RNA Relics and Origin of Life

Abstract
:1. Introduction
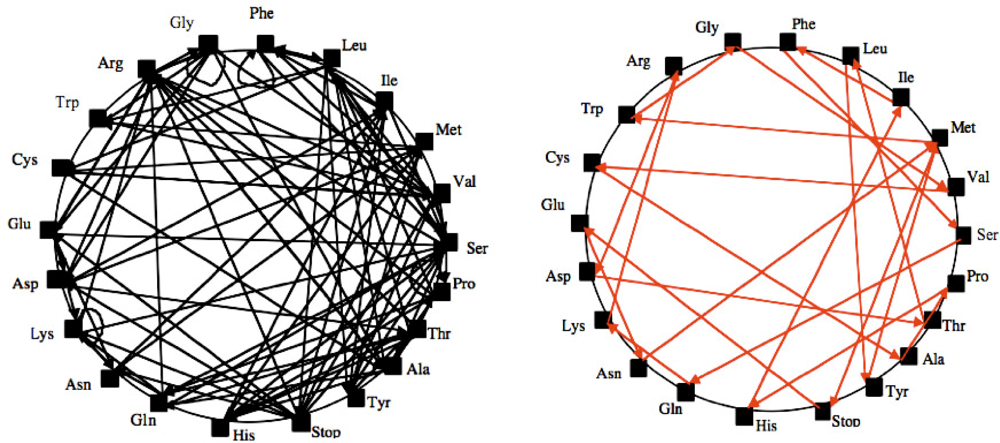
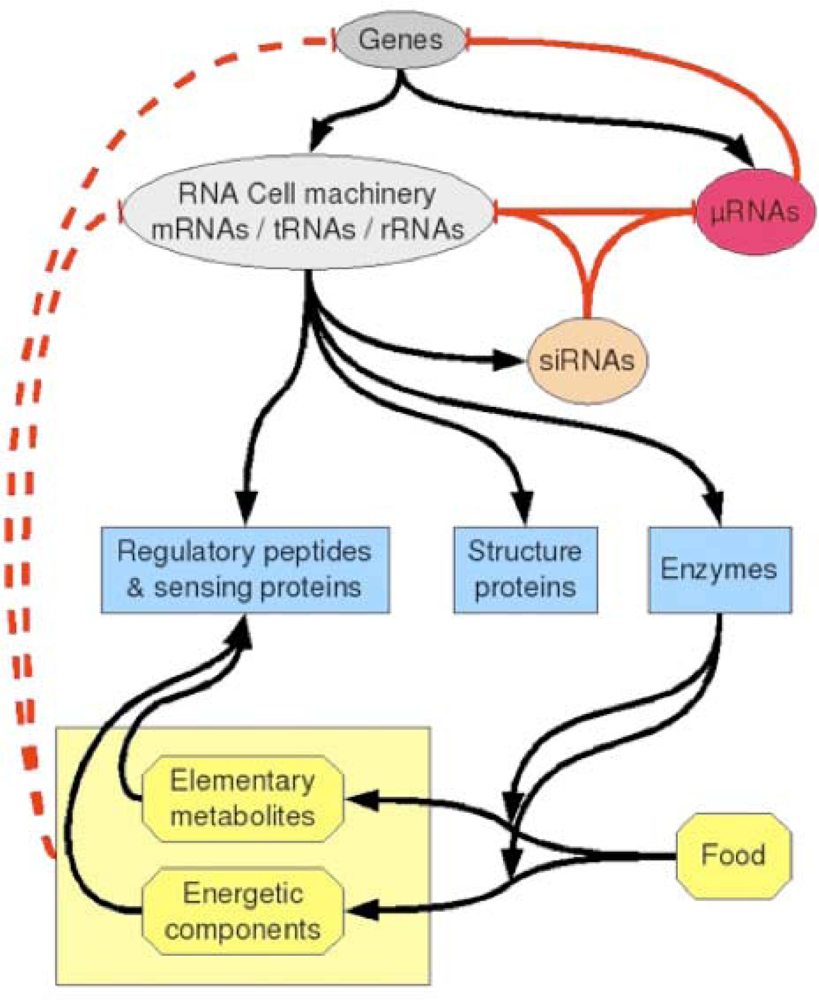
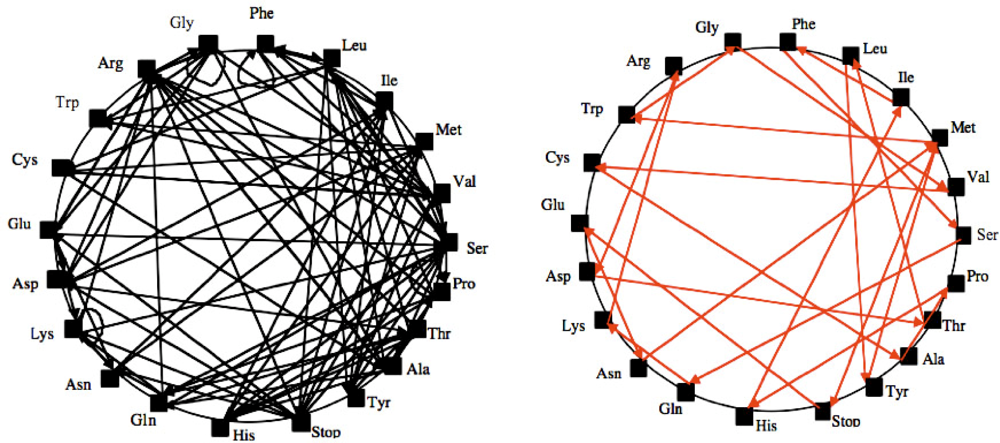
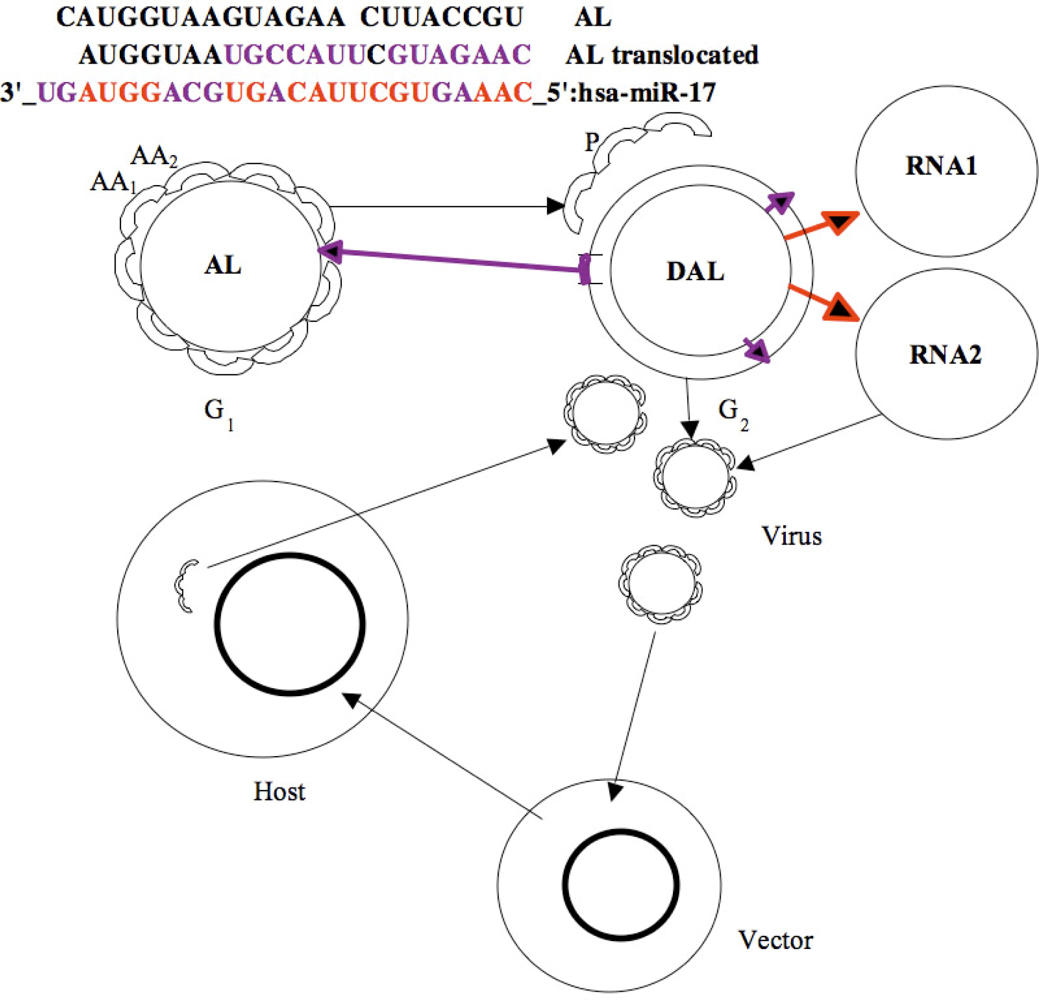
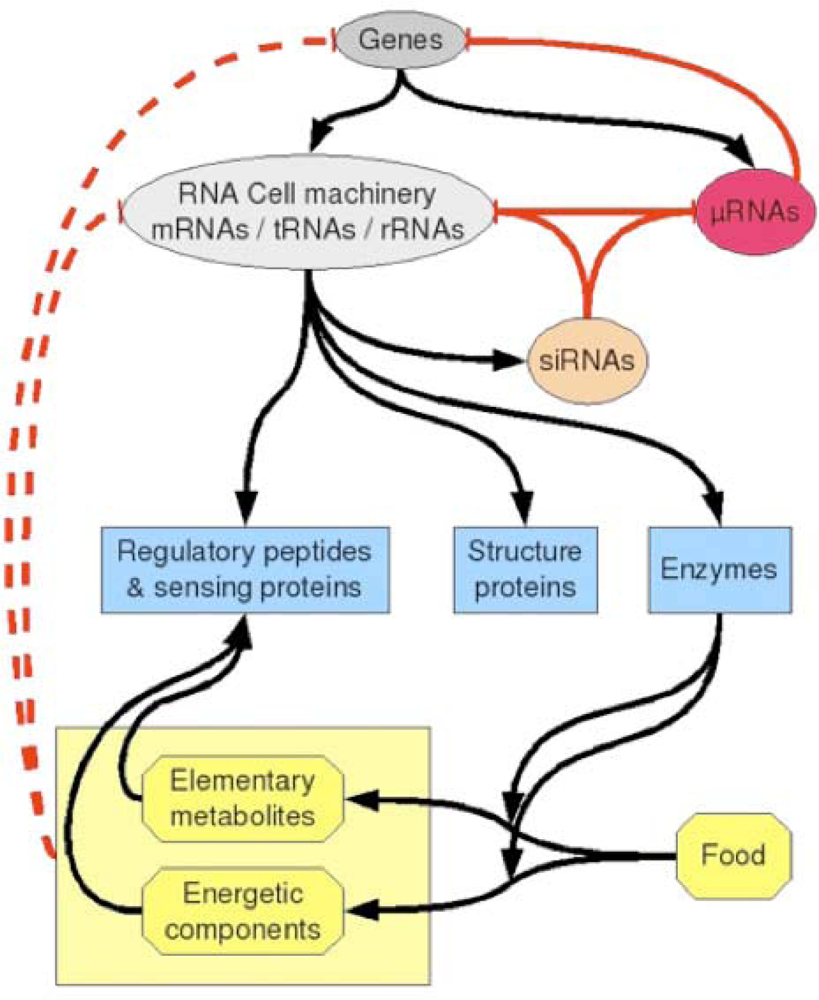
2. An Archetypal Genome
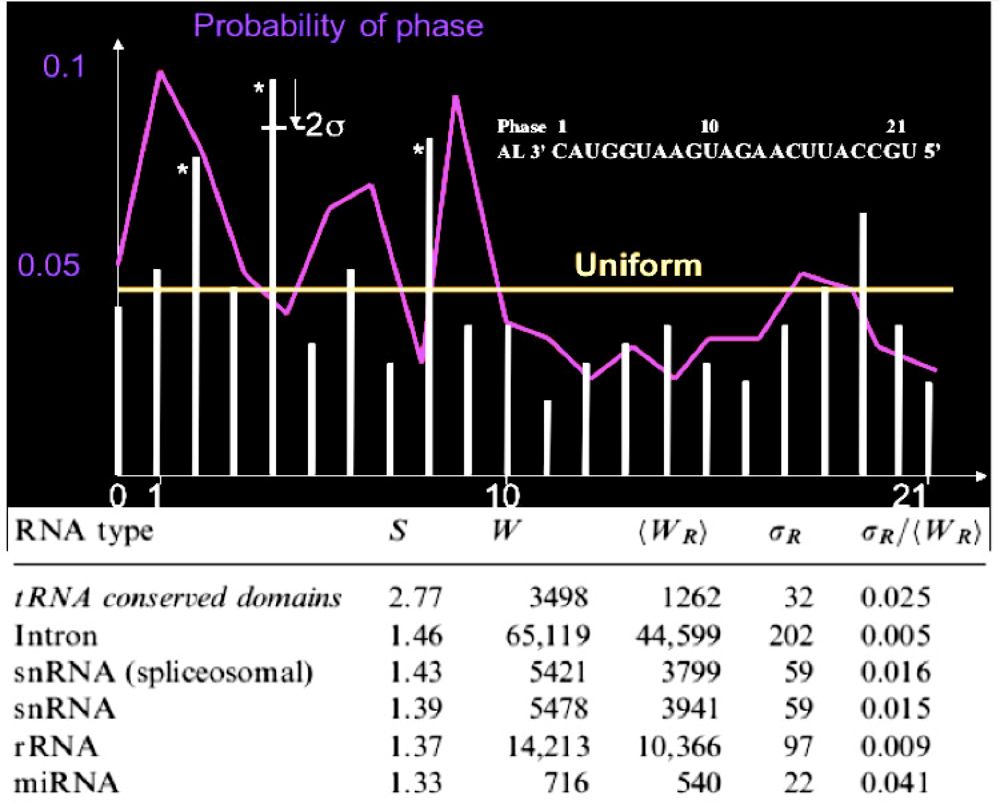
3. RNA Relics
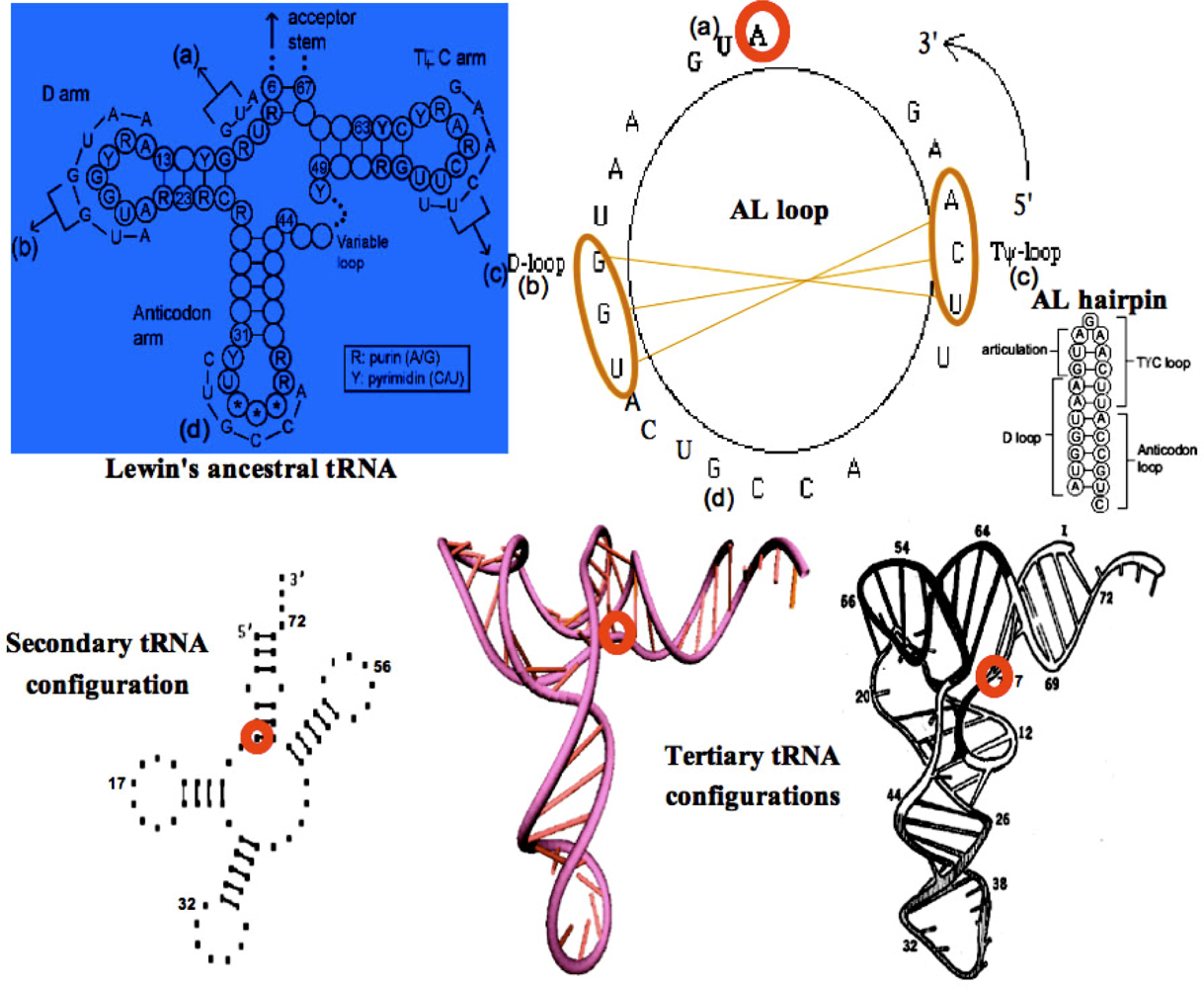
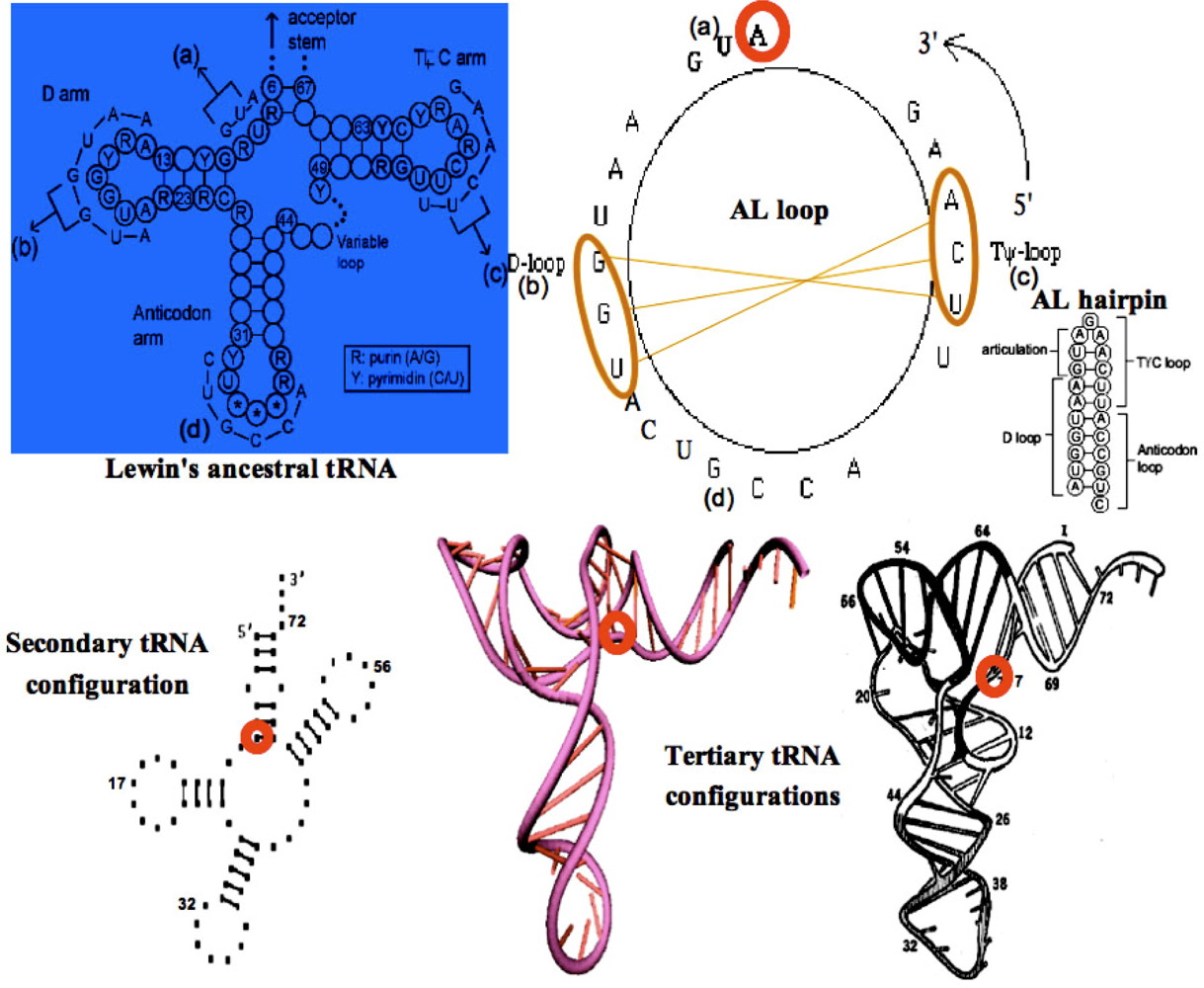
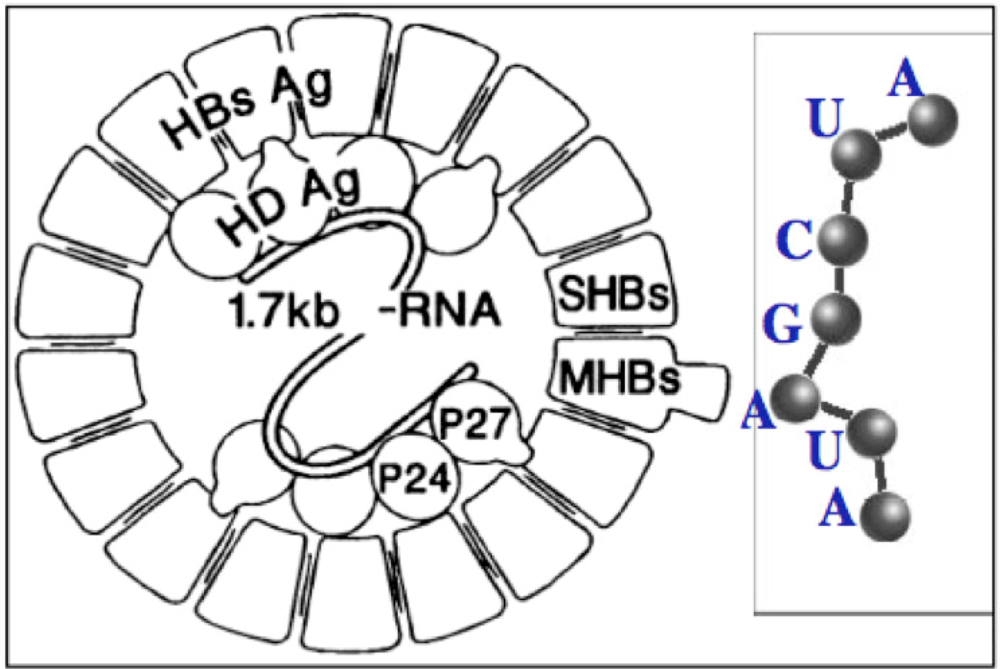
3.1. tRNAs Loops
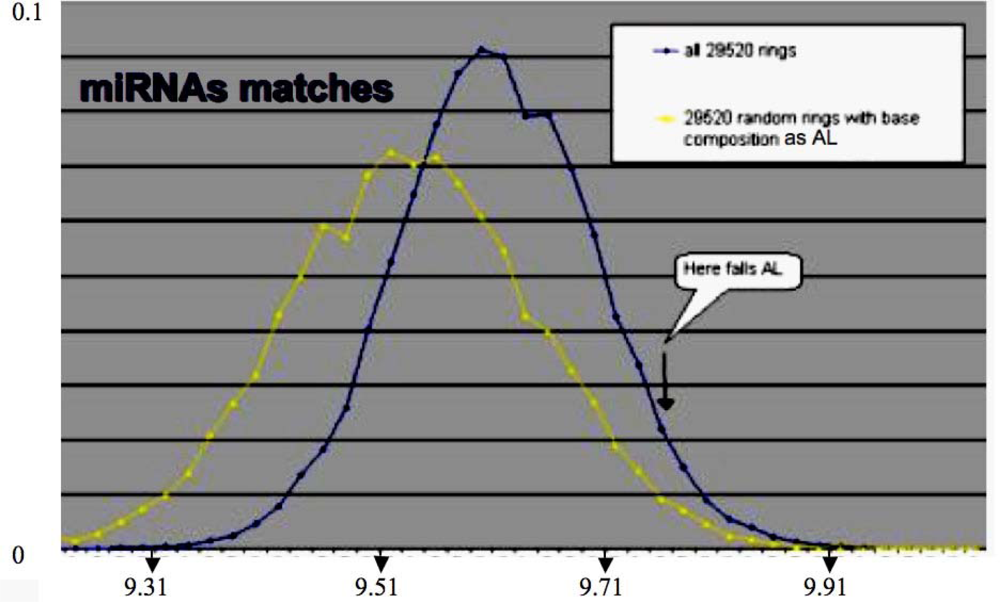
3.2. Small RNAs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
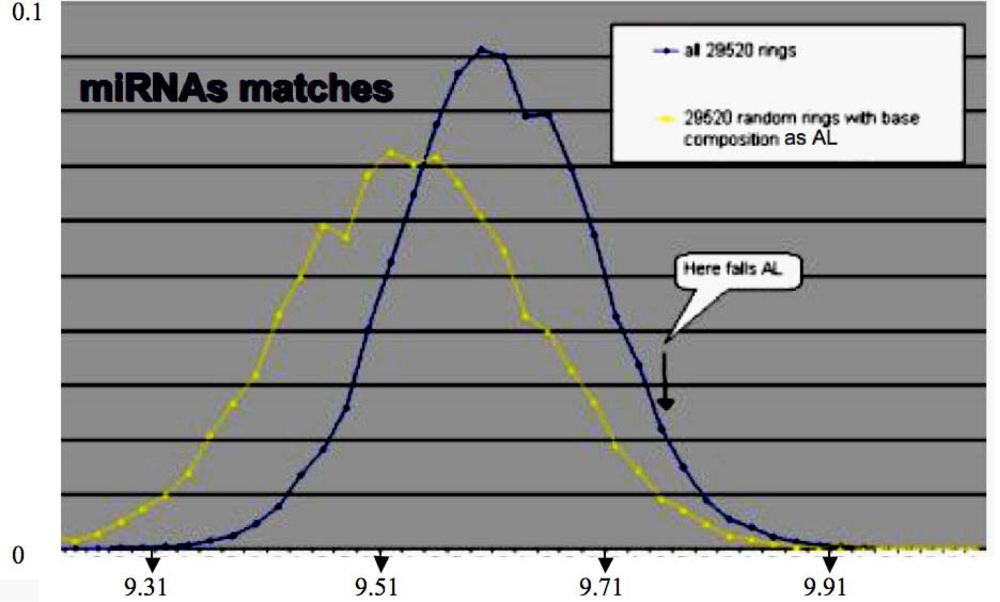
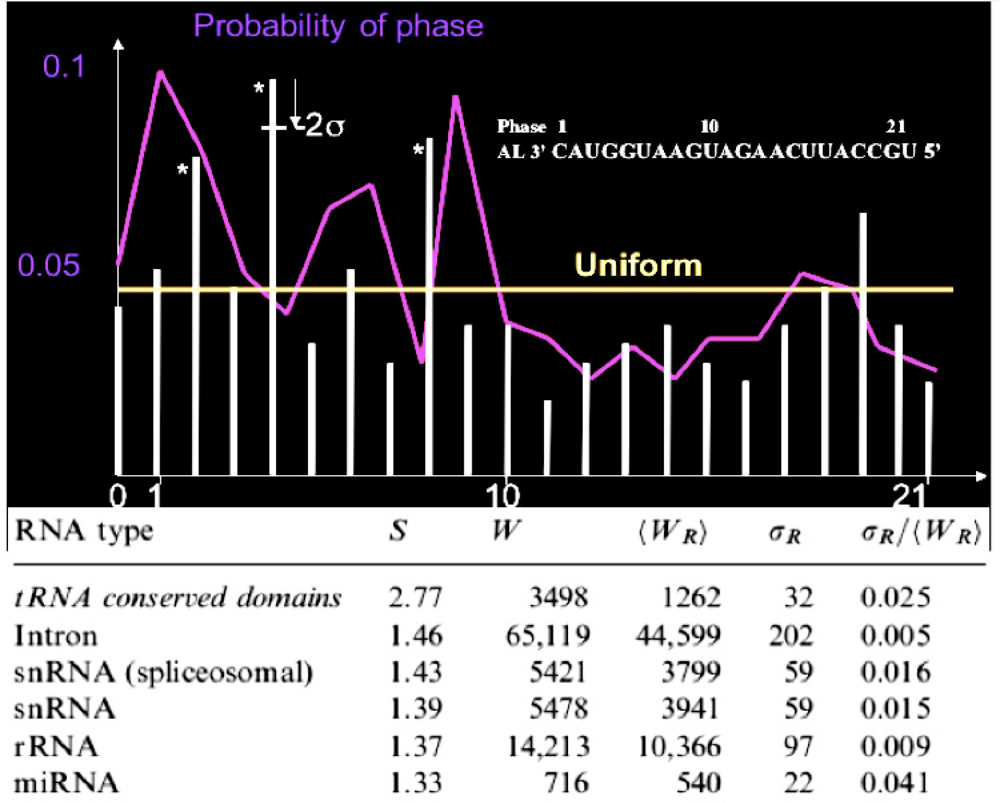
| Match value with AL: 11/22 | 5′_ACAACCAGCUAAGACACUGCCA_3′:hsa-miR-34 a UUCAAGAUGAAUGGUACUGCCA |
| Match value with AL: 9/22 | 5′_UGAGAUGAAGCACUGUAGCUCA_5′:hsa-miR-143 UUCAAGAUGAAUGGUACUGCCA |
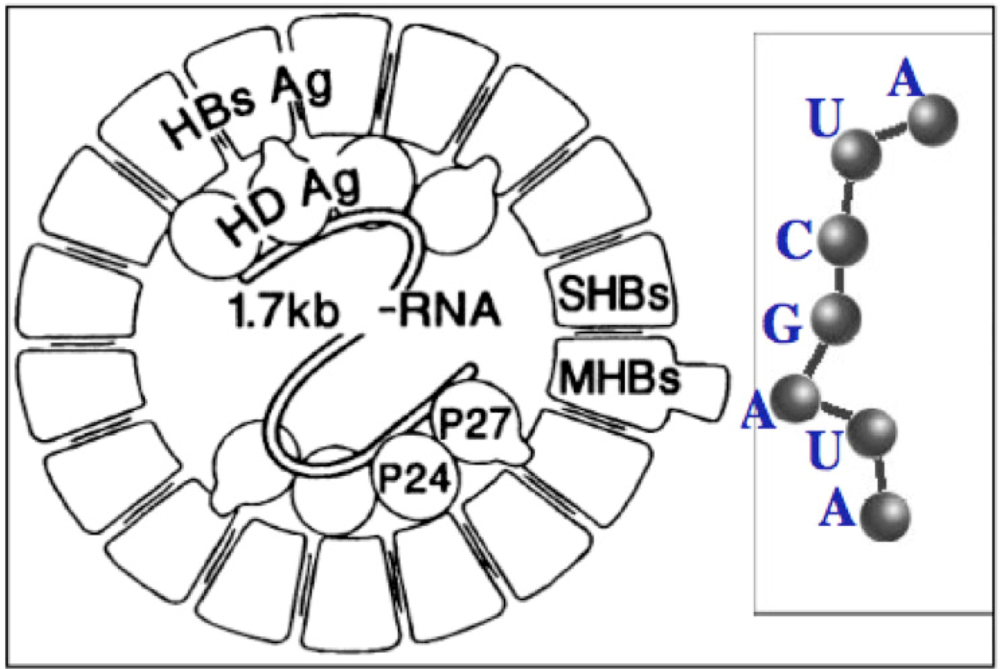
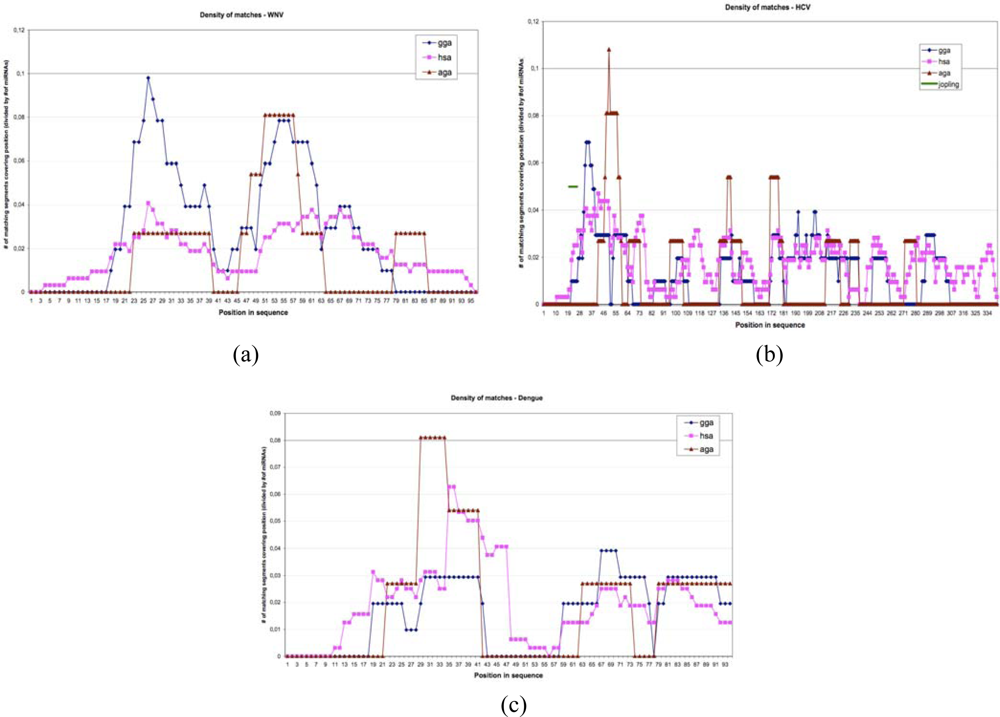
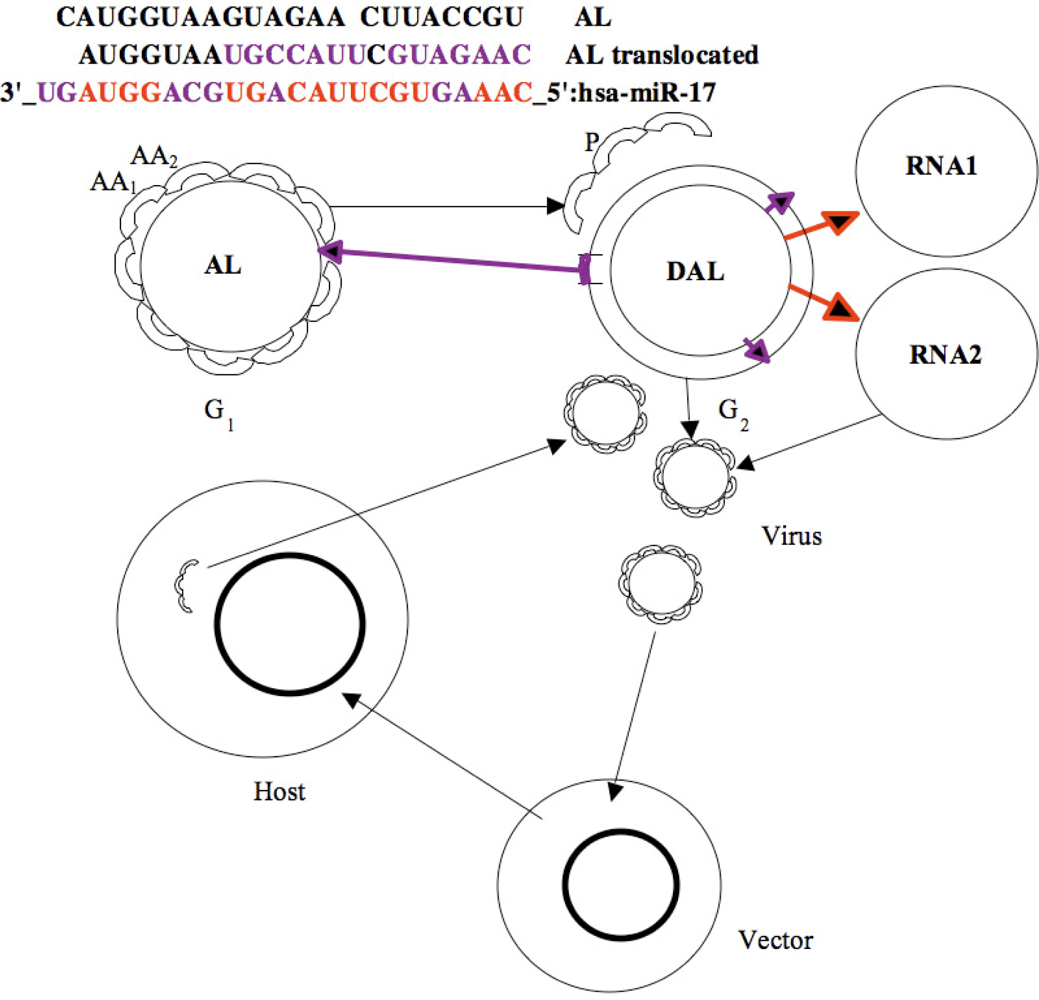
4. Similarities between Archetypal Genome, Viral Genomes and RNA Relics
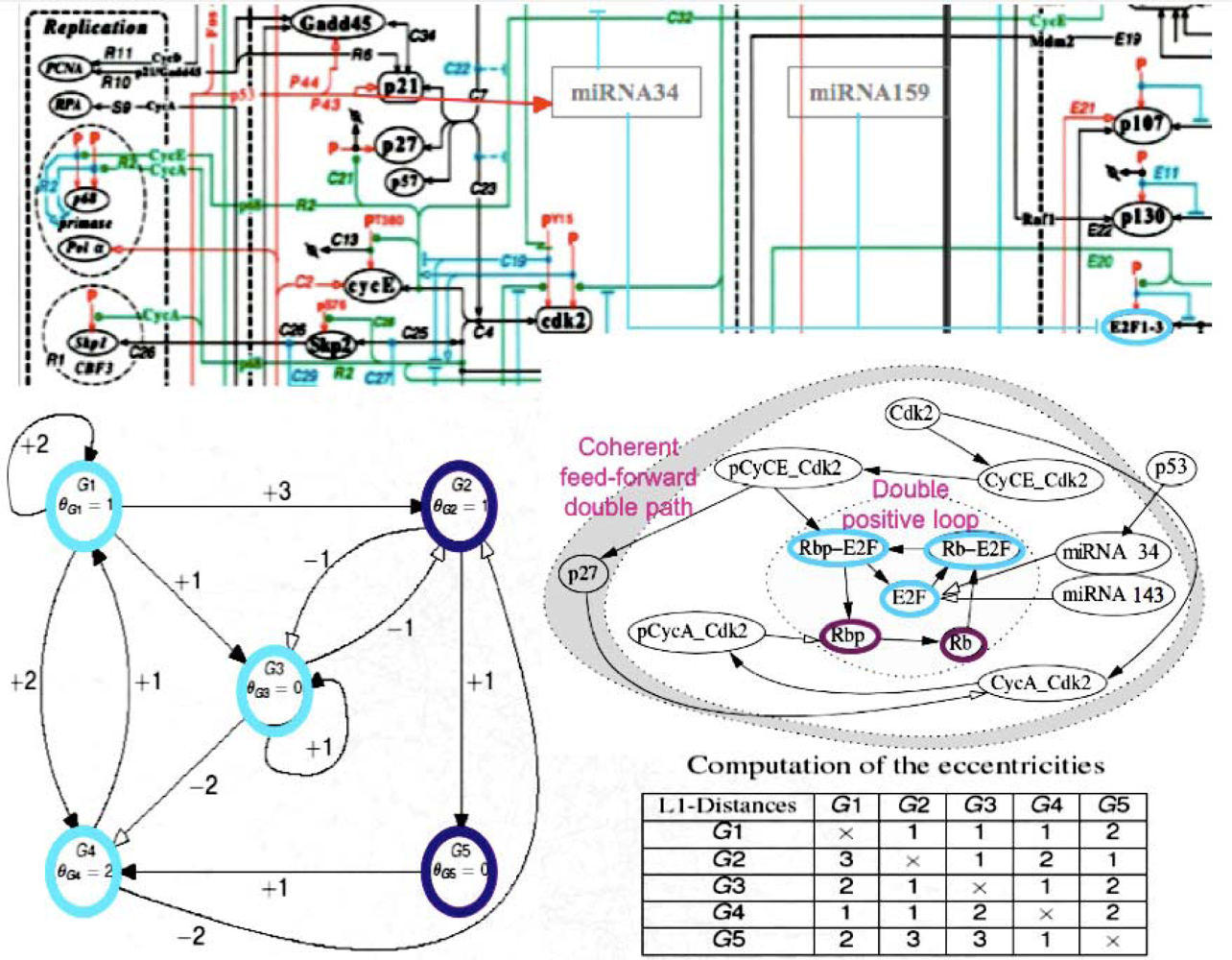
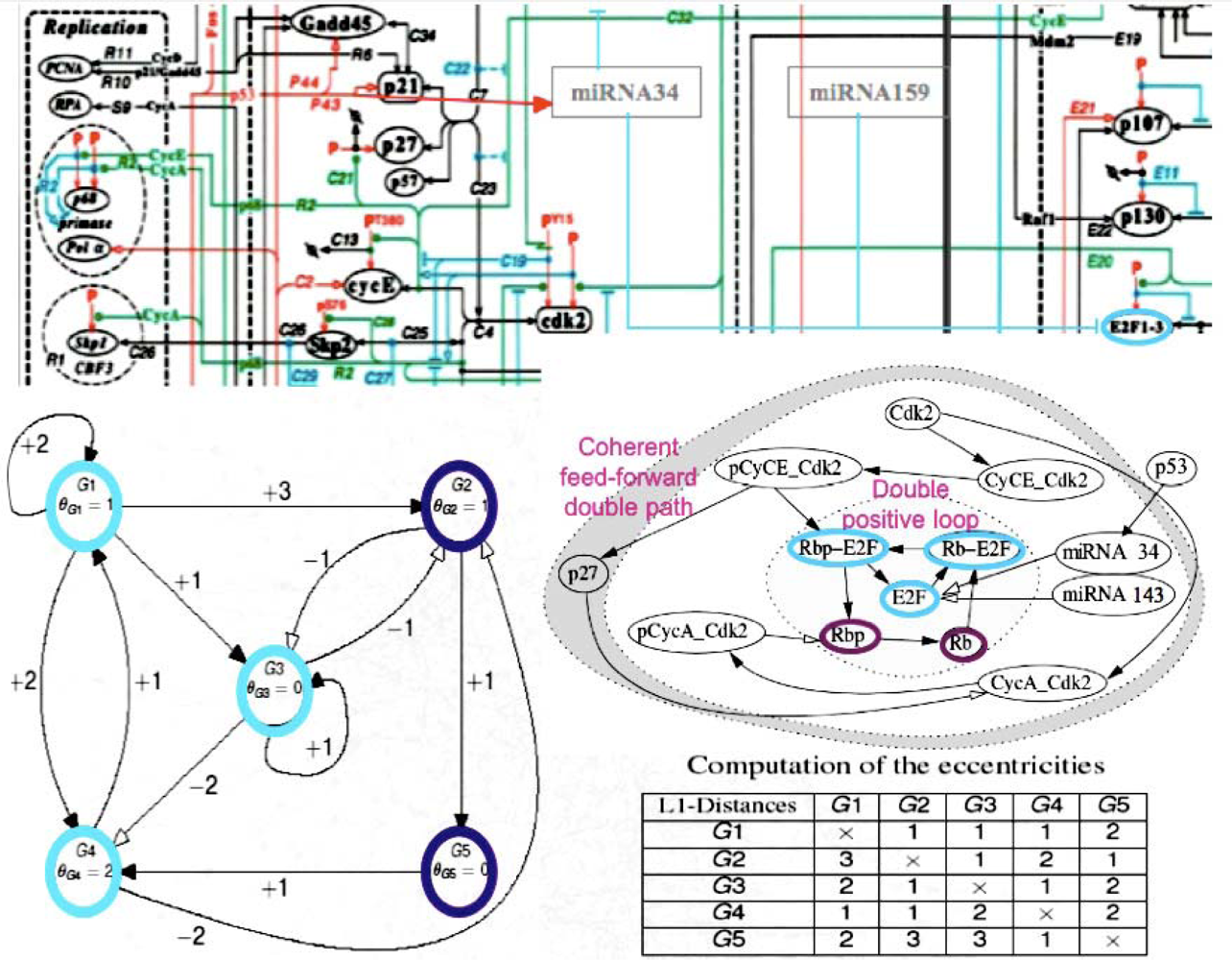
5. Co-Evolution
6. Ancestral Processes Inherited from the Origins of Life
6.1. From the Discovery of the Genetic Code to the Birth of the Stereochemical Model
6.2. Models for the Emergence of a Genetic Code
6.3. Experimentally Testing the Stereochemical Model for the Origin of the Genetic Code
6.4. Implication of the Stereochemical Model in the Existing Living Systems
7. Conclusions
Acknowledgments
References
- Hertel, J; Lindemeyer, M; Missal, K; Fried, C; Tanzer, A; Flamm, C; Hofacker, IL; Stadler, PF. Students of Bioinformatics Computer Labs 2004 and 2005. The expansion of the metazoan microRNA repertoire. BMC Genomics 2006, 7, 25. [Google Scholar]
- Tanzer, A; Stadler, PF. Molecular evolution of a microRNA cluster. J. Mol. Biol 2004, 339, 327–335. [Google Scholar]
- Lai, EC; Weil, C; Rubin, GM. Complementary miRNA pairs suggest a regulatory role for miRNA:miRNA duplexes. RNA 2007, 10, 171–175. [Google Scholar]
- Jouanneau, J; Larsen, CJ. Les microARN: un « bras armé » du suppresseur de tumeur p53. Bull. Cancer 2007, 94, 634–635. [Google Scholar]
- Bartel, DP; Chen, CZ. Micromanagers of gene expression: The potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet 2004, 5, 396–400. [Google Scholar]
- Lim, LP; Lau, NC; Garett-Engele, P; Grimson, A; Schelter, JM; Castle, J; Bartel, DP; Linsley, PS; Johnson, JM. Microarray analysis shows that some microRNAs downregulate large numbers of targets mRNAs. Nature 2005, 433, 769–773. [Google Scholar]
- Sullivan, CS; Don Ganem, D. MicroRNAs & viral infection. Mol. Cell 2005, 20, 3–7. [Google Scholar]
- Gatignol, A; Lainé, S; Clerzius, G. Dual role of TRBP in HIV replication and RNA interference: Viral diversion of a cellular pathway or evasion from antiviral immunity? Retrovirology 2005, 2, 65. [Google Scholar]
- Appel, N; Bartenschlager, R. A novel function for a miR: Negative regulators can do positive for the hepatitis C virus. Hepatology 2006, 43, 612–615. [Google Scholar]
- Jopling, CL; Yi, M; Lancaster, AM; Lemon, SM; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific miR. Science 2005, 309, 1577–1581. [Google Scholar]
- Lecellier, CH; Dunoyer, P; Arar, K; Lehmann-Che, J; Eyquem, S; Himber, C; Saïb, A; Voinnet, O. A cellular microRNA mediates antiviral defense in human cells. Science 2005, 308, 557–560. [Google Scholar]
- Demongeot, J. Sur la possibilité de considérer le code génétique comme un code à enchaînement. Revue de Biomaths 1978, 62, 61–66. [Google Scholar]
- Demongeot, J; Besson, J. Code génétique et codes à enchaînement I. C. R. Acad. Sci. Paris Ser. III 1983, 296, 807–810. [Google Scholar]
- Demongeot, J; Besson, J. Genetic code and cyclic codes II. C. R. Acad. Sci. Paris Ser. III 1996, 319, 520–528. [Google Scholar]
- Moreira, A. Particles and Simple Agents in Cellular Automata and Other Discrete Systems. Ph.D. Dissertation; Universidad de Chile: Santiago de Chile, Chile, 2003. [Google Scholar]
- Weil, G; Heus, K; Faraut, T; Demongeot, J. An archetypal basic code for the primitive genome. Theoret. Comp. Sc 2004, 322, 313–334. [Google Scholar]
- Demongeot, J; Elena, A; Weil, G. Potential automata. Application to the genetic code III. Comptes Rendus Biologies 2006, 329, 953–962. [Google Scholar]
- Demongeot, J; Moreira, A. A circular RNA at the origin of life. J. Theor. Biol 2007, 249, 314–324. [Google Scholar]
- Demongeot, J; Moreira, A. A circular Hamming distance, circular Gumbel distribution, RNA relics and primitive genome. AINA’07, Advanced Information Networking and Applications, IEEE Proc., Piscataway, NJ, USA; 2007; pp. 719–726. [Google Scholar]
- Hartman, H. Speculations on the evolution of the genetic code III. Origin of Life 1984, 14, 643–648. [Google Scholar]
- di Giulio, M. On the origin of the tRNA molecule. J. Theor. Biol 1992, 159, 199–214. [Google Scholar]
- di Giulio, M. On the origin of the genetic code. J. Theor. Biol 1997, 187, 573–581. [Google Scholar]
- Hopfield, J. Origin of the genetic code: A testable hypothesis based on tRNA structure, sequence, and kinetic proofreading. Proc. Natl. Acad. Sci. USA 1978, 75, 4334–4338. [Google Scholar]
- Lewin, B. Genes IX; Jones and Bartlett Publishers: Sudbury, MA, USA, 2008. [Google Scholar]
- Binder, S; Schuster, W; Grienenberger, JM; Weil, JH; Brennicke, A. Genes for Gly-, His, Lys-, Phe-, Ser- and Tyr-tRNA are encoded in Oenothera mitochondrial DNA. Curr. Genet 1990, 17, 353–358. [Google Scholar]
- Doi, M; Tarui, M; Ishida, T. Crystal structure of hybrid dipeptide, uracil-1-yl-(2-carboxyethyl)-glycine. Anal. Sci 2000, 16, 557–558. [Google Scholar]
- He, M; Petoukhov, S; Ricci, P. Genetic code, hamming distance and stochastic matrices. Bull. Math. Biol 2004, 66, 1405–1421. [Google Scholar]
- Scherrer, K; Jost, J. The gene and the genon concept: A functional and information-theoretic analysis. Mol. Syst. Biol 2007, 3, 87. [Google Scholar]
- Elena, A; Ben-Amor, H; Glade, N; Demongeot, J. Motifs in regulatory networks and their structural robustness. IEEE BIBE’08; IEEE Proc., Piscataway, NJ, USA; 2008; pp. 234–242. [Google Scholar]
- Elena, A; Demongeot, J. Interaction motifs in regulatory networks and structural robustness. IEEE ARES-CISIS’08 & IIBM’08; IEEE Proc., Piscataway, NJ, USA; 2008; pp. 682–686. [Google Scholar]
- Ben Amor, H; Demongeot, J; Sené, S. Structural sensitivity of neural and genetic networks. In Lecture Notes in Computer Science, MICAI 2008: Advances in Artificial Intelligence, 7th Mexican International Conference on Artificial Intelligence, Atizapán de Zaragoza, Mexico; 27–31 October 2008; pp. 973–986. [Google Scholar]
- Demongeot, J; Morvan, M; Sené, S. Robustness of dynamical systems attraction basins against state perturbations: Theoretical protocol and application in systems biology. IEEE ARES-CISIS’08 & IIBM’08; IEEE Proc., Piscataway, NJ, USA; 2008; pp. 675–681. [Google Scholar]
- Kohn, KW. Molecular interaction map of the mammalian cell cycle control and DNA repair systems. Mol. Biol. Cell 1999, 10, 2703–2734. [Google Scholar]
- Demongeot, J; Ben Amor, H; Elena, A; Gillois, P; Sené, S. Robustness in regulatory interaction networks. A generic approach with applications at different levels: Physiologic, metabolic and genetic. Int J Mol Sci. submitted for publication, 2009.
- Bellini, R; Casali, B; Carrieri, M; Zambonelli, C; Rivasi, P; Rivasi, F. Aedes albopictus is incompetent as a vector of hepatitis C virus. APMIS 1997, 105, 299–302. [Google Scholar]
- Pfeffer, S; Zavolan, M; Grasser, FA; Chien, M; Russo, JJ; Ju, J; John, B; Enright, AJ; Marks, D; Sander, C; Tuschl, T. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar]
- Sullivan, CS; Grundhoff, AT; Tevethia, S; Pipas, JM; Ganem, D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 2005, 435, 682–686. [Google Scholar]
- Forterre, P. The origin of viruses and their possible rôles in major evolutionary transitions. Virus Res 2006, 117, 5–16. [Google Scholar]
- Witzany, G. Natural genome-editing competences of viruses. Acta Bio 2006, 54, 235–253. [Google Scholar]
- Stoltz, DB; Whitfield, JB. Viruses and virus-like entities in the parasitic Hymenoptera. J. Hymenoptera Res 1992, 1, 125–139. [Google Scholar]
- Harwood, SH; Grosovsky, AJ; Cowles, EA; Davis, JW; Beckage, NE. An abundantly expressed hemolymph glycoprotein isolated from newly parasitized Manduca sexta larvae is a polydnavirus gene product. Virology 1994, 205, 381–392. [Google Scholar]
- Knibbe, C; Mazet, O; Chaudier, F; Fayard, JM; Beslon, G. Evolutionary coupling between the deleteriousness of gene mutations and the amount of non-coding sequences. J. Theor. Biol 2007, 244, 621–630. [Google Scholar]
- Varetto, L. Typogenetics: An artificial genetic system. J. Theor. Biol 1993, 160, 185–205. [Google Scholar]
- Ray, TS. Evolution, complexity, entropy, and artificial reality. Physica D 1994, 75, 239–263. [Google Scholar]
- Gillespie, DT. A General Method for Numerically Simulating the Stochastic Time Evolution of Coupled Chemical Reactions. J. Comp. Phys 1976, 22, 403–434. [Google Scholar]
- Demongeot, J. A stochastic model for the cellular metabolism. In Recent Developments in Statistics; Barra, JR, Brodeau, F, Romier, G, Eds.; North Holland: Amsterdam, The Netherlands, 1977; pp. 655–662. [Google Scholar]
- Pasqual, N; Gallagher, M; Aude-Garcia, C; Loiodice, M; Thuderoz, F; Demongeot, J; Ceredig, R; Marche, PN; Jouvin-Marche, N. Quantitative and qualitative changes in ADV-AJ rearrangements during mouse thymocytes differentiation: Implication for a limited TCR ALPHA chain repertoire. J. Exp. Medicine 2002, 196, 1163–1174. [Google Scholar]
- Baum, TP; Pasqual, N; Thuderoz, F; Hierle, V; Chaume, D; Lefranc, MP; Jouvin-Marche, E; Marche, PN; Demongeot, J. IMGT/GeneInfo: Enhancing V(D)J recombination database accessibility. Nucleic Acids Res 2004, 32, 51–54. [Google Scholar]
- Baum, TP; Pasqual, N; Hierle, V; Bellahcene, F; Chaume, D; Lefranc, MP; Jouvin-Marche, E; Marche, P; Demongeot, J. IMGT/GeneInfo: New gamma and delta chains for database V(D)J recombination. BMC Bioinformatics 2006, 7, 224–228. [Google Scholar]
- Simonet, MA; Thuderoz, F; Hansen, O; Jouvin-Marche, E; Marche, PN; Demongeot, J. Modelling the Rearrangements Mechanisms in Immune Genome Toward a prediction tool of human immune specificity, IEEE ARES-CISIS’ 09 & BT’ 09; IEEE Proc., Piscataway, NJ, USA; 2009; pp. 943–948.
- Wilkins, MHF; Stokes, AR; Wilson, HR. Molecular structure of deoxypentose nucleic acids. Nature 1953, 171, 738–740. [Google Scholar]
- Franklin, RE; Gosling, RG. Molecular configuration in sodium thymonucleate. Nature 1953, 171, 740–741. [Google Scholar]
- Watson, J; Crick, FH. A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar]
- Alberti, S. Origins of the genetic code and protein synthesis. J. Mol. Evol 1997, 45, 352–358. [Google Scholar]
- Commeyras, A; Taillades, J; Collet, H; Boiteau, L; Vandenabeele-Trambouze, O; Pascal, R; Rousset, A; Garrel, L; Rossi, C; Cottet, H; Biron, JP; Lagrille, O; Plasson, R; Souaid, E; Selsis, F; Dobrijevic, M. Approche dynamique de la synthèse des peptides et leurs précurseurs sur la Terre primitive. In Les Traces du Vivant; Gargaud, M, Despois, D, Parisot, JP, Reisse, J, Eds.; Presses Un de Bordeaux: Bordeaux, France, 2003; pp. 115–162. [Google Scholar]
- Demongeot, J; Glade, N; Moreira, A. Evolution and RNA relics. A systems biology view. Acta Bio 2008, 56, 5–25. [Google Scholar]
- Gamow, G. Possible relation between deoxyribonucleic acid and protein structures. Nature 1954, 173, 318. [Google Scholar]
- Nirenberg, M; Leder, P; Bernfield, M; Brimacombe, R; Trupin, J; Rottman, F; O’Neal, C. RNA codewords and protein synthesis, VII. On the general nature of the RNA code. Proc. Natl. Acad. Sci. USA 1965, 53, 1161–1168. [Google Scholar]
- Pelc, SR. Correlation between coding-triplets and amino-acids. Nature 1965, 207, 597–599. [Google Scholar]
- Knight, RD; Landweber, LF. Rhyme or reason: RNA-arginine interactions and the genetic code. Chem. & Biol 1998, 5, 215–220. [Google Scholar]
- Knight, RD; Landweber, LF. Early evolution of the genetic code. Cell 2000, 101, 569–572. [Google Scholar]
- Mannironi, C; Scerch, C; Fruscolini, P; Tocchini-Valentini, GP. Molecular recognition of amino-acids by RNA aptamers: The evolution into an L-tyrosine binder of a dopamine-binding RNA motif. RNA 2000, 6, 520–527. [Google Scholar]
- Yarus, M. A specific amino-acid binding site composed of RNA. Science 1988, 240, 1751–1758. [Google Scholar]
- Yarus, M. RNA-ligand chemistry, testable source for genetic code. RNA 2000, 6, 475–484. [Google Scholar]
- Pelc, SR; Welton, MGE. Stereochemical relationship between coding triplets and amino-acids. Nature 1966, 209, 868–870. [Google Scholar]
- Reisse, J; Cronin, J. Chiralité et origine de l’homochiralité. In Les Traces du Vivant; Gargaud, M, Despois, D, Parisot, JP, Reisse, J, Eds.; Presses Un de Bordeaux: Bordeaux, France, 2003; pp. 83–114. [Google Scholar]
- Welton, MGE; Pelc, SR. Specificity of the stereochemical relationship between ribonucleic acid-triplets and amino-acids. Nature 1966, 209, 870–872. [Google Scholar]
- Hendry, LB; Bransome, ED; Hutson, MS; Campbell, LK. First approximation of the stereochemical rationale for the genetic code based on the topography and physicochemical properties of “cavities” constructed from models of DNA. Proc. Natl. Acad. Sci. USA 1981, 78, 7440–7444. [Google Scholar]
- Johnson, AP; Cleaves, HJ; Dworkin, JP; Glavin, DP; Lazcano, A; Bada, JL. The Miller volcanic spark discharge experiment. Science 2008, 322, 404. [Google Scholar]
- Hunding, A; Képès, F; Lancet, D; Minsky, A; Norris, V; Raine, D; Sriram, K; Root-Bernstein, R. Hypothesis: Compositional complementarily and prebiotic ecology in the origin of life. Bioessays 2006, 28, 399–412. [Google Scholar]
- Norris, V; Hunding, A; Képès, F; Lancet, D; Minsky, A; Raine, D; Root-Bernstein, R; Sriram, K. Question 7: The first units of life were not simple cells. Orig. Life Evol. Biosph 2007, 37, 429–443. [Google Scholar]
- Corbett, PT; Leclaire, J; Vial, L; West, KR; Wietor, J-L; Sanders, JKM; Otto, S. Dynamic combinational chemistry. Chem. Rev 2006, 106, 3652–3711. [Google Scholar]
- Berezikov, E; Plasterk, RHA. Camels and zebrafish, viruses and cancer: A microRNA update. Hum. Mol. Genet 2005, 14, 183–190. [Google Scholar]
- Yang, J; Mani, SA; Donaher, JL; Ramaswamy, S; Itzykson, RA; Come, C; Savagner, P; Gitelman, I; Richardson, A; Weinberg, RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar]
- Tao, K; Fang, M; Alroy, J; Sahagian, GG. Imagable 4T1 model for the study of late stage breast cancer. BMC Cancer 2008, 8, 22. [Google Scholar]
- Brown, D; Shingara, J; Keiger, K; Shelton, J; Lew, K; Cannon, B; Wowk, S; Byrom, M; Cheng, A; Wang, X. Cancer-related miRNAs uncovered by the mirVana miRNA microarray platform. Ambion TechNotes Newsletter 2005, 12, 8–11. [Google Scholar]
- Brock, A; Chang, H; Huang, S. Non-genetic heterogeneity - A mutation-independent driving force for the somatic evolution of tumours. Nat. Rev. Genet 2009, 10, 336–342. [Google Scholar]
- Chen, K; Rajewsky, N. Natural selection on human microRNA binding sites inferred from SNP data. Nat. Genet 2006, 38, 1452–1456. [Google Scholar]
- Berezikov, E; Thuemmler, F; van Laake, LW; Kondova, I; Bontrop, R; Cuppen, E; Plasterk, RHA. Diversity of microRNAs in human and chimpanzee brain. Nat. Genet 2006, 38, 1375–1377. [Google Scholar]
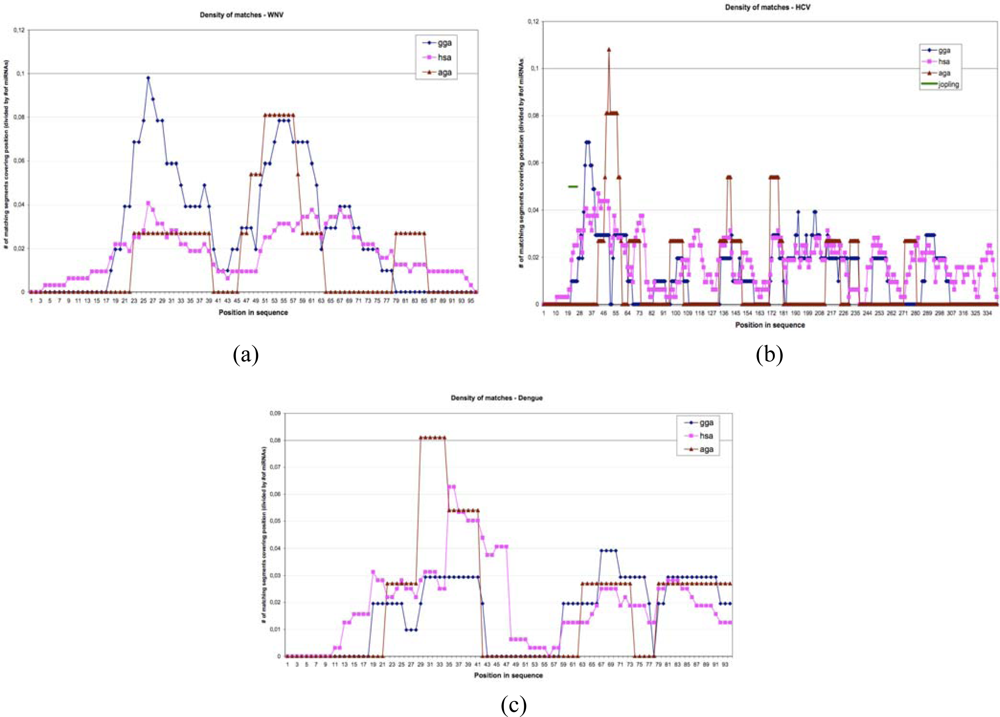
| miR | Virus | Number | Extr | Match | start | end | start | end | Aligned sequence |
|---|---|---|---|---|---|---|---|---|---|
| miR-34 | Jap. encephalitis | - | 3− | 8 | 3 | 10 | 184 | 177 | aatcagct |
| miR-34 | Jap. encephalitis | rAT | 3+ | 8 | 4 | 11 | 522 | 529 | atcagcta |
| miR-34 | Jap. encephalitis | - | 3− | 7 | 9 | 15 | 19 | 13 | ctaacta |
| miR-34 | Yellow fever | - | 3− | 8 | 16 | 23 | 234 | 227 | cactgcct |
| miR-34 | Pestivirus | - | 3+ | 7 | 6 | 12 | 101 | 107 | cagctaa |
| miR-34 | Pestivirus | - | 3− | 7 | 6 | 12 | 170 | 164 | cagctaa |
| miR-34 | Pestivirus | - | 3+ | 8 | 12 | 19 | 145 | 152 | actacact |
| miR-34 | Pestivirus | - | 3− | 7 | 6 | 12 | 174 | 168 | cagctaa |
| miR-34 | Hepacivirus | H77-pH21 | 5+ | 7 | 17 | 23 | 288 | 294 | actgcct |
| miR-34 | Hepacivirus | 1b | 5+ | 7 | 17 | 23 | 288 | 294 | actgcct |
| miR-34 | Hepacivirus | JHF-1 | 5+ | 7 | 17 | 23 | 287 | 293 | actgcct |
| miR-34 | Hepacivirus | JHF-1 | 3+ | 8 | 7 | 14 | 27 | 34 | agctaact |
| miR-34 | Uncl. Hepacivirus | JPUT971017 | 5+ | 7 | 17 | 23 | 288 | 294 | actgcct |
| miR-34 | Uncl. Hepacivirus | JPUT971017 | 3+ | 7 | 13 | 19 | 16 | 22 | ctacact |
| miR-34 | Hepacivirus | 3a | 5+ | 7 | 17 | 23 | 286 | 292 | actgcct |
| miR-34 | Flaviviridae | b | 5+ | 7 | 17 | 23 | 392 | 398 | actgcct |
| miR-34 | Flaviviridae | c | 3+ | 7 | 16 | 22 | 165 | 171 | cactgcc |
| miR-34 | Phlebovirus | segM | 3+ | 7 | 7 | 13 | 43 | 49 | agctaac |
| miR-34 | Phlebovirus | segM | 3− | 7 | 6 | 12 | 230 | 224 | cagctaa |
| miR-34 | Hepatovirus | - | 5− | 7 | 15 | 21 | 353 | 347 | acactgc |
| miR-143 | Dengue | 2 | 3− | 10 | 1 | 10 | 147 | 138 | tgagctacag |
| miR-143 | Dengue | 2 | 3− | 11 | 12 | 22 | 280 | 270 | gcttcatctca |
| miR-143 | Dengue | 3 | 3− | 7 | 8 | 14 | 273 | 267 | cagtgct |
| miR-143 | Dengue | 4 | 3− | 9 | 2 | 10 | 82 | 74 | gagctacag |
| miR-143 | Jap. encephalitis | - | 5+ | 7 | 14 | 20 | 6 | 12 | ttcatct |
| miR-143 | Jap. encephalitis | - | 3− | 10 | 12 | 21 | 252 | 243 | gcttcatctc |
| miR-143 | Jap. encephalitis | rAT | 3− | 7 | 3 | 9 | 558 | 552 | agctaca |
| miR-143 | Jap. encephalitis | - | 5+ | 7 | 7 | 13 | 63 | 69 | acagtgc |
| miR-143 | Jap. encephalitis | - | 3− | 7 | 3 | 9 | 248 | 242 | agctaca |
| miR-143 | Jap. encephalitis | - | 3+ | 8 | 4 | 13 | 305 | 314 | gctacagtgc (miR)
gcgacagtgc (virus) |
| miR-143 | Jap. encephalitis | - | 3− | 8 | 1 | 10 | 462 | 453 | tgagctacag (miR)
tgacctacag (virus) |
| miR-143 | Jap. encephalitis | - | 3− | 7 | 3 | 17 | 51 | 37 | agctacagtgcttca (miR)
agctaaacttctaca (virus) |
| miR-143 | Tick-borne enc. | w | 3+ | 7 | 1 | 7 | 236 | 242 | tgagcta |
| miR-143 | Tick-borne enc. | w | 3+ | 8 | 11 | 18 | 262 | 269 | tgcttcat |
| miR-143 | Tick-borne enc. | w | 3− | 7 | 10 | 16 | 590 | 584 | gtgcttc |
| miR-143 | Tick-borne enc. | w | 3− | 8 | 11 | 18 | 379 | 372 | tgcttcat |
| miR-143 | Pestivirus | - | 5− | 7 | 6 | 12 | 353 | 347 | tacagtg |
| miR-143 | Hepacivirus | JPUT | 3+ | 7 | 3 | 9 | 14 | 20 | agctaca |
| miR-143 | Uncl. Flaviviridae | a | 5+ | 7 | 4 | 10 | 444 | 450 | gctacag |
| miR-143 | Uncl. Flaviviridae | b | 5− | 9 | 5 | 13 | 268 | 260 | ctacagtgc |
| miR-143 | Uncl. Flaviviridae | c | 3+ | 7 | 7 | 13 | 248 | 254 | acagtgc |
| miR-143 | Uncl. Flaviviridae | c | 3+ | 7 | 14 | 20 | 7 | 13 | ttcatct |
| miR-143 | Uncl. Flaviviridae | c | 3− | 7 | 7 | 13 | 259 | 253 | acagtgc |
| miR-143 | Phlebovirus | segM | 3+ | 7 | 9 | 15 | 180 | 186 | agtgctt |
| miR-143 | Phlebovirus | segS | 5+ | 7 | 9 | 15 | 16 | 22 | agtgctt |
| miR | Virus | Number | Extr | Match | start | end | start | end | Aligned sequence |
|---|---|---|---|---|---|---|---|---|---|
| miR-21 | Jap. encephalitis | - | 3+ | 11 | 2 | 12 | 549 | 559 | caacatcagtc |
| miR-422 | Uncl. Flaviviridae | c | 5+ | 12 | 9 | 20 | 420 | 431 | gactccaagtcc |
| miR-490 | Yellow fever | - | 3+ | 11 | 12 | 22 | 252 | 262 | cctccaggttg |
| miR-495 | Tick-borne enc. | w | 3− | 11 | 8 | 18 | 565 | 555 | tgcaccatgtt |
| miR-500 | Uncl. Flaviviridae | c | 5− | 11 | 9 | 19 | 315 | 305 | ttgcccaggtg |
| miR-511 | Hepacivirus | H77-pH21 | 3+ | 11 | 1 | 11 | 180 | 190 | tgactgcagag |
| miR-511 | Hepacivirus | 1b | 3+ | 11 | 1 | 11 | 190 | 200 | tgactgcagag |
| miR-511 | Hepacivirus | JHF-1 | 3+ | 11 | 1 | 11 | 194 | 204 | tgactgcagag |
| miR-511 | Hepacivirus | JPUT9710 | 3+ | 11 | 1 | 11 | 169 | 179 | tgactgcagag |
| miR-520 | Dengue | 4 | 3+ | 11 | 5 | 15 | 15 | 25 | caccaaagaga |
| miR-9 | Pestivirus | - | 3− | 11 | 1 | 11 | 175 | 165 | tcatacagcta |
| miR-93 | Pestivirus | - | 3+ | 11 | 12 | 22 | 159 | 169 | aacagcacttt |
| Let-7 | Pestivirus | csfv | 3+ | 11 | 4 | 14 | 121 | 131 | gtacaaactac |
| miR-143 | Dengue | 2 | 3− | 11 | 12 | 22 | 280 | 270 | gcttcatctca |
| miR-453 | Pestivirus | bvdb | 5− | 12 | 1 | 12 | 160 | 149 | cgaactcaccac |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Demongeot, J.; Glade, N.; Moreira, A.; Vial, L. RNA Relics and Origin of Life. Int. J. Mol. Sci. 2009, 10, 3420-3441. https://doi.org/10.3390/ijms10083420
Demongeot J, Glade N, Moreira A, Vial L. RNA Relics and Origin of Life. International Journal of Molecular Sciences. 2009; 10(8):3420-3441. https://doi.org/10.3390/ijms10083420
Chicago/Turabian StyleDemongeot, Jacques, Nicolas Glade, Andrés Moreira, and Laurent Vial. 2009. "RNA Relics and Origin of Life" International Journal of Molecular Sciences 10, no. 8: 3420-3441. https://doi.org/10.3390/ijms10083420