Hedgehog Signaling Regulates the Survival of Gastric Cancer Cells by Regulating the Expression of Bcl-2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
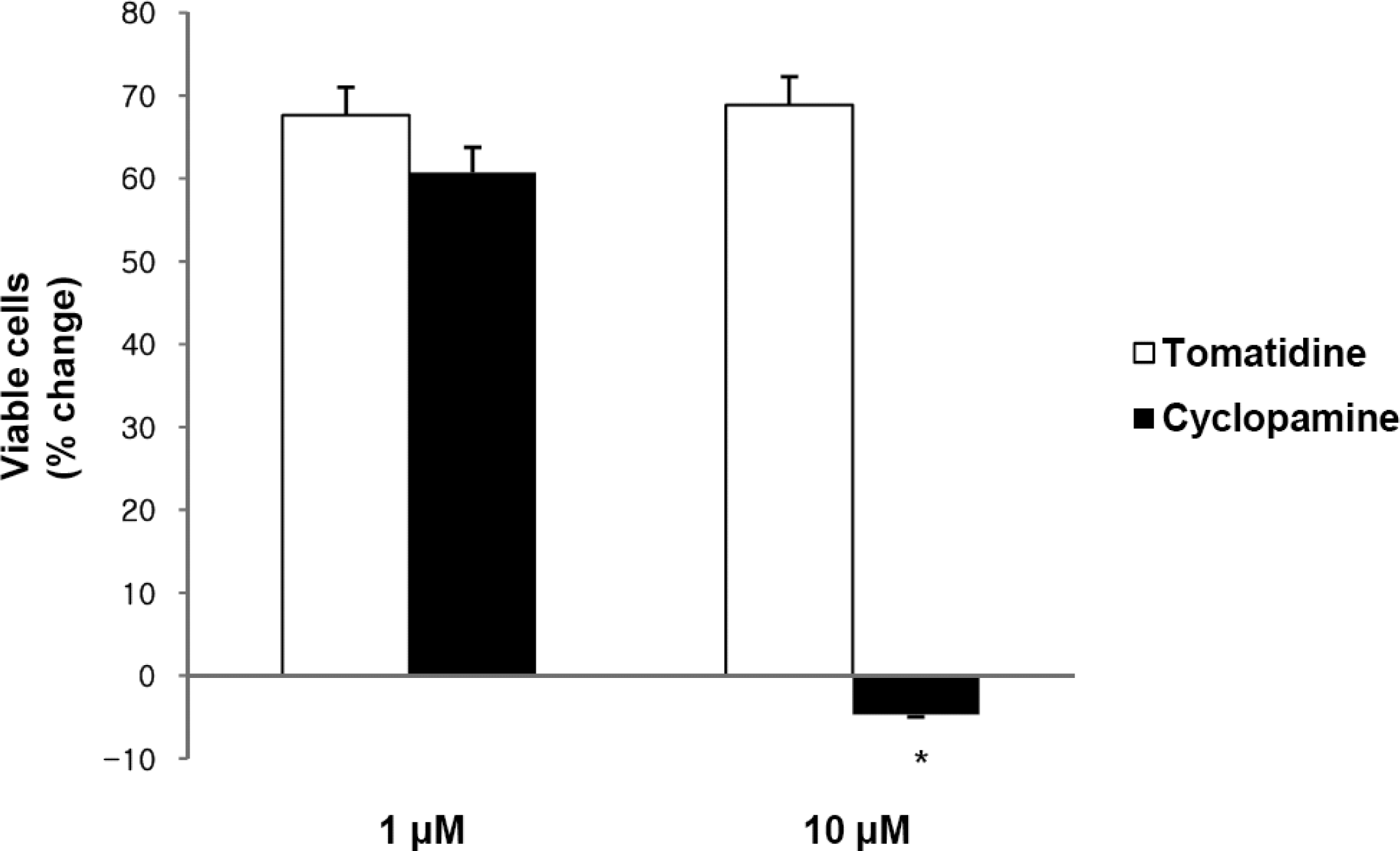
2.1. Hh Signaling Regulates the Proliferation of Gastric Cancer Cells
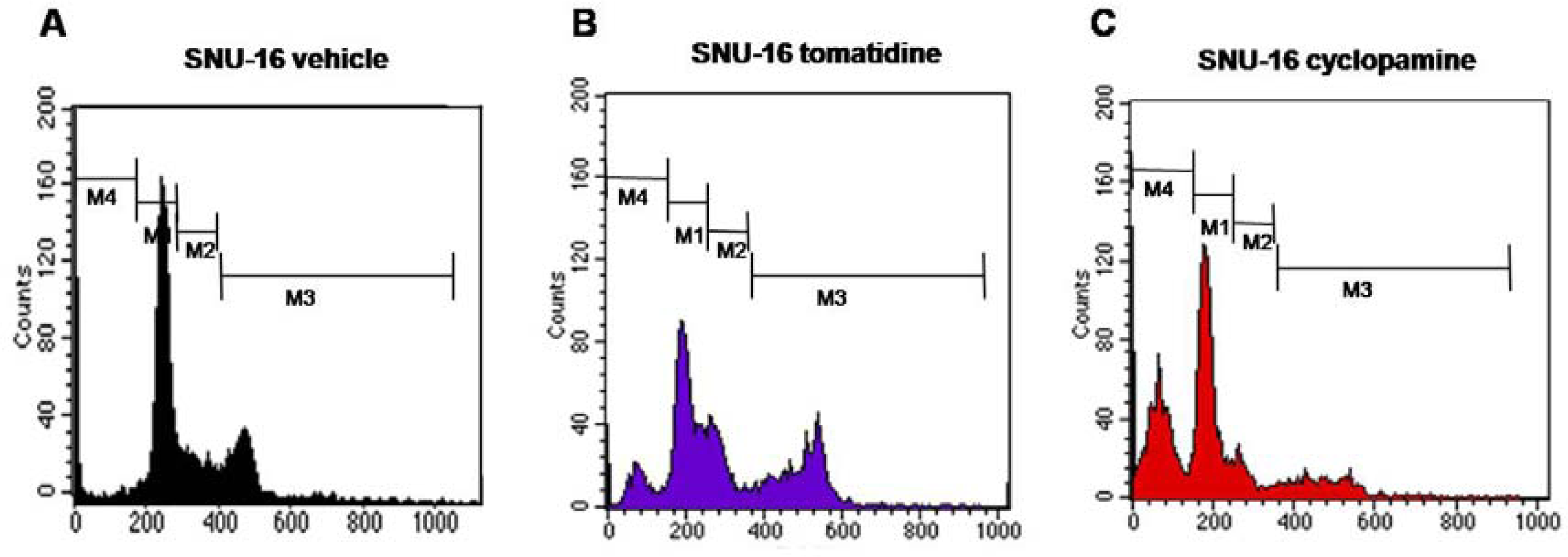
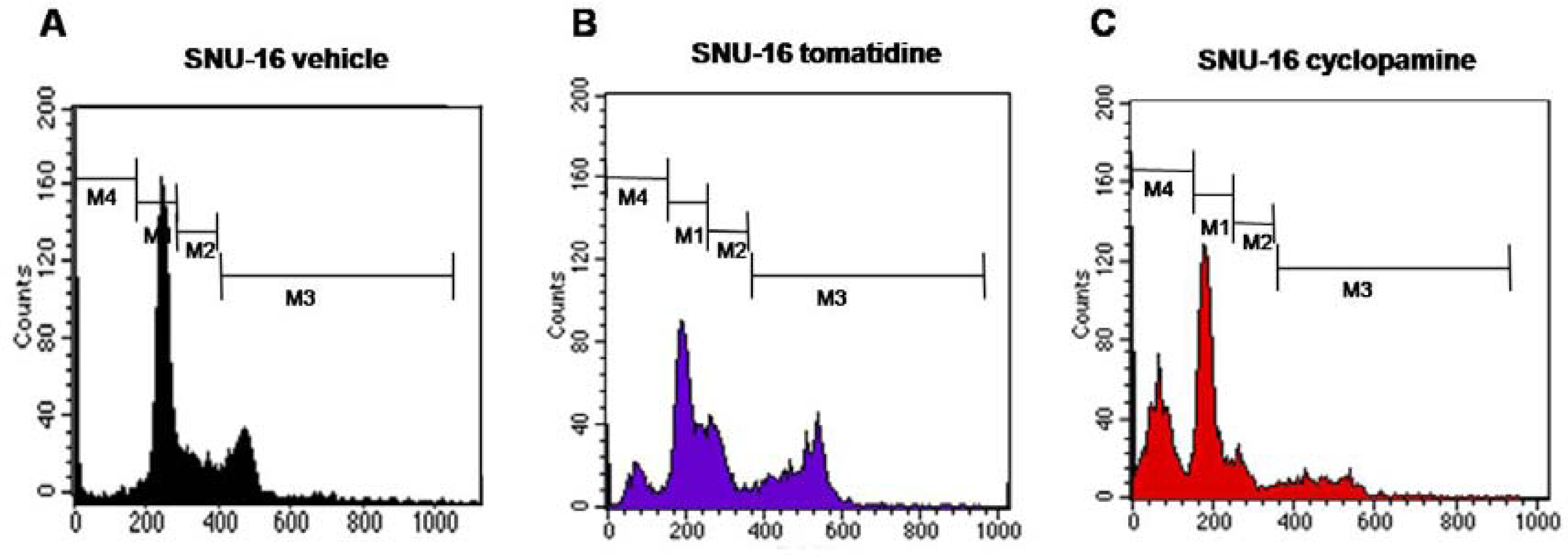
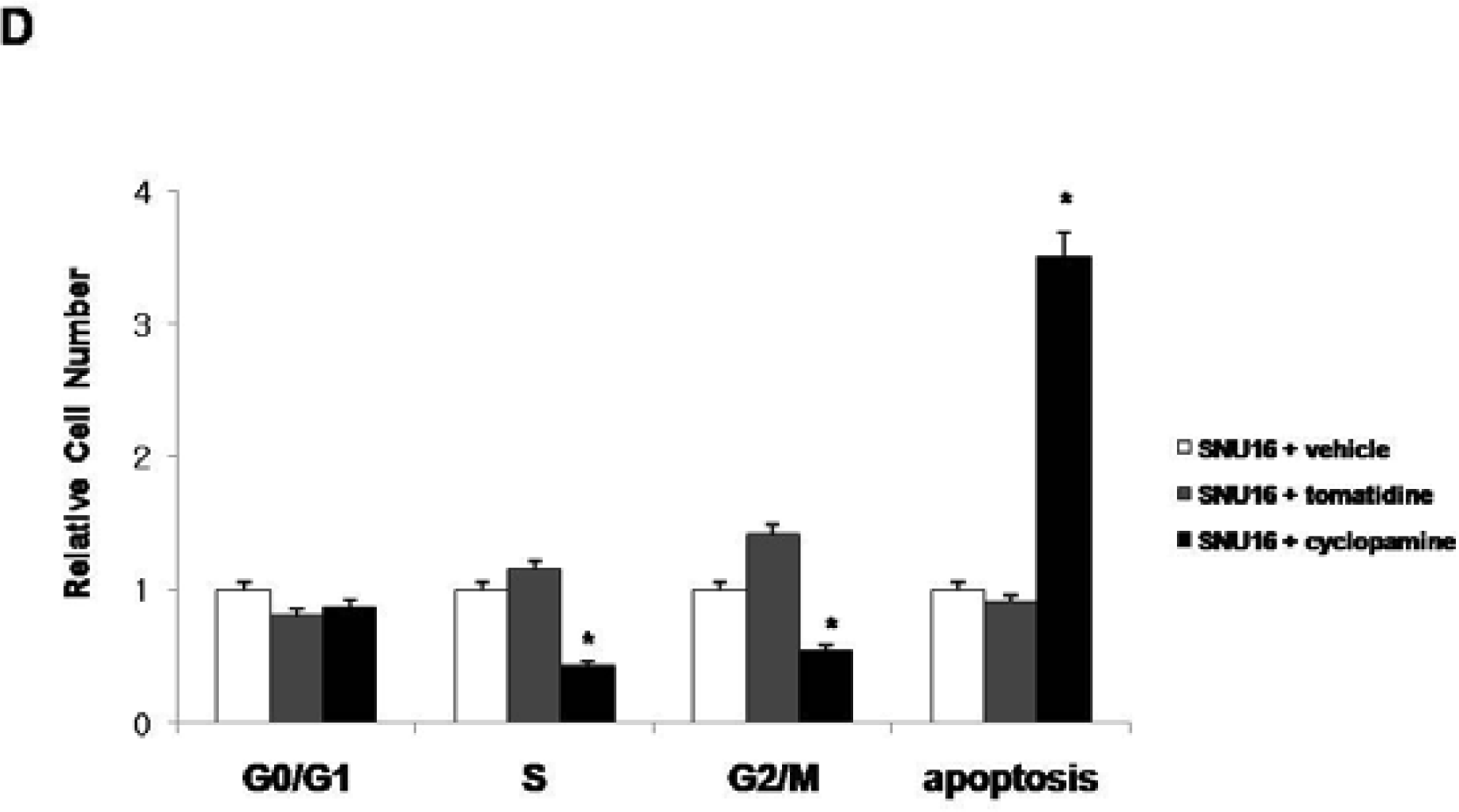
2.2. Blockade of Hh Signaling Induces Apoptosis of Gastric Cancer Cells
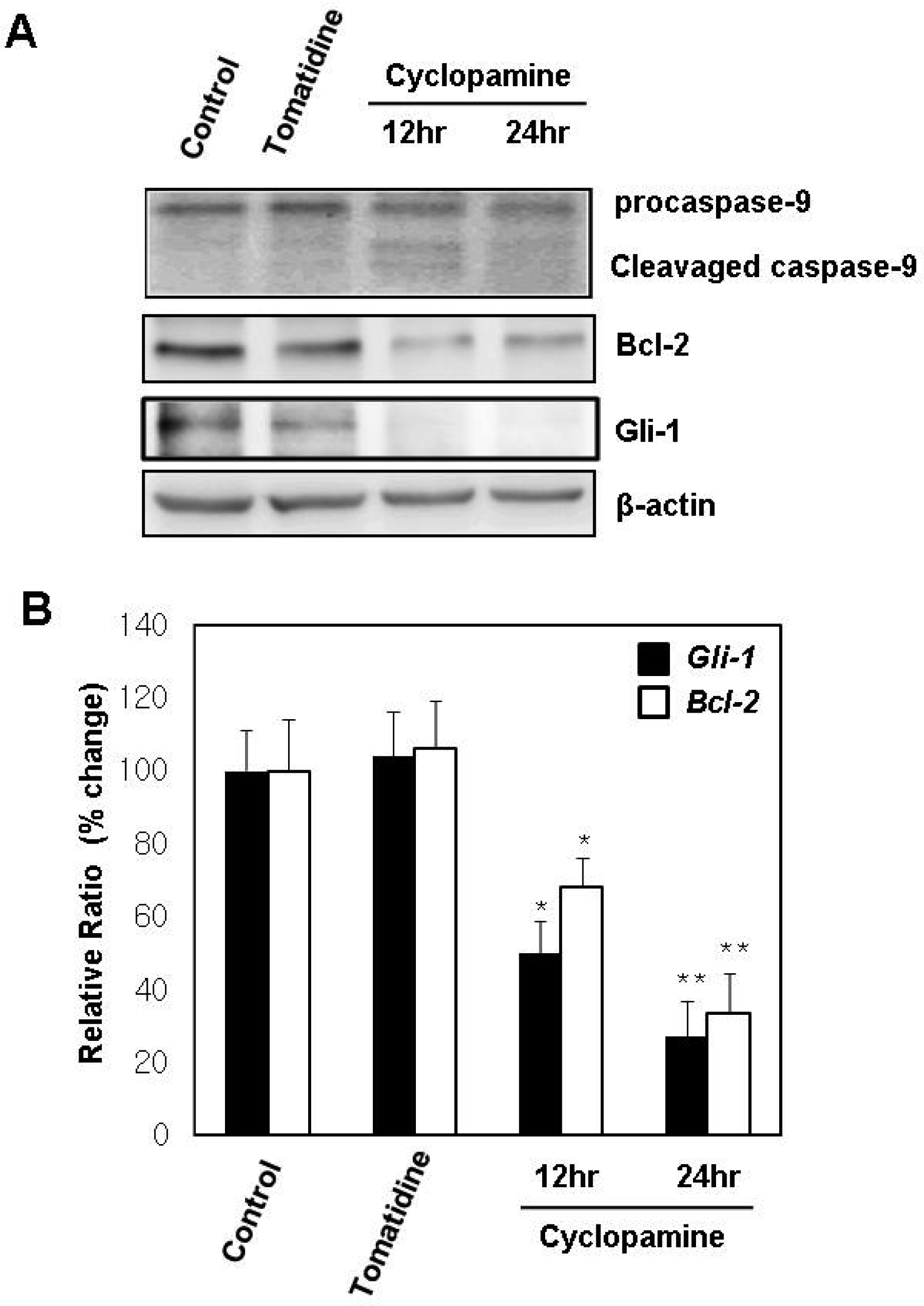
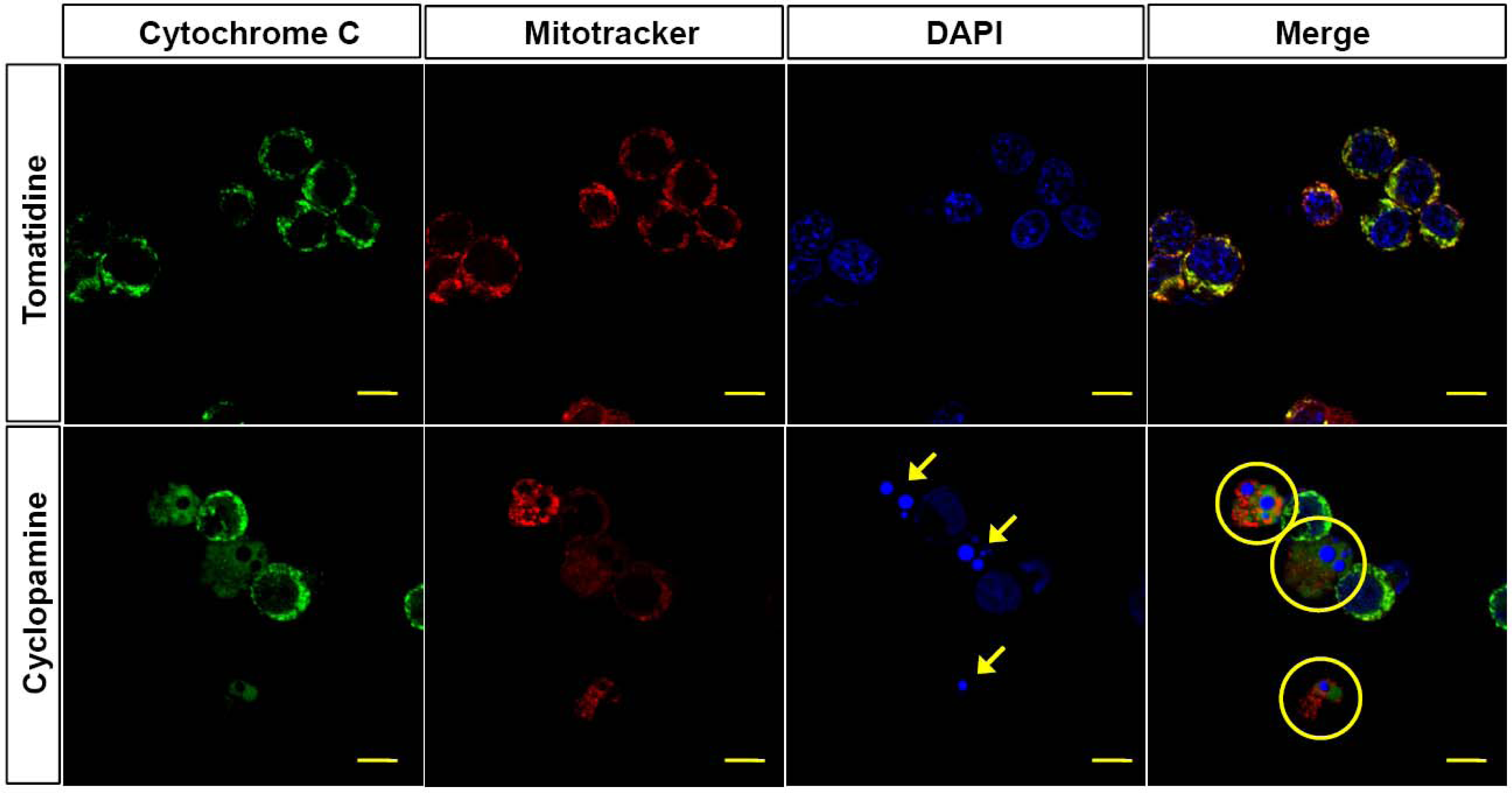
2.3. Mitochondria are Involved in the Cyclopamine-Induced Apoptosis of Gastric Cancer Cells
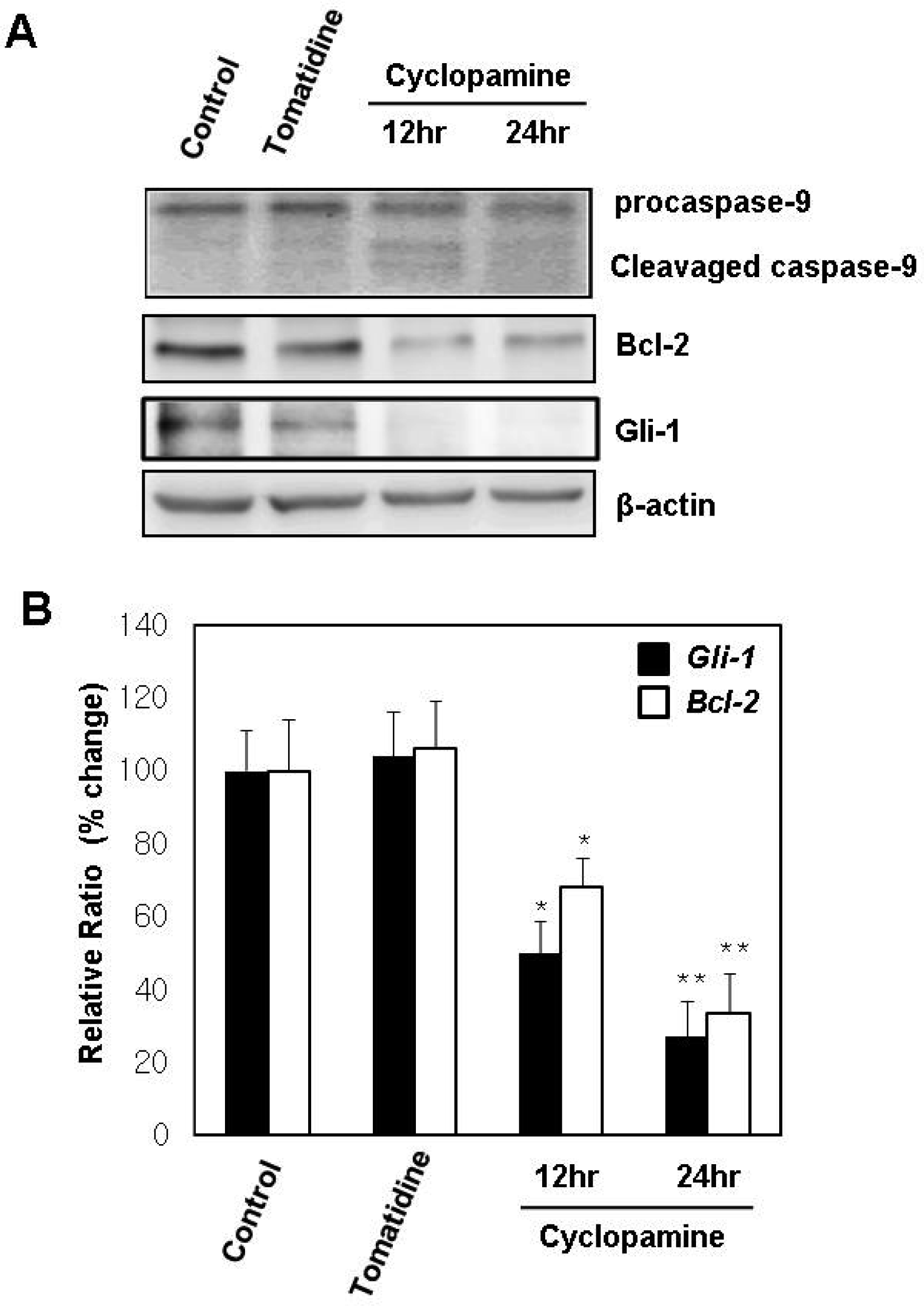
2.4. Hh Signaling Regulates the Expression of Bcl-2
3. Discussion
4. Experimental Section
4.1. Cell Line
4.2. Proliferation Assays
4.3. Flow Cytometry
4.4. Nuclear Morphology and Immunocytochemistry
4.5. Western Blot
4.6. Real-Time RT-PCR
4.7. Data Analysis
5. Conclusions
Acknowledgments
References and Notes
- Yuasa, Y. Control of gut differentiation and intestinal-type gastric carcinogenesis. Nat. Rev. Cancer 2003, 3, 592–600. [Google Scholar]
- Vogiatzi, P; Vindigni, C; Roviello, F; Renieri, A; Giordano, A. Deciphering the underlying genetic and epigenetic events leading to gastric carcinogenesis. J. Cell. Physiol 2007, 211, 287–295. [Google Scholar]
- Ahn, J; Murphy, M; Kratowicz, S; Wang, A; Levine, AJ; George, DL. Down-regulation of the stathmin/Op18 and FKBP25 genes following p53 induction. Oncogene 1999, 18, 5954–5958. [Google Scholar]
- Wu, CW; Hsieh, MC; Lo, SS; Lui, WY; P'Eng, FK. Results of curative gastrectomy for carcinoma of the distal third of the stomach. J. Am. Coll. Surg 1996, 183, 201–207. [Google Scholar]
- Wu, CW; Hsieh, MC; Lo, SS; Tsay, SH; Li, AF; Lui, WY; P'Eng, FK. Prognostic indicators for survival after curative resection for patients with carcinoma of the stomach. Digest. Dis. Sci 1997, 42, 1265–1269. [Google Scholar]
- Berman, DM; Karhadkar, SS; Hallahan, AR; Pritchard, JI; Eberhart, CG; Watkins, DN; Chen, JK; Cooper, MK; Taipale, J; Olson, JM; Beachy, PA. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002, 297, 1559–1561. [Google Scholar]
- Berman, DM; Karhadkar, SS; Maitra, A; Montes De Oca, R; Gerstenblith, MR; Briggs, K; Parker, AR; Shimada, Y; Eshleman, JR; Watkins, DN; Beachy, PA. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–581. [Google Scholar]
- Karhadkar, SS; Bova, GS; Abdallah, N; Dhara, S; Gardner, D; Maitra, A; Isaacs, JT; Berman, DM; Beachy, PA. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar]
- Thayer, SP; di Magliano, MP; Heiser, PW; Nielsen, CM; Roberts, DJ; Lauwers, GY; Qi, YP; Gysin, S; Fernandez-del Castillo, C; Yajnik, V; Antoniu, B; McMahon, M; Warshaw, AL; Hebrok, M. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar]
- Dahmane, N; Ruiz, I; Altaba, A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 1999, 126, 3089–3100. [Google Scholar]
- Wechsler-Reya, RJ; Scott, MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 1999, 22, 103–114. [Google Scholar]
- Chen, JK; Taipale, J; Cooper, MK; Beachy, PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev 2002, 16, 2743–2748. [Google Scholar]
- Taipale, J; Chen, JK; Cooper, MK; Wang, B; Mann, RK; Milenkovic, L; Scott, MP; Beachy, PA. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar]
- Iannolo, G; Conticello, C; Memeo, L; De Maria, R. Apoptosis in normal and cancer stem cells. Crit Rev Oncol Hematol 2008, 66, 42–51. [Google Scholar]
- Lorenzo, HK; Susin, SA. Therapeutic potential of AIF-mediated caspase-independent programmed cell death. Drug Resist. Updat 2007, 10, 235–255. [Google Scholar]
- Klein, S; McCormick, F; Levitzki, A. Killing time for cancer cells. Nat. Rev. Cancer 2005, 5, 573–580. [Google Scholar]
- Oliver, L; Vallette, FM. The role of caspases in cell death and differentiation. Drug Resist. Updat 2005, 8, 163–170. [Google Scholar]
- Qiao, L; Wong, BC. Targeting apoptosis as an approach for gastrointestinal cancer therapy. Drug Resist. Updat 2009, 12, 55–64. [Google Scholar]
- Degterev, A; Boyce, M; Yuan, J. A decade of caspases. Oncogene 2003, 22, 8543–8567. [Google Scholar]
- Green, DR. Apoptotic pathways: ten minutes to dead. Cell 2005, 121, 671–674. [Google Scholar]
- Boise, LH; Gonzalez-Garcia, M; Postema, CE; Ding, L; Lindsten, T; Turka, LA; Mao, X; Nunez, G; Thompson, CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar]
- Yang, E; Zha, J; Jockel, J; Boise, LH; Thompson, CB; Korsmeyer, SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar]
- Guo, B; Godzik, A; Reed, JC. Bcl-G, a novel pro-apoptotic member of the Bcl-2 family. J. Biol. Chem 2001, 276, 2780–2785. [Google Scholar]
- Antonsson, B; Martinou, JC. The Bcl-2 protein family. Exp. Cell. Res 2000, 256, 50–57. [Google Scholar]
- Romer, JT; Kimura, H; Magdaleno, S; Sasai, K; Fuller, C; Baines, H; Connelly, M; Stewart, CF; Gould, S; Rubin, LL; Curran, T. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer Cell 2004, 6, 229–240. [Google Scholar]
- Bigelow, RL; Chari, NS; Unden, AB; Spurgers, KB; Lee, S; Roop, DR; Toftgard, R; McDonnell, TJ. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. J. Biol. Chem 2004, 279, 1197–1205. [Google Scholar]
- Regl, G; Kasper, M; Schnidar, H; Eichberger, T; Neill, GW; Philpott, MP; Esterbauer, H; Hauser-Kronberger, C; Frischauf, AM; Aberger, F. Activation of the BCL2 promoter in response to Hedgehog/GLI signal transduction is predominantly mediated by GLI2. Cancer Res 2004, 64, 7724–7731. [Google Scholar]
- Rodriguez-Villanueva, J; Colome, MI; Brisbay, S; McDonnell, TJ. The expression and localization of bcl-2 protein in normal skin and in non-melanoma skin cancers. Pathol. Res. Pract 1995, 191, 391–398. [Google Scholar]
- Delehedde, M; Cho, SH; Sarkiss, M; Brisbay, S; Davies, M; El-Naggar, AK; McDonnell, TJ. Altered expression of bcl-2 family member proteins in nonmelanoma skin cancer. Cancer 1999, 85, 1514–1522. [Google Scholar]
- Rodriguez-Villanueva, J; Greenhalgh, D; Wang, XJ; Bundman, D; Cho, S; Delehedde, M; Roop, D; McDonnell, TJ. Human keratin-1.bcl-2 transgenic mice aberrantly express keratin 6, exhibit reduced sensitivity to keratinocyte cell death induction, and are susceptible to skin tumor formation. Oncogene 1998, 16, 853–863. [Google Scholar]
- Guo, JY; Huo, HR; Zhao, BS; Liu, HB; Li, LF; Ma, YY; Guo, SY; Jiang, TL. Cinnamaldehyde reduces IL-1beta-induced cyclooxygenase-2 activity in rat cerebral microvascular endothelial cells. Eur. J. Pharmacol 2006, 537, 174–180. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Han, M.-E.; Lee, Y.-S.; Baek, S.-Y.; Kim, B.-S.; Kim, J.-B.; Oh, S.-O. Hedgehog Signaling Regulates the Survival of Gastric Cancer Cells by Regulating the Expression of Bcl-2. Int. J. Mol. Sci. 2009, 10, 3033-3043. https://doi.org/10.3390/ijms10073033
Han M-E, Lee Y-S, Baek S-Y, Kim B-S, Kim J-B, Oh S-O. Hedgehog Signaling Regulates the Survival of Gastric Cancer Cells by Regulating the Expression of Bcl-2. International Journal of Molecular Sciences. 2009; 10(7):3033-3043. https://doi.org/10.3390/ijms10073033
Chicago/Turabian StyleHan, Myoung-Eun, Young-Suk Lee, Sun-Yong Baek, Bong-Seon Kim, Jae-Bong Kim, and Sae-Ock Oh. 2009. "Hedgehog Signaling Regulates the Survival of Gastric Cancer Cells by Regulating the Expression of Bcl-2" International Journal of Molecular Sciences 10, no. 7: 3033-3043. https://doi.org/10.3390/ijms10073033