Phylogenetics Applied to Genotype/Phenotype Association and Selection Analyses with Sequence Data from Angptl4 in Humans

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Phylogenetic and Treescanning Results
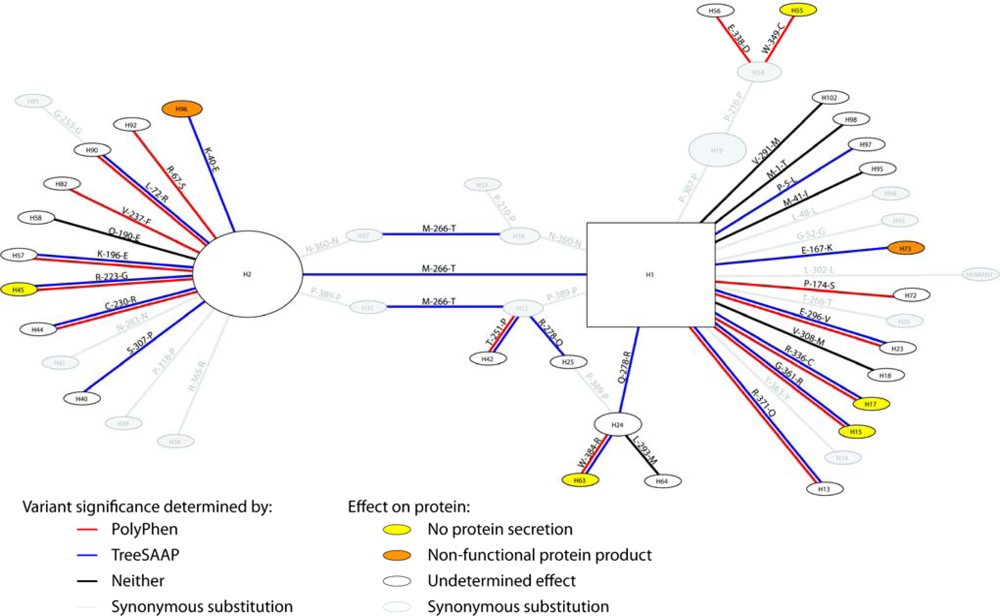
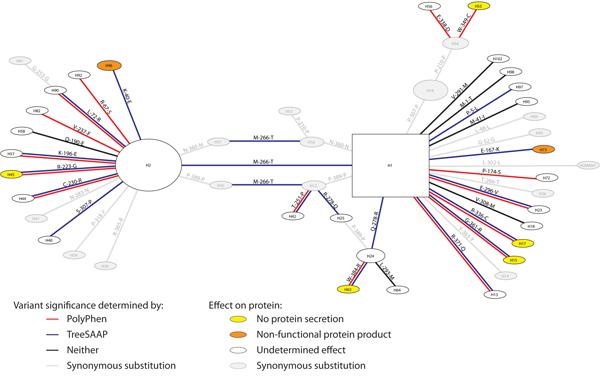
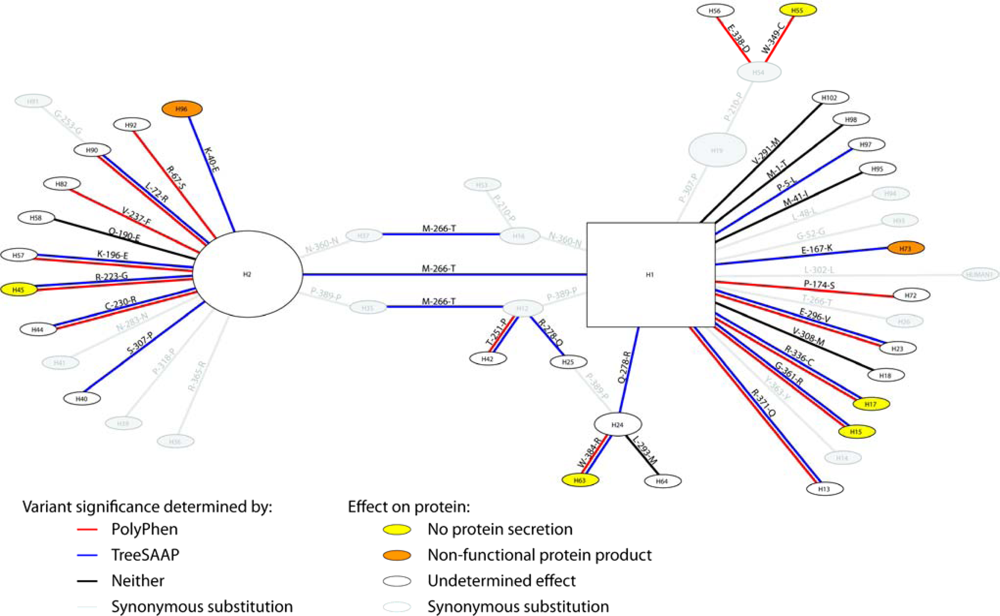
2.1.1. Variants, Haplotypes, Networks, and Phylogenetic Trees
2.1.2. Treescanning Results
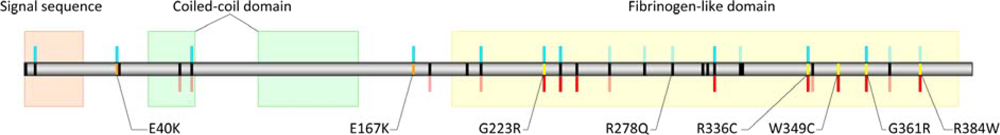
2.2. Bioinformatics and Site Prediction Analysis Results
2.2.1. PolyPhen and TreeSAAP Results
2.2.2. PAML and HyPhy results
2.3. Comparison of PolyPhen and TreeSAAP
3. Materials and Methods
3.1. Study Description and Genetic Data
3.2. Haplotype Networks and Phylogenetic Trees
3.3. Genotype/Phenotype Association via Treescan
3.4. Bioinformatics, Site Prediction, and Selection Analyses
3.4.1. PolyPhen Analysis
3.4.2. TreeSAAP Analysis
3.4.3. Likelihood Selection Analysis
3.5. Comparison of TreeSAAP and PolyPhen
4. Conclusions
Acknowledgments
References and Notes
- Templeton, AR. Population Genetics and Microevolutionary Theory; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Page, RDM; Holmes, EC. Molecular Evolution: A Phylogenetic Approach; Blackwell Science Ltd: Osney Mead, Oxford, UK, 1998. [Google Scholar]
- Romeo, S; Pennancchio, LA; Fu, Y-X; Boerwinkle, E; Tybjaerg-Hansen, A; Hobbs, HH; Cohen, JC. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat. Genet 2007, 39, 513–516. [Google Scholar]
- Romeo, S; Yin, W; Kozlitina, J; Pennacchio, LA; Boerwinkle, E; Hobbs, HH; Cohen, JC. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J. Clin. Invest 2009, 119, 70–79. [Google Scholar]
- Victor, RG; Haley, RW; Willett, DL; Peshock, MD; Vaith, PC; Leonard, D; Basit, M; Cooper, RS; Iannacchione, VG; Visscher, WA; Staab, JM; Hobbs, HH; Dallas Heart Study Investigators. The Dallas heart study: A population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health. Am. J. Cardiol 2004, 93, 1473–1480. [Google Scholar]
- Ramensky, V; Bork, P; Sunyaev, S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res 2002, 30, 3894–4900. [Google Scholar]
- Woolley, S; Johnson, J; Smith, MJ; Crandall, KA; McClellan, DA. TreeSAAP: Selection on amino acid properties using phylogenetic trees. Bioinformatics 2003, 19, 671–672. [Google Scholar]
- Templeton, AR; Maxwell, T; Posada, D; Stengård, JH; Boerwinkle, E; Sing, CF. Tree scanning: A method for using haplotype trees in phenotype/genotype association studies. Genetics 2005, 169, 441–453. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Bio. Evol 2007, 24, 1586–1591. [Google Scholar]
- Kosakovsky Pond, SL; Frost, SD; Muse, SV. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar]
- Templeton, AR; Crandall, KA; Sing, CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 1992, 132, 619–633. [Google Scholar]
- Talmud, PJ; Smart, M; Presswood, E; Cooper, JA; Nicaud, V; Drenos, F; Palmen, J; Marmot, MG; Boekholdt, SM; Wareham, NJ; Khaw, K; Kumari, M; Humphries, SE; On behalf of the EARSII Consortium and the HIFMECH Consortium. ANGPTL4 E40K and T266M: Effects on Plasma Triglyceride and HDL Levels; Postprandial Responses; and CHD Risk. Arterioscler. Thromb. Vasc. Biol 2008, 28, 2319–2325. [Google Scholar]
- McClellan, DA; Palfreyman, EJ; Smith, MJ; Moss, JL; Christensen, RG; Sailsbery, JK. Physicochemical evolution and molecular adaption of the cetacean and artiodactyls cytochrome b proteins. Mol. Bio. Evol 2005, 22, 437–455. [Google Scholar]
- Pérez-Losada, M; Viscidi, RP; Demma, JC; Zenilman, J; Crandall, KA. Population genetics of Neisseria gonorrhoeae in a high-prevalence community using a hypervariable outer membrane porB and 13 slowly evolving housekeeping genes. Mol. Biol. Evol 2005, 22, 1887–1902. [Google Scholar]
- Crandall, KA; Kelsey, CR; Imamichi, H; Lane, HC; Salzman, NP. Parallel evolution of drug resistance in HIV: Failure of nonsynonymous/synonymous substitution rate ratio to detect selection. Mol. Bio. Evol 1999, 16, 372–382. [Google Scholar]
- Tartaglia, M; Pennacchio, LA; Zhao, C; Yadav, KK; Fodale, V; Sarkozy, A; Pandit, B; Oishi, K; Martinelli, S; Schackwitz, W; Ustaszewska, A; Martin, J; Bristow, J; Carta, C; Lepri, F; Neri, C; Vasta, I; Gibson, K; Curry, CJ; Siguero, JPL; Digilio, MC; Zampino, G; Dallapiccola, B; Bar-Sagi, D; Gelb, BD. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat. Genet 2007, 39, 75–79. [Google Scholar]
- Stephens, M; Donnelly, P. Inference in molecular population genetics. J. R. Stat. Soc. Ser. B 2000, 62, 605–655. [Google Scholar]
- Stephens, M; Donnelly, P. A comparison of Bayesian methods for haplotypes reconstruction. Am. J. Hum. Genet 2003, 73, 1162–1169. [Google Scholar]
- Clement, M; Posada, D; Crandall, KA. TCS: A computer program to estimate gene genealogies. Mol. Ecol 2000, 9, 1657–1659. [Google Scholar]
- Crandall, KA. Templeton A.R. Statistical methods for detecting recombination. In The Evolution of HIV; Crandall, KA, Ed.; The Johns Hopkins University Press: Baltimore, MD, USA, 1999; pp. 153–176. [Google Scholar]
- Castelloe, J; Templeton, AR. Root probabilities for intraspecific gene trees under neutral coalescent theory. Mol. Phylogenet. Evol 1994, 3, 102–113. [Google Scholar]
- Crandall, KA; Templeton, AR. Empirical tests of some predictions from coalescent theory with applications to intraspecific phylogeny reconstruction. Genetics 1993, 134, 959–969. [Google Scholar]
- Swofford, DL. PAUP*. In Phylogenetic Analysis Using Parsimony (*and Other Methods); Version 4Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Posada, D; Crandall, KA. Modeltest: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar]
- Hasegawa, M; Kishino, H; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol 1985, 22, 160–174. [Google Scholar]
- Guindon, S; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol 2003, 52, 696–704. [Google Scholar]
- Nowotny, P; Hinrichs, AL; Smemo, S; Kauwe, JSK; Maxwell, T; Holmans, P; Hamshere, M; Turic, D; Jehu, L; Hollingsworth, P; Moore, L; Bryden, P; Myers, A; Doil, LM; Tacey, KM; Gibson, AM; McKeith, IG; Perry, RH; Morris, CM; Thal, L; Morris, JC; O’Donovan, MC; Lovestone, S; Grupe, A; Hardy, J; Owen, MJ; Williams, J; Goate, A. Association studies between risk for late-onset alzheimer’s disease (LOAD) and variants in Insulin Degrading Enzyme. Am. J. Med. Genet. B 2005, 136B, 62–68. [Google Scholar]
- Grupe, A; Li, Y; Rowland, C; Nowotny, P; Hinrichs, AL; Smemo, S; Kauwe, JSK; Maxwell, TJ; Cherny, S; Doil, L; Tacey, K; van Luchene, R; Myers, A; Vriexe, FW; Kaleem, M; Hollingworth, P; Jehu, L; Foy, C; Archer, N; Hamilton, G; Homans, P; Morris, CM; Catanese, J; Sninsky, J; White, TJ; Powell, J; Hardy, J; O’Donovan, M; Lovestone, S; Jones, L; Morris, JC; Thal, L; Owen, M; Williams, J; Goate, A. A scan of chromosome 10 identifies a novel locus showing strong association with Late-Onset alzheimer disease. Am. J. Hum. Genet 2006, 78, 78–88. [Google Scholar]
- Westfall, P; Young, SS. Resampling-Based Multiple Testing: Examples and Methods for p-Value Adjustments; Wiley-Interscience: New York, NY, USA, 1993. [Google Scholar]
- Rencher, AC. Methods of Multivariate Analysis; Wiley: New York, NY, USA, 1995; p. 316. [Google Scholar]
- McClellan, DA; McCracken, KG. Estimating the influence of selection on the variable amino acid sites of the cytochrome B protein functional domains. Mol. Biol. Evol 2001, 18, 917–925. [Google Scholar]
- Goldman, N; Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol 1994, 11, 725–736. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
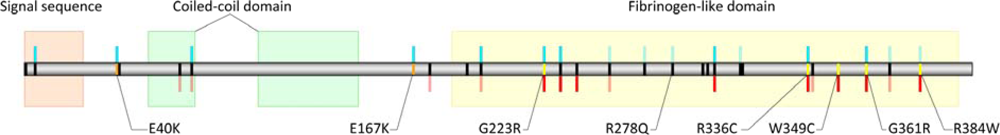{kind=link}
| Haplotype | All | EA | AA | MA | Other |
|---|---|---|---|---|---|
| h1 | 0.51242 | 0.54341 | 0.47735 | 0.54052 | 0.68493 |
| h2 | 0.26020 | 0.28447 | 0.21706 | 0.34828 | 0.23288 |
| h12 | 0.06475 | 0.14402 | 0.02102 | 0.05862 | 0.06164 |
| h13 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h14 | 0.00015 | 0.00051 | 0 | 0 | 0 |
| h15 | 0.00030 | 0.00102 | 0 | 0 | 0 |
| h16 | 0.04186 | 0.00153 | 0.07936 | 0.00517 | 0.00685 |
| h17 | 0.00105 | 0.00255 | 0.00030 | 0.00086 | 0 |
| h18 | 0.00045 | 0.00102 | 0 | 0.00086 | 0 |
| h19 | 0.01220 | 0 | 0.02399 | 0 | 0 |
| h23 | 0.00015 | 0.00051 | 0 | 0 | 0 |
| h24 | 0.02997 | 0.00051 | 0.05774 | 0.00259 | 0 |
| h25 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h26 | 0.00030 | 0 | 0.00059 | 0 | 0 |
| h35 | 0.00045 | 0 | 0.00089 | 0 | 0 |
| h36 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h37 | 0.00211 | 0 | 0.00415 | 0 | 0 |
| h39 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h40 | 0.00015 | 0.00051 | 0 | 0 | 0 |
| h41 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h42 | 0.00015 | 0.00051 | 0 | 0 | 0 |
| h44 | 0.00015 | 0 | 0 | 0.00086 | 0 |
| h45 | 0.00015 | 0.00051 | 0 | 0 | 0 |
| h53 | 0.00030 | 0 | 0.00059 | 0 | 0 |
| h54 | 0.05285 | 0.00204 | 0.10127 | 0.00431 | 0 |
| h55 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h56 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h57 | 0.00015 | 0.00051 | 0 | 0 | 0 |
| h58 | 0.00467 | 0.00153 | 0.00651 | 0.00517 | 0 |
| h63 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h64 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h72 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h73 | 0.00015 | 0.00051 | 0 | 0 | 0 |
| h82 | 0.00015 | 0.00051 | 0 | 0 | 0 |
| h90 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h91 | 0.00030 | 0 | 0.00059 | 0 | 0 |
| h92 | 0.00030 | 0.00051 | 0.00000 | 0 | 0.00685 |
| h93 | 0.00060 | 0 | 0.00118 | 0 | 0 |
| h94 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h95 | 0.00407 | 0 | 0 | 0.02328 | 0 |
| h96 | 0.00708 | 0.01277 | 0.00296 | 0.00948 | 0.00685 |
| h97 | 0.00030 | 0 | 0.00059 | 0 | 0 |
| h98 | 0.00015 | 0 | 0.00030 | 0 | 0 |
| h102 | 0.00015 | 0.00051 | 0 | 0 | 0 |
| Missense Variant | Phenotype Distribution | Biological Assay | PolyPhen Score | PolyPhen Prediction | TreeSAAP Property |
|---|---|---|---|---|---|
| M-1-T | M | - | NA | benign | |
| P-5-L | M | - | NA | benign | αc, αn, K0, Hp |
| E-40-K | Significant | - | 1.424 | benign | pHi |
| M-41-I | NonSig | - | 1.16 | benign | |
| S-67-R | M | - | 1.563 | possibly damaging | |
| R-72-L | M | - | 1.958 | possibly damaging | H, Hnc, αn |
| E-167-K | L | LPL Inhib | 0.194 | benign | pHi |
| P-174-S | M | - | 1.715 | possibly damaging | |
| E-190-Q | NonSig | - | 0.243 | benign | |
| E-196-K | M | - | 1.541 | possibly damaging | pHi, El |
| G-223-R | L | Secretion | 2.065 | probably damaging | E’sm |
| R-230-C | M | - | 2.792 | probably damaging | pHi, E’sm, Et, Br, Ns, C |
| F-237-V | M | - | 2.51 | probably damaging | |
| P-251-T | H | Nothing | 1.781 | possibly damaging | |
| T-266-M | NonSig | - | 0.783 | benign | K0, Ht |
| R-278-Q | Significant | - | 0.644 | benign | pHi |
| V-291-M | M | - | 1.012 | benign | |
| L-293-M | M | - | 1.236 | benign | |
| E-296-V | M | - | 2.057 | probably damaging | Ns, Pβ, Br, H, Ra |
| P-307-S | M | - | 0.955 | benign | αc |
| V-308-M | M | - | 1.199 | benign | |
| R-336-C | L | Secretion | 2.255 | probably damaging | Br, pHi, Et, Ns, C, Ca, Hnc |
| D-338-E | M | - | 1.626 | possibly damaging | |
| W-349-C | L | Secretion | 3.677 | probably damaging | |
| Ca, E’sm, Mv, Mw, Hnc, V0, | |||||
| G-361-R | L | Secretion | 2.274 | probably damaging | μ |
| R-371-Q | H | Nothing | 1.558 | possibly damaging | pHi |
| R-384-W | L | Secretion | 2.304 | probably damaging | Br, Ht |
| TreeSAAP Property Key | |||
|---|---|---|---|
| Alpha-helical tendency | Pα | Molecular weight | Mw |
| Average # of surrounding residues | Ns | Normalized hydrophobicity | Hnc |
| Beta-structure tendency | Pβ | Partial specific volume | V0 |
| Buriedness | Br | Power to be at the C-terminal | αc |
| Composition | C | Power to be at the N-terminal | αn |
| Compressibility | K0 | Refractive index | μ |
| Helical contact | Ca | Short-range & medium-range nonbonded energy | E’sm |
| Hydropathy | H | Solvent accessible reduction ratio | Ra |
| Isoelectric point | pHi | Surrounding hydrophobicity | Hp |
| Long-range nonbonded energy | El | Thermodyn. transfer hydrophobicity | Ht |
| Molecular volume | Mv | Total non-bonded energy | Et |
| Significance Criteria | Functional or Significant | Tested Not Functional | Middle or Not Sig | p-val | Odds Ratio | Lower 95 CI | Upper 95 CI | |
|---|---|---|---|---|---|---|---|---|
| PolyPhen | Significant | 5 | 2 | 8 | 0.673 | 1.828 | 0.254 | 15.766 |
| Not Significant | 3 | 0 | 9 | |||||
| Sensitivity | 0.625 | Specificity | 0.529 | alpha | 0.471 | beta | 0.375 | |
| TreeSAAP | 493.08 | |||||||
| Significant | 7 | 2 | 7 | 0.042 | 9.130 | 0.859 | 8 | |
| Not Significant | 1 | 0 | 10 | |||||
| Sensitivity | 0.875 | Specificity | 0.588 | alpha | 0.412 | beta | 0.125 | |
| Strict PolyPhen | Significant | 5 | 0 | 3 | 0.061 | 7.012 | 0.846 | 77.356 |
| Not Significant | 3 | 2 | 14 | |||||
| Sensitivity | 0.625 | Specificity | 0.824 | alpha | 0.176 | beta | 0.375 | |
| Strict TreeSAAP | Significant | 5 | 0 | 5 | 0.194 | 3.762 | 0.505 | 34.675 |
| Not Significant | 3 | 2 | 12 | |||||
| Sensitivity | 0.625 | Specificity | 0.706 | alpha | 0.294 | beta | 0.375 | |
| PolyPhen & TreeSAAP | Significant | 4 | 0 | 4 | 0.359 | 3.084 | 0.385 | 27.020 |
| Not Significant | 4 | 2 | 13 | |||||
| Sensitivity | 0.5 | Specificity | 0.765 | alpha | 0.235 | beta | 0.5 | |
| Strict PolyPhen & Strict TreeSAAP | Significant | 3 | 0 | 2 | 0.283 | 4.192 | 0.369 | 64.438 |
| Not Significant | 5 | 2 | 15 | |||||
| Sensitivity | 0.375 | Specificity | 0.882 | alpha | 0.118 | beta | 0.625 | |
| Strict PolyPhen OR Strict TreeSAAP | 11.52 | 626.87 | ||||||
| Significant | 7 | 0 | 6 | 0.030 | 6 | 1.077 | 1 | |
| Not Significant | 1 | 2 | 11 | |||||
| Sensitivity | 0.875 | Specificity | 0.647 | alpha | 0.353 | beta | 0.125 | |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maxwell, T.J.; Bendall, M.L.; Staples, J.; Jarvis, T.; Crandall, K.A. Phylogenetics Applied to Genotype/Phenotype Association and Selection Analyses with Sequence Data from Angptl4 in Humans. Int. J. Mol. Sci. 2010, 11, 370-385. https://doi.org/10.3390/ijms11010370
Maxwell TJ, Bendall ML, Staples J, Jarvis T, Crandall KA. Phylogenetics Applied to Genotype/Phenotype Association and Selection Analyses with Sequence Data from Angptl4 in Humans. International Journal of Molecular Sciences. 2010; 11(1):370-385. https://doi.org/10.3390/ijms11010370
Chicago/Turabian StyleMaxwell, Taylor J., Matthew L. Bendall, Jeffrey Staples, Todd Jarvis, and Keith A. Crandall. 2010. "Phylogenetics Applied to Genotype/Phenotype Association and Selection Analyses with Sequence Data from Angptl4 in Humans" International Journal of Molecular Sciences 11, no. 1: 370-385. https://doi.org/10.3390/ijms11010370