Physiological and Pathological Role of Alpha-synuclein in Parkinson’s Disease Through Iron Mediated Oxidative Stress; The Role of a Putative Iron-responsive Element

Abstract
:1. Parkinson’s Disease: Clinical Profile, Pathophysiology and Treatments
1.1. Clinical profile
1.2. Pathophysiology
1.3. Treatments (see Table 1)
2. alpha-Synuclein: Relationship with PD. Biochemical and Biological Properties
2.1. Relationship with PD
2.2. Biochemical and biological properties
2.2.1. Posttranslational modifications
2.2.1.1. Phosphorylation
2.2.1.2. Nitration
2.2.1.3. Ubiquitination
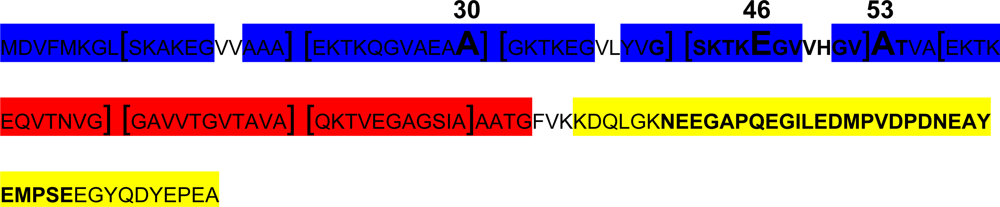
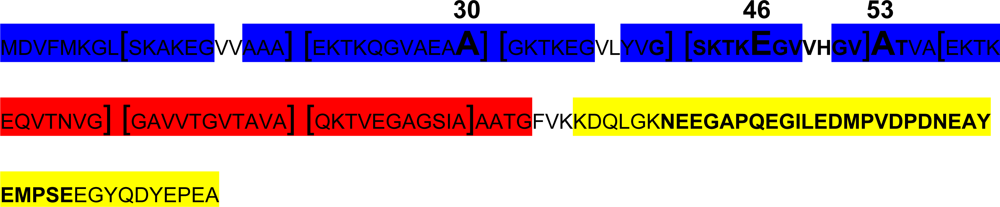
2.2.2. α-Synuclein alternative splicing
2.2.3. Vesicle trafficking regulation
2.2.4. Interaction with other proteins
2.2.5. Chaperone activity
3. Pathogenic Mechanisms of Alpha-Synuclein. Factors Affecting Fibrillization. Role of Iron in Oxidative Stress
3.1. Conformational states of α-synuclein
- The intrinsic unfolded state under physiologic conditions, both in vitro and in vivo.
- The pre-molten globule state, a compact but incompletely folded state of proteins that contains most of the secondary structure but lacks tertiary interactions [97], and is predominant under conditions such as low pH, high temperature, several metal ions [98], several salts [98], and several common pesticides/herbicides [99]. It is stabilized as result of spontaneous oligomerization both in vivo and in vitro [100]. It is thought that the negative electrostatical potential and locally low pH in the vicinity of the membrane surface induces protein conformation to molten globules. Early stages of fibrillization involve the partial folding of α-syn into the highly fibrillization-prone pre-molten globule conformation, which represents a key intermediate along the fibrillization pathway [101].
- The α-helical membrane-bound form in the N-terminal fragment, while the glutamate-rich C-terminal region remains unstructured.
- The β-sheet state: it has been observed that under certain conditions α-syn acquires a β-pleated sheet, which is very prone to form amorphous aggregates [102].
- Dimers: α-syn is able to form morphologically distinct oligomers, for example under high temperature [100], where dimers are formed first, and aggregates. The formation of oxidative dimers and high-order oligomers with covalent di-tyrosine cross-links under conditions of oxidative stress has also been reported [74].
- Oligomers: nitrated α-syn assembles into spherical oligomers [100]. Incubation of α-syn with several metals gave rise to different classes of oligomers: Cu2+, Fe3+ and Ni2+ yielded 0.8–4 nm spherical particles, similar to those formed by incubation of α-syn alone; Mg2+, Cd2+ and Zn2+ yielded larger (5–8 nm) spherical oligomers; and Co2+ and Ca2+ produced ring oligomers with diameters between 70–90 nm for the former and 22–30 nm in the case of the latter [103]. It has been observed that the earliest form of α-syn protofibrils appeared to be mainly spherical [35]. The incubation of spherical α-syn oligomers with brain-derived membranes has been shown to produce pore-like ring-type protofibrils [103], which may disturb ionic gradients in cells. This conjecture was supported by showing that α-syn oligomers (and not monomeric or filamentous α-syn) enhanced the membrane permeability for Ca2+, an important subcellular messenger in liposomes [104]. The role for membrane permeabilization by α-syn in vivo is not clear and a large number of pores could lead to cell lysis, but even a subtle ionic disturbance could lead to neuronal dysfunction, death and degeneration.
- Insoluble aggregates: finally, α-syn have been shown to assemble into large, insoluble aggregates with two distinct morphologies (amorphous aggregates and fibrils), with a high amount of β-sheet structure.
3.2. Factors affecting α-syn fibrillization
- Oxidation: the exposure of α-syn to oxidative agents induces the formation of high-order oligomers [74], and both the familial Parkininsonian A30P and A53T mutants have shown an even higher rate of self-assembly [108], providing support for the hypothesis that an impairment of cellular antioxidative mechanisms and/or overproduction of reactive oxygen species (ROS) may cause the initiation and progression of neurodegenerative synucleinopathies [109]. However, all amino acids are susceptible to oxidation [110], methionine being one of the easiest to undergo oxidation to form methionine sulfoxide (MetO). Thus, contrary to expectations, under mild oxidative conditions, when all four Met in α-syn are oxidized to MetO, the oxidized α-syn was found to be more unfolded than the non-oxidized form and less prone to oligomerize and fibrillate. Indeed, it even proved able to inhibit the fibrillization of non-modified α-syn [111].DA metabolism in nigrostratial neurons can produce ROS and contribute to lipid peroxidation, DNA damage, impairment of mitochondrial function, depletion of reduced glutathione (GSH), and, finally, cell death [112]. Over-expression of α-syn, and especially its mutant forms, enhances the vulnerability of neurons to DA-induced cell death through massive generation of ROS [113]. It has been proved that conjugation of DA with α-syn impedes the protofibril-to-fiber transition and, therefore, potentially more cytotoxic protofibrils may accumulate [114]. Transfection of wt- α-syn, A30P or A53T mutants has been reported to trigger apoptosis of cultured dopaminergic neurons, whereas there was an increase in survival of non-dopaminergic neurons [65]. Inhibition of DA synthesis by blocking TH activity prevented α-syn induced apoptosis.Damage caused by DA species is mediated by DA auto-oxidation catalyzed by free metals (especially Fe) [115], yielding 6-hydroxydopamine (6-OHDA) or through enzymatic deamination by MAOs to form toxic DA metabolites and hydrogen peroxide (H2O2) [116]. Levels of MAO-B appear to be highest in the substantia nigra. H2O2 produced as a by-product of DA oxidation and as normal oxygen reduction by MAOs is highly permeable and cannot be converted to water by GSH peroxidase due the low level of available GSH as a reducer [117], allowing H2O2 to potentially diffuses out of dopaminergic neurons and damage the neighbouring neurons. Under normal conditions, ROS are kept under control by an efficient antioxidant cascade. This includes the cytosolic copper-zinc superoxide dismutase and the mitochondrial manganese superoxide dismutase, which convert superoxide to oxygen and H2O2. The latter, in turn, is removed by catalases and peroxidases. These enzymes are key to scavenge ROS generated by oxidative insults. For example, in a transgenic murine model that overexpressed Cu/Zn superoxide dismutase or GSH peroxidase and was treated with pesticide paraquat, which causes a PD-like profile, the transgenic animals did not show alterations as reductions in locomotor activity, levels of striatal DA and metabolites, or dopaminergic neurons in the substantia nigra, unlike non-transgenic controls in which all of these were affected [118].Soluble α-syn is able to interact with the DA transporter (DAT) through the NAC domain [119], decreasing the amount of DAT in the PM, to allow for an optimal moderate level of synaptic DA reuptake to be accumulated into vesicles. In the event of α-syn aggregation, a decrease in the level of soluble α-syn results, and leads to the increased PM accumulation of DAT, giving rise to a massive entry of DA into the cell and consequent potential generation of ROS [116]. In accordance, the much more neurotoxic A53T mutant (but not A30P) interacts very weakly with DAT and causes an impairment of vesicular DA storage and release [119].Interestingly, the dopaminergic neurotoxin MPTP, which causes a PD-like neurodegeneration in rodents, humans and primates, enters into the cell through the DAT as its ionic metabolite MPP+. It then targets the mitochondria, inhibits complex I of the electron transport chain, impairs ATP production, induces a loss of mitochondrial membrane potential allowing the release of cytochrome c and generation of ROS, and additionally, increases α-syn mRNA and protein levels. The A53T mutant can enhance the vulnerability of cells to MPP+, while α-syn null-mice are resistant to MPP+-induced degeneration [120], signifying a fundamental role of α-syn in drug-mediated neurotoxicity.
- Interactions with polyanions: different glycosaminoglycans (GAGs) are involved in the formation of amyloid plaques in a variety of neurological disorders [121] and some highly sulphated GAGs (heparin and heparan-sulphate) as well as the proteoglycan agrin are able to bind to α-syn and stimulate its fibrillization in vitro. Furthermore, agrin and α-syn co-localize in LBs and LNs [122,123].
- Interaction with polycations: polycations, such as polyamines, are cellular stabilizers of nucleic acids and membranes, and are essential for growth and differentiation. Interaction with α-syn induced the partial folding of α-syn and, consequently, its oligomerization and fibrillization [124] by binding to the negatively charged C-terminus.
- Interaction with histones: some animal models of PD are created by administration of the pesticide paraquat to mice where it elevates mouse brain α-syn levels [125]. Additionally, paraquat promoted in vitro the overexpression and translocation of α-syn into the cell nucleus, where it can interact with highly basic histones to form complexes that trigger its aggregation, and may reflect aspects of the in vivo situation [124].
- α-Syn--α-syn crosslinking: tissue transglutaminase (tTG) catalyzes covalent crosslinking between reactive Lys and Glu residues [126]. Substrates for tTG include Ap, tau (another hallmark of AD) and the NAC fragment of α-syn, all of them are proteins that undergo aggregation in several neurodegenerative disorders. tTG catalyzes α-syn cross-linking, leading to the formation of high molecular weight aggregates in vitro [127], mainly associated with the membrane fraction. Increased levels of tTG have been reported in the substantia nigra of PD postmortem brains [128].
- Interaction with membranes: interaction with synaptic vesicles is one of the biological functions of α-syn. The membrane-bound fraction (about a 15% of total), was shown to have a high aggregation propensity and was able to seed aggregation of the cytosolic form of α-syn [55]. Furthermore, fatty acids and anionic lipids are potent inducers of α-syn fibrillization. In vitro association of soluble α-syn with lipid bilayers resulted in the formation of amorphous aggregates and filaments [129].
- Interaction with other proteins: several proteins have been found to interact with α-syn and some of them were shown to stimulate α-syn aggregation in vitro, including tau [130], brain-specific protein p25α [131], MAP-1B [132] and tubulin [133], all of which are components of LBs and/or GCIs, leading to cytoskeleton impairment. The mechanism of interaction remains unknown, but all contain basic motifs, suggesting that interaction could be through ionic bonds. On the other hand, transcriptional factors, such as NF-κB or Elk-1, have been found in LBs [134], suggesting that the sequestration of these factors in the cytosol may compromise the coordination of gene expression in degenerating cells. Similarly, high mobility group B-1 protein (HMGB-1), which is a nuclear DNA-binding protein that facilitates the interaction between DNA and transcriptional factors, has been demonstrated to bind directly with filamentous α-syn in vitro and it too is present in LBs [135], potentially disturbing gene expression. α-syn also interacts with other important signalling proteins, epitomized by PKC or ERKs, which can affect cellular viability.
- Proteins inhibiting α-synuclein aggregation:
- Chaperones: heat shock proteins (HSPs) are a family of chaperones induced by stress conditions. These proteins suppress protein aggregation and participate in refolding and/or degradation. Hsp70 and Hsp40 are components of LBs and/or GCIs and co-localize with α-syn. Overexpression of HSPs is able to suppress α-syn aggregation in vitro [136].
- β- and γ-synucleins: these proteins share some features with α-syn, but lack others. The α-and β-syn have a conserved C-terminus; however β-syn is deficient of 11 aa within the NAC region [137]. On the other hand, γ-syn lacks the Tyr rich C-terminal. All the three behave as typical natively unfolded proteins, but there is a little structural variability. β-syn has properties of a typical random coil, whereas α- and γ-syn are slightly more compact and structured [138]. Both are able to form fibrils, while β-syn is not, when incubated under the same conditions, however even β-syn can be forced to fibrillate in the presence of specific metals (Zn2+, Pb2+, Cu2+), pesticides [139] or by addition of GAGs. Interestingly, the addition of β- or γ-syn in a 1:1 ratio with α-syn increased the time duration of the lag phase and decreased the elongation phase of α-syn fibrillization [138], and was completely inhibited at a 4:1 ratio of an excess of β- or γ-syn over α-syn. This suggests that β- and γ-syn may be regulators of α-syn fibrillization in vivo [140], potentially acting as chaperones.
- Phosphorylation: as discussed above, α-syn undergoes extensive phosphorylation at Ser-129 in synucleinopathies and in ageing brains [141] and it is mostly unphosphorylated under normal conditions [71]. The specific phosphorylation at Ser-129 by CK2 resulted in oligomerization and fibrillization. Furthermore, oxidative stress has been described to enhance α-syn phosphorylation [72].
- Ubiquitin proteasome system (UPS) malfunction: mutated, misfolded or unassembled proteins are ubiquitinated to be degraded. There is evidence that UPS is impaired in several neurodegenerative diseases, including PD. For example, proteosomal subunits and ubiquitinated proteins have been found in LBs [142]. An inhibitory effect of α-syn aggregates on the hydrolytic activity of the 26S proteosome subunit in vitro has been reported [143], and a direct interaction between filaments and the 20S subunit has been shown. Accordingly, in transgenic animals, the inhibition of 20/26 S proteasome in substantia nigra led to α-syn accumulation and inclusion body formation, and resulted in a relatively selective degeneration of dopaminergic neurons [144]. Hsp70 expression can attenuate α-syn aggregation toxicity, by binding to α-syn filaments, abrogating its proteasomal inhibitory effect [145].
- Effect of A30P, A53T and E46K mutations: all these three mutants have been shown to accelerate α-syn oligomerization. While A53T and E46K increase fibril formation more rapidly than wt-α-syn and do not alter lipid-vesicle binding, suggesting that enhanced polymerization induces the disease in patients harbouring these mutations [129]. On the other hand, it has been reported that A30P fibrillates slowly, retarding significantly the formation of mature fibrils [35] and binds poorly to vesicles compared with the wt, maybe hindering axonal transport, leading to accumulation and aggregation, and accelerating the initial oligomerization of α-syn.
- C-terminal truncation: C-terminal truncated α-syn can increase α-syn-induced toxicity and aggregation ratio. Additionally, co-expression of full-length α-syn and the C-truncated form induced the formation of cytoplasmic inclusions and increased the susceptibility of cells to oxidative stress. This suggests that the C-terminus can play a role of an intramolecular chaperone by preventing α-syn from fibrillization [62]. Interstingly, C-terminally truncated A53T α-syn has been shown to induce the aggregation of full-length A53T protein faster than its wt counterpart, demonstrating that the mutation increases the accelerating effect that the truncated protein has on the aggregation of full-length α-syn [146].
- Interaction with metals: postmortem analysis of brain tissues from patients with PD confirm the presence of considerable amounts of metals, such as Fe, Zn and Al, in the substantia nigra as well as in LBs when compared with healthy age-matched controls [147,148]. α-syn can interact with several polycations, including Fe2+, Al3+, Zn2+, Cu2+, Mg2+ and Ca2+ [149], through the C-terminal domain, and this binding can catalyze protein aggregation. Additionally, some metals, such as Al [150], can induce a conformational change of α-syn from an unstructured to partially folded β-sheet structure intermediates and, lately, to fibrils [98]. Moreover, there is a shift in the Fe2+/Fe3+ ratio in favour of Fe3+, and a significant increase in the Fe3+-binding protein ferritin, together with a decrease in GSH content. Nevertheless, the concentration of metals necessary to induce aggregation is controversial. In general, the required concentration above the physiological values of the metals [151]. Hence, it is probable that metal-induced aggregation is carried out by oxidation of redox metals, rather than specific binding [152].
3.3. Overexpression of α-synuclein
3.4. Role of oxidative damage induced by Fe in PD
- Injection of Fe3Cl into the substantia nigra of rats has been reported to result in a selective decrease of striatal DA, which supports the assumption that Fe initiates dopaminergic neurodegeneration in PD. This decrease was prevented by infusion of the Fe chelator, desferrioxiamine [171].
- Neurodegeneration by 6-OHDA is selective for catecholamine neurons, and it is rapidly oxidized to yield cytotoxic catecholaminergic semiquinones and quinones with production of H2O2 and hydroxyl radicals. Iron-deficient rats are resistant to 6-OHDA neurotoxicity, suggesting that Fe could be a trigger [172]. Moreover, 6-OHDA-induced toxicity has been reversed by the Fe chelator, desferal [173]. These studies indicate that an enhancement of Fe concentration in substantia nigra may be upstream in neurodegenerative process associated with PD.
- By using MPTP injection in monkeys, a number of Fe chelators have been shown to attenuate MPTP oxidative toxicity, suggesting that Fe mediates or accentuates MPTP effects [174].
- Targeted deletion of the gene encoding IRP2 in mice causes a misregulation of Fe metabolism and neurodegeneration, leading to abnormal brain Fe deposition, ataxia, bradykinesia and tremors [175].
- Mutation in the gene that codifies for a ferritin subunit, resulted in disease similar to PD, termed neuroferritinopathy, which is characterized by Fe deposits, progressive neurodegeneration and axonal cyst formation with neurofilaments, ubiquitin and tau protein at the periphery, all of them components of LBs [176].
- Polymorphisms in five genes related to Fe homeostasis (Tf, TfR, frataxin, lactoferrin and haemochromatosis-related protein gene) have been linked to sporadic PD incidence, suggesting that there are variations in proteins involved in Fe metabolism that contribute to PD pathogenesis [177].
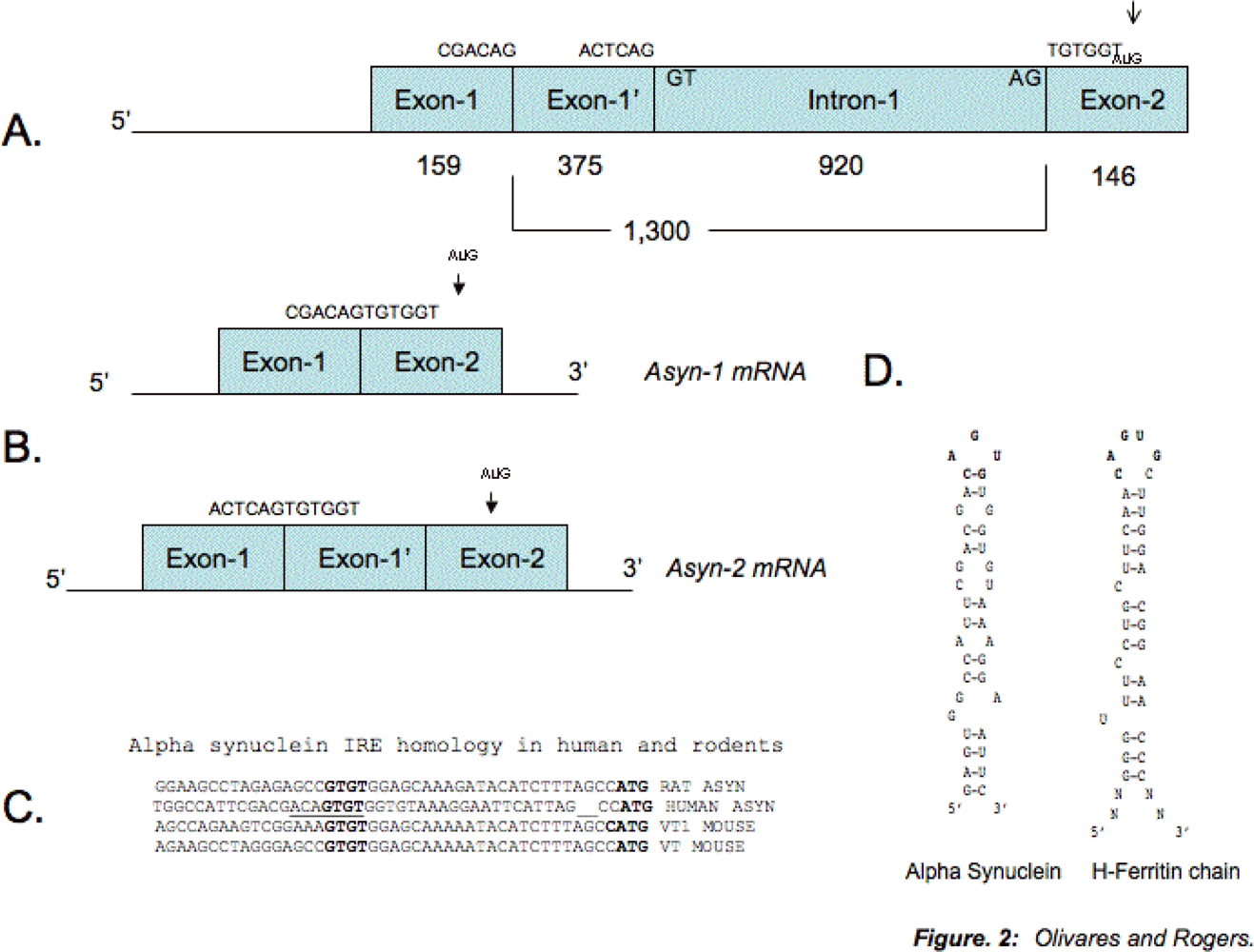
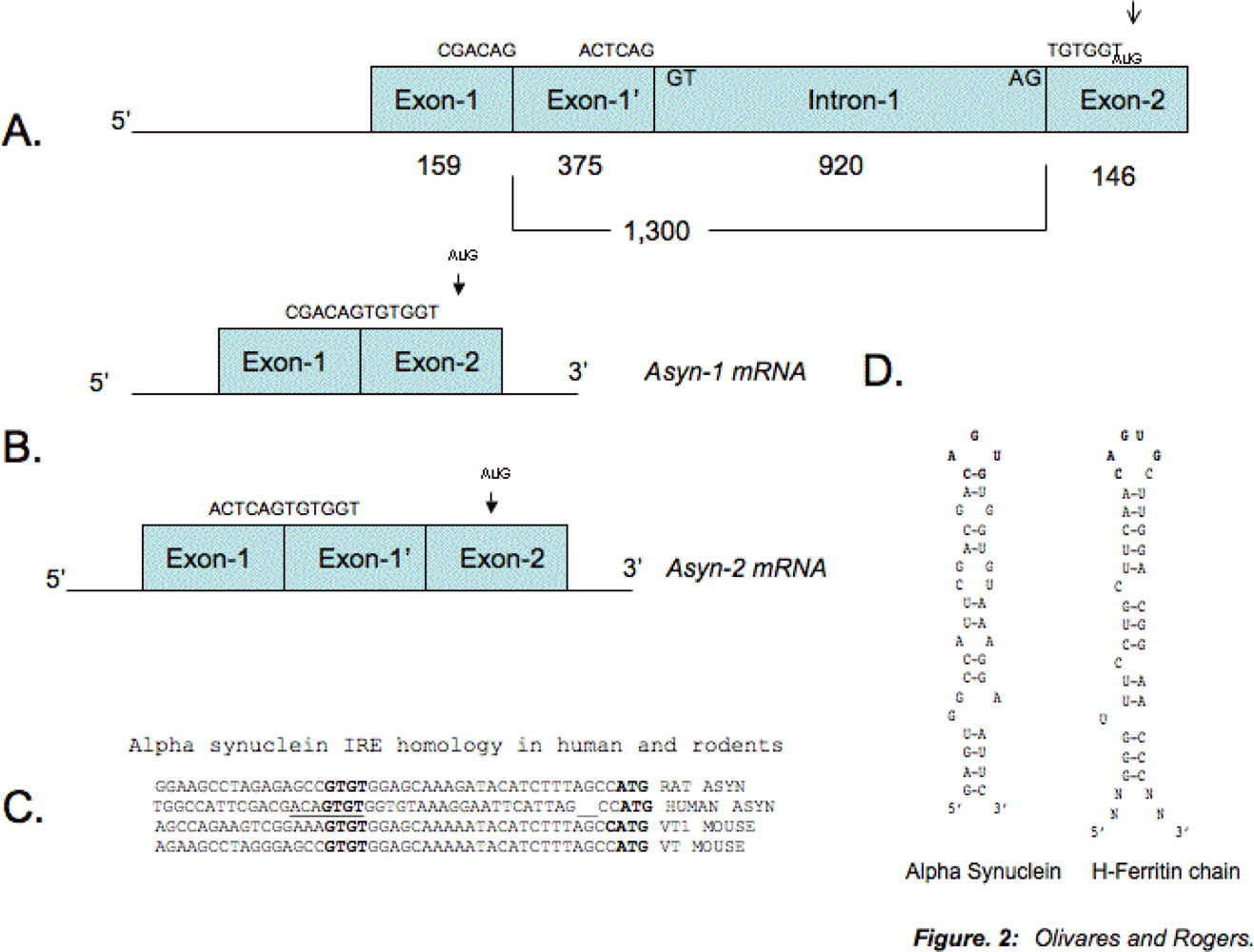
4. Protein-binding Control Sequences within the 5-UTR of the Alpha Synuclein Transcript
5. Conclusions
Acknowledgments
References
- de Rijk, MC; Tzourio, C; Breteler, MM; Dartigues, JF; Amaducci, L; Lopez-Pousa, S; Manubens-Bertran, JM; Alperovitch, A; Rocca, WA. Prevalence of parkinsonism and Parkinson’s disease in Europe: the EUROPARKINSON Collaborative Study. European Community Concerted Action on the Epidemiology of Parkinson’s disease. J Neurol Neurosurg Psychiat 1997, 62(1), 10–15. [Google Scholar]
- Guttman, M; Slaughter, PM; Theriault, ME; DeBoer, DP; Naylor, CD. Parkinsonism in Ontario: increased mortality compared with controls in a large cohort study. Neurology 2001, 57(12), 2278–2282. [Google Scholar]
- Inamdar, N; Arulmozhi, D; Tandon, A; Bodhankar, S. Parkinson’s disease: genetics and beyond. Curr Neuropharmacol 2007, 5(2), 99–113. [Google Scholar]
- Bennett, MC. The role of alpha-synuclein in neurodegenerative diseases. Pharmacol Ther 2005, 105(3), 311–331. [Google Scholar]
- Forman, MS; Lee, VM; Trojanowski, JQ. Nosology of Parkinson’s disease: looking for the way out of a quagmire. Neuron 2005, 47(4), 479–482. [Google Scholar]
- Yamamura, Y; Hattori, N; Matsumine, H; Kuzuhara, S; Mizuno, Y. Autosomal recessive early-onset parkinsonism with diurnal fluctuation: clinicopathologic characteristics and molecular genetic identification. Brain Dev 2000, 22(Suppl 1), S87–S91. [Google Scholar]
- Lee, VM; Trojanowski, JQ. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 2006, 52(1), 33–38. [Google Scholar]
- Miyasaki, JM; Martin, W; Suchowersky, O; Weiner, WJ; Lang, AE. Practice parameter: initiation of treatment for Parkinson’s disease: an evidence-based review: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2002, 58(1), 11–17. [Google Scholar]
- Mercuri, NB; Bernardi, G. The ‘magic’ of L-dopa: Why is it the gold standard Parkinson’s disease therapy? Trends Pharmacol Sci 2005, 26(7), 341–344. [Google Scholar]
- Guttman, M; Kish, SJ; Furukawa, Y. Current concepts in the diagnosis and management of Parkinson’s disease. CMAJ 2003, 168(3), 293–301. [Google Scholar]
- Verhagen Metman, L; Del Dotto, P; van den Munckhof, P; Fang, J; Mouradian, MM; Chase, TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson’s disease. Neurology 1998, 50(5), 1323–1326. [Google Scholar]
- Quinn, N; Parkes, D; Janota, I; Marsden, CD. Preservation of the substantia nigra and locus coeruleus in a patient receiving levodopa (2 kg) plus decarboxylase inhibitor over a four-year period. Mov Disord 1986, 1(1), 65–68. [Google Scholar]
- Rajput, AH; Fenton, M; Birdi, S; Macaulay, R. Is levodopa toxic to human substantia nigra? Mov Disord 1997, 12(5), 634–638. [Google Scholar]
- Ludin, HP. [Long-term therapy of the Parkinson syndrome]. Schweiz Med Wochenschr 1984, 114(33), 1131–1136. [Google Scholar]
- Stern, MB. The early treatment of Parkinson’s disease: levodopa, dopamine agonists or both. Parkinsonism Relat Disord 2000, 7(1), 27–33. [Google Scholar]
- Weiner, WJ; Factor, SA; Sanchez-Ramos, JR; Singer, C; Sheldon, C; Cornelius, L; Ingenito, A. Early combination therapy (bromocriptine and levodopa) does not prevent motor fluctuations in Parkinson’s disease. Neurology 1993, 43(1), 21–27. [Google Scholar]
- Rascol, O; Brooks, DJ; Korczyn, AD; De Deyn, PP; Clarke, CE; Lang, AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000, 342(20), 1484–1491. [Google Scholar]
- Carvey, PM; Pieri, S; Ling, ZD. Attenuation of levodopa-induced toxicity in mesencephalic cultures by pramipexole. J Neural Transm 1997, 104(2–3), 209–28. [Google Scholar]
- Clow, A; Freestone, C; Lewis, E; Dexter, D; Sandler, M; Glover, V. The effect of pergolide and MDL 72974 on rat brain CuZn superoxide dismutase. Neurosci Lett 1993, 164(1–2), 41–43. [Google Scholar]
- Felten, DL; Felten, SY; Fuller, RW; Romano, TD; Smalstig, EB; Wong, DT; Clemens, JA. Chronic dietary pergolide preserves nigrostriatal neuronal integrity in aged-Fischer-344 rats. Neurobiol Aging 1992, 13(2), 339–351. [Google Scholar]
- Ogawa, N; Tanaka, K; Asanuma, M; Kawai, M; Masumizu, T; Kohno, M; Mori, A. Bromocriptine protects mice against 6-hydroxydopamine and scavenges hydroxyl free radicals in vitro. Brain Res 1994, 657(1–2), 207–213. [Google Scholar]
- Leegwater-Kim, J; Waters, C. Role of tolcapone in the treatment of Parkinson’s disease. Expert Rev Neurother 2007, 7(12), 1649–1657. [Google Scholar]
- Arnold, G; Kupsch, A. [Inhibition of catechol-O-methyltransferase. Optimizing dopaminergic therapy in idiopathic Parkinson syndrome with entacapone]. Nervenarzt 2000, 71(2), 78–83. [Google Scholar]
- Ravina, BM; Fagan, SC; Hart, RG; Hovinga, CA; Murphy, DD; Dawson, TM; Marler, JR. Neuroprotective agents for clinical trials in Parkinson’s disease: a systematic assessment. Neurology 2003, 60(8), 1234–1240. [Google Scholar]
- Gomez-Tortosa, E; Newell, K; Irizarry, MC; Albert, M; Growdon, JH; Hyman, BT. Clinical and quantitative pathologic correlates of dementia with Lewy bodies. Neurology 1999, 53(6), 1284–1291. [Google Scholar]
- Spillantini, MG; Crowther, RA; Jakes, R; Hasegawa, M; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci USA 1998, 95(11), 6469–6473. [Google Scholar]
- Spillantini, MG; Schmidt, ML; Lee, VM; Trojanowski, JQ; Jakes, R; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388(6645), 839–840. [Google Scholar]
- Duda, JE; Lee, VM; Trojanowski, JQ. Neuropathology of synuclein aggregates. J Neurosci Res 2000, 61(2), 121–127. [Google Scholar]
- Polymeropoulos, MH. Autosomal dominant Parkinson’s disease and alpha-synuclein. Ann Neurol 1998, 44(Suppl 1), S63–S64. [Google Scholar]
- Polymeropoulos, MH; Lavedan, C; Leroy, E; Ide, SE; Dehejia, A; Dutra, A; Pike, B; Root, H; Rubenstein, J; Boyer, R; Stenroos, ES; Chandrasekharappa, S; Athanassiadou, A; Papapetropoulos, T; Johnson, WG; Lazzarini, AM; Duvoisin, RC; Di Iorio, G; Golbe, LI; Nussbaum, RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276(5321), 2045–2047. [Google Scholar]
- Kruger, R; Kuhn, W; Muller, T; Woitalla, D; Graeber, M; Kosel, S; Przuntek, H; Epplen, JT; Schols, L; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 1998, 18(2), 106–108. [Google Scholar]
- Zarranz, JJ; Alegre, J; Gomez-Esteban, JC; Lezcano, E; Ros, R; Ampuero, I; Vidal, L; Hoenicka, J; Rodriguez, O; Atares, B; Llorens, V; Gomez Tortosa, E; del Ser, T; Munoz, DG; de Yebenes, JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004, 55(2), 164–173. [Google Scholar]
- Brandis, KA; Holmes, IF; England, SJ; Sharma, N; Kukreja, L; DebBurman, SK. alpha-Synuclein fission yeast model: concentration-dependent aggregation without plasma membrane localization or toxicity. J Mol Neurosci 2006, 28(2), 179–191. [Google Scholar]
- Conway, KA; Harper, JD; Lansbury, PT, Jr. Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 2000, 39(10), 2552–2563. [Google Scholar]
- Conway, KA; Lee, SJ; Rochet, JC; Ding, TT; Williamson, RE; Lansbury, PT, Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci USA 2000, 97(2), 571–576. [Google Scholar]
- Kotzbauer, PT; Giasson, BI; Kravitz, AV; Golbe, LI; Mark, MH; Trojanowski, JQ; Lee, VM. Fibrillization of alpha-synuclein and tau in familial Parkinson’s disease caused by the A53T alpha-synuclein mutation. Exp. Neurol 2004, 187(2), 279–288. [Google Scholar]
- Winkler, S; Hagenah, J; Lincoln, S; Heckman, M; Haugarvoll, K; Lohmann-Hedrich, K; Kostic, V; Farrer, M; Klein, C. alpha-Synuclein and Parkinson disease susceptibility. Neurology 2007, 69(18), 1745–1750. [Google Scholar]
- Kobayashi, H; Ujike, H; Hasegawa, J; Yamamoto, M; Kanzaki, A; Sora, I. Identification of a risk haplotype of the alpha-synuclein gene in Japanese with sporadic Parkinson’s disease. Mov Disord 2006, 21(12), 2157–2164. [Google Scholar]
- Singleton, AB; Farrer, M; Johnson, J; Singleton, A; Hague, S; Kachergus, J; Hulihan, M; Peuralinna, T; Dutra, A; Nussbaum, R; Lincoln, S; Crawley, A; Hanson, M; Maraganore, D; Adler, C; Cookson, MR; Muenter, M; Baptista, M; Miller, D; Blancato, J; Hardy, J; Gwinn-Hardy, K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302(5646), 841. [Google Scholar]
- Savitt, JM; Dawson, VL; Dawson, TM. Diagnosis and treatment of Parkinson disease: Molecules to medicine. J Clin Invest 2006, 116(7), 1744–1754. [Google Scholar]
- Dawson, T; Mandir, A; Lee, M. Animal models of PD: Pieces of the same puzzle? Neuron 2002, 35(2), 219–222. [Google Scholar]
- Abeliovich, A; Schmitz, Y; Farinas, I; Choi-Lundberg, D; Ho, WH; Castillo, PE; Shinsky, N; Verdugo, JM; Armanini, M; Ryan, A; Hynes, M; Phillips, H; Sulzer, D; Rosenthal, A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25(1), 239–252. [Google Scholar]
- El-Agnaf, OM; Salem, SA; Paleologou, KE; Cooper, LJ; Fullwood, NJ; Gibson, MJ; Curran, MD; Court, JA; Mann, DM; Ikeda, S; Cookson, MR; Hardy, J; Allsop, D. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J 2003, 17(13), 1945–1947. [Google Scholar]
- El-Agnaf, OM; Salem, SA; Paleologou, KE; Curran, MD; Gibson, MJ; Court, JA; Schlossmacher, MG; Allsop, D. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J 2006, 20(3), 419–425. [Google Scholar]
- von Bohlen Und Halbach, O. Synucleins and their relationship to Parkinson’s disease. Cell Tissue Res 2004, 318(1), 163–174. [Google Scholar]
- Iwai, A; Masliah, E; Yoshimoto, M; Ge, N; Flanagan, L; de Silva, HA; Kittel, A; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14(2), 467–475. [Google Scholar]
- Nakajo, S; Shioda, S; Nakai, Y; Nakaya, K. Localization of phosphoneuroprotein 14 (PNP 14) and its mRNA expression in rat brain determined by immunocytochemistry and in situ hybridization. Brain Res Mol Brain Res 1994, 27(1), 81–86. [Google Scholar]
- Lavedan, C. The synuclein family. Genome Res 1998, 8(9), 871–80. [Google Scholar]
- Eliezer, D; Kutluay, E; Bussell, R, Jr; Browne, G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol 2001, 307(4), 1061–1073. [Google Scholar]
- Li, J; Uversky, VN; Fink, AL. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry 2001, 40(38), 11604–11613. [Google Scholar]
- Fiebig, KM; Rice, LM; Pollock, E; Brunger, AT. Folding intermediates of SNARE complex assembly. Nat Struct Biol 1999, 6(2), 117–123. [Google Scholar]
- George, JM. The synucleins. Genome Biol 2002, 3(1). Reviews3002. [Google Scholar]
- Kahle, PJ; Haass, C; Kretzschmar, HA; Neumann, M. Structure/function of alpha-synuclein in health and disease: Rational development of animal models for Parkinson’s and related diseases. J Neurochem 2002, 82(3), 449–457. [Google Scholar]
- Davidson, WS; Jonas, A; Clayton, DF; George, JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 1998, 273(16), 9443–9449. [Google Scholar]
- Lee, HJ; Choi, C; Lee, SJ. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem 2002, 277(1), 671–678. [Google Scholar]
- Bussell, R, Jr; Eliezer, D. A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J Mol Biol 2003, 329(4), 763–778. [Google Scholar]
- Ueda, K; Fukushima, H; Masliah, E; Xia, Y; Iwai, A; Yoshimoto, M; Otero, DA; Kondo, J; Ihara, Y; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA 1993, 90(23), 11282–11286. [Google Scholar]
- el-Agnaf, OM; Irvine, GB. Aggregation and neurotoxicity of alpha-synuclein and related peptides. Biochem Soc Trans 2002, 30(4), 559–565. [Google Scholar]
- Han, H; Weinreb, PH; Lansbury, PT, Jr. The core Alzheimer’s peptide NAC forms amyloid fibrils which seed and are seeded by beta-amyloid: Is NAC a common trigger or target in neurodegenerative disease? Chem Biol 1995, 2(3), 163–169. [Google Scholar]
- El-Agnaf, OM; Jakes, R; Curran, MD; Middleton, D; Ingenito, R; Bianchi, E; Pessi, A; Neill, D; Wallace, A. Aggregates from mutant and wild-type alpha-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of beta-sheet and amyloid-like filaments. FEBS Lett 1998, 440(1–2), 71–75. [Google Scholar]
- Negro, A; Brunati, AM; Donella-Deana, A; Massimino, ML; Pinna, LA. Multiple phosphorylation of alpha-synuclein by protein tyrosine kinase Syk prevents eosin-induced aggregation. FASEB J 2002, 16(2), 210–212. [Google Scholar]
- Kim, TD; Paik, SR; Yang, CH. Structural and functional implications of C-terminal regions of alpha-synuclein. Biochemistry 2002, 41(46), 13782–13790. [Google Scholar]
- Ostrerova, N; Petrucelli, L; Farrer, M; Mehta, N; Choi, P; Hardy, J; Wolozin, B. alpha-Synuclein shares physical and functional homology with 14–3–3 proteins. J Neurosci 1999, 19(14), 5782–5791. [Google Scholar]
- Yuan, J; Yankner, BA. Apoptosis in the nervous system. Nature 2000, 407(6805), 802–809. [Google Scholar]
- Xu, J; Kao, SY; Lee, FJ; Song, W; Jin, LW; Yankner, BA. Dopamine-dependent neurotoxicity of alpha-synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nat Med 2002, 8(6), 600–606. [Google Scholar]
- Perez, RG; Waymire, JC; Lin, E; Liu, JJ; Guo, F; Zigmond, MJ. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neurosci 2002, 22(8), 3090–3099. [Google Scholar]
- Beyer, K. Alpha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol 2006, 112(3), 237–251. [Google Scholar]
- Krantz, DE; Peter, D; Liu, Y; Edwards, RH. Phosphorylation of a vesicular monoamine transporter by casein kinase II. J Biol Chem 1997, 272(10), 6752–6759. [Google Scholar]
- Pronin, AN; Morris, AJ; Surguchov, A; Benovic, JL. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem 2000, 275(34), 26515–26522. [Google Scholar]
- Chen, L; Feany, MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci 2005, 8(5), 657–663. [Google Scholar]
- Fujiwara, H; Hasegawa, M; Dohmae, N; Kawashima, A; Masliah, E; Goldberg, MS; Shen, J; Takio, K; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 2002, 4(2), 160–164. [Google Scholar]
- Smith, WW; Margolis, RL; Li, X; Troncoso, JC; Lee, MK; Dawson, VL; Dawson, TM; Iwatsubo, T; Ross, CA. Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J Neurosci 2005, 25(23), 5544–5552. [Google Scholar]
- Yamada, M; Iwatsubo, T; Mizuno, Y; Mochizuki, H. Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: Resemblance to pathogenetic changes in Parkinson’s disease. J Neurochem 2004, 91(2), 451–461. [Google Scholar]
- Souza, JM; Giasson, BI; Chen, Q; Lee, VM; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem 2000, 275(24), 18344–18349. [Google Scholar]
- Hodara, R; Norris, EH; Giasson, BI; Mishizen-Eberz, AJ; Lynch, DR; Lee, VM; Ischiropoulos, H. Functional consequences of alpha-synuclein tyrosine nitration: Diminished binding to lipid vesicles and increased fibril formation. J Biol Chem 2004, 279(46), 47746–47753. [Google Scholar]
- Giasson, BI; Duda, JE; Murray, IV; Chen, Q; Souza, JM; Hurtig, HI; Ischiropoulos, H; Trojanowski, JQ; Lee, VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290(5493), 985–989. [Google Scholar]
- Takahashi, T; Yamashita, H; Nakamura, T; Nagano, Y; Nakamura, S. Tyrosine 125 of alpha-synuclein plays a critical role for dimerization following nitrative stress. Brain Res 2002, 938(1–2), 73–80. [Google Scholar]
- Shimura, H; Schlossmacher, MG; Hattori, N; Frosch, MP; Trockenbacher, A; Schneider, R; Mizuno, Y; Kosik, KS; Selkoe, DJ. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 2001, 293(5528), 263–269. [Google Scholar]
- Tofaris, GK; Layfield, R; Spillantini, MG. alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett 2001, 509(1), 22–26. [Google Scholar]
- Leroy, E; Boyer, R; Auburger, G; Leube, B; Ulm, G; Mezey, E; Harta, G; Brownstein, MJ; Jonnalagada, S; Chernova, T; Dehejia, A; Lavedan, C; Gasser, T; Steinbach, PJ; Wilkinson, KD; Polymeropoulos, MH. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395(6701), 451–452. [Google Scholar]
- Sampathu, DM; Giasson, BI; Pawlyk, AC; Trojanowski, JQ; Lee, VM. Ubiquitination of alpha-synuclein is not required for formation of pathological inclusions in alpha-synucleinopathies. Am J Pathol 2003, 163(1), 91–100. [Google Scholar]
- Lim, KL; Dawson, VL; Dawson, TM. The cast of molecular characters in Parkinson’s disease: Felons, conspirators, and suspects. Ann. N. Y. Acad. Sci 2003, 99, 80–92. [Google Scholar]
- Rechsteiner, M; Rogers, SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci 1996, 21(7), 267–271. [Google Scholar]
- Beyer, K; Domingo-Sabat, M; Lao, JI; Carrato, C; Ferrer, I; Ariza, A. Identification and characterization of a new alpha-synuclein isoform and its role in Lewy body diseases. Neurogenetics 2008, 9(1), 15–23. [Google Scholar]
- Perrin, RJ; Woods, WS; Clayton, DF; George, JM. Interaction of human alpha-Synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J Biol Chem 2000, 275(44), 34393–34398. [Google Scholar]
- Crowther, RA; Jakes, R; Spillantini, MG; Goedert, M. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett 1998, 436(3), 309–312. [Google Scholar]
- Beyer, K; Lao, JI; Carrato, C; Mate, JL; Lopez, D; Ferrer, I; Ariza, A. Differential expression of alpha-synuclein isoforms in dementia with Lewy bodies. Neuropathol Appl Neurobiol 2004, 30(6), 601–607. [Google Scholar]
- McLean, PJ; Kawamata, H; Ribich, S; Hyman, BT. Membrane association and protein conformation of alpha-synuclein in intact neurons. Effect of Parkinson’s disease-linked mutations. J Biol Chem 2000, 275(12), 8812–8816. [Google Scholar]
- Scherzer, CR; Jensen, RV; Gullans, SR; Feany, MB. Gene expression changes presage neurodegeneration in a Drosophila model of Parkinson’s disease. Hum Mol Genet 2003, 12(19), 2457–2466. [Google Scholar]
- Sharon, R; Bar-Joseph, I; Frosch, MP; Walsh, DM; Hamilton, JA; Selkoe, DJ. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 2003, 37(4), 583–595. [Google Scholar]
- Cooper, AA; Gitler, AD; Cashikar, A; Haynes, CM; Hill, KJ; Bhullar, B; Liu, K; Xu, K; Strathearn, KE; Liu, F; Cao, S; Caldwell, KA; Caldwell, GA; Marsischky, G; Kolodner, RD; Labaer, J; Rochet, JC; Bonini, NM; Lindquist, S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313(5785), 324–328. [Google Scholar]
- Larsen, KE; Schmitz, Y; Troyer, MD; Mosharov, E; Dietrich, P; Quazi, AZ; Savalle, M; Nemani, V; Chaudhry, FA; Edwards, RH; Stefanis, L; Sulzer, D. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci 2006, 26(46), 11915–11922. [Google Scholar]
- Dev, KK; Hofele, K; Barbieri, S; Buchman, VL; van der Putten, H. Part II: alpha-Synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology 2003, 45(1), 14–44. [Google Scholar]
- Jenco, JM; Rawlingson, A; Daniels, B; Morris, AJ. Regulation of phospholipase D2: Selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry 1998, 37(14), 4901–4909. [Google Scholar]
- Chen, YG; Siddhanta, A; Austin, CD; Hammond, SM; Sung, TC; Frohman, MA; Morris, AJ; Shields, D. Phospholipase D stimulates release of nascent secretory vesicles from the trans-Golgi network. J Cell Biol 1997, 138(3), 495–504. [Google Scholar]
- Chandra, S; Gallardo, G; Fernandez-Chacon, R; Schluter, OM; Sudhof, TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123(3), 383–396. [Google Scholar]
- Dolgikh, DA; Gilmanshin, RI; Brazhnikov, EV; Bychkova, VE; Semisotnov, GV; Venyaminov, S; Ptitsyn, OB. Alpha-Lactalbumin: Compact state with fluctuating tertiary structure? FEBS Lett 1981, 136(2), 311–315. [Google Scholar]
- Uversky, VN; Li, J; Fink, AL. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J Biol Chem 2001, 276(47), 44284–44296. [Google Scholar]
- Uversky, VN; Li, J; Fink, AL. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: A possible factor in Parkinson’s disease. FEBS Lett 2001, 500(3), 105–108. [Google Scholar]
- Uversky, VN; Lee, HJ; Li, J; Fink, AL; Lee, SJ. Stabilization of partially folded conformation during alpha-synuclein oligomerization in both purified and cytosolic preparations. J Biol Chem 2001, 276(47), 43495–43498. [Google Scholar]
- Uversky, VN; Li, J; Fink, AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem 2001, 276(14), 10737–10744. [Google Scholar]
- Munishkina, LA; Henriques, J; Uversky, VN; Fink, AL. Role of protein-water interactions and electrostatics in alpha-synuclein fibril formation. Biochemistry 2004, 43(11), 3289–3300. [Google Scholar]
- Ding, TT; Lee, SJ; Rochet, JC; Lansbury, PT, Jr. Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 2002, 41(32), 10209–10217. [Google Scholar]
- Volles, MJ; Lansbury, PT, Jr. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002, 41(14), 4595–4602. [Google Scholar]
- Uversky, VN. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J Neurochem 2007, 103(1), 17–37. [Google Scholar]
- Cappai, R; Leck, SL; Tew, DJ; Williamson, NA; Smith, DP; Galatis, D; Sharples, RA; Curtain, CC; Ali, FE; Cherny, RA; Culvenor, JG; Bottomley, SP; Masters, CL; Barnham, KJ; Hill, AF. Dopamine promotes alpha-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J 2005, 19(10), 1377–1379. [Google Scholar]
- Kowall, NW; Hantraye, P; Brouillet, E; Beal, MF; McKee, AC; Ferrante, RJ. MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. Neuroreport 2000, 11(1), 211–213. [Google Scholar]
- Krishnan, S; Chi, EY; Wood, SJ; Kendrick, BS; Li, C; Garzon-Rodriguez, W; Wypych, J; Randolph, TW; Narhi, LO; Biere, AL; Citron, M; Carpenter, JF. Oxidative dimer formation is the critical rate-limiting step for Parkinson’s disease alpha-synuclein fibrillogenesis. Biochemistry 2003, 42(3), 829–837. [Google Scholar]
- Beal, MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol 1995, 38(3), 357–366. [Google Scholar]
- Stadtman, ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu. Rev. Biochem 1993, 62, 797–821. [Google Scholar]
- Uversky, VN; Yamin, G; Souillac, PO; Goers, J; Glaser, CB; Fink, AL. Methionine oxidation inhibits fibrillation of human alpha-synuclein in vitro. FEBS Lett 2002, 517(1–3), 239–244. [Google Scholar]
- Jones, DC; Gunasekar, PG; Borowitz, JL; Isom, GE. Dopamine-induced apoptosis is mediated by oxidative stress and Is enhanced by cyanide in differentiated PC12 cells. J Neurochem 2000, 74(6), 2296–2304. [Google Scholar]
- Junn, E; Mouradian, MM. Human alpha-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci Lett 2002, 320(3), 146–150. [Google Scholar]
- Conway, KA; Rochet, JC; Bieganski, RM; Lansbury, PT, Jr. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 2001, 294(5545), 1346–1349. [Google Scholar]
- Youdim, MB. What have we learnt from CDNA microarray gene expression studies about the role of iron in MPTP induced neurodegeneration and Parkinson’s disease? J. Neural. Transm. Suppl 2003, 65, 73–88. [Google Scholar]
- Sidhu, A; Wersinger, C; Vernier, P. alpha-Synuclein regulation of the dopaminergic transporter: A possible role in the pathogenesis of Parkinson’s disease. FEBS Lett 2004, 565(1–3), 1–5. [Google Scholar]
- Kaur, D; Andersen, J. Does cellular iron dysregulation play a causative role in Parkinson’s disease? Ageing Res Rev 2004, 3(3), 327–343. [Google Scholar]
- Thiruchelvam, M; Prokopenko, O; Cory-Slechta, DA; Buckley, B; Mirochnitchenko, O. Overexpression of superoxide dismutase or glutathione peroxidase protects against the paraquat + maneb-induced Parkinson disease phenotype. J Biol Chem 2005, 280(23), 22530–22539. [Google Scholar]
- Wersinger, C; Prou, D; Vernier, P; Sidhu, A. Modulation of dopamine transporter function by alpha-synuclein is altered by impairment of cell adhesion and by induction of oxidative stress. FASEB J 2003, 17(14), 2151–2153. [Google Scholar]
- Dauer, W; Kholodilov, N; Vila, M; Trillat, AC; Goodchild, R; Larsen, KE; Staal, R; Tieu, K; Schmitz, Y; Yuan, CA; Rocha, M; Jackson-Lewis, V; Hersch, S; Sulzer, D; Przedborski, S; Burke, R; Hen, R. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci USA 2002, 99(22), 14524–14529. [Google Scholar]
- Multhaup, G. Identification and regulation of the high affinity binding site of the Alzheimer’s disease amyloid protein precursor (APP) to glycosaminoglycans. Biochimie 1994, 76(3–4), 304–311. [Google Scholar]
- Cohlberg, JA; Li, J; Uversky, VN; Fink, AL. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry 2002, 41(5), 1502–1511. [Google Scholar]
- Liu, IH; Uversky, VN; Munishkina, LA; Fink, AL; Halfter, W; Cole, GJ. Agrin binds alpha-synuclein and modulates alpha-synuclein fibrillation. Glycobiology 2005, 15(12), 1320–1331. [Google Scholar]
- Goers, J; Uversky, VN; Fink, AL. Polycation-induced oligomerization and accelerated fibrillation of human alpha-synuclein in vitro. Protein. Sci 2003, 12(4), 702–707. [Google Scholar]
- Manning-Bog, AB; McCormack, AL; Li, J; Uversky, VN; Fink, AL; Di Monte, DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: Paraquat and alpha-synuclein. J Biol Chem 2002, 277(3), 1641–1644. [Google Scholar]
- Andringa, G; Lam, KY; Chegary, M; Wang, X; Chase, TN; Bennett, MC. Tissue transglutaminase catalyzes the formation of alpha-synuclein crosslinks in Parkinson’s disease. FASEB J 2004, 18(7), 932–934. [Google Scholar]
- Junn, E; Ronchetti, RD; Quezado, MM; Kim, SY; Mouradian, MM. Tissue transglutaminase-induced aggregation of alpha-synuclein: Implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 2003, 100(4), 2047–2052. [Google Scholar]
- Citron, BA; Suo, Z; SantaCruz, K; Davies, PJ; Qin, F; Festoff, BW. Protein crosslinking, tissue transglutaminase, alternative splicing and neurodegeneration. Neurochem Int 2002, 40(1), 69–78. [Google Scholar]
- Jo, E; McLaurin, J; Yip, CM; St George-Hyslop, P; Fraser, PE. alpha-Synuclein membrane interactions and lipid specificity. J Biol Chem 2000, 275(44), 34328–33434. [Google Scholar]
- Giasson, BI; Uryu, K; Trojanowski, JQ; Lee, VM. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem 1999, 274(12), 7619–7622. [Google Scholar]
- Lindersson, E; Lundvig, D; Petersen, C; Madsen, P; Nyengaard, JR; Hojrup, P; Moos, T; Otzen, D; Gai, WP; Blumbergs, PC; Jensen, PH. p25alpha Stimulates alpha-synuclein aggregation and is co-localized with aggregated alpha-synuclein in alpha-synucleinopathies. J Biol Chem 2005, 280(7), 5703–5715. [Google Scholar]
- Lundvig, D; Lindersson, E; Jensen, PH. Pathogenic effects of alpha-synuclein aggregation. Brain Res Mol Brain Res 2005, 134(1), 3–17. [Google Scholar]
- Alim, MA; Hossain, MS; Arima, K; Takeda, K; Izumiyama, Y; Nakamura, M; Kaji, H; Shinoda, T; Hisanaga, S; Ueda, K. Tubulin seeds alpha-synuclein fibril formation. J Biol Chem 2002, 277(3), 2112–2117. [Google Scholar]
- Iwata, A; Miura, S; Kanazawa, I; Sawada, M; Nukina, N. alpha-Synuclein forms a complex with transcription factor Elk-1. J Neurochem 2001, 77(1), 239–252. [Google Scholar]
- Lindersson, EK; Hojrup, P; Gai, WP; Locker, D; Martin, D; Jensen, PH. alpha-Synuclein filaments bind the transcriptional regulator HMGB-1. Neuroreport 2004, 15(18), 2735–2739. [Google Scholar]
- McLean, PJ; Kawamata, H; Shariff, S; Hewett, J; Sharma, N; Ueda, K; Breakefield, XO; Hyman, BT. TorsinA and heat shock proteins act as molecular chaperones: Suppression of alpha-synuclein aggregation. J Neurochem 2002, 83(4), 846–854. [Google Scholar]
- Clayton, DF; George, JM. Synucleins in synaptic plasticity and neurodegenerative disorders. J Neurosci Res 1999, 58(1), 120–129. [Google Scholar]
- Uversky, VN; Li, J; Souillac, P; Millett, IS; Doniach, S; Jakes, R; Goedert, M; Fink, AL. Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem 2002, 277(14), 11970–11978. [Google Scholar]
- Yamin, G; Munishkina, LA; Karymov, MA; Lyubchenko, YL; Uversky, VN; Fink, AL. Forcing nonamyloidogenic beta-synuclein to fibrillate. Biochemistry 2005, 44(25), 9096–9107. [Google Scholar]
- Hashimoto, M; Rockenstein, E; Mante, M; Mallory, M; Masliah, E. beta-Synuclein inhibits alpha-synuclein aggregation: A possible role as an anti-parkinsonian factor. Neuron 2001, 32(2), 213–223. [Google Scholar]
- Saito, Y; Kawashima, A; Ruberu, NN; Fujiwara, H; Koyama, S; Sawabe, M; Arai, T; Nagura, H; Yamanouchi, H; Hasegawa, M; Iwatsubo, T; Murayama, S. Accumulation of phosphorylated alpha-synuclein in aging human brain. J Neuropathol Exp Neurol 2003, 62(6), 644–654. [Google Scholar]
- Ii, K; Ito, H; Tanaka, K; Hirano, A. Immunocytochemical co-localization of the proteasome in ubiquitinated structures in neurodegenerative diseases and the elderly. J Neuropathol Exp Neurol 1997, 56(2), 125–131. [Google Scholar]
- Lindersson, E; Beedholm, R; Hojrup, P; Moos, T; Gai, W; Hendil, KB; Jensen, PH. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem 2004, 279(13), 12924–12934. [Google Scholar]
- Recchia, A; Debetto, P; Negro, A; Guidolin, D; Skaper, SD; Giusti, P. Alpha-synuclein and Parkinson’s disease. FASEB J 2004, 18(6), 617–626. [Google Scholar]
- Klucken, J; Shin, Y; Masliah, E; Hyman, BT; McLean, PJ. Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J Biol Chem 2004, 279(24), 25497–25502. [Google Scholar]
- Paleologou, KE; Irvine, GB; El-Agnaf, OM. Alpha-synuclein aggregation in neurodegenerative diseases and its inhibition as a potential therapeutic strategy. Biochem Soc Trans 2005, 33, 1106–1110. [Google Scholar]
- Hirsch, EC; Brandel, JP; Galle, P; Javoy-Agid, F; Agid, Y. Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: An X-ray microanalysis. J Neurochem 1991, 56(2), 446–451. [Google Scholar]
- Sofic, E; Riederer, P; Heinsen, H; Beckmann, H; Reynolds, GP; Hebenstreit, G; Youdim, MB. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm 1988, 74(3), 199–205. [Google Scholar]
- Paik, SR; Shin, HJ; Lee, JH. Metal-catalyzed oxidation of alpha-synuclein in the presence of Copper(II) and hydrogen peroxide. Arch Biochem Biophys 2000, 378(2), 269–277. [Google Scholar]
- Paik, SR; Lee, JH; Kim, DH; Chang, CS; Kim, J. Aluminum-induced structural alterations of the precursor of the non-A beta component of Alzheimer’s disease amyloid. Arch Biochem Biophys 1997, 344(2), 325–334. [Google Scholar]
- Brown, DR. Interactions between metals and alpha-synuclein--function or artefact? FEBS J 2007, 274(15), 3766–3774. [Google Scholar]
- Yamin, G; Glaser, CB; Uversky, VN; Fink, AL. Certain metals trigger fibrillation of methionine-oxidized alpha-synuclein. J Biol Chem 2003, 278(30), 27630–27635. [Google Scholar]
- Ostrerova-Golts, N; Petrucelli, L; Hardy, J; Lee, JM; Farer, M; Wolozin, B. The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci 2000, 20(16), 6048–6054. [Google Scholar]
- Chiba-Falek, O; Lopez, GJ; Nussbaum, RL. Levels of alpha-synuclein mRNA in sporadic Parkinson disease patients. Mov Disord 2006, 21(10), 1703–1708. [Google Scholar]
- Hsu, LJ; Sagara, Y; Arroyo, A; Rockenstein, E; Sisk, A; Mallory, M; Wong, J; Takenouchi, T; Hashimoto, M; Masliah, E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol 2000, 157(2), 401–410. [Google Scholar]
- Stefanis, L; Larsen, KE; Rideout, HJ; Sulzer, D; Greene, LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 2001, 21(24), 9549–9560. [Google Scholar]
- Gosavi, N; Lee, HJ; Lee, JS; Patel, S; Lee, SJ. Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J Biol Chem 2002, 277(50), 48984–48992. [Google Scholar]
- Lo Bianco, C; Ridet, JL; Schneider, BL; Deglon, N; Aebischer, P. alpha -Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc Natl Acad Sci USA 2002, 99(16), 10813–10818. [Google Scholar]
- Rockenstein, E; Hansen, LA; Mallory, M; Trojanowski, JQ; Galasko, D; Masliah, E. Altered expression of the synuclein family mRNA in Lewy body and Alzheimer’s disease. Brain Res 2001, 914(1–2), 48–56. [Google Scholar]
- Vila, M; Vukosavic, S; Jackson-Lewis, V; Neystat, M; Jakowec, M; Przedborski, S. Alpha-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J Neurochem 2000, 74(2), 721–729. [Google Scholar]
- Talpade, DJ; Greene, JG; Higgins, DS, Jr; Greenamyre, JT. In vivo labeling of mitochondrial complex I (NADH:ubiquinone oxidoreductase) in rat brain using [(3)H]dihydrorotenone. J Neurochem 2000, 75(6), 2611–2621. [Google Scholar]
- Greenamyre, JT; Hastings, TG. Biomedicine. Parkinson’s--divergent causes, convergent mechanisms. Science 2004, 304(5674), 1120–1122. [Google Scholar]
- Seo, JH; Rah, JC; Choi, SH; Shin, JK; Min, K; Kim, HS; Park, CH; Kim, S; Kim, EM; Lee, SH; Lee, S; Suh, SW; Suh, YH. Alpha-synuclein regulates neuronal survival via Bcl-2 family expression and PI3/Akt kinase pathway. FASEB J 2002, 16(13), 1826–1828. [Google Scholar]
- Pezzella, A; d’Ischia, M; Napolitano, A; Misuraca, G; Prota, G. Iron-mediated generation of the neurotoxin 6-hydroxydopamine quinone by reaction of fatty acid hydroperoxides with dopamine: A possible contributory mechanism for neuronal degeneration in Parkinson’s disease. J Med Chem 1997, 40(14), 2211–2216. [Google Scholar]
- Jellinger, K; Paulus, W; Grundke-Iqbal, I; Riederer, P; Youdim, MB. Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J Neural Transm Park Dis Dement Sect 1990, 2(4), 327–340. [Google Scholar]
- Sayre, LM; Moreira, PI; Smith, MA; Perry, G. Metal ions and oxidative protein modification in neurological disease. Ann Ist Super Sanita 2005, 41(2), 143–164. [Google Scholar]
- Kaur, D; Andersen, JK. Ironing out Parkinson’s disease: Is therapeutic treatment with iron chelators a real possibility? Aging Cell 2002, 1(1), 17–21. [Google Scholar]
- Andersen, JK. Iron dysregulation and Parkinson’s disease. J Alzheimers Dis 2004, 6(Suppl 6), S47–S52. [Google Scholar]
- Theil, EC; Eisenstein, RS. Combinatorial mRNA regulation: Iron regulatory proteins and iso-iron-responsive elements (Iso-IREs). J Biol Chem 2000, 275(52), 40659–40662. [Google Scholar]
- Cairo, G; Pietrangelo, A. Iron regulatory proteins in pathobiology. Biochem J 2000, 352, 241–250. [Google Scholar]
- Youdim, MB; Ben-Shachar, D; Riederer, P. Iron in brain function and dysfunction with emphasis on Parkinson’s disease. Eur Neurol 1991, 31(Suppl 1), 34–40. [Google Scholar]
- Glinka, Y; Tipton, KF; Youdim, MB. Nature of inhibition of mitochondrial respiratory complex I by 6-Hydroxydopamine. J Neurochem 1996, 66(5), 2004–2010. [Google Scholar]
- Ben-Shachar, D; Eshel, G; Finberg, JP; Youdim, MB. The iron chelator desferrioxamine (Desferal) retards 6-hydroxydopamine-induced degeneration of nigrostriatal dopamine neurons. J Neurochem 1991, 56(4), 1441–1444. [Google Scholar]
- Grunblatt, E; Mandel, S; Berkuzki, T; Youdim, MB. Apomorphine protects against MPTP-induced neurotoxicity in mice. Mov Disord 1999, 14(4), 612–618. [Google Scholar]
- LaVaute, T; Smith, S; Cooperman, S; Iwai, K; Land, W; Meyron-Holtz, E; Drake, SK; Miller, G; Abu-Asab, M; Tsokos, M; Switzer, R, 3rd; Grinberg, A; Love, P; Tresser, N; Rouault, TA. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet 2001, 27(2), 209–214. [Google Scholar]
- Crompton, DE; Chinnery, PF; Fey, C; Curtis, AR; Morris, CM; Kierstan, J; Burt, A; Young, F; Coulthard, A; Curtis, A; Ince, PG; Bates, D; Jackson, MJ; Burn, J. Neuroferritinopathy: A window on the role of iron in neurodegeneration. Blood Cells Mol Dis 2002, 29(3), 522–531. [Google Scholar]
- Borie, C; Gasparini, F; Verpillat, P; Bonnet, AM; Agid, Y; Hetet, G; Brice, A; Durr, A; Grandchamp, B. Association study between iron-related genes polymorphisms and Parkinson’s disease. J Neurol 2002, 249(7), 801–804. [Google Scholar]
- Balla, G; Jacob, HS; Balla, J; Rosenberg, M; Nath, K; Apple, F; Eaton, JW; Vercellotti, GM. Ferritin: A cytoprotective antioxidant strategem of endothelium. J Biol Chem 1992, 267(25), 18148–18153. [Google Scholar]
- Kwak, EL; Larochelle, DA; Beaumont, C; Torti, SV; Torti, FM. Role for NF-kappa B in the regulation of ferritin H by tumor necrosis factor-alpha. J Biol Chem 1995, 270(25), 15285–15293. [Google Scholar]
- Martins, EA; Robalinho, RL; Meneghini, R. Oxidative stress induces activation of a cytosolic protein responsible for control of iron uptake. Arch Biochem Biophys 1995, 316(1), 128–134. [Google Scholar]
- Nunez, MT; Nunez-Millacura, C; Tapia, V; Munoz, P; Mazariegos, D; Arredondo, M; Mura, C; Maccioni, RB. Iron-activated iron uptake: A positive feedback loop mediated by iron regulatory protein 1. Biometals 2003, 16(1), 83–90. [Google Scholar]
- Faucheux, BA; Martin, ME; Beaumont, C; Hunot, S; Hauw, JJ; Agid, Y; Hirsch, EC. Lack of up-regulation of ferritin is associated with sustained iron regulatory protein-1 binding activity in the substantia nigra of patients with Parkinson’s disease. J Neurochem 2002, 83(2), 320–330. [Google Scholar]
- Lin, E; Graziano, JH; Freyer, GA. Regulation of the 75-kDa subunit of mitochondrial complex I by iron. J Biol Chem 2001, 276(29), 27685–27692. [Google Scholar]
- Dandekar, T; Stripecke, R; Gray, NK; Goossen, B; Constable, A; Johansson, HE; Hentze, MW. Identification of a novel iron-responsive element in murine and human erythroid delta-aminolevulinic acid synthase mRNA. EMBO J 1991, 10(7), 1903–1909. [Google Scholar]
- Castellani, RJ; Siedlak, SL; Perry, G; Smith, MA. Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol 2000, 100(2), 111–114. [Google Scholar]
- Friedlich, AL; Tanzi, RE; Rogers, JT. The 5’-untranslated region of Parkinson’s disease alpha-synuclein messengerRNA contains a predicted iron responsive element. Mol Psychiatry 2007, 12(3), 222–223. [Google Scholar]
- Xia, Y; Saitoh, T; Ueda, K; Tanaka, S; Chen, X; Hashimoto, M; Hsu, L; Conrad, C; Sundsmo, M; Yoshimoto, M; Thal, L; Katzman, R; Masliah, E. Characterization of the human alpha-synuclein gene: Genomic structure, transcription start site, promoter region and polymorphisms. J Alzheimers Dis 2001, 3(5), 485–494. [Google Scholar]
- Preiss, T; Muckenthaler, M; Hentze, MW. Poly(A)-tail-promoted translation in yeast: Implications for translational control. RNA 1998, 4(11), 1321–1331. [Google Scholar]
- Bordeleau, ME; Matthews, J; Wojnar, JM; Lindqvist, L; Novac, O; Jankowsky, E; Sonenberg, N; Northcote, P; Teesdale-Spittle, P; Pelletier, J. Stimulation of mammalian translation initiation factor eIF4A activity by a small molecule inhibitor of eukaryotic translation. Proc Natl Acad Sci USA 2005, 102(30), 10460–10465. [Google Scholar]
- Hundsdoerfer, P; Thoma, C; Hentze, MW. Eukaryotic translation initiation factor 4GI and p97 promote cellular internal ribosome entry sequence-driven translation. Proc Natl Acad Sci USA 2005, 102(38), 13421–13426. [Google Scholar]
- Nie, M; Htun, H. Different modes and potencies of translational repression by sequence-specific RNA-protein interaction at the 5’-UTR. Nucleic Acids Res 2006, 34(19), 5528–5540. [Google Scholar]
- Rogers, JT; Randall, JD; Cahill, CM; Eder, PS; Huang, X; Gunshin, H; Leiter, L; McPhee, J; Sarang, SS; Utsuki, T; Greig, NH; Lahiri, DK; Tanzi, RE; Bush, AI; Giordano, T; Gullans, SR. An iron-responsive element type II in the 5’-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem 2002, 277(47), 45518–45528. [Google Scholar]
- Payton, S; Cahill, CM; Randall, JD; Gullans, SR; Rogers, JT. Drug discovery targeted to the Alzheimer’s APP mRNA 5’-untranslated region: The action of paroxetine and dimercaptopropanol. J Mol Neurosci 2003, 20(3), 267–275. [Google Scholar]
- Rogers, JT; Randall, JD; Eder, PS; Huang, X; Bush, AI; Tanzi, RE; Venti, A; Payton, SM; Giordano, T; Nagano, S; Cahill, CM; Moir, R; Lahiri, DK; Greig, N; Sarang, SS; Gullans, SR. Alzheimer’s disease drug discovery targeted to the APP mRNA 5’untranslated region. J Mol Neurosci 2002, 19(1–2), 77–82. [Google Scholar]
- Lahiri, DK; Chen, D; Maloney, B; Holloway, HW; Yu, QS; Utsuki, T; Giordano, T; Sambamurti, K; Greig, NH. The experimental Alzheimer’s disease drug posiphen [(+)-phenserine] lowers amyloid-beta peptide levels in cell culture and mice. J Pharmacol Exp Ther 2007, 320(1), 386–396. [Google Scholar]
- Shaw, KT; Utsuki, T; Rogers, J; Yu, QS; Sambamurti, K; Brossi, A; Ge, YW; Lahiri, DK; Greig, NH. Phenserine regulates translation of beta -amyloid precursor protein mRNA by a putative interleukin-1 responsive element, a target for drug development. Proc Natl Acad Sci USA 2001, 98(13), 7605–7610. [Google Scholar]
- Scherzer, CR; Grass, JA; Liao, Z; Pepivani, I; Zheng, B; Eklund, AC; Ney, PA; Ng, J; McGoldrick, M; Mollenhauer, B; Bresnick, EH; Schlossmacher, MG. GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha-synuclein. Proc Natl Acad Sci USA 2008, 105(31), 10907–10912. [Google Scholar]
- Lippa, CF; Schmidt, ML; Lee, VM; Trojanowski, JQ. Antibodies to alpha-synuclein detect Lewy bodies in many Down’s syndrome brains with Alzheimer’s disease. Ann Neurol 1999, 45(3), 353–357. [Google Scholar]

{kind=link}
{kind=link}
| Class drug | Mechanism of action | Side effects | Specific drug |
|---|---|---|---|
| Anticholinergics | Block acetylcholine receptors | Dry mouth, dry eyes, urinary retention, exacerbation of glaucoma, cognitive impairment | Trihexyphenidy
Benztropine Ethopropazine |
| Amantadine | Blocks NMDA and acetylcholine receptors and promotes release of DA | Cognitive dysfunction, peripheral edema and skin rash | Amantadine |
| l-dopa | Metabolism to DA in cells containing dopa-decarboxylase | Nausea, hypotension, hallucinations, psychosis, dystonic and choreiform dyskinesias | L-dopa/carbidopa
Sinemet CR L-dopa/benserazide |
| DA agonists | Stimulate DA receptors | Nausea, hypotension, hallucinations, psychosis peripheral edema, pulmonary fibrosis, insomnia | Bromocriptine
Pergolide Ropinirole Pramipexole |
| MAO inhibitors | Block MAO-B receptors to reduce DA metabolism | Nausea, dizziness, sleep disorder and impaired cognition | Selegiline |
| Catechol O-(COMT) inhibitors | Block peripheral COMT Methyltranferase to improve L-dopa pharmacokinetics | l-dopa related side-effect activity exacerbation, diarrhea, urine discoloration | Entacapone
Tolcapone |
| Agent | Mechanism | Comments |
|---|---|---|
| Caffeine | Adenosine antagonist | KW-6002, a specific A2A receptor antagonist in development |
| Coenzyme Q10 | Antioxidant/mitochondrial stabilizer | Dietary supplement; modest symptomatic benefit based on phase 2 studies |
| Creatine | Mitochondrial stabilizer | Dietary supplement |
| Estrogen (17 beta estradiol) | Undetermined | |
| GM-1 ganglioside | Trophic factor | |
| GPI-1485 | Trophic factor | Neuroimmunophilin ligand |
| Minocycline | Anti-inflammatory/anti-apoptotic | Antibiotic |
| Nicotine | Undetermined | |
| Pramipexole | Antioxidant | Dopamine agonist; clinical neuroimaging data demonstrate a possible disease-modifying effect; exact interpretation and clinical meaning of data remain unclear |
| Rasagiline | Antioxidant/anti-apoptotic | Selective MAO-B inhibitor; symptomatic benefit in early- and advanced-stage PD based on several phase 3 studies |
| Ropinirole | Antioxidant | Dopamine agonist; clinical neuroimaging data demonstrate a possible disease-modifying effect; exact interpretation and clinical meaning of data remain unclear |
| Selegiline | Antioxidant/anti-apoptotic | Selective MAO-B inhibitor; DATATOP study failed to demonstrate neuroprotective benefits |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/). This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Olivares, D.; Huang, X.; Branden, L.; Greig, N.H.; Rogers, J.T. Physiological and Pathological Role of Alpha-synuclein in Parkinson’s Disease Through Iron Mediated Oxidative Stress; The Role of a Putative Iron-responsive Element. Int. J. Mol. Sci. 2009, 10, 1226-1260. https://doi.org/10.3390/ijms10031226
Olivares D, Huang X, Branden L, Greig NH, Rogers JT. Physiological and Pathological Role of Alpha-synuclein in Parkinson’s Disease Through Iron Mediated Oxidative Stress; The Role of a Putative Iron-responsive Element. International Journal of Molecular Sciences. 2009; 10(3):1226-1260. https://doi.org/10.3390/ijms10031226
Chicago/Turabian StyleOlivares, David, Xudong Huang, Lars Branden, Nigel H. Greig, and Jack T. Rogers. 2009. "Physiological and Pathological Role of Alpha-synuclein in Parkinson’s Disease Through Iron Mediated Oxidative Stress; The Role of a Putative Iron-responsive Element" International Journal of Molecular Sciences 10, no. 3: 1226-1260. https://doi.org/10.3390/ijms10031226