Neuroprotective Effects of Ischemic Preconditioning on Global Brain Ischemia through Up-Regulation of Acid-Sensing Ion Channel 2a

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Physiological variables
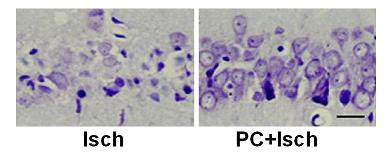
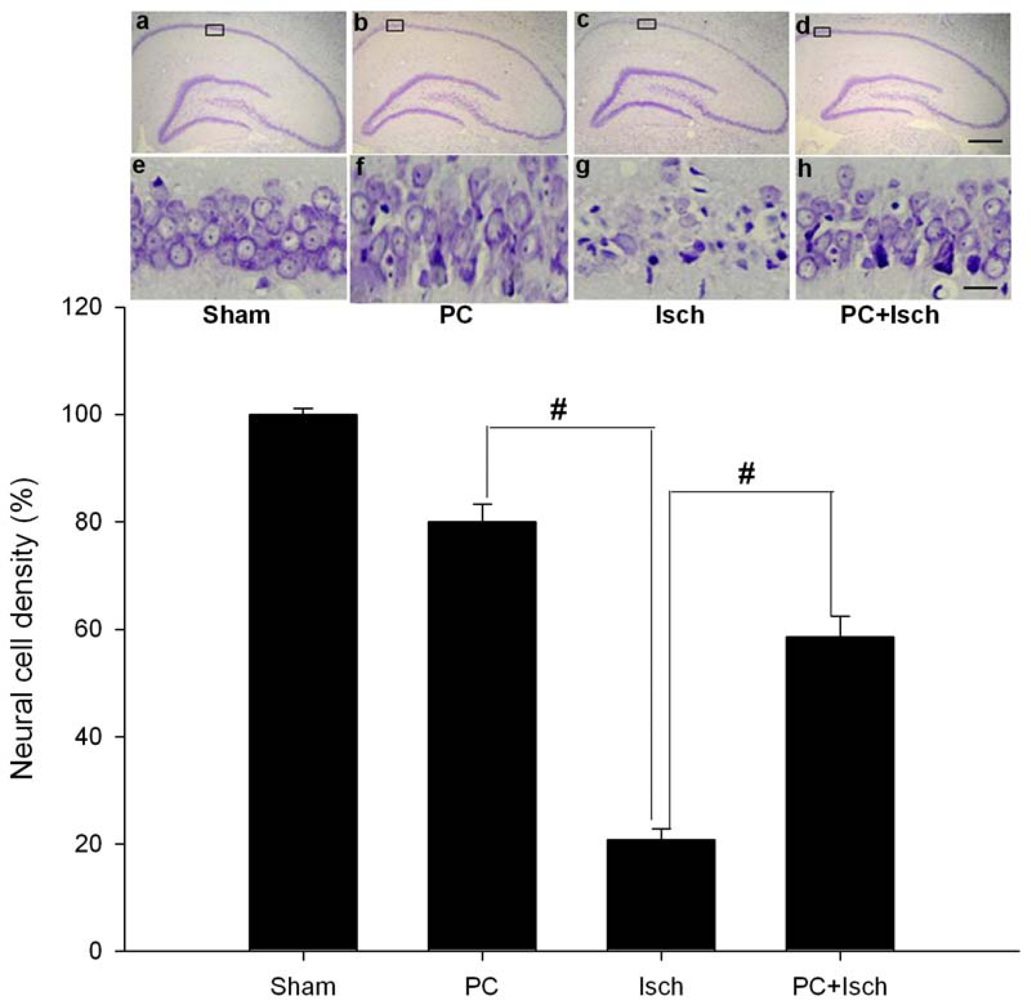
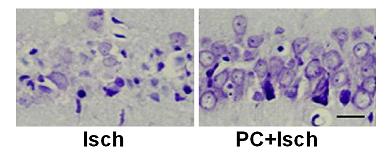
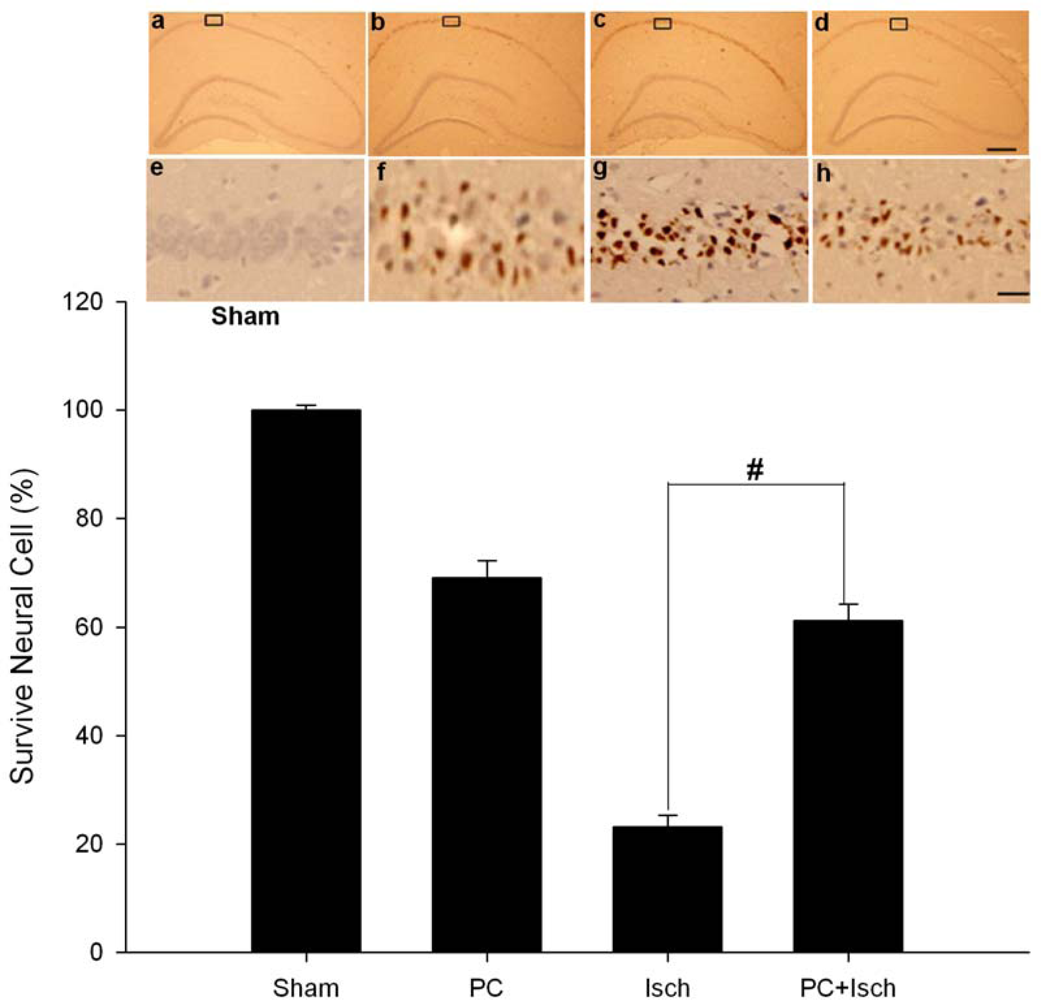
2.2. Cresyl violet stain
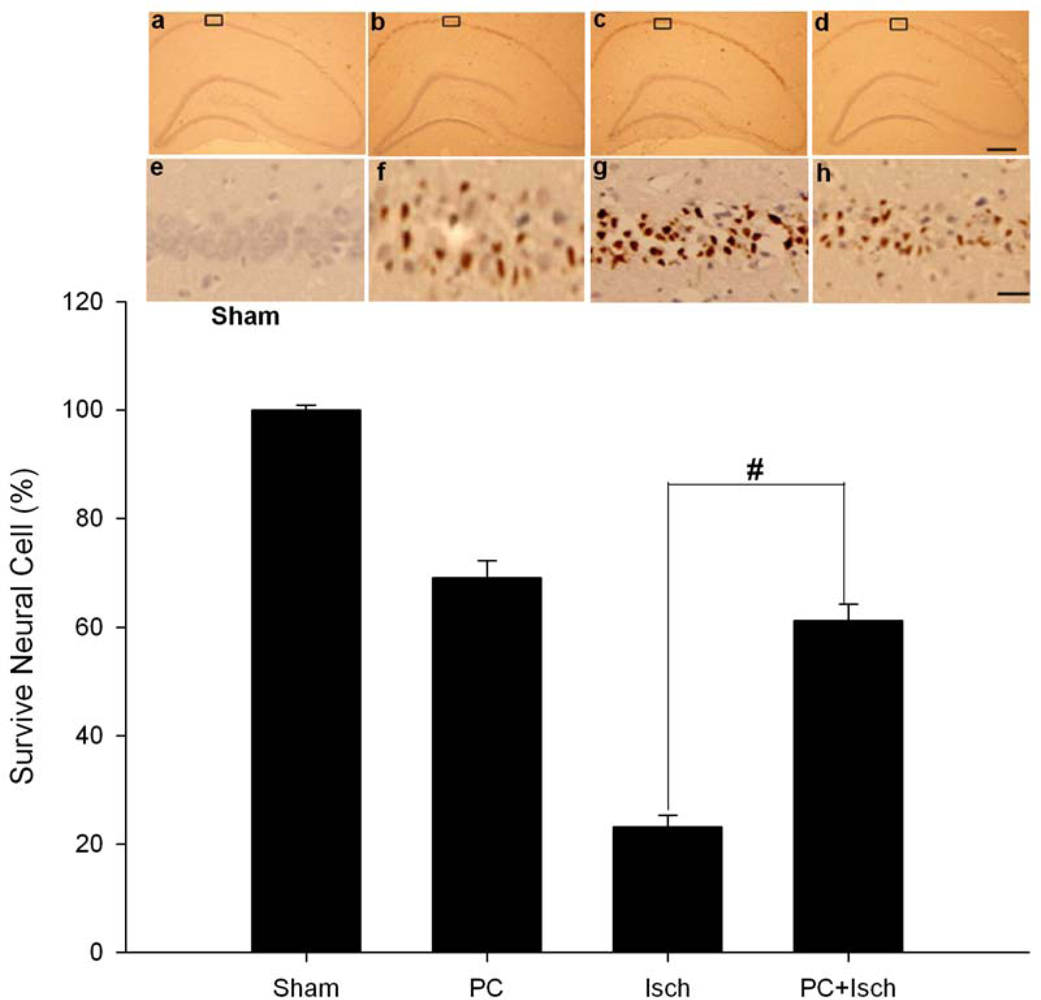
2.3. Tunel stain
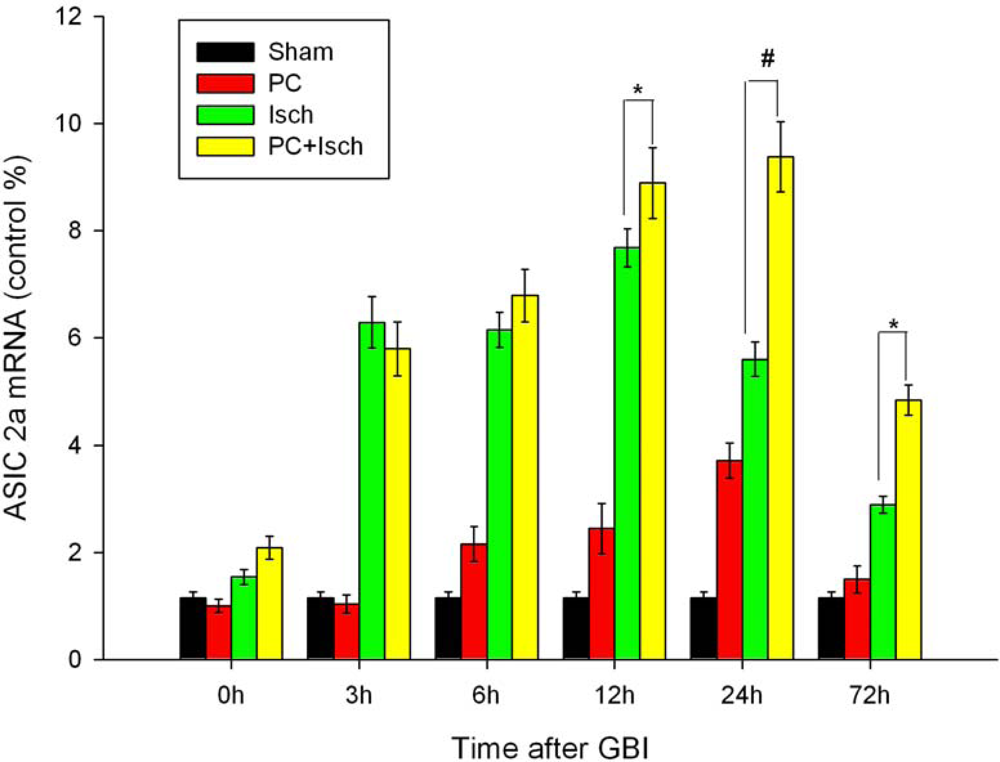
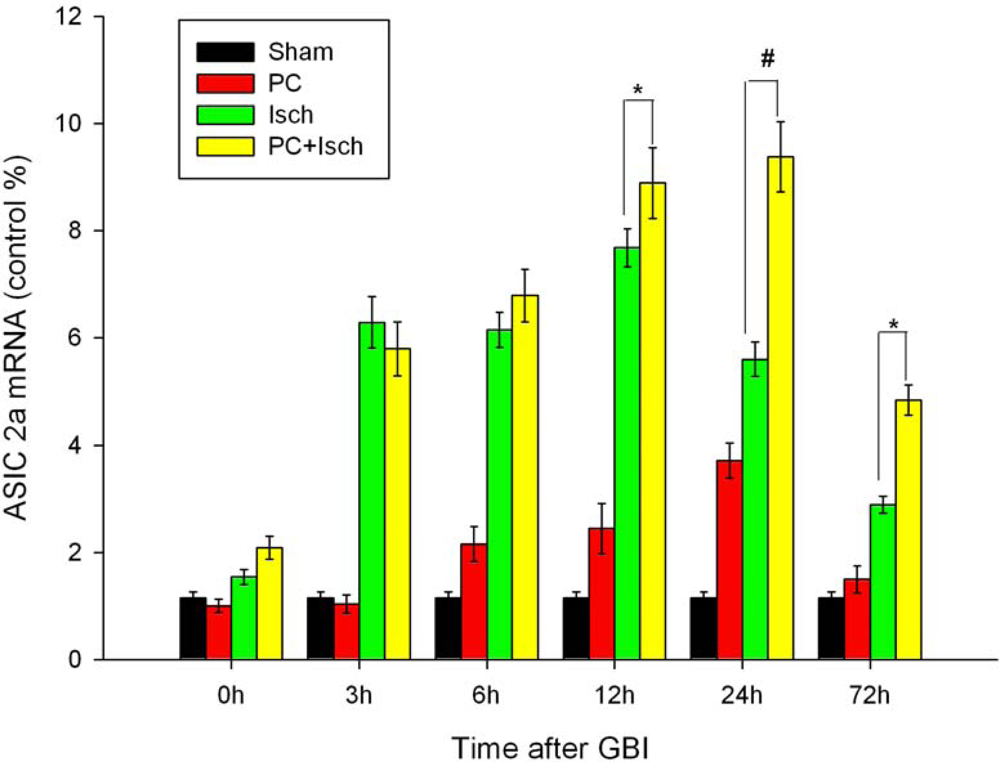
2.4. RT-PCR
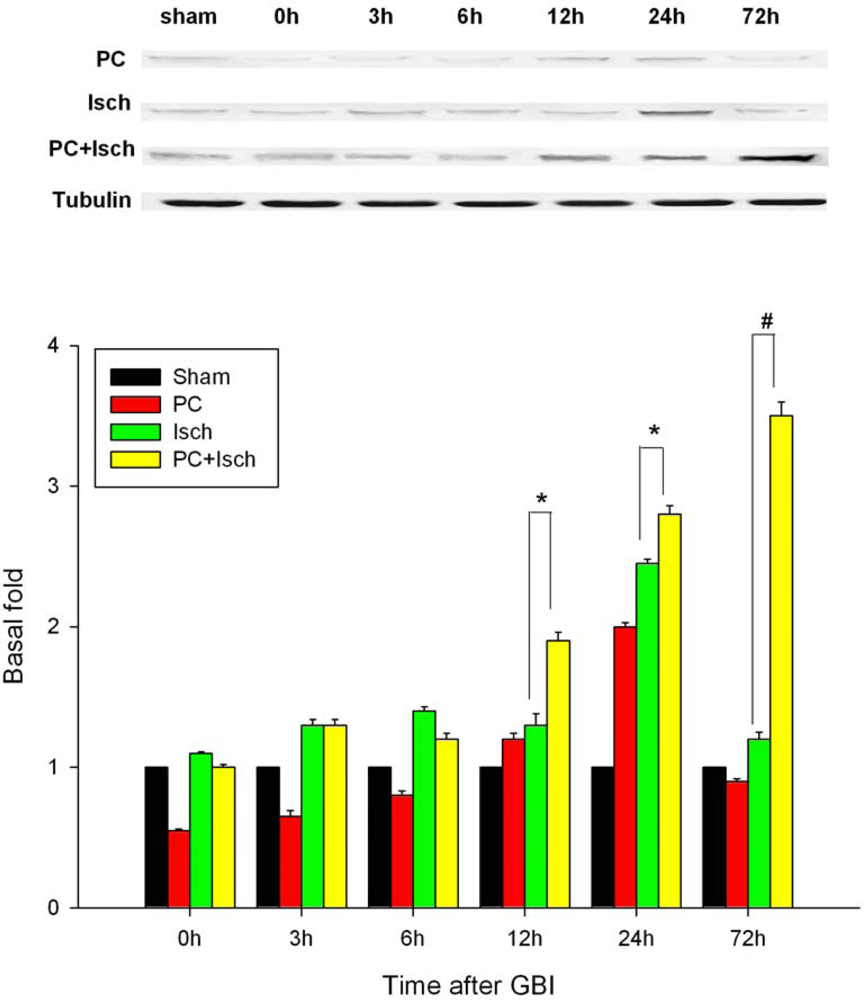
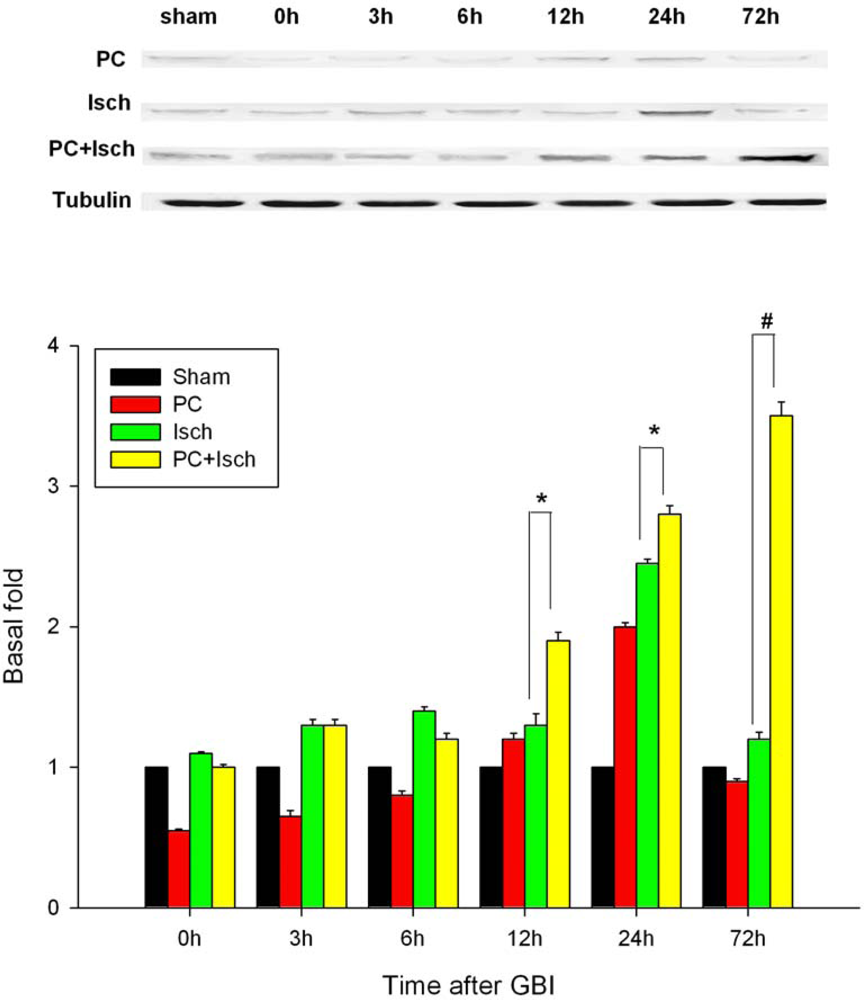
2.5. Western blot
2.6. Discussion
3. Experimental Section
3.1. Animals
3.2. Ischemic preconditioning and global ischemia
3.3. Histological analysis
3.4. In Situ labeling of DNA fragmentation by TUNEL
3.5. Reverse transcription—Polymerase chain reaction
- ASIC 2a _Forward:5′-ATGTTTAACTCAGGCGAGGATG-3′
- ASIC 2a _Reverse:5′-CCACGAAGGTCTGGAACCC-3′
3.6. Western blotting
3.7. Statistical analysis
4. Conclusions
Acknowledgments
References
- Siren, AL; Radyushkin, K; Boretius, S; Kammer, D; Riechers, CC; Natt, O; Sargin, D; Watanabe, T; Sperling, S; Michaelis, T; Price, J; Meyer, B; Frahm, J; Ehrenreich, H. Global brain atrophy after unilateral parietal lesion and its prevention by erythropoietin. Brain 2006, 129, 480–489. [Google Scholar]
- Yenari, M; Kitagawa, K; Lyden, P; Perez-Pinzon, M. Metabolic downregulation—A key to successful neuroprotection? Stroke 2008, 39, 2910–2917. [Google Scholar]
- Richards, EM; Fiskum, G; Rosenthal, RE; Hopkins, I; McKenna, MC. Hyperoxic reperfusion after global ischemia decreases hippocampal energy metabolism. Stroke 2007, 38, 1578–1584. [Google Scholar]
- Tanaka, R; Yamashiro, K; Mochizuki, H; Cho, N; Onodera, M; Mizuno, Y; Urabe, T. Neurogenesis after transient global ischemia in the adult hippocampus visualized by improved retroviral vector. Stroke 2004, 35, 1454–1459. [Google Scholar]
- Pignataro, G; Simon, RP; Xiong, ZG. Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain 2007, 130, 151–158. [Google Scholar]
- Gao, J; Duan, B; Wang, DG; Deng, XH; Zhang, GY; Xu, L; Xu, TL. Coupling between NMDA receptor and acid-sensing ion channel contributes to ischemic neuronal death. Neuron 2005, 48, 635–646. [Google Scholar]
- Xiong, ZG; Zhu, XM; Chu, XP; Minami, M; Hey, J; Wei, WL; MacDonald, JF; Wemmie, JA; Price, MP; Welsh, MJ; Simon, RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 2004, 118, 687–698. [Google Scholar]
- Huang, YZ; McNamara, JO. Ischemic stroke: “Acidotoxicity” is a perpetrator. Cell 2004, 118, 665–666. [Google Scholar]
- Nedergaard, M; Goldman, SA; Desai, S; Pulsinelli, WA. Acid-induced death in neurons and glia. J. Neurosci 1991, 11, 2489–2497. [Google Scholar]
- Hoshi, A; Nakahara, T; Kayama, H; Yamamoto, T. Ischemic tolerance in chemical preconditioning: possible role of astrocytic glutamine synthetase buffering glutamate-mediated neurotoxicity. J. Neurosci. Res 2006, 84, 130–141. [Google Scholar]
- Stenzel-Poore, MP; Stevens, SL; Xiong, Z; Lessov, NS; Harrington, CA; Mori, M; Meller, R; Rosenzweig, HL; Tobar, E; Shaw, TE; Chu, XP; Simon, RP. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 2003, 362, 1028–1037. [Google Scholar]
- Kirino, T. Ischemic tolerance. J. Cereb. Blood Flow Metab 2002, 22, 1283–1296. [Google Scholar]
- Jasti, J; Furukawa, H; Gonzales, EB; Gouaux, E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 2007, 449, 316–323. [Google Scholar]
- Yermolaieva, O; Leonard, AS; Schnizler, MK; Abboud, FM; Welsh, MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. USA 2004, 101, 6752–6757. [Google Scholar]
- Wemmie, JA; Chen, J; Askwith, CC; Hruska-Hageman, AM; Price, MP; Nolan, BC; Yoder, PG; Lamani, E; Hoshi, T; Freeman, JH, Jr; Welsh, MJ. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 2002, 34, 463–477. [Google Scholar]
- Waldmann, R; Champigny, G; Bassilana, F; Heurteaux, C; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nature 1997, 386, 173–177. [Google Scholar]
- Chu, XP; Close, N; Saugstad, JA; Xiong, ZG. ASIC1a-specific modulation of acid-sensing ion channels in mouse cortical neurons by redox reagents. J. Neurosci 2006, 26, 5329–5339. [Google Scholar]
- Askwith, CC; Wemmie, JA; Price, MP; Rokhlina, T; Welsh, MJ. Acid-sensing ion channel 2 (ASIC2) modulates ASIC1 H+-activated currents in hippocampal neurons. J. Biol. Chem 2004, 279, 18296–18305. [Google Scholar]
- Chen, CC; Zimmer, A; Sun, WH; Hall, J; Brownstein, MJ; Zimmer, A. A role for ASIC3 in the modulation of high-intensity pain stimuli. Proc. Natl. Acad. Sci. USA 2002, 99, 8992–8997. [Google Scholar]
- Krishtal, O. The ASICs: Signaling molecules? Modulators? Trends Neurosci 2003, 26, 477–483. [Google Scholar]
- Wemmie, J; Price, M; Welsh, M. Acid-sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci 2006, 29, 578–586. [Google Scholar]
- Alvarez de la Rosa, D; Krueger, SR; Kolar, A; Shao, D; Fitzsimonds, RM; Canessa, CM. Distribution, subcellular localization and ontogeny of ASIC1 in the mammalian central nervous system. J. Physiol 2003, 546, 77–87. [Google Scholar]
- Ichikawa, H; Sugimoto, T. The co-expression of ASIC3 with calcitonin gene-related peptide and parvalbumin in the rat trigeminal ganglion. Brain Res 2002, 943, 287–291. [Google Scholar]
- Xiong, ZG; Pignataro, G; Li, M; Chang, SY; Simon, RP. Acid-sensing ion channels (ASICs) as pharmacological targets for neurodegenerative diseases. Curr. Opin. Pharmacol 2008, 8, 25–32. [Google Scholar] [Green Version]
- Benveniste, M; Dingledine, R. Limiting stroke-induced damage by targeting an acid channel. N. Engl. J. Med 2005, 352, 85–86. [Google Scholar]
- Vila-Carriles, WH; Kovacs, GG; Jovov, B; Zhou, ZH; Pahwa, AK; Colby, G; Esimai, O; Gillespie, GY; Mapstone, TB; Markert, JM; Fuller, CM; Bubien, JK; Benos, DJ. Surface expression of ASIC2 inhibits the amiloride-sensitive current and migration of glioma cells. J. Biol. Chem 2006, 281, 19220–19232. [Google Scholar]
- Johnson, MB; Jin, K; Minami, M; Chen, D; Simon, RP. Global ischemia induces expression of acid-sensing ion channel 2a in rat brain. J. Cereb. Blood Flo. Metab 2001, 21, 734–740. [Google Scholar]
- Yin, XH; Zhang, QG; Miao, B; Zhang, GY. Neuroprotective effects of preconditioning ischaemia on ischaemic brain injury through inhibition of mixed-lineage kinase 3via NMDA receptor-mediated Akt1 activation. J. Neurochem 2005, 93, 1021–1029. [Google Scholar]
- Miao, B; Yin, XH; Pei, DS; Zhang, QG; Zhang, GY. Neuroprotective effects of preconditioning ischemia on ischemic brain injury through down-regulating activation of JNK1/2 via N-methyl-D-aspartate receptor-mediated Akt1 activation. J. Biol. Chem 2005, 280, 21693–21699. [Google Scholar]
- Nedergaard, M; Kraig, RP; Tanabe, J; Pulsinelli, WA. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am. J. Physiol 1991, 260, 581–588. [Google Scholar]
- Ginsberg, MD. Adventures in the pathophysiology of brain ischemia: penumbra, gene expression, neuroprotection: The 2002 Thomas Willis Lecture. Stroke 2003, 34, 214–223. [Google Scholar]
- Kikuchi, S; Ninomiya, T; Kawamata, T; Tatsumi, H. Expression of ASIC2 in ciliated cells and stereociliated cells. Cell Tissue Res 2008, 333, 217–224. [Google Scholar]
- Grifoni, SC; McKey, SE; Drummond, HA. Hsc70 regulates cell surface ASIC2 expression and vascular smooth muscle cell migration. Am. J. Physiol. Heart C 2008, 294, 2022–2030. [Google Scholar]
- Hughes, PA; Brierley, SM; Young, RL; Blackshaw, LA. Localization and comparative analysis of acid-sensing ion channel (ASIC1, 2, and 3) mRNA expression in mouse colonic sensory neurons within thoracolumbar dorsal root ganglia. J. Comp. Neurol 2007, 500, 863–875. [Google Scholar]
- Bano, D; Young, KW; Guerin, CJ; Lefeuvre, R; Rothwell, NJ; Naldini, L; Rizzuto, R; Carafoli, E; Nicotera, P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 2005, 120, 275–285. [Google Scholar]
- Paschen, W. Dependence of vital cell function on endoplasmic reticulum calcium levels: implications for the mechanisms underlying neuronal cell injury in different pathological states. Cell Calcium 2001, 29, 1–11. [Google Scholar]
- Nakamura, T; Minamisawa, H; Katayama, Y; Ueda, M; Terashi, A; Nakamura, K; Kudo, Y. Increased intracellular Ca2+ concentration in the hippocampal CA1 area during global ischemia and reperfusion in the rat: A possible cause of delayed neuronal death. Neuroscience 1999, 88, 57–67. [Google Scholar]
- Schaller, B; Graf, R. Cerebral ischemic preconditioning: An experimental phenomenon or a clinical important entity of stroke prevention? J. Neurol 2002, 249, 1503–1511. [Google Scholar]
- Froyland, E; Skjaeret, C; Wright, MS; Dalen, ML; Cvancarova, M; Kasi, C; Rootwelt, T. Inflammatory receptors and pathways in human NT2-N neurons during hypoxia and reoxygenation. Impact of acidosis. Brain Res 2008, 1217, 37–49. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham | Isch | PC | PC + Isch | |
|---|---|---|---|---|
| Before Ischemia (n = 5) | ||||
| pH | 7.44 ± 0.04 | 7.45 ± 0.07 | 7.43 ± 0.03 | 7.44 ± 0.05 |
| pCO2 (mmHg) | 35.6 ± 1.6 | 36.2 ± 4.2 | 38.2 ± 5.2 | 37.6 ± 4.9 |
| pO2 (mmHg) | 106.2 ± 10.9 | 110.2 ± 10.2 | 108.4 ± 8.3 | 105.4 ± 6.7 |
| Glucose (mg/dL) | 118.9 ± 7.4 | 121.1 ± 12.4 | 119.4 ± 7.0 | 129.9 ± 8.4 |
| Hemoglobin (g/dL) | 16.5 ± 1.2 | 15.6 ± 0.9 | 16.7 ± 0.5 | 16.2 ± 0.6 |
| MABP (mmHg) | 86.2 ± 7.1 | 85.3 ± 11.1 | 79.6 ± 12.3 | 84.2 ± 11.4 |
| Temp rect (°C) | 36.6 ± 0.3 | 36.7 ± 0.16 | 36.6 ± 0.21 | 36.6 ± 0.25 |
| During Ischemia (n = 5) | ||||
| pH | 7.41 ± 0.05 | 7.38 ± 0.12 | 7.39 ± 0.11 | 7.40 ± 0.08 |
| pCO2 (mmHg) | 41.2 ± 6.1 | 40.1 ± 3.5 | 41.8 ± 3.9 | 41.1 ± 5.1 |
| pO2 (mmHg) | 93.1 ± 5.6 | 98.1 ± 7.2 | 96.1 ± 5.4 | 99.2 ± 10.2 |
| Glucose (mg/dL) | 109.1 ± 5.6 | 104.7 ± 7.7 | 98.4 ± 10.4 | 116.9 ± 11.2 |
| Hemoglobin (g/dL) | 18.4 ± 2.1 | 19.1 ± 1.2 | 17.2 ± 5.1 | 19.2 ± 3.6 |
| MABP (mmHg) | 71.8 ± 8.6 | 75.1 ± 10.5 | 74.7 ± 8.7 | 79.2 ± 7.2 |
| Temp rect (°C) | 36.7 ± 0.12 | 36.9 ± 0.05 | 36.8 ± 0.03 | 36.8 ± 0.08 |
| After Ischemia (n = 5) | ||||
| pH | 7.41 ± 0.05 | 7.40 ± 0.03 | 7.39 ± 0.06 | 7.41 ± 0.03 |
| pCO2 (mmHg) | 38.0 ± 3.8 | 35.9 ± 4.2 | 40.1 ± 3.5 | 36.1 ± 1.8 |
| pO2 (mmHg) | 106.7 ± 8.4 | 115.8 ± 5.9 | 109.4 ± 9.2 | 116.4 ± 10.3 |
| Glucose (mg/dL) | 110.6 ± 7.9 | 109.2 ± 10.3 | 115.4 ± 13.2 | 117.9 ± 12.9 |
| Hemoglobin (g/dL) | 15.3 ± 1.1 | 16.4 ± 1.6 | 16.1 ± 1.7 | 15.7 ± 0.9 |
| MABP (mmHg) | 73.1 ± 12.3 | 77.3 ± 11.3 | 72.9 ± 4.9 | 73.1 ± 11.2 |
| Temp rect (°C) | 36.5 ± 0.11 | 36.9 ± 0.12 | 36.7 ± 0.05 | 36.9 ± 0.03 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Miao, Y.; Zhang, W.; Lin, Y.; Lu, X.; Qiu, Y. Neuroprotective Effects of Ischemic Preconditioning on Global Brain Ischemia through Up-Regulation of Acid-Sensing Ion Channel 2a. Int. J. Mol. Sci. 2010, 11, 140-153. https://doi.org/10.3390/ijms11010140
Miao Y, Zhang W, Lin Y, Lu X, Qiu Y. Neuroprotective Effects of Ischemic Preconditioning on Global Brain Ischemia through Up-Regulation of Acid-Sensing Ion Channel 2a. International Journal of Molecular Sciences. 2010; 11(1):140-153. https://doi.org/10.3390/ijms11010140
Chicago/Turabian StyleMiao, Yifeng, Weiqiao Zhang, Yuchang Lin, Xiaojie Lu, and Yongming Qiu. 2010. "Neuroprotective Effects of Ischemic Preconditioning on Global Brain Ischemia through Up-Regulation of Acid-Sensing Ion Channel 2a" International Journal of Molecular Sciences 11, no. 1: 140-153. https://doi.org/10.3390/ijms11010140