; Habermann, A; Griffiths, G. Selected lipids activate phagosome actin assembly and maturation resulting in killing of pathogenic mycobacteria. Nat. Cell Biol 2003, 5, 793–802. [Google Scholar]
- Gutierrez, MG; Gonzalez, AP; Anes, E; Griffiths, G. Role of lipids in killing mycobacteria by macrophages: Evidence for NF-kappaB-dependent and -independent killing induced by different lipids. Cell Microbiol 2009, 11, 406–420. [Google Scholar]
- Stremmel, W; Merle, U; Zahn, A; Autschbach, F; Hinz, U; Ehehalt, R. Retarded release phosphatidylcholine benefits patients with chronic active ulcerative colitis. Gut 2005, 54, 966–971. [Google Scholar]
- Tromm, A; Griga, T; May, B. Oral mesalazine for the treatment of Crohn's disease: Clinical efficacy with respect to pharmacokinetic properties. Hepatogastroenterology 1999, 46, 3124–3135. [Google Scholar]
- Stremmel, W; Braun, A; Hanemann, A; Ehehalt, R; Autschbach, F; Karner, M. Delayed release phosphatidylcholine in chronic-active ulcerative colitis: A randomized, double-blinded, dose finding study. J. Clin. Gastroenterol 2010, 44, e101–107. [Google Scholar]
- Stremmel, W; Ehehalt, R; Autschbach, F; Karner, M. Phosphatidylcholine for steroid-refractory chronic ulcerative colitis: A randomized trial. Ann. Int. Med 2007, 147, 603–610. [Google Scholar]
- Parlesak, A; Schaeckeler, S; Moser, L; Bode, C. Conjugated primary bile salts reduce permeability of endotoxin through intestinal epithelial cells and synergize with phosphatidylcholine in suppression of inflammatory cytokine production. Crit. Care Med 2007, 35, 2367–2374. [Google Scholar]
- Dial, EJ; Tran, DM; Romero, JJ; Zayat, M; Lichtenberger, LM. A direct role for secretory phospholipase A2 and lyso-phosphatidylcholine in the mediation of lipopolysaccharide-induced gastric injury. Shock 2010, 33, 634–638. [Google Scholar]
- Hills, BA. Surface-active phospholipid: A Pandora's box of clinical applications. Part II. Barrier and lubricating properties. Int. Med. J 2002, 32, 242–251. [Google Scholar]
- Braun, A; Schoenfeld, U; Welsch, T; Kadmon, M; Funke, B; Autschbach, F; Grunze, M; Stremmel, W; Kienle, P; Ehehalt, R. The surface hydrophobicity of the colonic mucosa is reduced in ulcerative colitis. J. Crohn's Colitis 2010, 4, S13–S120. [Google Scholar]
- Hurley, JH; Tsujishita, Y; Pearson, MA. Floundering about at cell membranes: A structural view of phospholipid signaling. Curr. Opin. Struct. Biol 2000, 10, 737–743. [Google Scholar]
- Alpy, F; Tomasetto, C. Give lipids a START: The StAR-related lipid transfer (START) domain in mammals. J. Cell Sci 2005, 118, 2791–2801. [Google Scholar]
- Medina-Gomez, G; Gray, SL; Yetukuri, L; Shimomura, K; Virtue, S; Campbell, M; Curtis, RK; Jimenez-Linan, M; Blount, M; Yeo, GS; Lopez, M; Seppanen-Laakso, T; Ashcroft, FM; Oresic, M; Vidal-Puig, A. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet 2007, 3, e64. [Google Scholar]
- Chakravarthy, MV; Lodhi, IJ; Yin, L; Malapaka, RR; Xu, HE; Turk, J; Semenkovich, CF. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell 2009, 138, 476–488. [Google Scholar]
- Chen, X; Xun, K; Chen, L; Wang, Y. TNF-alpha, a potent lipid metabolism regulator. Cell Biochem. Funct 2009, 27, 407–416. [Google Scholar]
- Targan, SR; Hanauer, SB; van Deventer, SJ; Mayer, L; Present, DH; Braakman, T; DeWoody, KL; Schaible, TF; Rutgeerts, PJ. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N. Engl. J. Med 1997, 337, 1029–1035. [Google Scholar]
- van Deventer, SJ. Tumour necrosis factor and Crohn's disease. Gut 1997, 40, 443–448. [Google Scholar]
- Rutgeerts, P; Sandborn, WJ; Feagan, BG; Reinisch, W; Olson, A; Johanns, J; Travers, S; Rachmilewitz, D; Hanauer, SB; Lichtenstein, GR; de Villiers, WJ; Present, D; Sands, BE; Colombel, JF. Infliximab for induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med 2005, 353, 2462–2476. [Google Scholar]
- Hueber, AO. Role of membrane microdomain rafts in TNFR-mediated signal transduction. Cell Death Differ 2003, 10, 7–9. [Google Scholar]
- Simons, K; Ehehalt, R. Cholesterol, lipid rafts, and disease. J. Clin. Invest 2002, 110, 597–603. [Google Scholar]
- Lingwood, D; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar]
- Triantafilou, M; Morath, S; Mackie, A; Hartung, T; Triantafilou, K. Lateral diffusion of Toll-like receptors reveals that they are transiently confined within lipid rafts on the plasma membrane. J. Cell Sci 2004, 117, 4007–4014. [Google Scholar]
- Legler, DF; Micheau, O; Doucey, MA; Tschopp, J; Bron, C. Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity 2003, 18, 655–664. [Google Scholar]
- Cottin, V; Doan, JE; Riches, DW. Restricted localization of the TNF receptor CD120a to lipid rafts: A novel role for the death domain. J. Immunol 2002, 168, 4095–4102. [Google Scholar]
- Treede, I; Braun, A; Jeliaskova, P; Giese, T; Fullekrug, J; Griffiths, G; Stremmel, W; Ehehalt, R. TNF-alpha-induced up-regulation of pro-inflammatory cytokines is reduced by phosphatidylcholine in intestinal epithelial cells. BMC Gastroenterol 2009, 9, 53. [Google Scholar]
- Nishida, T; Miwa, H; Shigematsu, A; Yamamoto, M; Iida, M; Fujishima, M. Increased arachidonic acid composition of phospholipids in colonic mucosa from patients with active ulcerative colitis. Gut 1987, 28, 1002–1007. [Google Scholar]
- Ehehalt, R; Braun, A; Karner, M; Fullekrug, J; Stremmel, W. Phosphatidylcholine as a constituent in the colonic mucosal barrier--physiological and clinical relevance. Biochim. Biophys. Acta 2010, 1801, 983–993. [Google Scholar]
- Clark, JD; Lin, LL; Kriz, RW; Ramesha, CS; Sultzman, LA; Lin, AY; Milona, N; Knopf, JL. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca2+-dependent translocation domain with homology to PKC and GAP. Cell 1991, 65, 1043–1051. [Google Scholar]
- Hirabayashi, T; Murayama, T; Shimizu, T. Regulatory mechanism and physiological role of cytosolic phospholipase A2. Biol. Pharm. Bull 2004, 27, 1168–1173. [Google Scholar]
- Yu, W; Bozza, PT; Tzizik, DM; Gray, JP; Cassara, J; Dvorak, AM; Weller, PF. Co-compartmentalization of MAP kinases and cytosolic phospholipase A2 at cytoplasmic arachidonate-rich lipid bodies. Am. J. Pathol 1998, 152, 759–769. [Google Scholar]
- Gentile, LB; Piva, B; Capizzani, BC; Furlaneto, LG; Moreira, LS; Zamith-Miranda, D; Diaz, BL. Hypertonic environment elicits cyclooxygenase-2-driven prostaglandin E2 generation by colon cancer cells: Role of cytosolic phospholipase A2-alpha and kinase signaling pathways. Prostagl. Leukot Essent Fat. Ac 2010, 82, 131–139. [Google Scholar]
- Takaku, K; Sonoshita, M; Sasaki, N; Uozumi, N; Doi, Y; Shimizu, T; Taketo, MM. Suppression of intestinal polyposis in Apc(delta 716) knockout mice by an additional mutation in the cytosolic phospholipase A(2) gene. J. Biol. Chem 2000, 275, 34013–34016. [Google Scholar]
- Krimsky, M; Yedgar, S; Aptekar, L; Schwob, O; Goshen, G; Gruzman, A; Sasson, S; Ligumsky, M. Amelioration of TNBS-induced colon inflammation in rats by phospholipase A2 inhibitor. Am. J. Physiol. Gastrointest. Liver Physiol 2003, 285, G586–592. [Google Scholar]
- Adler, DH; Cogan, JD; Phillips, JA, 3rd; Schnetz-Boutaud, N; Milne, GL; Iverson, T; Stein, JA; Brenner, DA; Morrow, JD; Boutaud, O; Oates, JA. Inherited human cPLA(2alpha) deficiency is associated with impaired eicosanoid biosynthesis, small intestinal ulceration, and platelet dysfunction. J. Clin. Invest 2008, 118, 2121–2131. [Google Scholar]
- Yedgar, S; Cohen, Y; Shoseyov, D. Control of phospholipase A2 activities for the treatment of inflammatory conditions. Biochim. Biophys. Acta 2006, 1761, 1373–1382. [Google Scholar]
- Sawai, T; Lampman, R; Hua, Y; Segura, B; Drongowski, RA; Coran, AG; Harmon, CM. Lysophosphatidylcholine alters enterocyte monolayer permeability via a protein kinase C/Ca2+ mechanism. Pediatr. Surg. Int 2002, 18, 591–594. [Google Scholar]
- Tries, S; Neupert, W; Laufer, S. The mechanism of action of the new antiinflammatory compound ML3000: Inhibition of 5-LOX and COX-1/2. Inflamm. Res 2002, 51, 135–143. [Google Scholar]
- Panel, V; Boelle, PY; Ayala-Sanmartin, J; Jouniaux, AM; Hamelin, R; Masliah, J; Trugnan, G; Flejou, JF; Wendum, D. Cytoplasmic phospholipase A2 expression in human colon adenocarcinoma is correlated with cyclooxygenase-2 expression and contributes to prostaglandin E2 production. Cancer Lett 2006, 243, 255–263. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schneider, H.; Braun, A.; Füllekrug, J.; Stremmel, W.; Ehehalt, R. Lipid Based Therapy for Ulcerative Colitis—Modulation of Intestinal Mucus Membrane Phospholipids as a Tool to Influence Inflammation. Int. J. Mol. Sci. 2010, 11, 4149-4164. https://doi.org/10.3390/ijms11104149
Schneider H, Braun A, Füllekrug J, Stremmel W, Ehehalt R. Lipid Based Therapy for Ulcerative Colitis—Modulation of Intestinal Mucus Membrane Phospholipids as a Tool to Influence Inflammation. International Journal of Molecular Sciences. 2010; 11(10):4149-4164. https://doi.org/10.3390/ijms11104149
Chicago/Turabian StyleSchneider, Hannah, Annika Braun, Joachim Füllekrug, Wolfgang Stremmel, and Robert Ehehalt. 2010. "Lipid Based Therapy for Ulcerative Colitis—Modulation of Intestinal Mucus Membrane Phospholipids as a Tool to Influence Inflammation" International Journal of Molecular Sciences 11, no. 10: 4149-4164. https://doi.org/10.3390/ijms11104149