Dye-Sensitized Solar Cells Based on the Principles and Materials of Photosynthesis: Mechanisms of Suppression and Enhancement of Photocurrent and Conversion Efficiency

Abstract
:1. Introduction
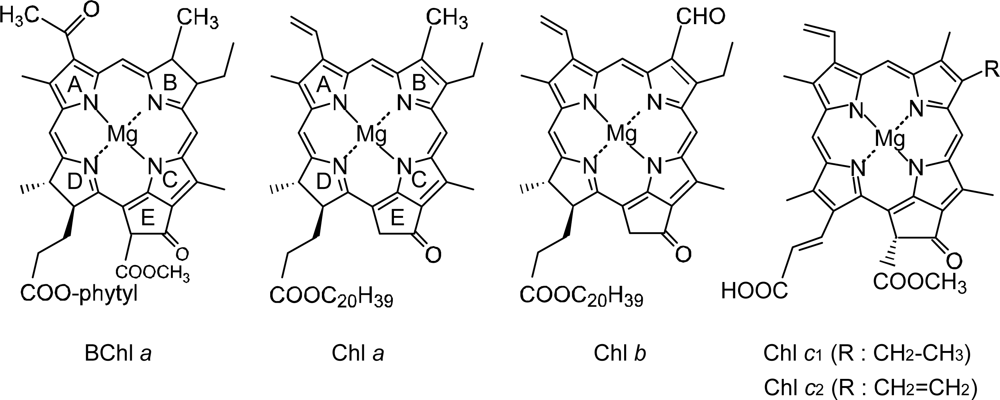
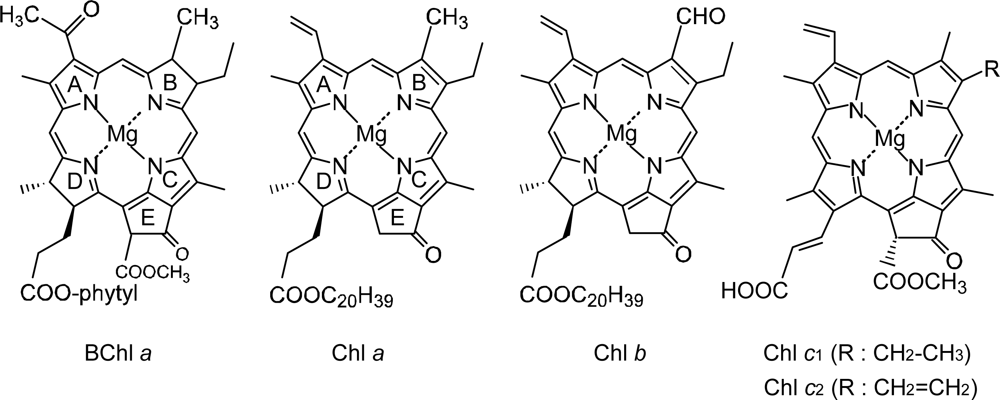
1.1. Photosynthetic Pigments
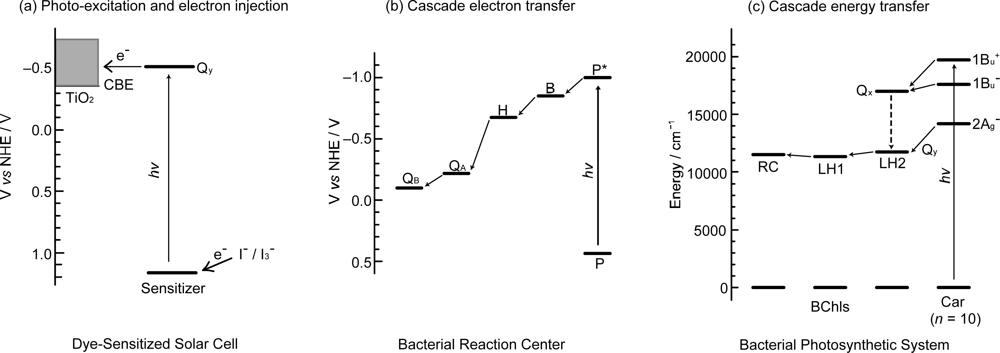
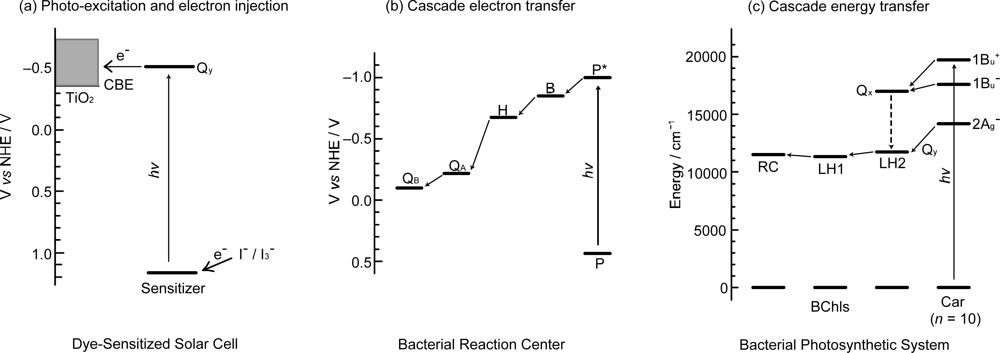
1.2. How to Apply the Principles of Photosynthesis to DSSCs
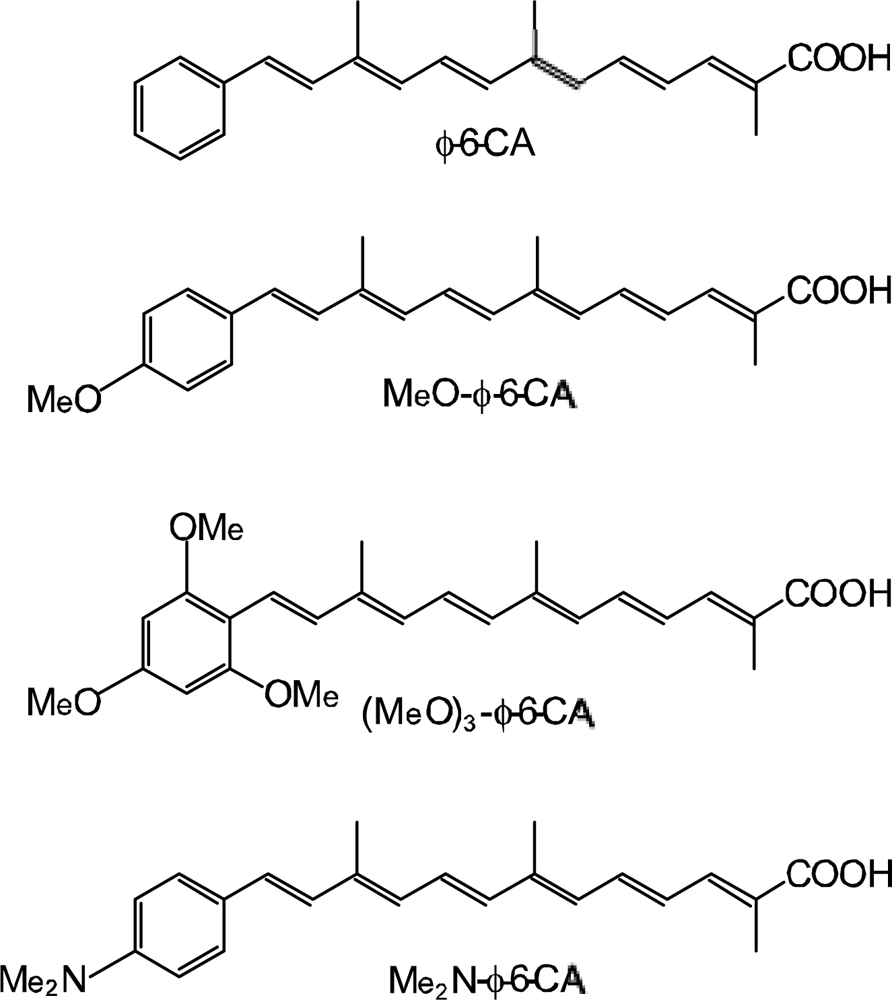
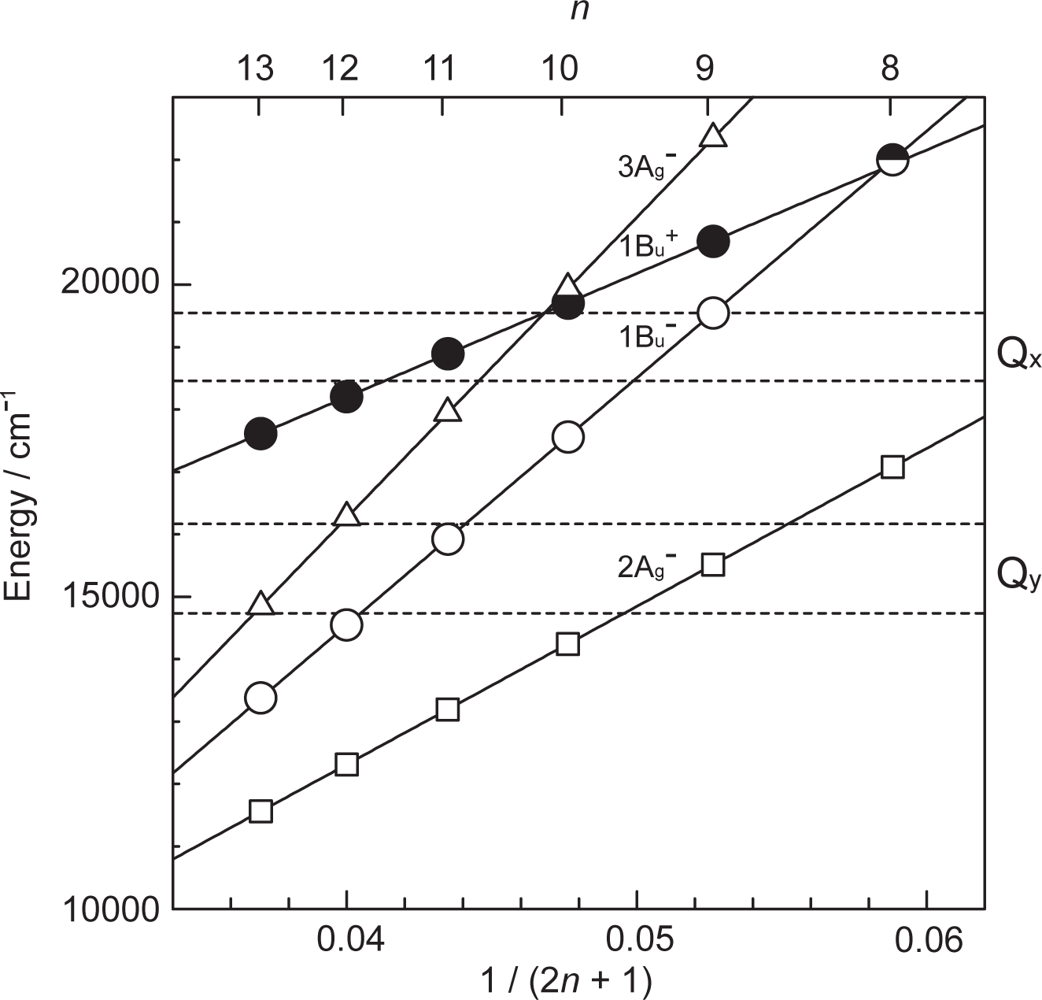
2. Polyene Sensitizers
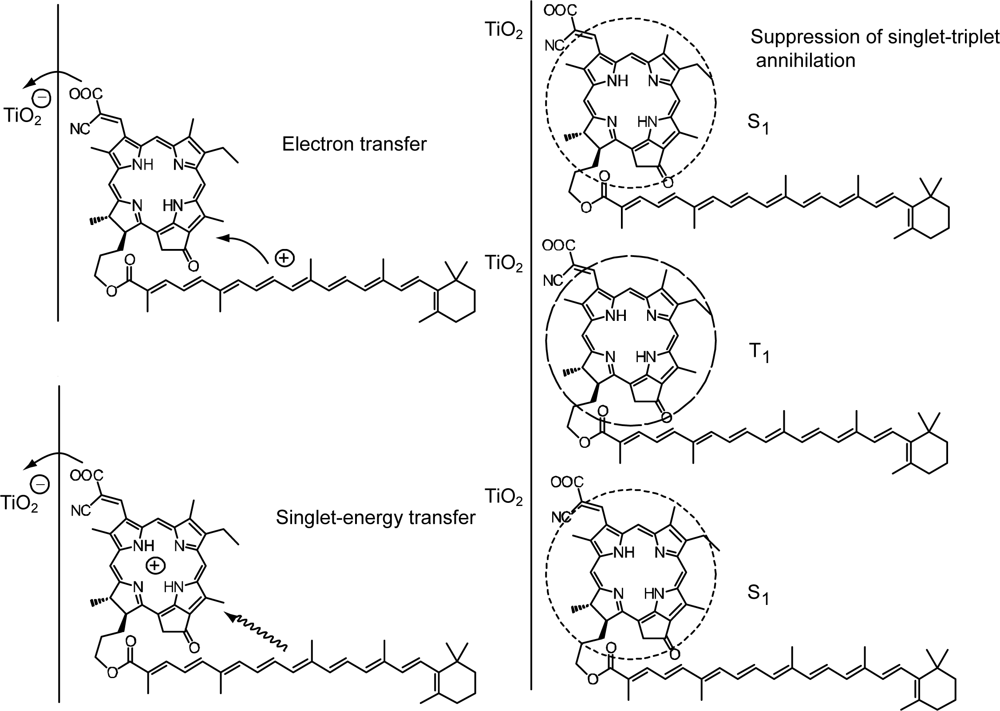
2.1. Mechanisms of Electron Injection and Charge Recombination Generating Radical Cation and Triplet Species
2.2. Mechanisms of Singlet-Triplet Annihilation Suppressing Photocurrent and Conversion Efficiency
3. Pheophorbide Sensitizers Combined with Polyene Spacers
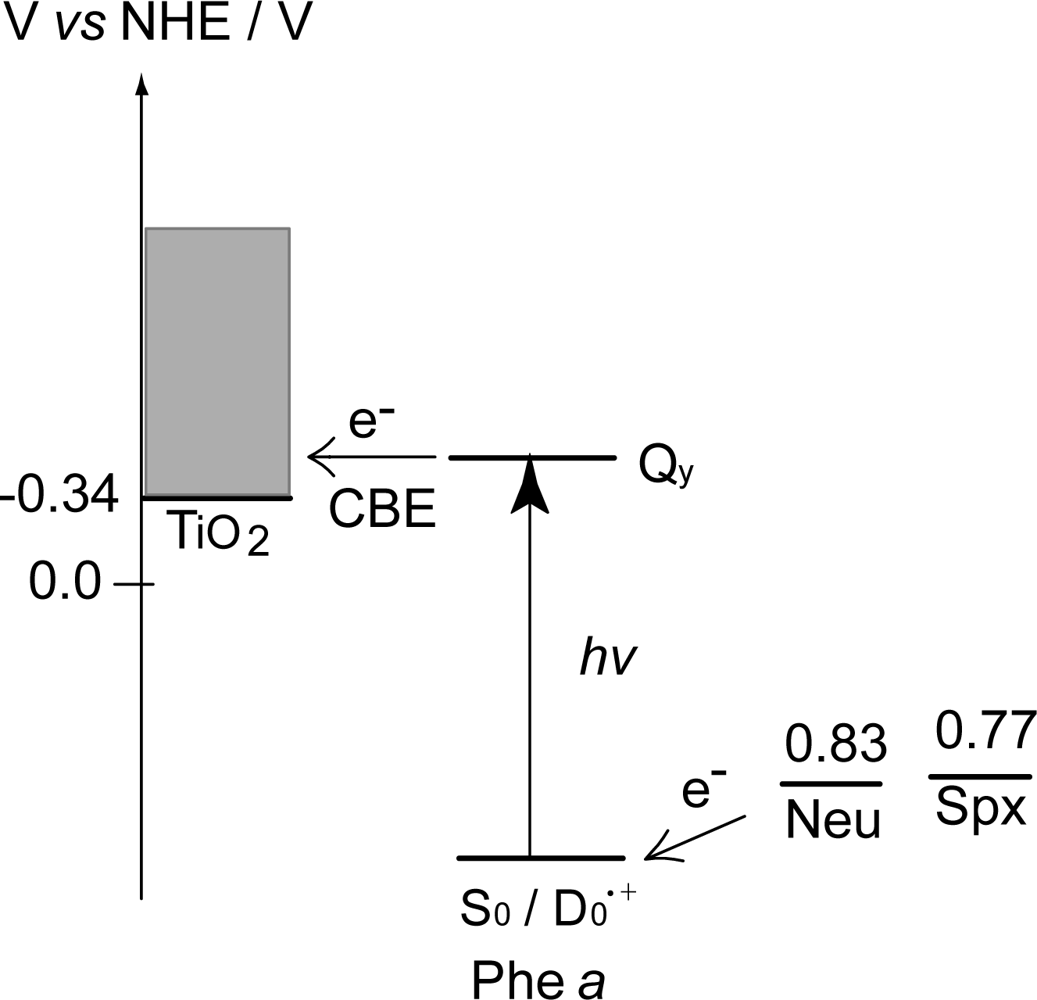
3.1. Mechanisms of Electron Transfer from Carotenoid Spacers to Pheophorbide a Sensitizer
3.2. Pheophorbide–Car adduct: Energy Transfer and Electron Transfer from Car to Phe Moiety
4. Bacteriochlorin, Chlorin and Porphyrin Sensitizers
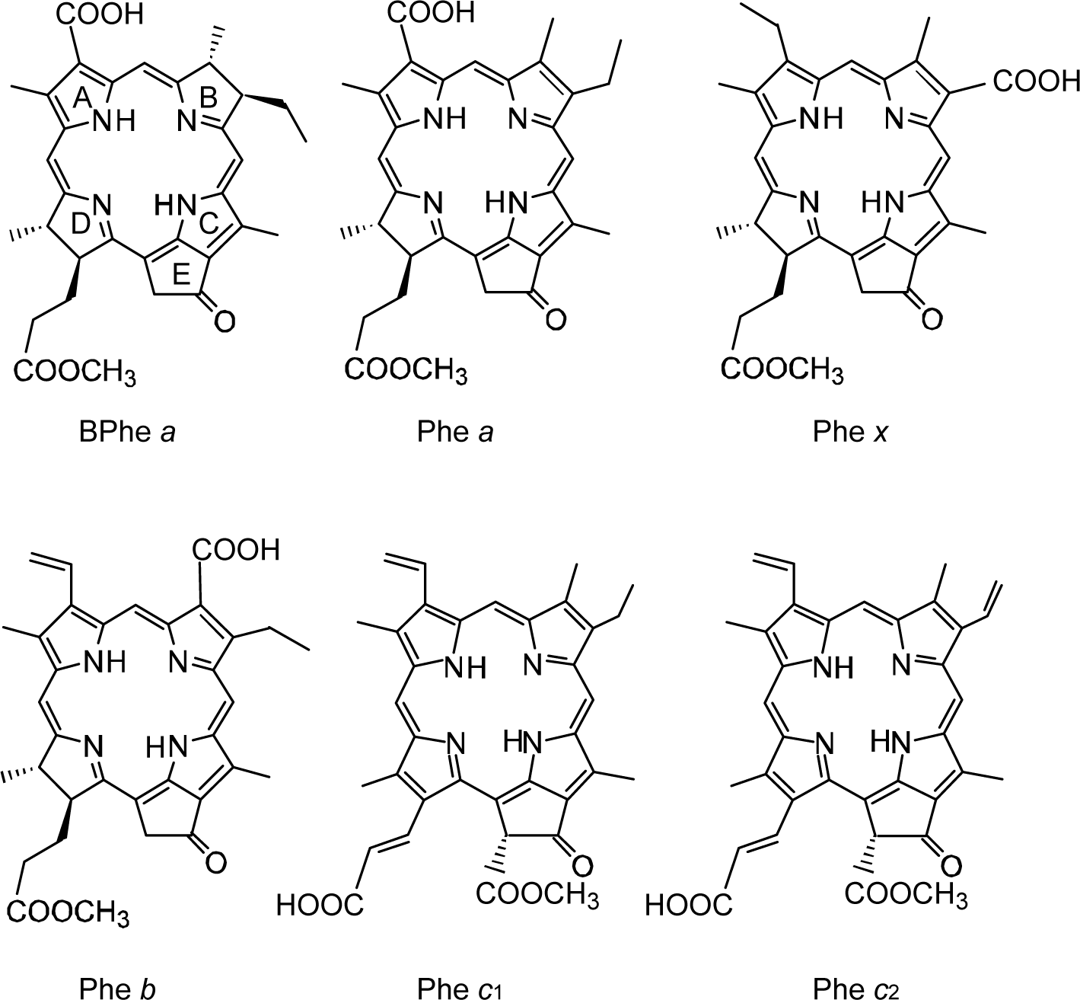
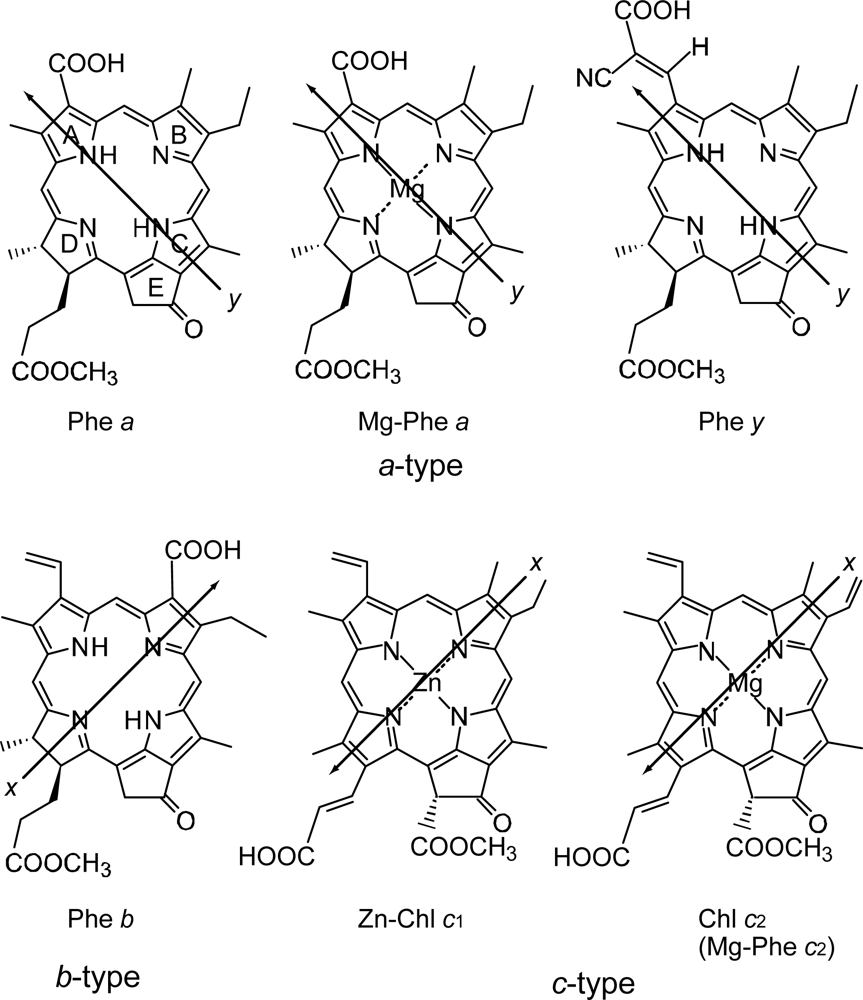
4.1. Pheophorbide Sensitizers Having Bacteriochlorin, Chlorin and Porphyrin Skeletons
4.2. Chl c (Mg-Pheophorbide) Sensitizers Having Porphyrin Skelton
4.3. Pheophorbide and Metal-Pheophorbide Sensitizers Having Chlorin and Porphyrin Macrocycles
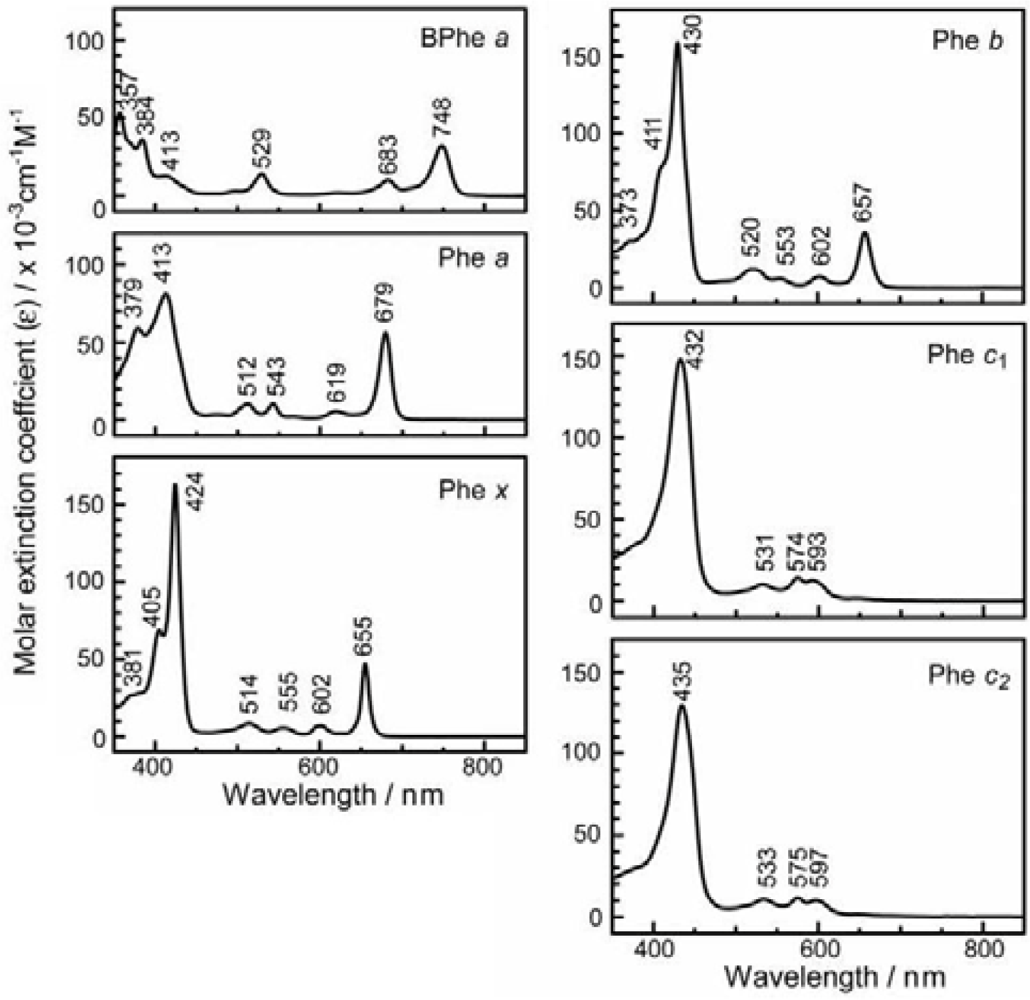
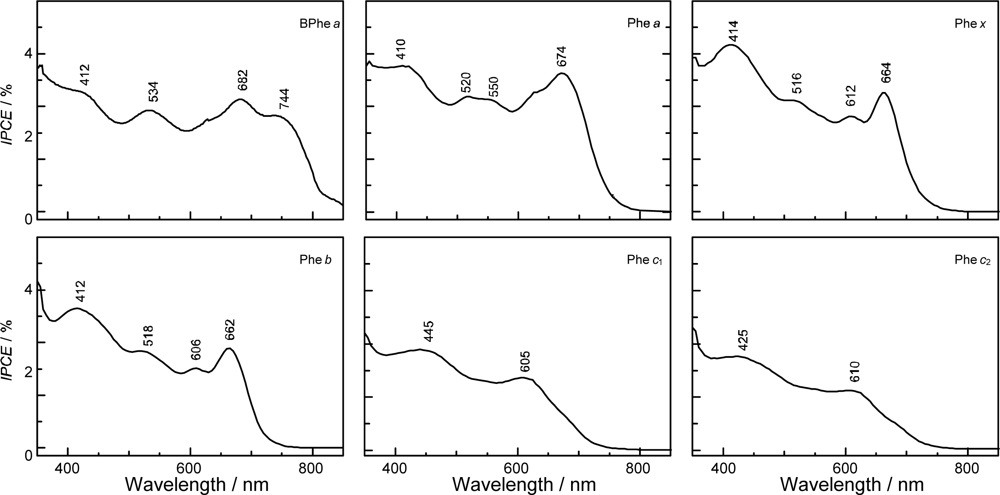
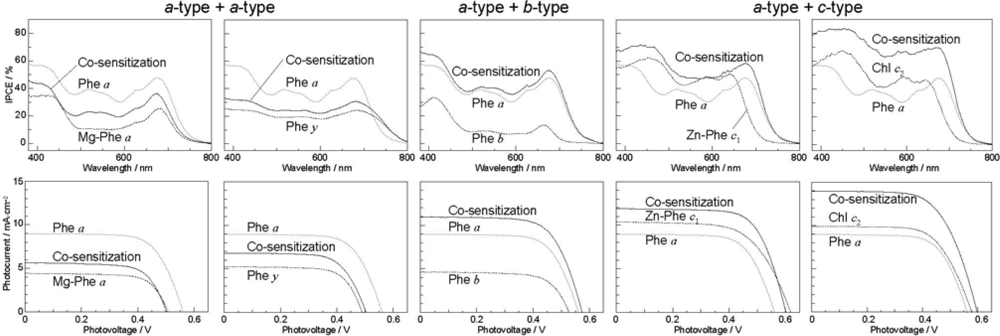
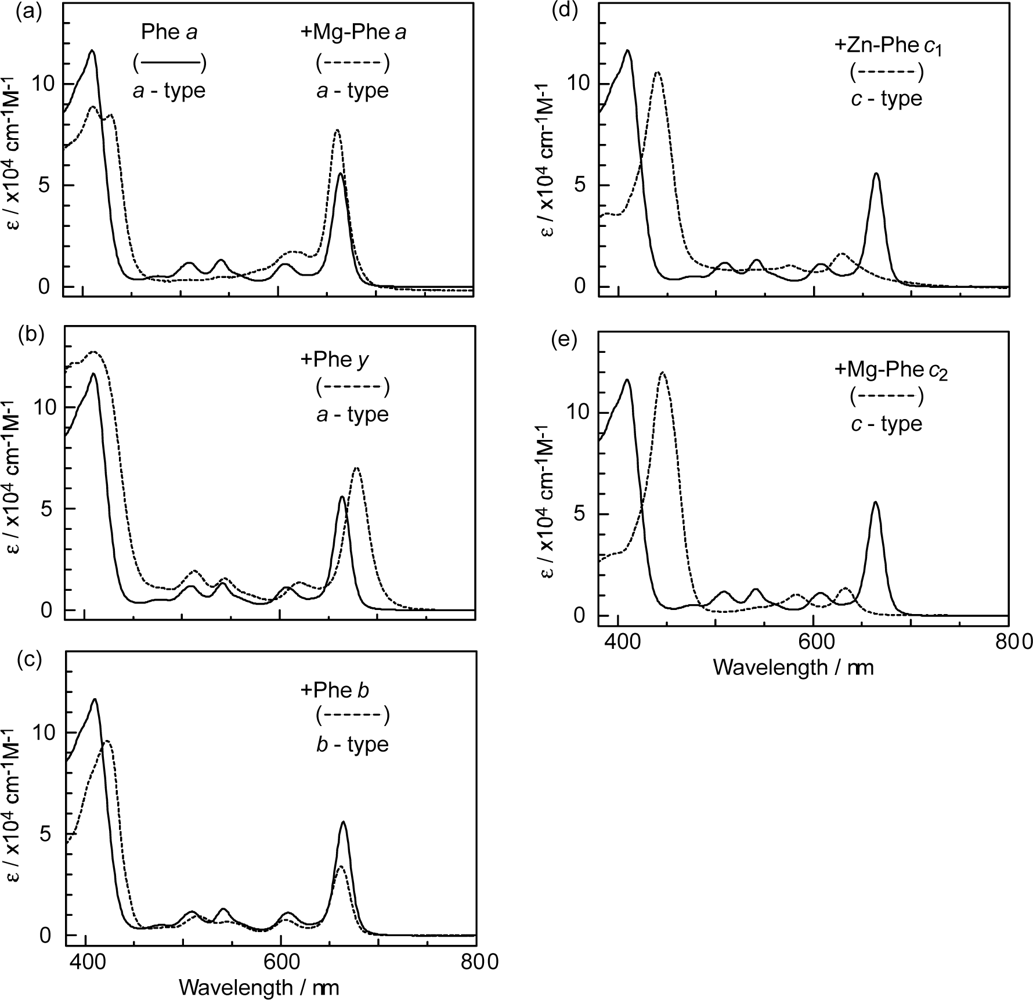
- The absorption spectra of the sensitizers (in Figure 45) show that the major light absorption through the Soret bands is highly competitive in the a-type + a-type pair, complementary rather than competitive in the a-type + b-type pair, and absolutely complementary in the a-type + c-type pair. Therefore, the highest enhancement in the a-type + b-type co-sensitization and the next highest enhancement in the a-type + c-type co-sensitization can be rationalized in terms of complementary absorption not by the Qx and Qy levels but by the Soret levels.
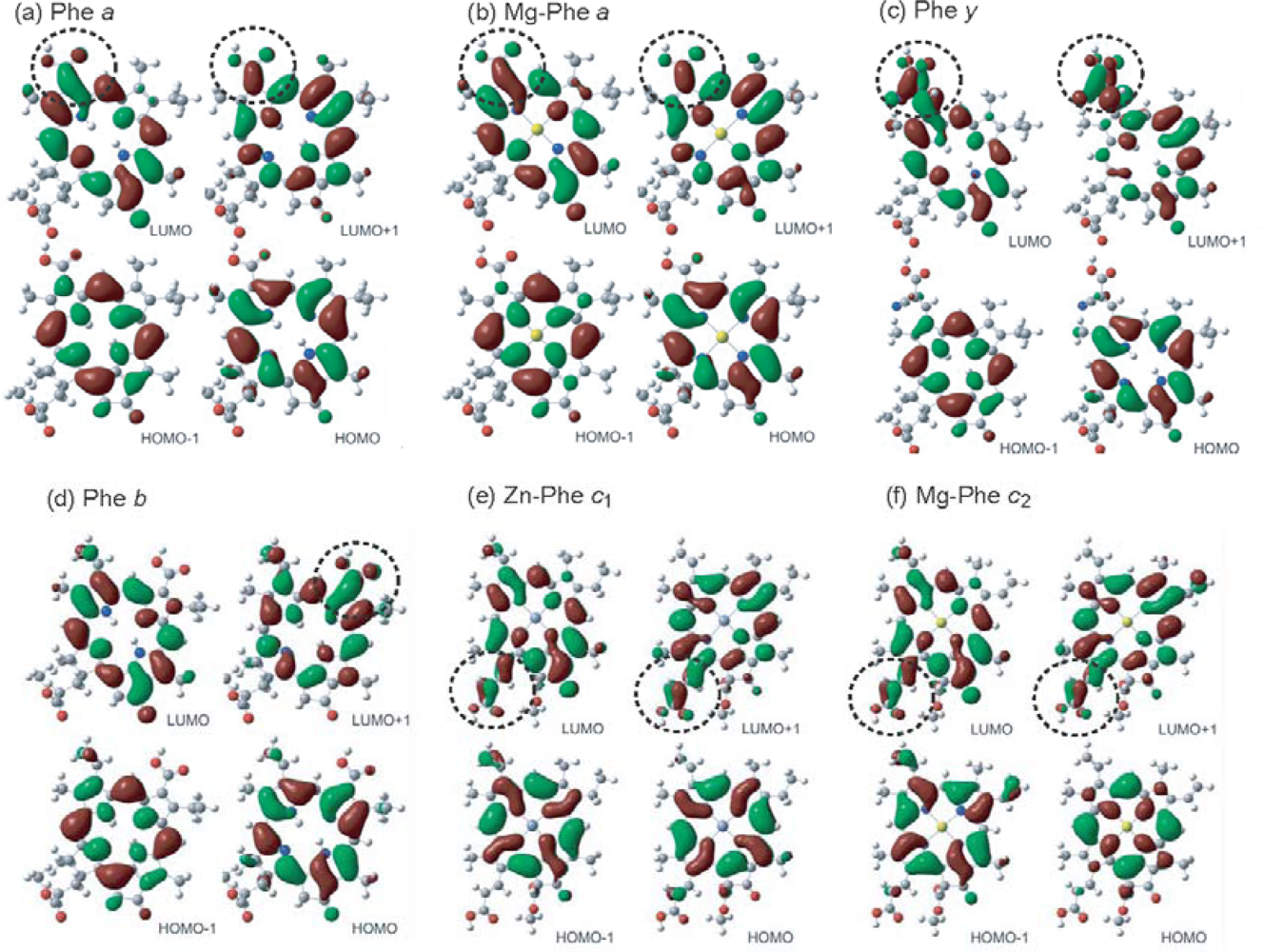
- The combination of the a-type sensitizer having the carboxyl group in the y-direction and the b-type or c-type sensitizer having the carboxyl group in the x-direction should give rise to the highest enhancement of photocurrent and conversion efficiency, because of the minimum interference of the transition dipoles between the pair of co-sensitizers. Polarization and electron-injection along the orthogonal directions must prevent the interference between the intermolecular transition dipole–transition dipole interactions that can trigger intermolecular energy transfer and the resultant dissipation of the singlet energy.
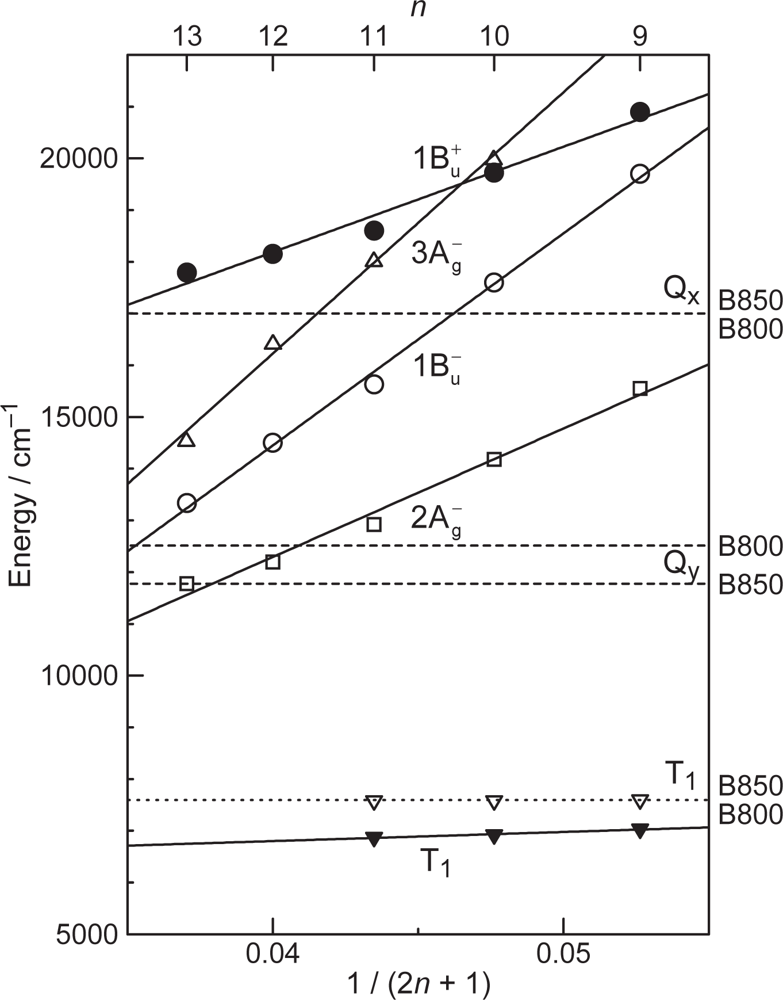
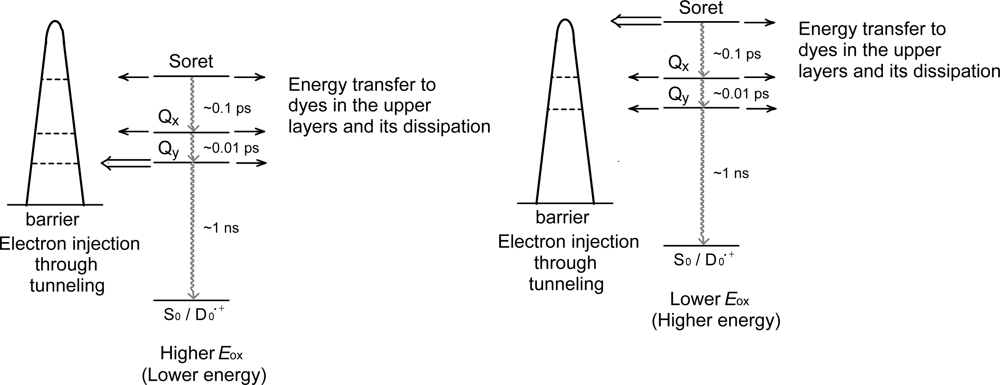
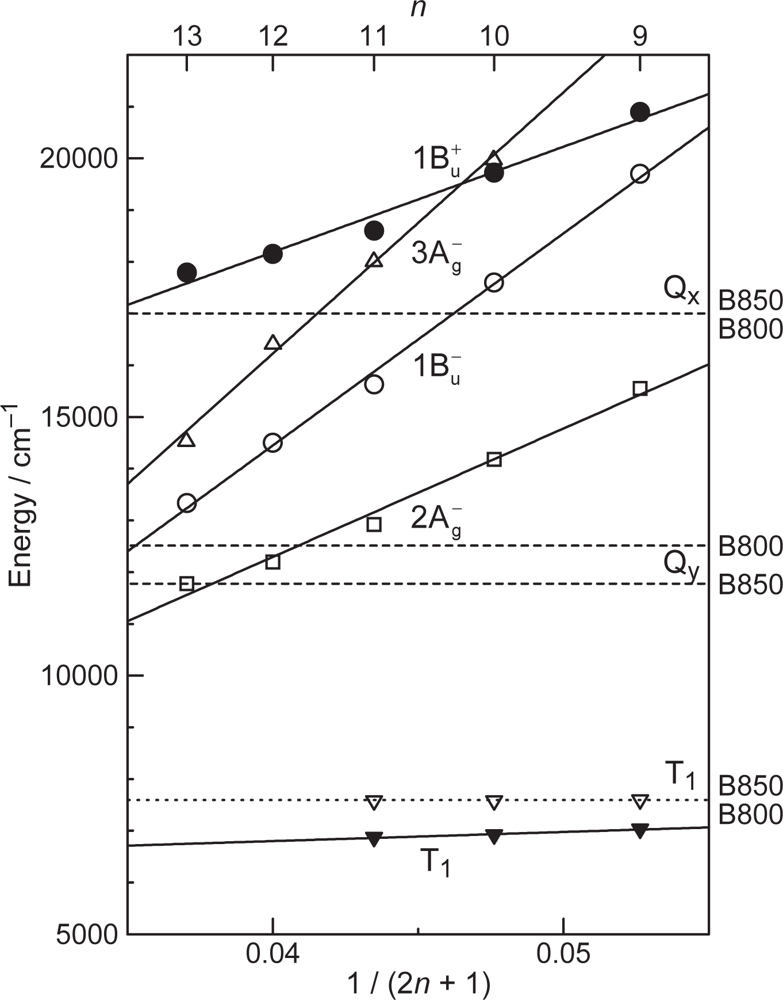
- The different pathways of internal conversion, Soret → Qx → Qy in the a-type sensitizer and Soret → Qy in the b-type or c-type sensitizer may also prevent interaction in the internal-conversion processes because of the different time scales of internal conversion.
5. Conclusions and Future Perspective
5.1. Conclusions
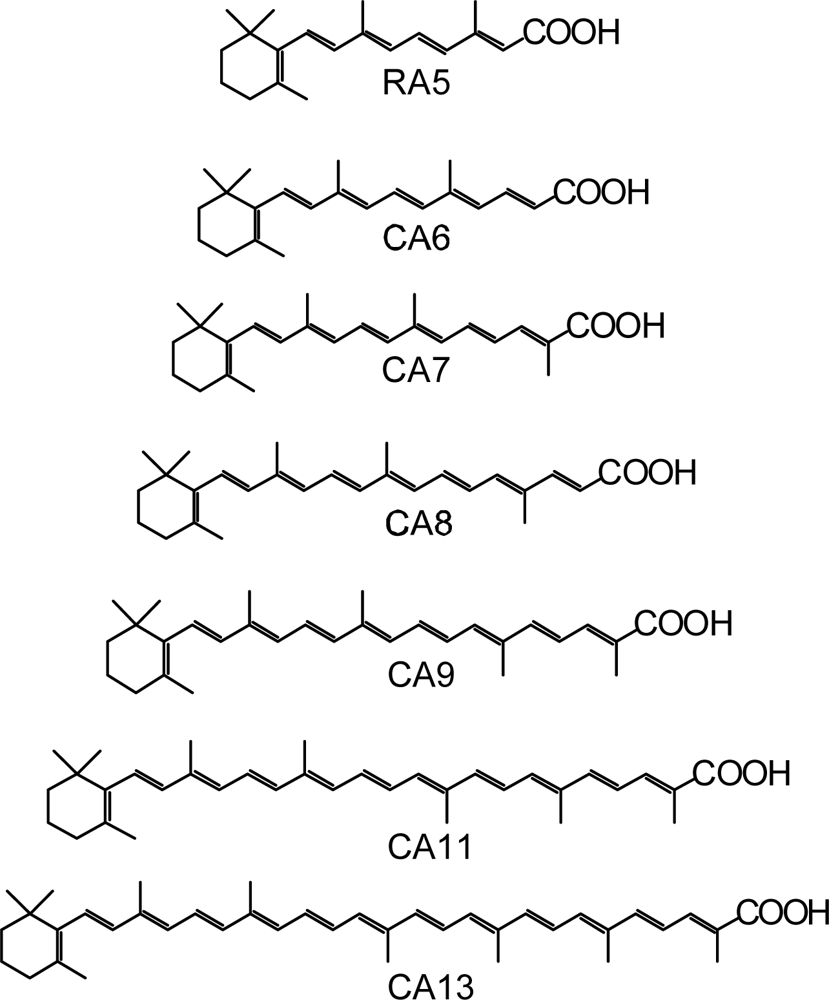
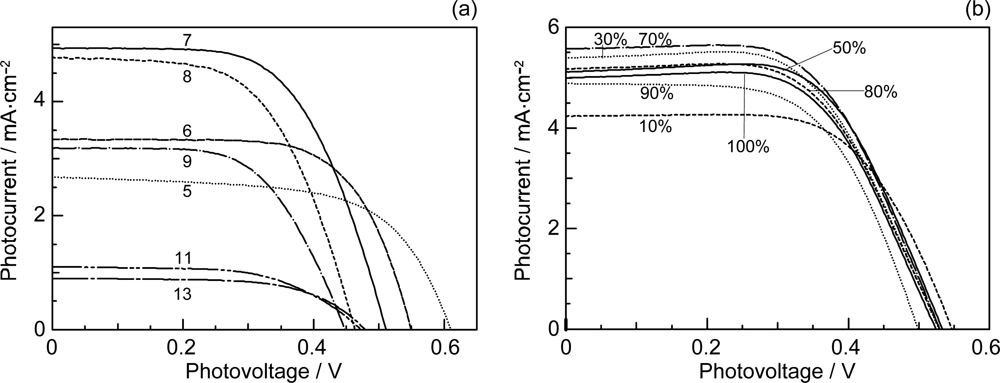
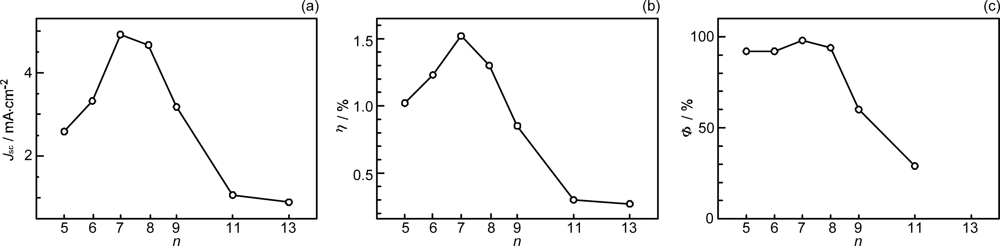
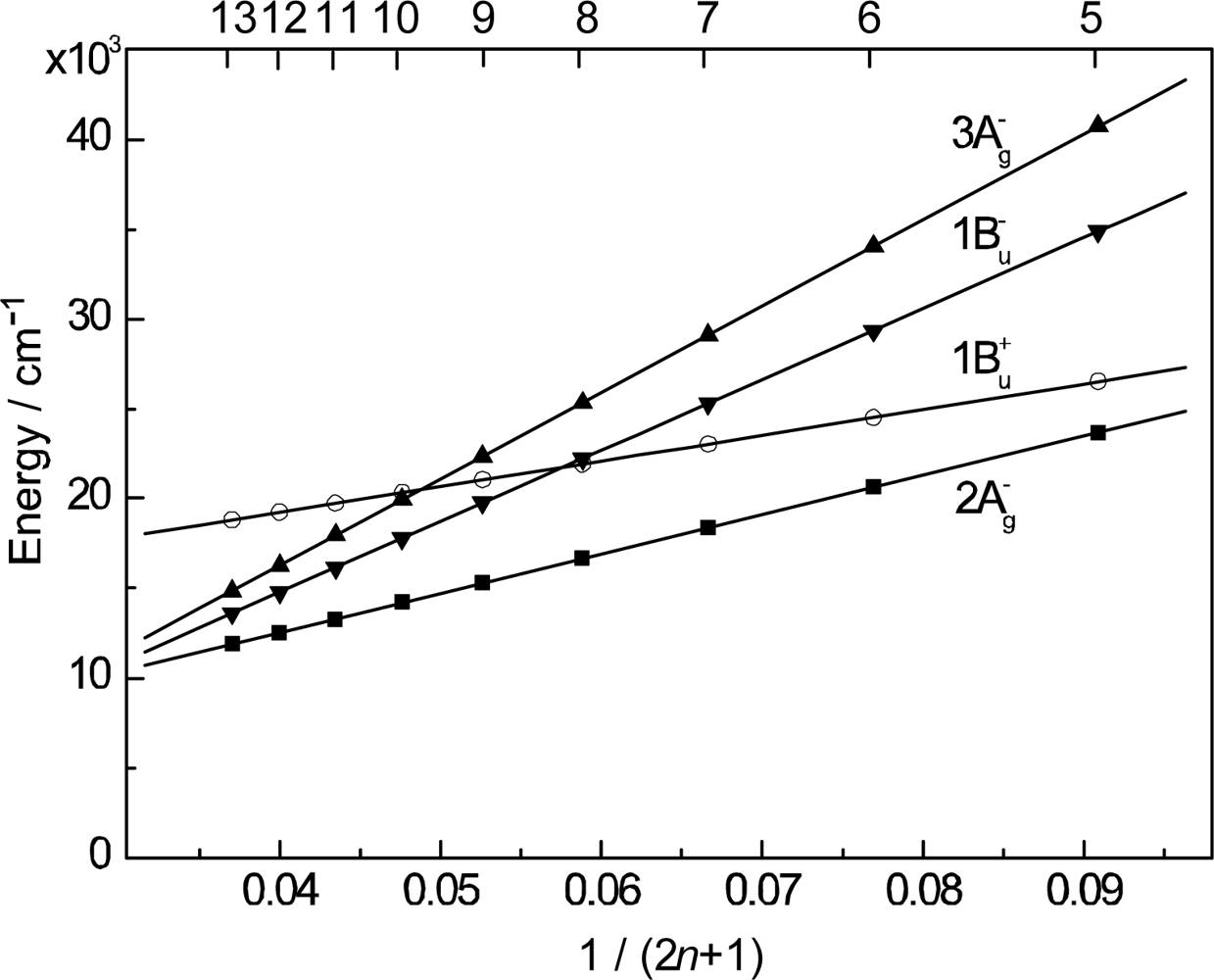
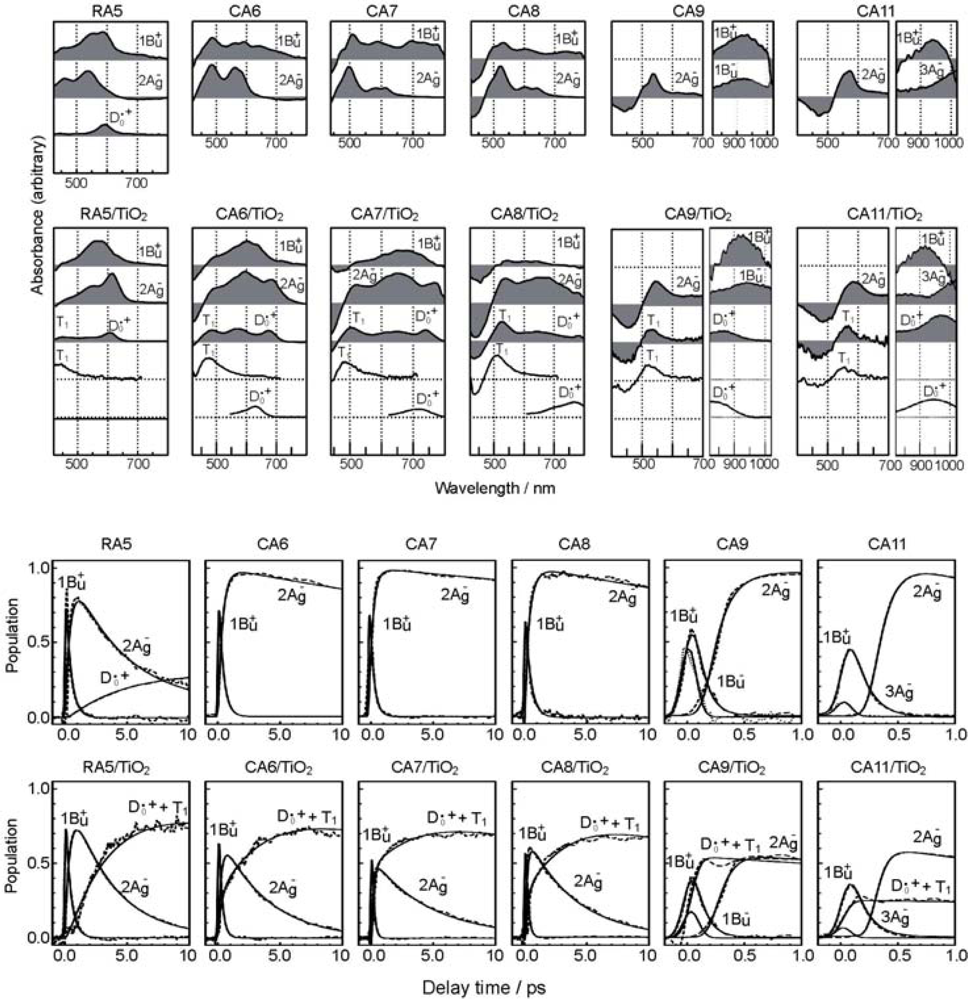
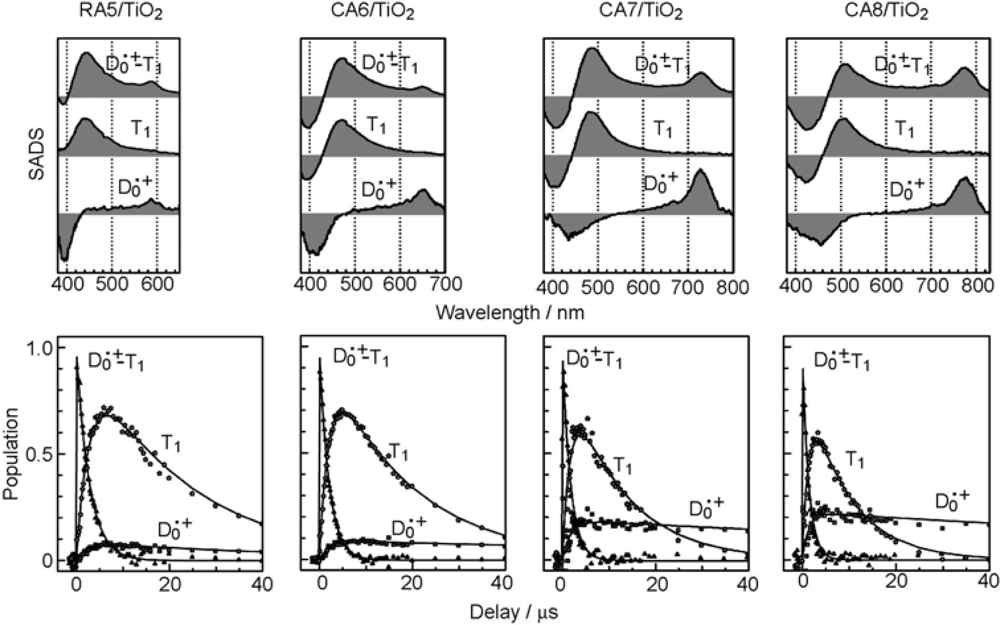
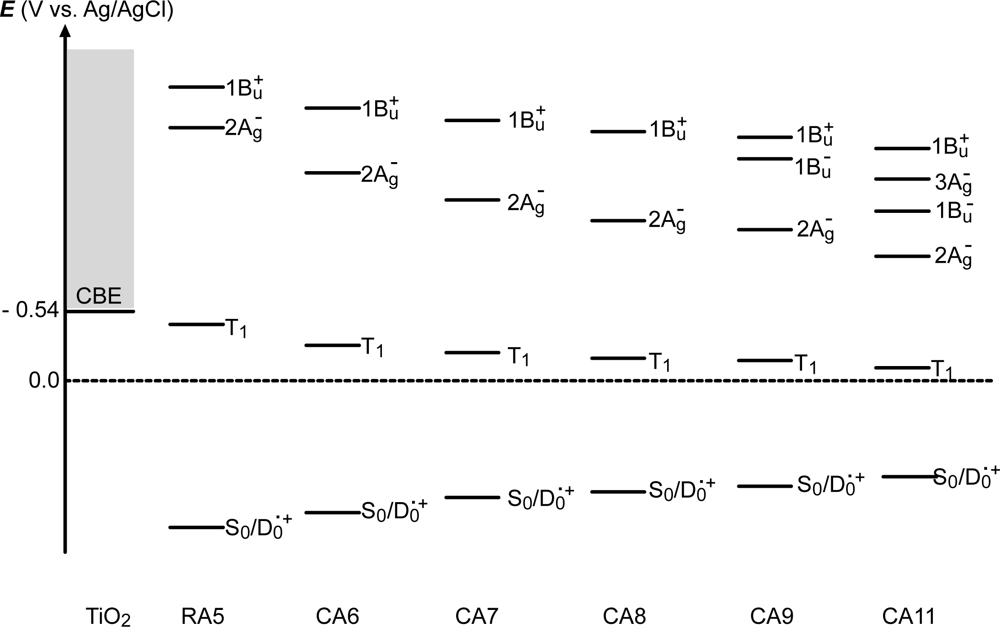
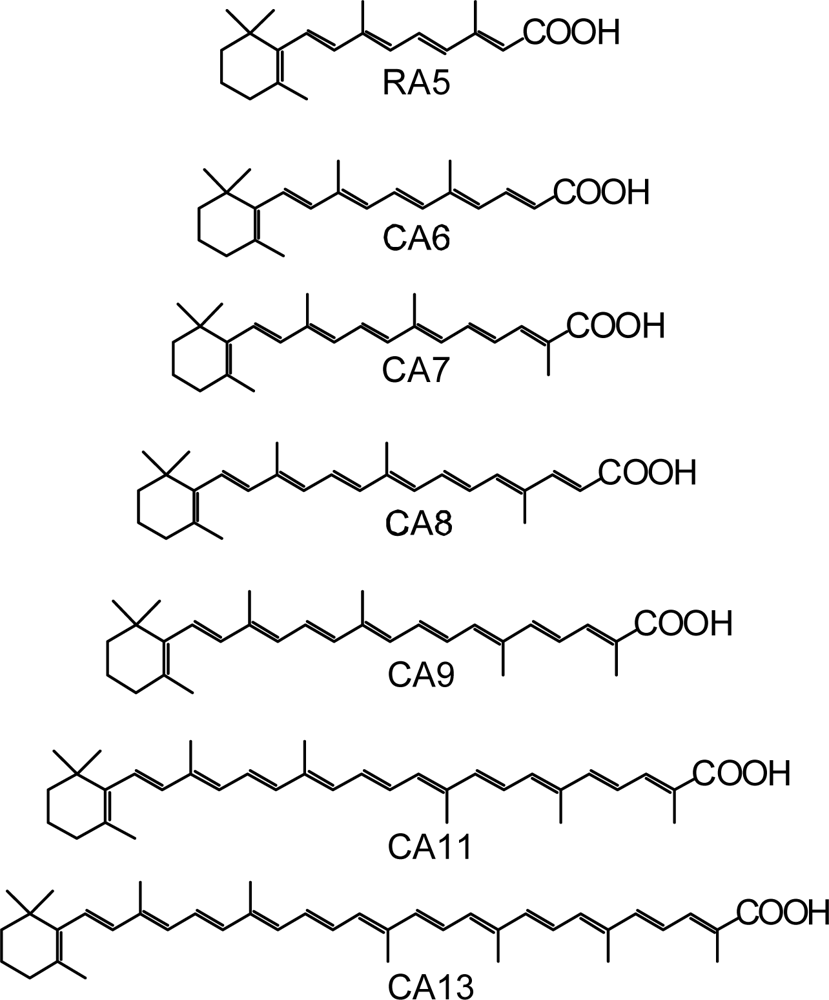
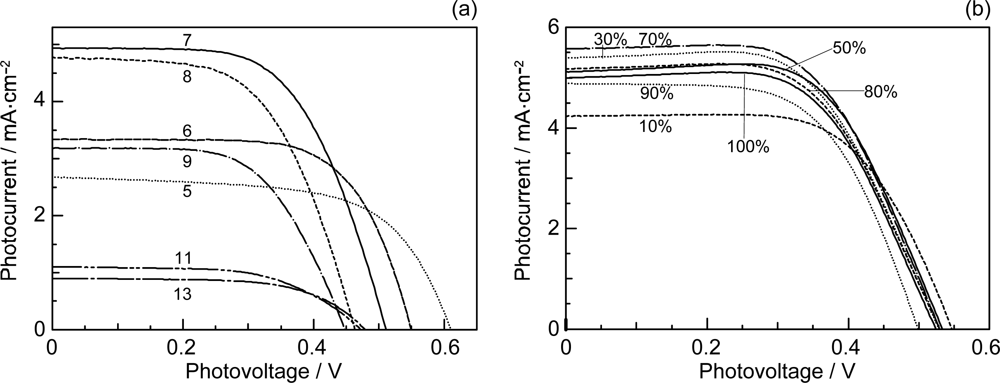
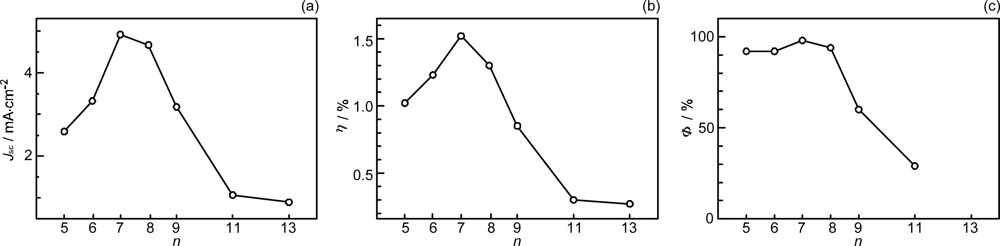
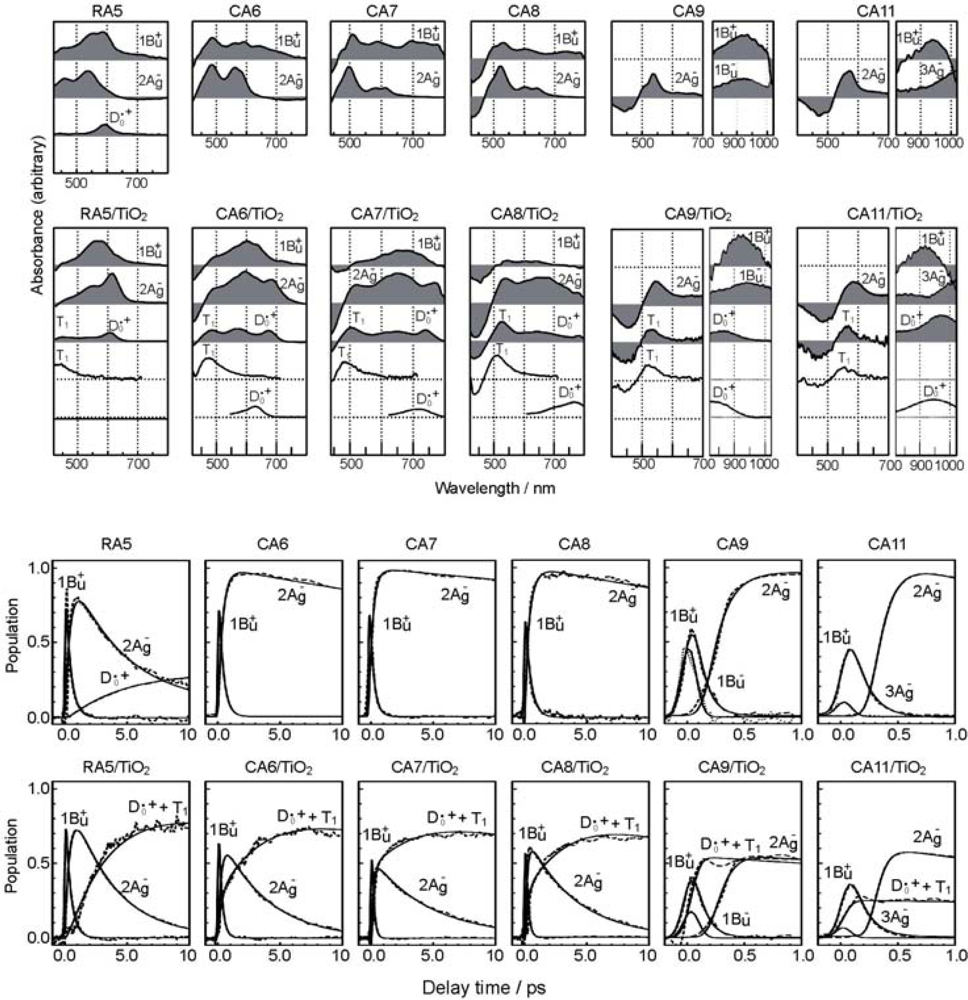
- By the use of a set of RA and CA sensitizers (n = 5~13), the dependence of photocurrent and conversion efficiency of DSSC on the conjugation-length of the sensitizer was determined to be, in the order, RA5 < CA6 < CA7 > CA8 > CA9 > CA11 > CA13. For comparison, the electron-injection efficiencies for RA5–CA11 bound to TiO2 nanoparticles in suspension were determined by means of subpicosecond time-resolved pump-probe spectroscopy. The maximum for CA7 and the decline toward CA11 were explained in terms of excited-state dynamics of the sensitizers. On the other hand, the decline toward RA5 was explained by the increasing efficiency of triplet generation and, as a result, the enhanced singlet-triplet annihilation due to the aggregate formation of the dye sensitizers on the TiO2 surface.
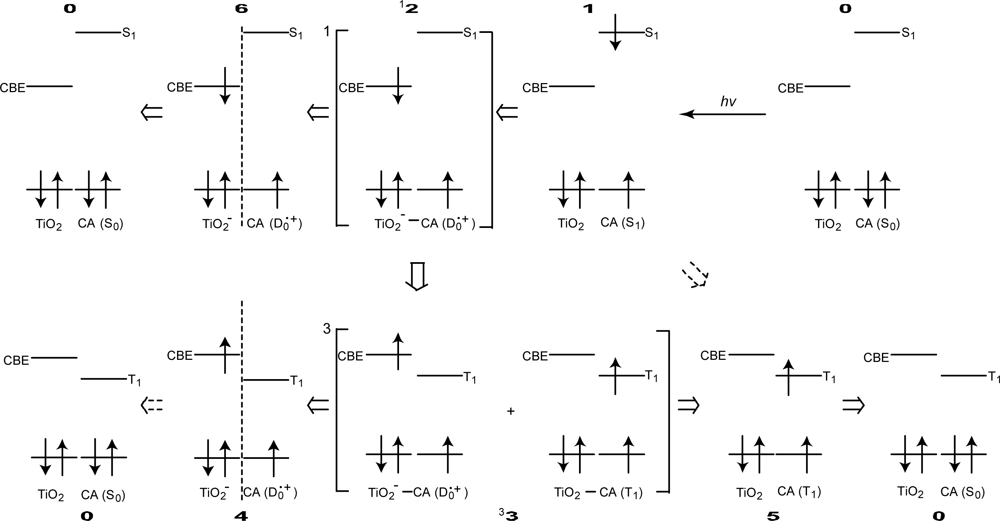
- Excited-state dynamics including the formation of a charge-transfer complex, what we call ‘the combined D0•+ + T1 state’, consisting of a charge-separated (TiO2−–CA (D0•+) and a neutral (TiO2–CA (T1)) states, and its subsequent splitting into the D0•+ plus T1 Car species, was identified by subpicosecond and microsecond time-resolved pump-probe spectroscopy, respectively.
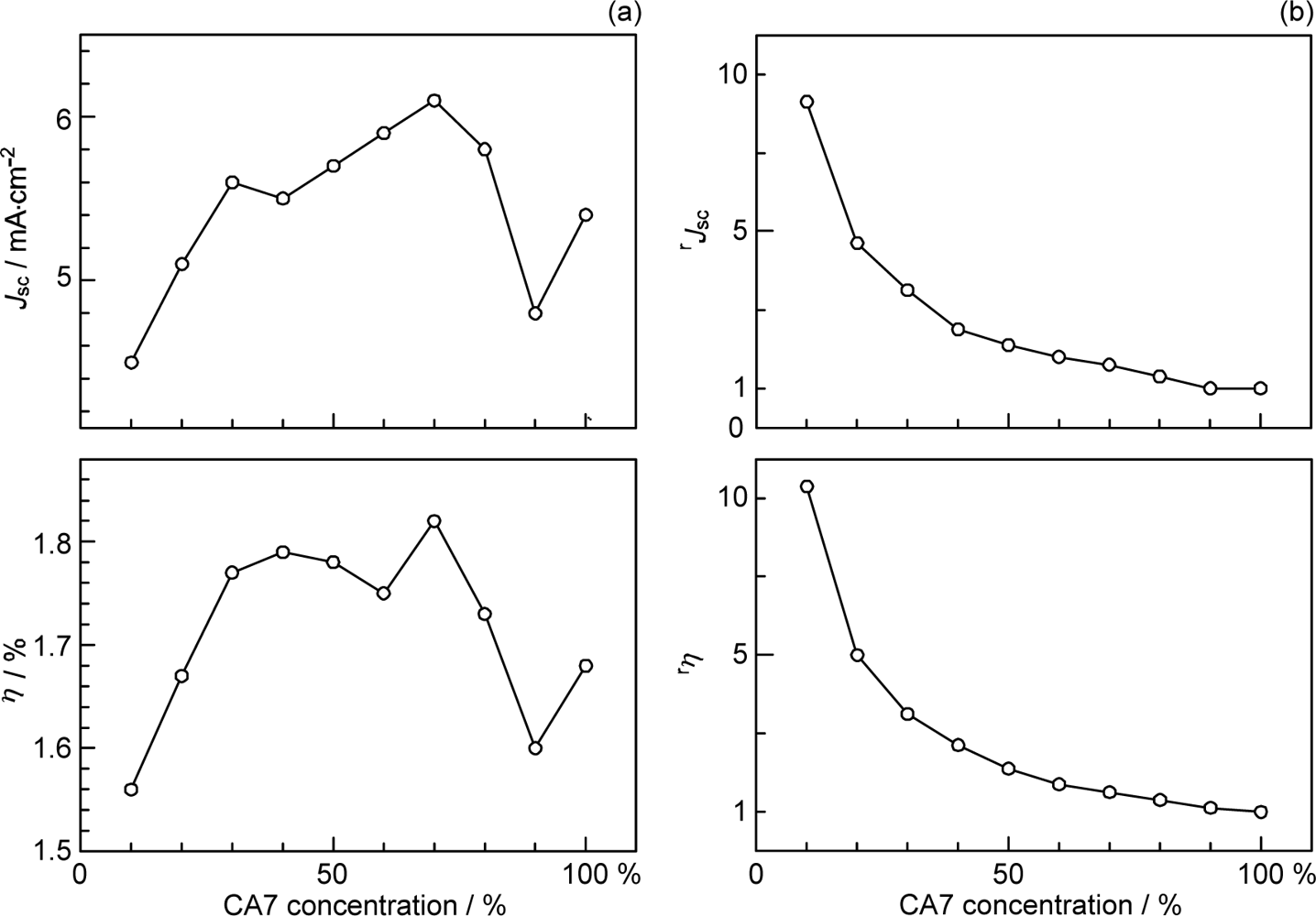
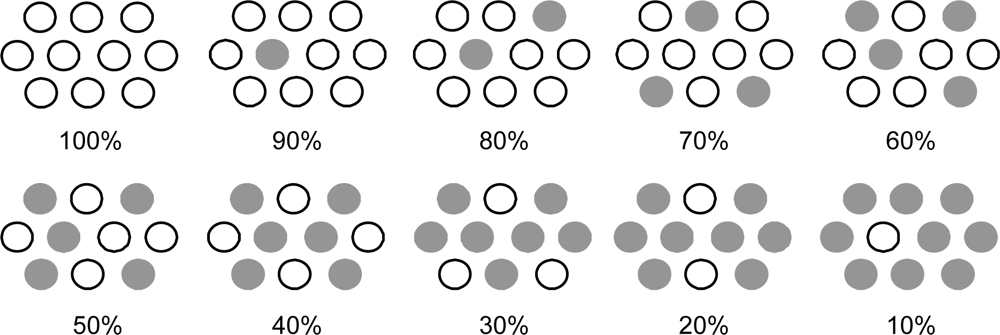
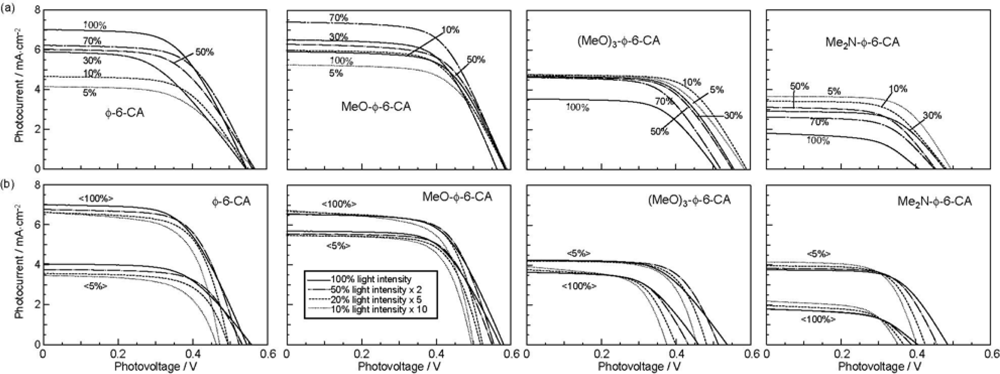
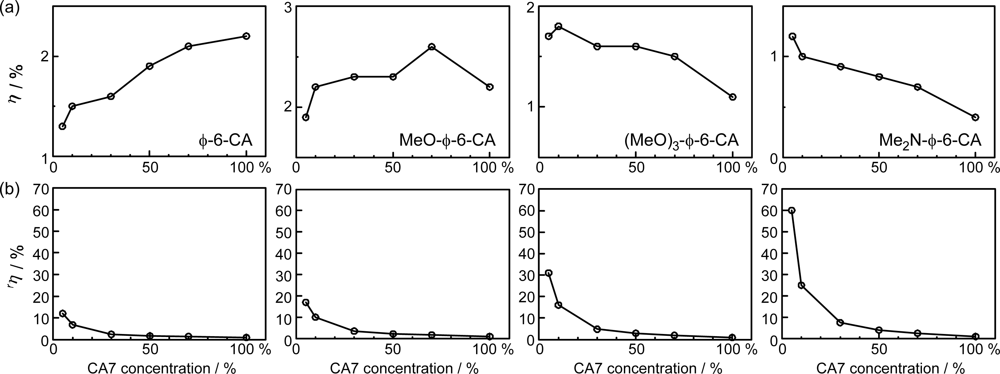
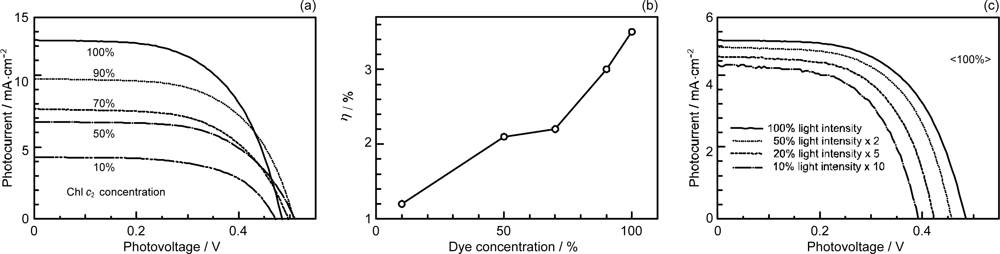
- The mechanism of singlet-triplet annihilation to suppress the photocurrent and conversion efficiency was first identified by their dependence on the dye concentration in the CA7-sensitized solar cell. This mechanism was confirmed by the use of sensitizers having the increasing transition-dipole moments and, as a result, the increasing trend of aggregate formation. The least polarizable (the least aggregate-forming) sensitizer gave rise to the decreasing conversion efficiency, whereas the most polarizable (the most aggregate-forming) sensitizer gave rise to the increasing conversion efficiency, both with the decreasing dye concentration and light intensity.
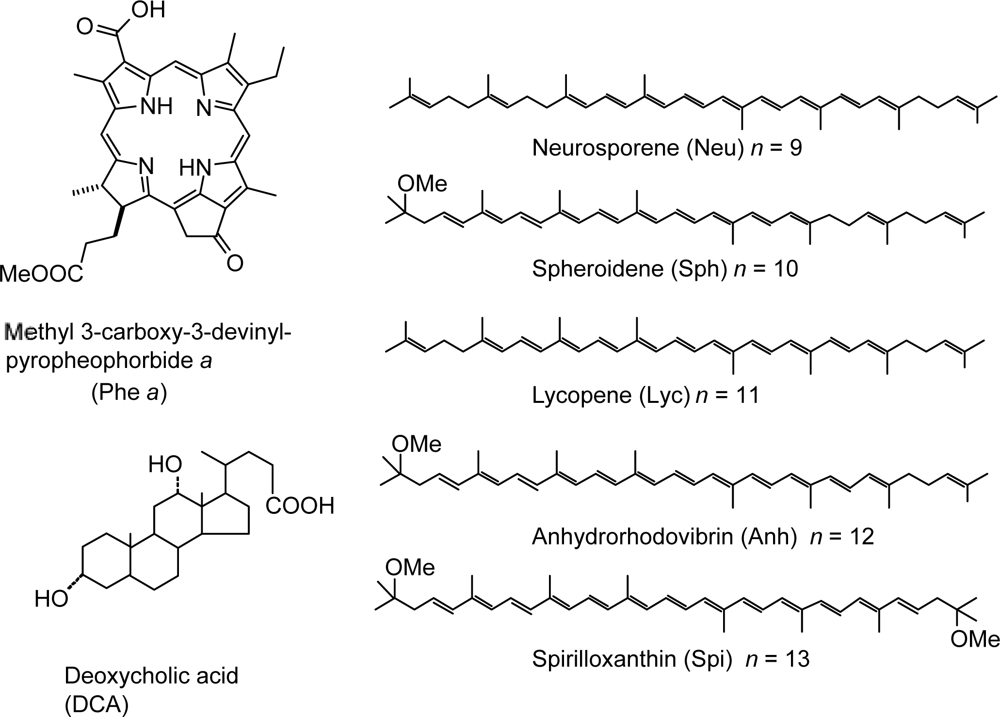
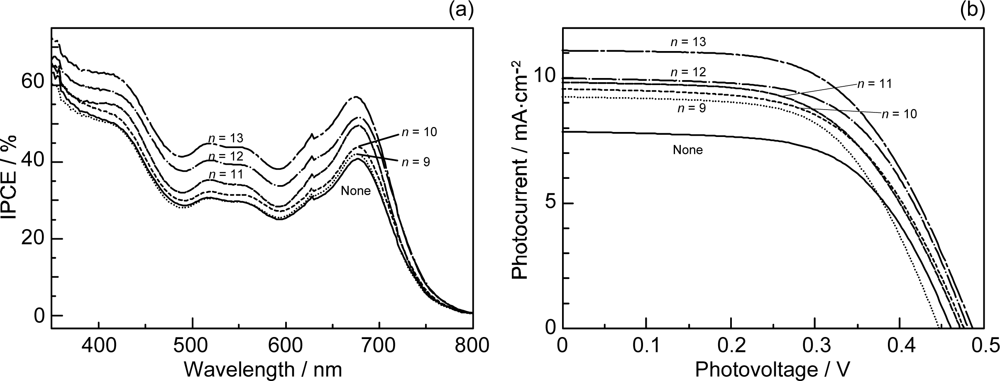
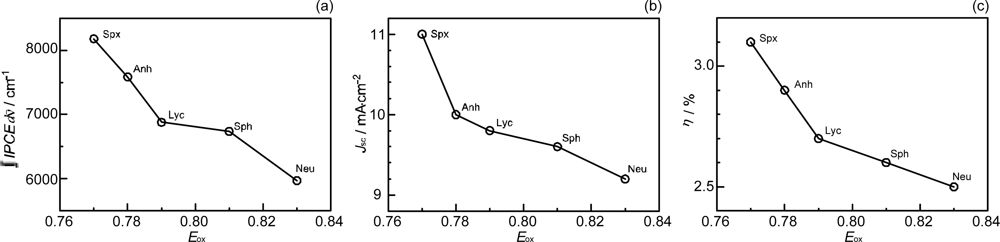
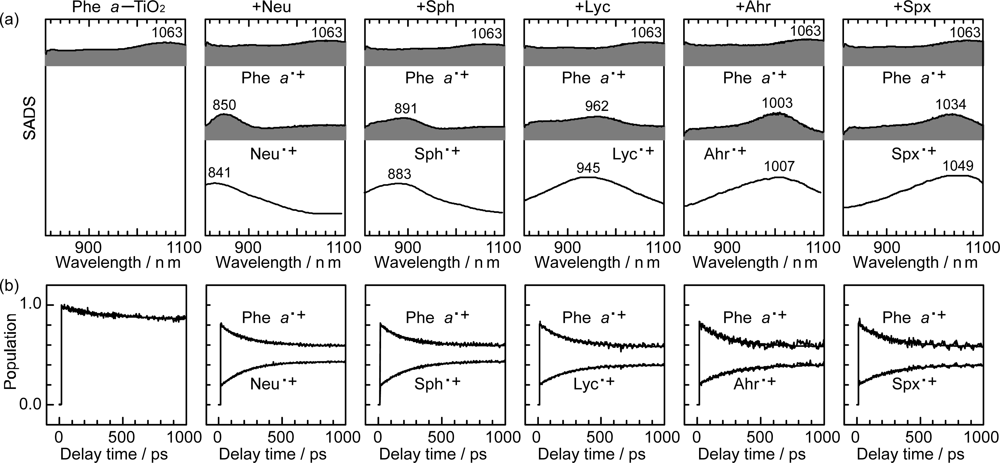
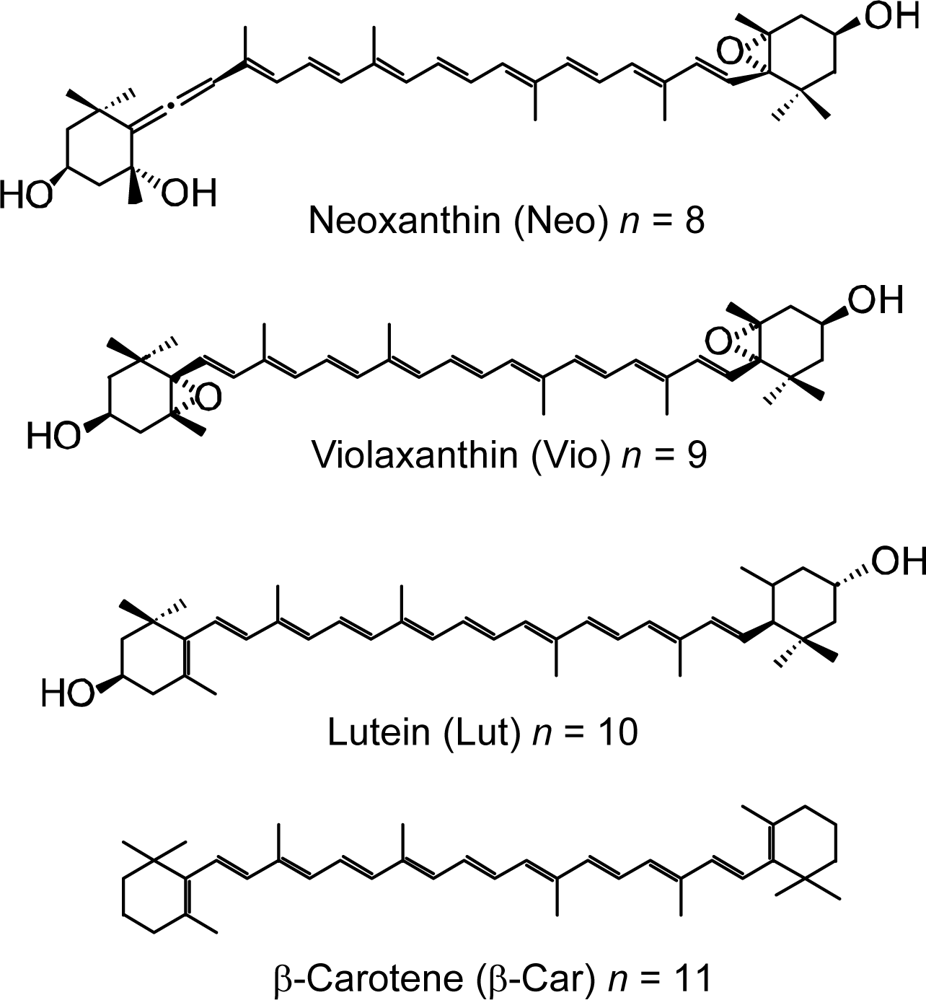
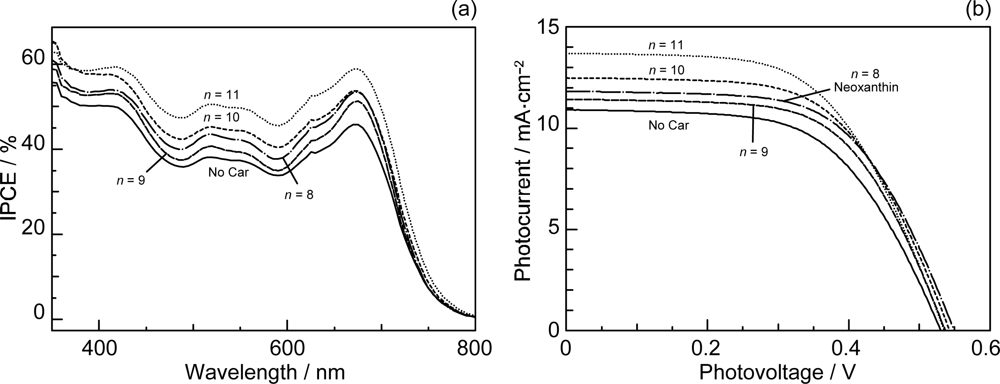
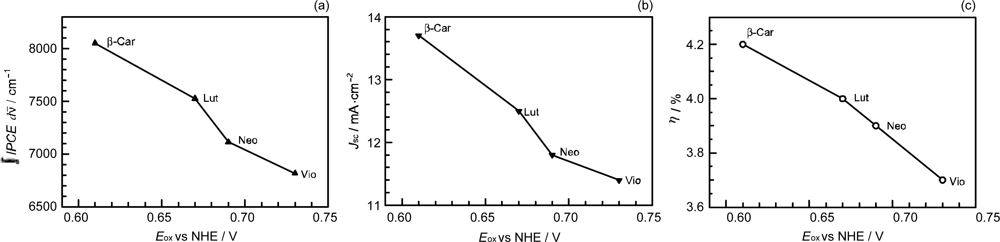
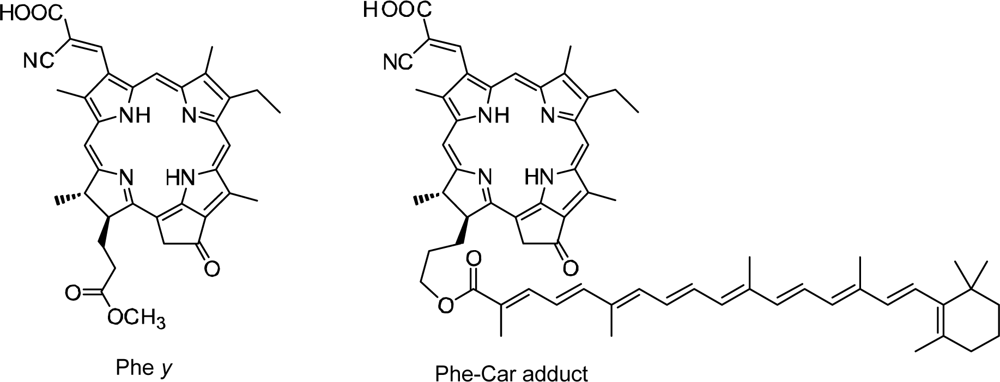
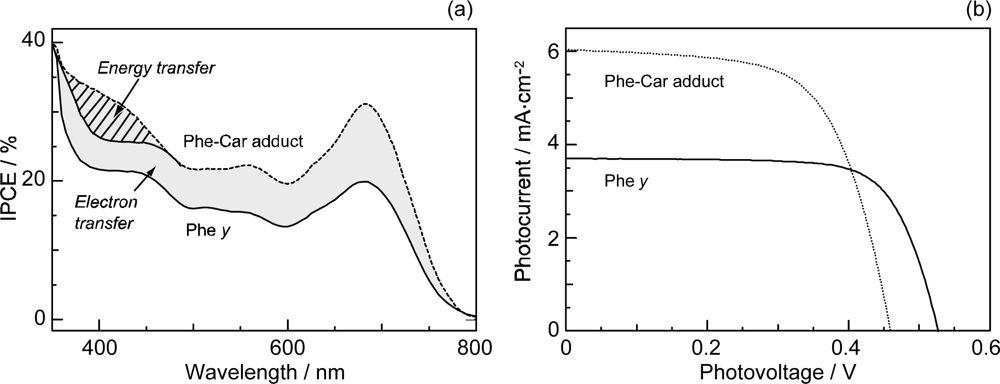
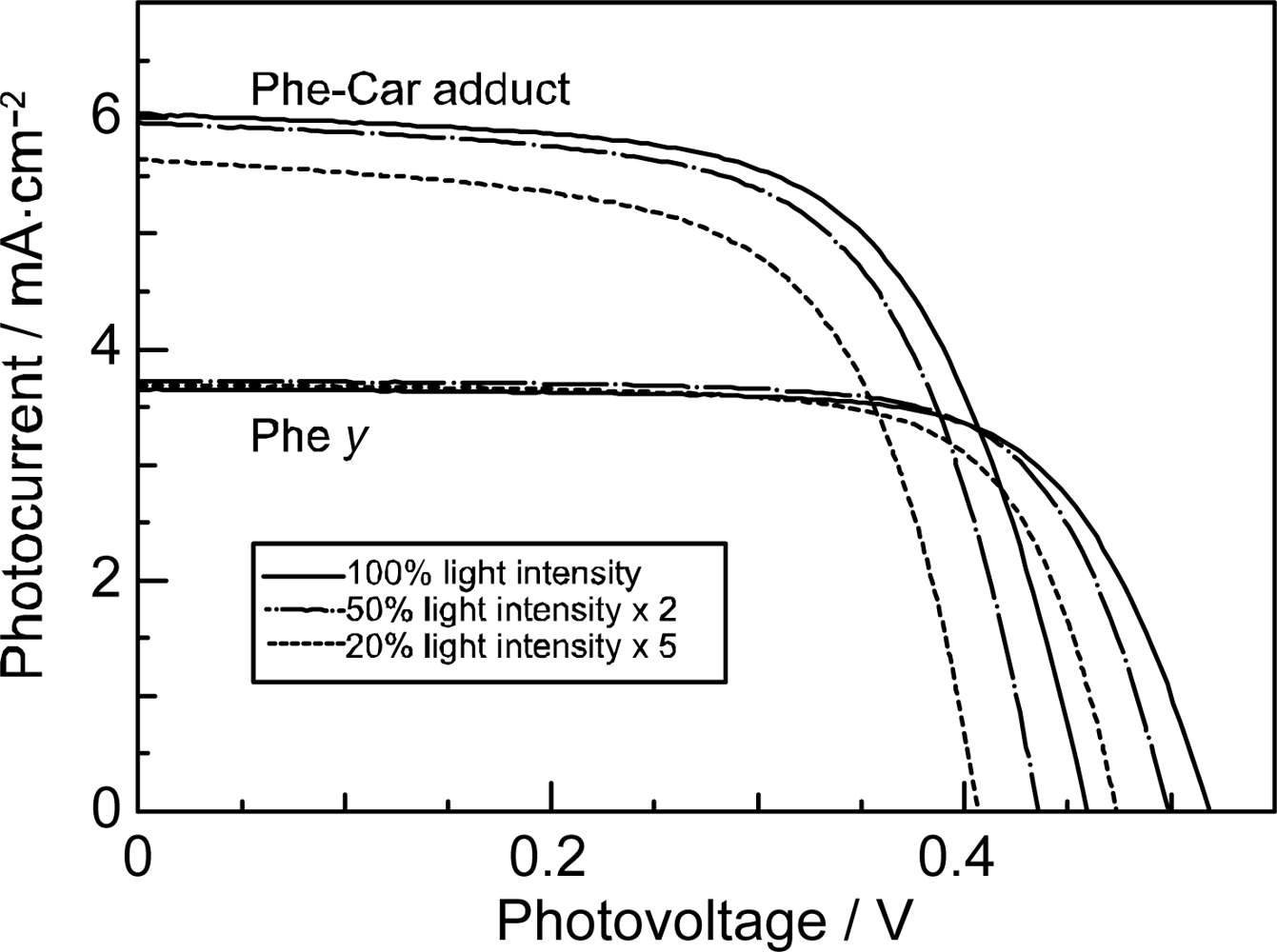
- Sets of bacterial (n = 9~13) and plant (n = 8~11) Cars were used as redox spacers for the Phe a–sensitized solar cell. The idea behind this attempt is to induce electron transfer from Car to Phe a radical cation (Phe a•+) to stabilize the charge-separated TiO2−– Car•+ state to prevent immediate charge recombination of the TiO2−–Phe a•+ pair. Rapid electron injection into TiO2 to generate Phe a•+ (20–40 fs) followed by electron transfer from bacterial Cars to Phe a•+ (200–240 ps) was evidenced by subpicosecond pump-probe spectroscopy of each Phe a−bacterial Car pair bound to TiO2 nanoparticles in suspension. Among the two set of Cars, β-carotene having the lowest one-electron oxidation potential (Eox = 0.61 V) exhibited the maximum enhancement of conversion efficiency (η = 3.4 → 4.2%). In the above mixture of Car and Phe a, no singlet-energy transfer was observed. However, in Phe–Car adduct sensitizer, both singlet-energy transfer and electron transfer from the Car to the Phe moiety were identified in the solar cell. No sign of singlet-triplet annihilation due to aggregate formation was seen in this particular sensitizer.
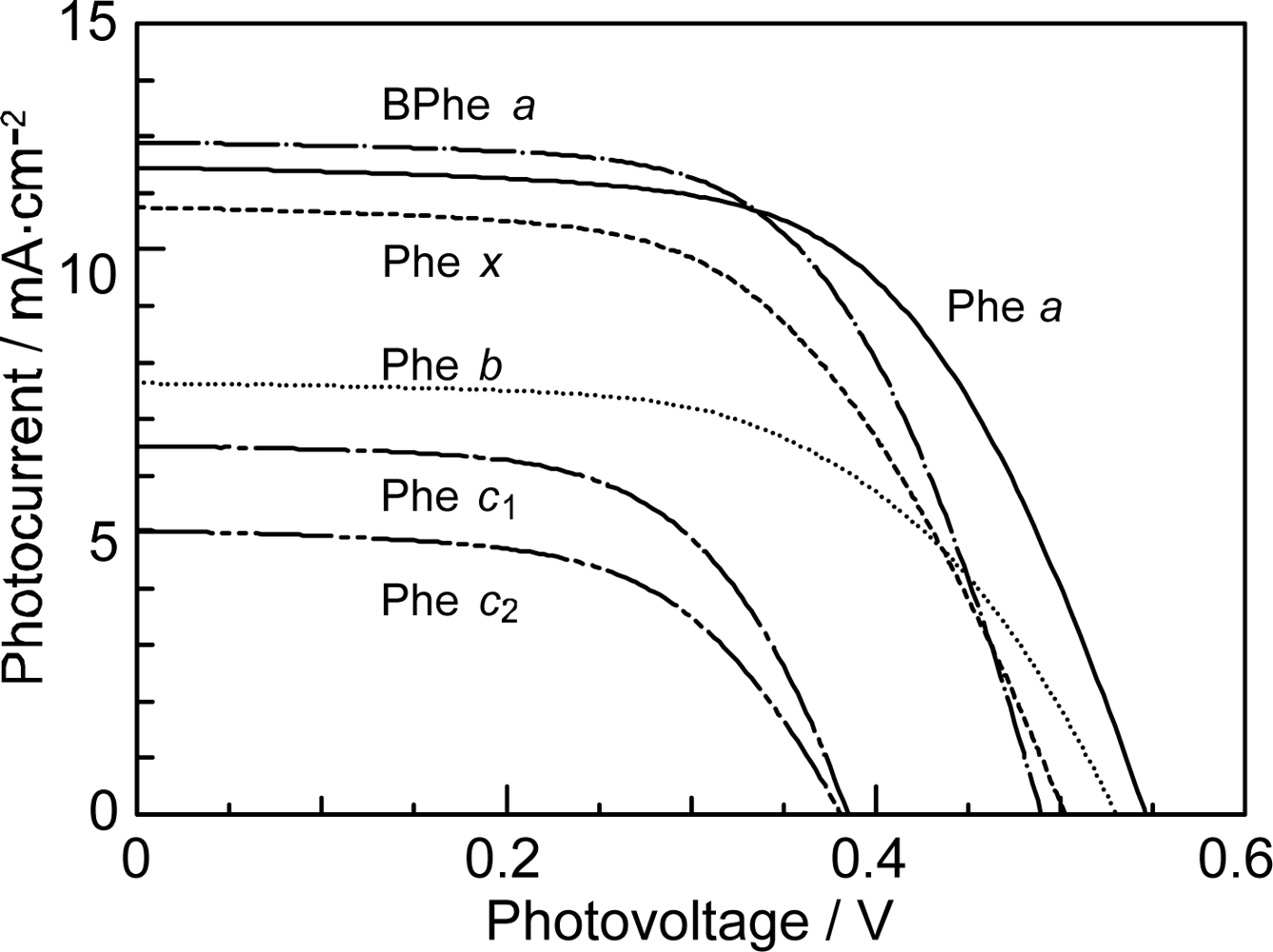
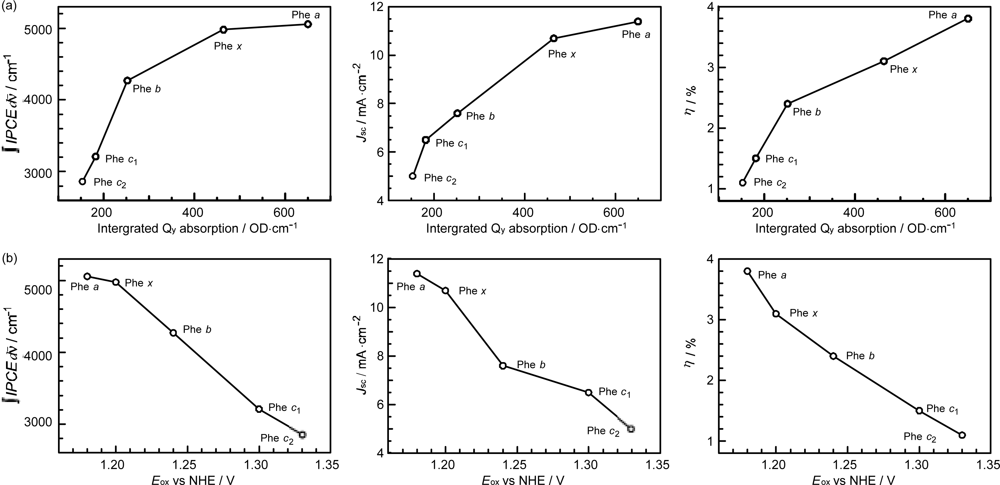
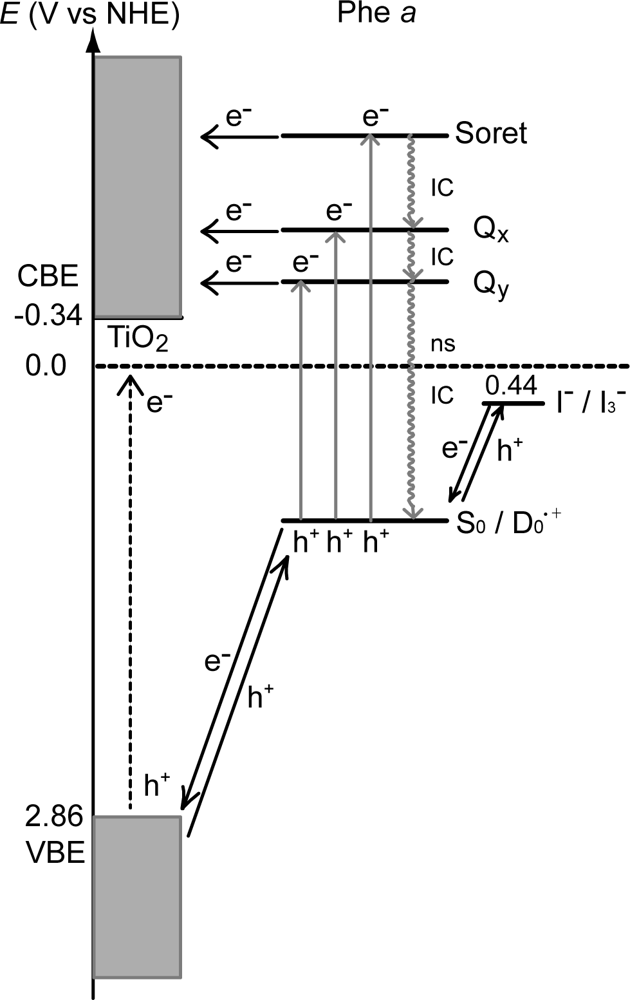
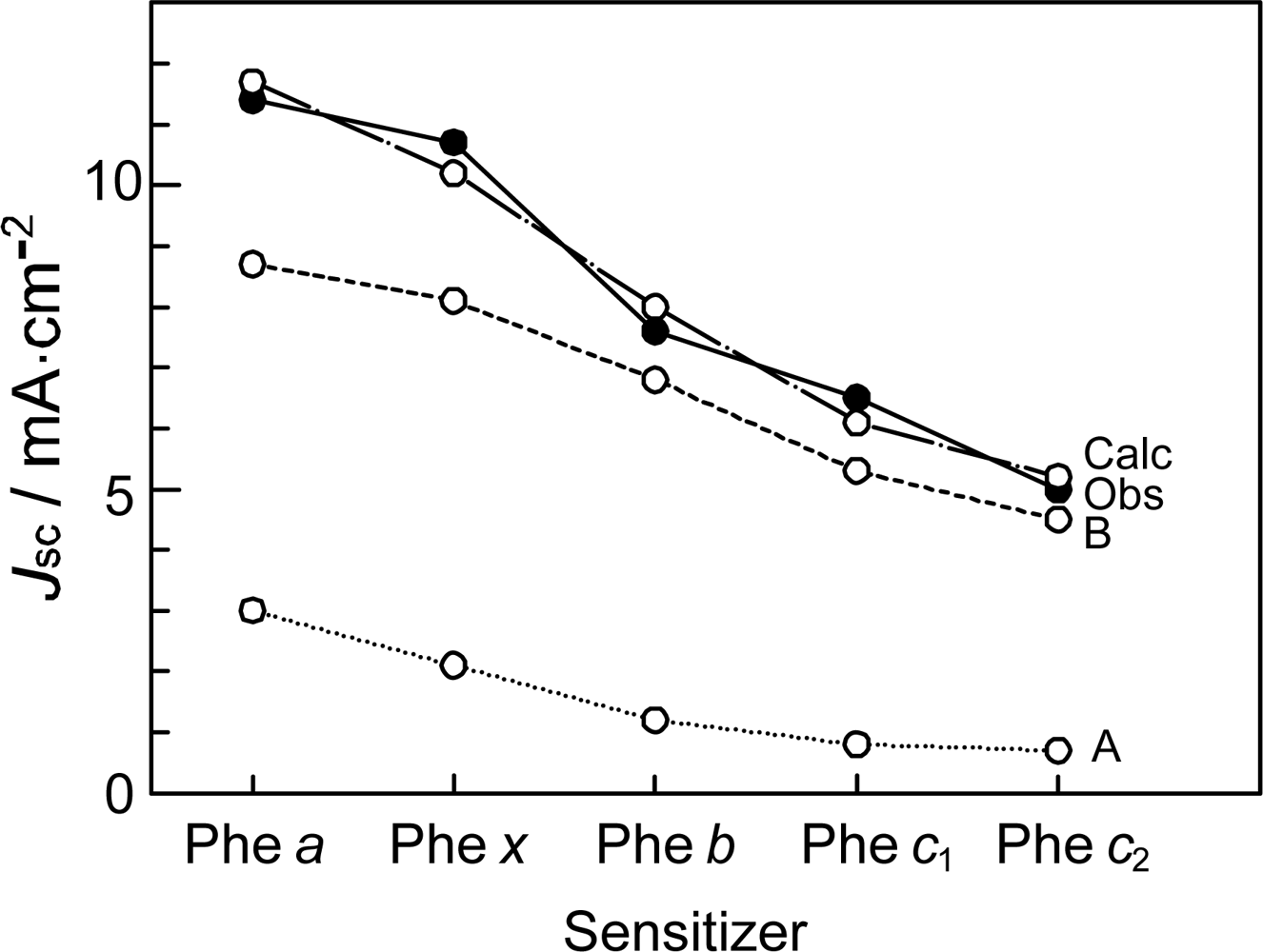
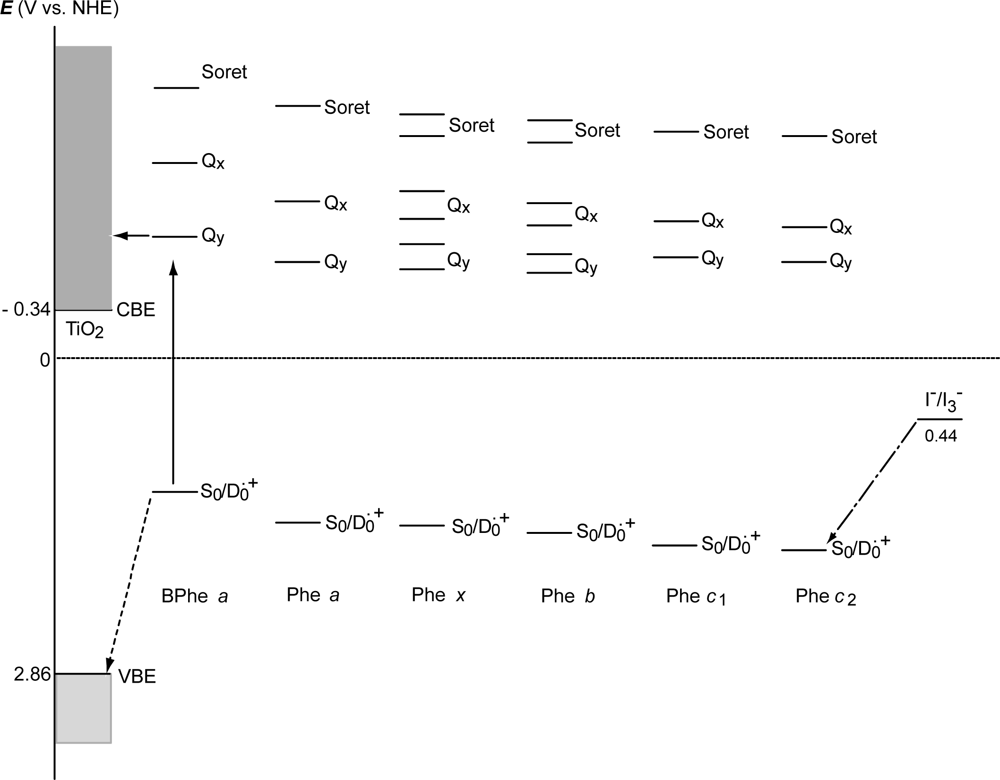
- In a set of Phe sensitizers having the chlorin and porphyrin macrocycles, photocurrent (Jsc) was found to be the functions of the integrated Qy absorption and one-electron oxidation potential (Eox). Phe c2 having the highest one-electron oxidation potential (Eox = 1.33 V) exhibited the lowest conversion efficiency (η = 1.1%) among the Phe sensitizers. On the other hand, Chl c2 (Mg-Phe c2) having low one-electron oxidation potential (Eox = 1.06 V) exhibited the highest conversion efficiency (η = 4.6%) among all the sensitizers we have tested. The extremely-low conversion efficiency in Phe c2 was ascribed to the high Eox value and electron injection via the Qy level, whereas the high conversion efficiency in Chl c2 was ascribed to the low Eox value and electron injection via the Soret level, which is stabilizer by the absence of the Qx level.
- By co-sensitization using the Phe a and Chl c2 sensitizers of the second-best and the best performance, we have succeeded in enhancing the photocurrent and conversion efficiency to 14.0 mA·cm−2 and η = 5.4%, respectively. The enhancement was ascribed to the supplementary light absorption, the orthogonal directions of transition-dipoles and the independent internal conversion processes between the pair of sensitizers.
5.2. Future Perspective
- Pump-probe subpicosecond time-resolved spectroscopy of the single Car sensitizer as well as the Chl a sensitizer plus Car redox spacer, both bound to TiO2 nanoparticles in suspension, has turned out to be very powerful in elucidating the initial electron-injection and electron-transfer mechanisms, respectively. This technique should be applied to determine the mechanisms of excitation, energy-transfer and electron injection in each chlorin or porphyrin sensitizer as well as the pairs of these sensitizers used for co-sensitization.
- In the case of the well-characterized CA and RA sensitizers, it is time to start pump-probe time-resolved spectroscopy of fabricated DSSCs, in various time regions, to elucidate the real electron flow processes in the cell.
- To establish the mechanism of singlet-triplet annihilation, which is a key issue to enhance the performance of DSSCs in general, other spectroscopic methods such as time-resolved fluorescence (up-conversion or Kerr-gate) and Raman, in the subpicosecond time region, will be most useful (see Ref. [9], for example).
6. Relevant Work by Other Investigators
Acknowledgments
- Notice: The clarity of large figures may depend on the computer and the print-out conditions.
References and Notes
- Furuichi, K; Sashima, T; Koyama, Y. The first detection of the 3Ag− state in carotenoids using resonance-Raman excitation profiles. Chem. Phys. Lett 2002, 356, 547–555. [Google Scholar]
- Rondonuwu, FS; Taguchi, T; Fujii, R; Yokoyama, K; Koyama, Y; Watanabe, Y. The energies and kinetics of triplet carotenoids in the LH2 antenna complexes as determined by phosphorescence spectroscopy. Chem. Phys. Lett 2004, 384, 364–371. [Google Scholar]
- Zhang, J-P; Fujii, R; Koyama, Y; Rondonuwu, FS; Watanabe, Y; Mortensen, A; Skibsted, LH. The 1Bu–type singlet state of β-carotene as a precursor of the radical cation found in chloroform solution by sub-picosecond time-resolved absorption spectroscopy. Chem. Phys. Lett 2001, 348, 235–241. [Google Scholar]
- Hoff, AJ; Amesz, J. Visible absorption spectroscopy of chlorophylls. In Chlorophylls; Scheer, H, Ed.; CRC Press, Inc: Boca Raton, FL, USA, 1991; pp. 723–738. [Google Scholar]
- Gouterman, M. Spectra of porphyrins. J. Mol. Spectrosc 1961, 6, 138–163. [Google Scholar]
- Gouterman, M; Wagnière, GH; Snyder, LC. Spectra of porphyrins: Part II. Four orbital model. J. Mol. Spectrosc 1963, 11, 108–127. [Google Scholar]
- Hanson, LK. Molecular orbital theory of monomer pigments. In Chlorophylls; Scheer, H, Ed.; CRC Press, Inc: Boca Raton, FL, USA, 1991; pp. 993–1014. [Google Scholar]
- Koyama, Y; Rondonuwu, FS; Fujii, R; Watanabe, Y. Light-harvesting function of carotenoids in photosynthesis: The roles of the newly found 11Bu− state. Biopolymer 2004, 74, 2–18. [Google Scholar]
- Koyama, Y; Kakitani, Y; Watanabe, Y. Photophysical properties and light-harvesting and photoprotective functions of carotenoids in bacterial photosynthesis: Structural selections. In Primary Processes of Photosynthesis, Part 1: Principles and Apparatus (Comprehensive Series in Photochemical and Photobiology vol 8); Renger, G, Ed.; RSC Publishing: Cambridge, UK, 2008; pp. 151–201. [Google Scholar]
- Wang, X-F; Fujii, R; Ito, S; Koyama, Y; Yamano, Y; Ito, M; Kitamura, T; Yanagida, S. Dye-sensitized solar cells using retinoic acid and carotenoic acids: Dependence of performance on the conjugation length and the dye concentration. Chem. Phys. Lett 2005, 416, 1–6. [Google Scholar]
- Xiang, J-F; Rondonuwu, FS; Kakitani, Y; Fujii, R; Watanabe, Y; Koyama, Y. Mechanisms of electron injection from retinoic acid and carotenoic acids to TiO2 nanoparticles and charge recombination via the T1 state as determined by subpicosecond to microsecond time-resolved absorption spectroscopy: Dependence on the conjugation length. J. Phys. Chem. B 2005, 109, 17066–17077. [Google Scholar]
- Fujii, R; Inaba, T; Watanabe, Y; Koyama, Y; Zhang, J-P. Two different pathways of internal conversion in carotenoids depending on the length of the conjugated chain. Chem. Phys. Lett 2003, 369, 165–172. [Google Scholar]
- Tavan, P; Schulten, K. Electronic excitations in finite and infinite polyenes. Phys. Rev. B 1987, 36, 4337–4358. [Google Scholar]
- Dung, D; Ramsden, J; Graetzel, M. Dynamics of interfacial electron-transfer processes in colloidal semiconductor systems. J. Am. Chem. Soc 1982, 104, 2977–2985. [Google Scholar]
- Wang, X-F; Koyama, Y; Nagae, H; Yamano, Y; Ito, M; Wada, Y. Photocurrents of solar cells sensitized by aggregate-forming polyenes: Enhancement due to suppression of singlet-triplet annihilation by lowering of dye concentration or light intensity. Chem. Phys. Lett 2006, 420, 309–315. [Google Scholar]
- Wang, X-F; Xiang, J-F; Wang, P; Koyama, Y; Yanagida, S; Wada, Y; Hamada, K; Sasaki, S; Tamiaki, H. Dye-sensitized solar cells using a chlorophyll a derivative as the sensitizer and carotenoids having different conjugation lengths as redox spacers. Chem. Phys. Lett 2005, 408, 409–414. [Google Scholar]
- Wang, X-F; Kakitani, Y; Xiang, J-F; Koyama, Y; Rondonuwu, FS; Nagae, H; Sasaki, S; Tamiaki, H. Generation of carotenoid radical cation in the vicinity of a chlorophyll derivative bound to titanium oxide, upon excitation of the chlorophyll derivative to the Qy state, as identified by time-resolved absorption spectroscopy. Chem. Phys. Lett 2005, 416, 229–233. [Google Scholar]
- Wang, X-F; Matsuda, A; Koyama, Y; Nagae, H; Sasaki, S; Tamiaki, H; Wada, Y. Effects of plant carotenoid spacers on the performance of a dye-sensitized solar cell using a chlorophyll derivative: Enhancement of photocurrent determined by one electron-oxidation potential of each carotenoid. Chem. Phys. Lett 2006, 423, 470–475. [Google Scholar]
- Wang, X-F; Koyama, Y; Wada, Y; Sasaki, S; Tamiaki, H. A dye-sensitized solar cell using pheophytin-carotenoid adduct: Enhancement of photocurrent by electron and singlet-energy transfer and by suppression of singlet-triplet annihilation due to the presence of the carotenoid moiety. Chem. Phys. Lett 2007, 439, 115–120. [Google Scholar]
- Wang, X-F; Koyama, Y; Nagae, H; Wada, Y; Sasaki, S; Tamiaki, H. Dependence of photocurrent and conversion efficiency of titania-based solar cell on the Qy absorption and one electron-oxidation potential of pheophorbide sensitizer. J. Phys. Chem. C 2008, 112, 4418–4426. [Google Scholar]
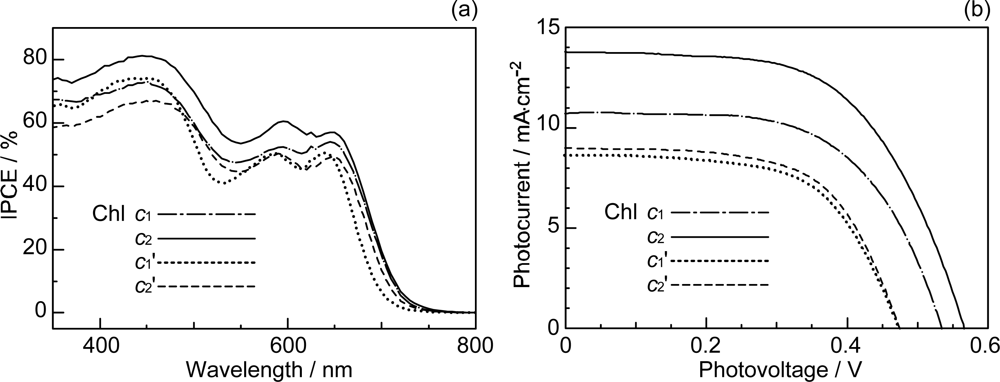
- Wang, X-F; Zhan, C-H; Maoka, T; Wada, Y; Koyama, T. Fabrication of dye-sensitized solar cells using chlorophylls c1 and c2 and their oxidized forms c1’ and c2’ from Undaria pinnatifida (Wakame). Chem. Phys. Lett 2007, 447, 79–85. [Google Scholar]
- Grätzel, M. The artificial leaf, molecular photovoltaics achieve efficient generation of electricity from sunlight. Comment. Inorganic. Chem 1991, 12, 93–111. [Google Scholar]
- Grätzel, M. The artificial leaf, bio-mimetic photocatalysis. CATTECH 1999, 3, 4–17. [Google Scholar]
- Cogdell, RJ; Lindsay, JG. Can photosynthesis provide a ‘biological blueprint’ for the design of novel solar cells? TIBTECH 1998, 16, 521–527. [Google Scholar]
- Gao, FG; Bard, AJ; Kispert, LD. Photocurrent generated on a carotenoid-sensitized TiO2 nanocrystalline mesoporuos electrode. J. Photochem. Photobiol. A: Chem 2000, 130, 49–56. [Google Scholar]
- Pan, J; Benkō, G; Xu, Y; Pascher, T; Sun, L; Sundström, V; Polívka, T. Photoinduced electron transfer between a carotenoid and TiO2 nanoparticle. J. Am. Chem. Soc 2002, 124, 13949–13957. [Google Scholar]
- Zhang, L; Yang, J; Wang, L; Yang, G-Z; Weng, Y-X. Direct observation of interfacial charge recombination to the excited-triplet state in all-trans-retinoic acid sensitized TiO2 nanoparticles by femtosecond time-resolved difference absorption spectroscopy. J. Phys. Chem. B 2003, 107, 13688–13697. [Google Scholar]
- Noguchi, T; Mitsuka, T; Inoue, Y. Fourier transform infrared spectrum of the radical cation of β-carotene photoinduced in photosystem II. FEBS Lett 1994, 356, 179–182. [Google Scholar]
- Hanley, J; Deligiannakís, Y; Pascal, A; Faller, P; Rutherford, AW. Carotenoid oxidation in photosystem II. Biochemistry 1999, 38, 8189–8195. [Google Scholar]
- Polívka, T; Zigmantas, D; Herek, JL; He, Z; Pascher, T; Pullerits, T; Cogdell, RJ; Frank, HA; Sundström, V. The carotenoid S1 state in LH2 complexes from purple bacteria Rhodobacter sphaeroides and Rhodopseudomonas acidophila: S1 energies, dynamics, and carotenoid radical formation. J. Phys. Chem. B 2002, 106, 11016–11025. [Google Scholar]
- Frank, HA; Brudvig, GW. Redox functions of carotenoids in photosynthesis. Biochemistry 2004, 43, 8607–8615. [Google Scholar]
- Debreczeny, MP; Wasielewski, MR; Shinoda, S; Osuka, A. Singlet-singlet energy transfer mechanisms in covalently-linked fucoxanthin- and zeaxanthin-pyropheophorbide molecules. J. Am. Chem. Soc 1997, 199, 6407–6414. [Google Scholar]
- Macpherson, AN; Liddell, PA; Kuciauskas, D; Tatman, D; Gillbro, T; Gust, D; Moore, TA; Moore, AL. Ultrafast energy transfer from a carotenoid to a chlorin in a simple artificial photosynthetic antenna. J. Phys. Chem. B 2002, 106, 9424–9433. [Google Scholar]
- Polívka, T; Pellnor, M; Melo, E; Pascher, T; Sundström, V; Osuka, A; Naqvi, KR. Polarity-tuned energy transfer efficiency in artificial light-harvesting antenna containing carbonyl carotenoids peridinin and fucoxanthin. J. Phys. Chem. B 2007, 111, 467–476. [Google Scholar]
- Kay, A; Grätzel, M. Artificial photosynthesis. 1. Photosensitization of TiO2 solar cells with chlorophyll derivatives and related natural porphyrins. J. Phys. Chem 1993, 97, 6272–6277. [Google Scholar]
- Nazeeruddin, MdK; Humphry-Baker, R; Officer, DL; Campbell, WM; Burrell, AK; Grätzel, M. Application of metalloporphyrins in nanocrystalline dye-sensitized solar cells for conversion of sunlight into electricity. Langmuir 2004, 20, 6514–6517. [Google Scholar]
- Campbell, WM; Burrell, AK; Officer, DL; Jolley, KW. Porphyrins as light harvesters in the dye-sensitised TiO2 solar cell. Coord. Chem. Rev 2004, 248, 1363–1379. [Google Scholar]
- Wang, Q; Campbell, WM; Bonfantani, EE; Jolley, KW; Officer, DL; Walsh, PJ; Gordon, K; Humphry-Baker, R; Nazeeruddin, MK; Grätzel, M. Efficient light harvesting by using green Zn-porphyrin-sensitized nanocrystalline TiO2 films. J. Phys. Chem. B 2005, 109, 15397–15409. [Google Scholar]
- Campbell, WM; Jolley, KW; Wagner, P; Wagner, K; Walsh, PJ; Gordon, KC; Schmidt-Mende, L; Nazeeruddin, MK; Wang, Q; Grätzel, M; Officer, DL. Highly efficient porphyrin sensitizers for dye-sensitized solar cells. J. Phys. Chem. C 2007, 111, 11760–11762. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RA5 | CA6 | CA7 | CA8 | CA9 | CA11 | |
|---|---|---|---|---|---|---|
| 1Bu+ channel | 0.04 | 0.31 | 0.46 | 0.31 | 0.60 | 0.29 |
| 2Ag− channel | 0.88 | 0.61 | 0.52 | 0.63 | — | — |
| Sum | 0.92 | 0.92 | 0.98 | 0.94 | 0.60 | 0.29 |
| RA5 | CA6 | CA7 | CA8 | CA9 | CA11 | |
|---|---|---|---|---|---|---|
| Eox (vs. Ag/AgCl) | 1.08 | 0.97 | 0.87 | 0.80 | 0.77 | 0.71 |
| RA5-TiO2 | CA6-TiO2 | CA7-TiO2 | CA8-TiO2 | |
|---|---|---|---|---|
| kd−1(μs) | 34 | 22 | 9.4 | 5.9 |
| kt−1 (μs) | 3.1 | 2.7 | 2.1 | 2.0 |
| kt0−1 (μs) | 22 | 18 | 12 | 9.0 |
| kd0−1 (μs) | ~50 | ~150 | ~150 | ~150 |
| ϕD (%) | 8 | 11 | 18 | 25 |
| ϕT (%) | 92 | 89 | 82 | 75 |
| Time constant | no Car | +Neu | +Sph | +Lyc | +Ahr | +Spx |
|---|---|---|---|---|---|---|
| Phe a• + rise | 0.03 | 0.04 | 0.03 | 0.03 | 0.02 | 0.02 |
| Phe a• + decay | 338 | 203 | 210 | 220 | 235 | 241 |
| Principal and Co-sensitizers | Voc/V | FF | Jsc (Av) r Jsc/mA·cm−2 | η (Av) rη/% | S | Eox/Vvs | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Phe a (a-type) a-type | 0.56 | 0.68 | 9.0 | 3.4 | 1.16 | ||||||
| Mg-Phe a | 0.51 | 0.70 | 4.4 | 1.6 | 0.79 | ||||||
| co-sensitization | 0.50 | 0.69 | 5.6 | (6.7) | 0.83 | 1.9 | (2.5) | 0.79 | 0.8 | 41 | |
| Phe y | 0.49 | 0.70 | 5.2 | 1.8 | 1.19 | ||||||
| co-sensitization b-type | 0.50 | 0.68 | 6.8 | (7.1) | 0.36 | 2.3 | (2.6) | 0.92 | 0.9 | 62 | |
| Phe b | 0.53 | 0.70 | 4.6 | 1.7 | 1.24 | ||||||
| co-sensitization c-type | 0.57 | 0.68 | 10.9 | (6.8) | 1.60 | 4.3 | (2.6) | 1.65 | 1.6 | 39 | |
| Zn-Phe c1 | 0.62 | 0.63 | 10.4 | 4.0 | 1.16 | ||||||
| co-sensitization | 0.60 | 0.69 | 11.9 | (9.7) | 1.23 | 5.0 | (3.7) | 1.35 | 1.3 | 80 | |
| Mg-Phe c2 (Chl c2) | 0.58 | 0.66 | 9.9 | 3.8 | 1.06 | ||||||
| co-sensitization | 0.60 | 0.64 | 14.0 | (9.5) | 1.47 | 5.4 | (3.6) | 1.50 | 1.5 | 95 | |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Koyama, Y.; Miki, T.; Wang, X.-F.; Nagae, H. Dye-Sensitized Solar Cells Based on the Principles and Materials of Photosynthesis: Mechanisms of Suppression and Enhancement of Photocurrent and Conversion Efficiency. Int. J. Mol. Sci. 2009, 10, 4575-4622. https://doi.org/10.3390/ijms10114575
Koyama Y, Miki T, Wang X-F, Nagae H. Dye-Sensitized Solar Cells Based on the Principles and Materials of Photosynthesis: Mechanisms of Suppression and Enhancement of Photocurrent and Conversion Efficiency. International Journal of Molecular Sciences. 2009; 10(11):4575-4622. https://doi.org/10.3390/ijms10114575
Chicago/Turabian StyleKoyama, Yasushi, Takeshi Miki, Xiao-Feng Wang, and Hiroyoshi Nagae. 2009. "Dye-Sensitized Solar Cells Based on the Principles and Materials of Photosynthesis: Mechanisms of Suppression and Enhancement of Photocurrent and Conversion Efficiency" International Journal of Molecular Sciences 10, no. 11: 4575-4622. https://doi.org/10.3390/ijms10114575