3.1. Modelling of Metal-Support Interactions
Our first example for the combination of DFT calculation with experimental studies of heterogeneous catalysts deals with the production of methane from synthesis gas. The effect of the composition of synthesis gas, starting with pure CO + H
2 and approaching in stepwise fashion the composition as delivered from an existing wood gasifier, on the surface properties of a commercial Ni/Al
2O
3 catalyst and on its activity under methanation conditions was studied on an atomic level by
quasi in-situ X-ray photoelectron spectroscopy (XPS) [
1]. One of the conclusions was that the stability of the Ni particles on the γ-Al
2O
3 support can be influenced by cluster growth phenomena, which influence both size and distribution of the metal particles.
In order to shed light on the involved metal-support interactions, a theoretical study has been performed [
2,
3]. It was shown that the deposition of a very low number (Θ
Ni < 0.4 mL) of metal atoms on γ-Al
2O
3 only influences the local surface structure, in particular the neighbouring centres of Ni, such as: Al(4), Al(5), O(3) and O’(3) (see previous paper [
3]). The stabilization energy of deposited nickel particles on Al
15O
40H
35 cluster was calculated as difference between the total energy of the metal deposited on the γ-Al
2O
3 surface and sum of the total energies of pure Al
15O
40H
35 and the metal atoms, respectively. In all cases, the first layer of nickel on Al
2O
3(100) deposits in positions closer to octahedral O(3) centres (see
Table 1) with different stabilization energies per Ni atom (for Ni
2: −0.82 eV, for Ni
7: −0.90 eV and for Ni
9: −0.71 eV). The calculated stabilization energies for the different Ni clusters are comparable with published energies obtained for the adsorption of different metals, mainly Pd, on γ-Al
2O
3 [
19,
20]. Being strongly bound to O(3) centres, the deposited nickel influences the electronic structure of the γ-Al
2O
3, too. The largest investigated nickel cluster (Ni
9) creates many interesting structures at the support surface.
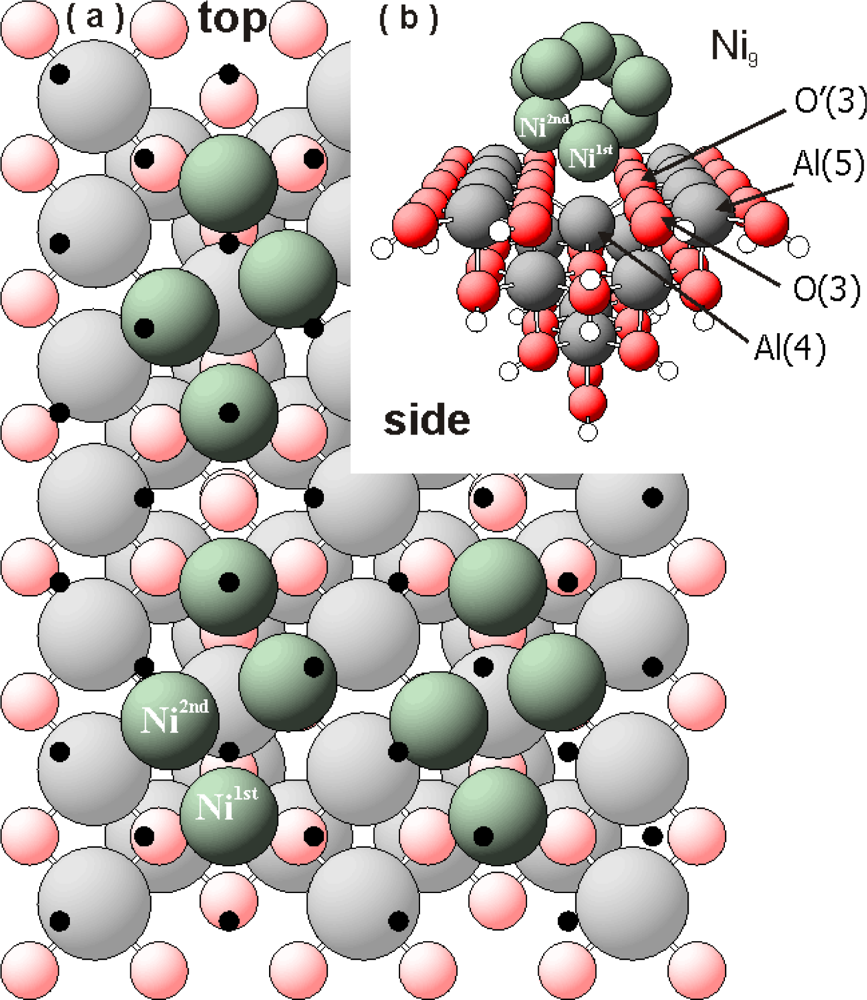
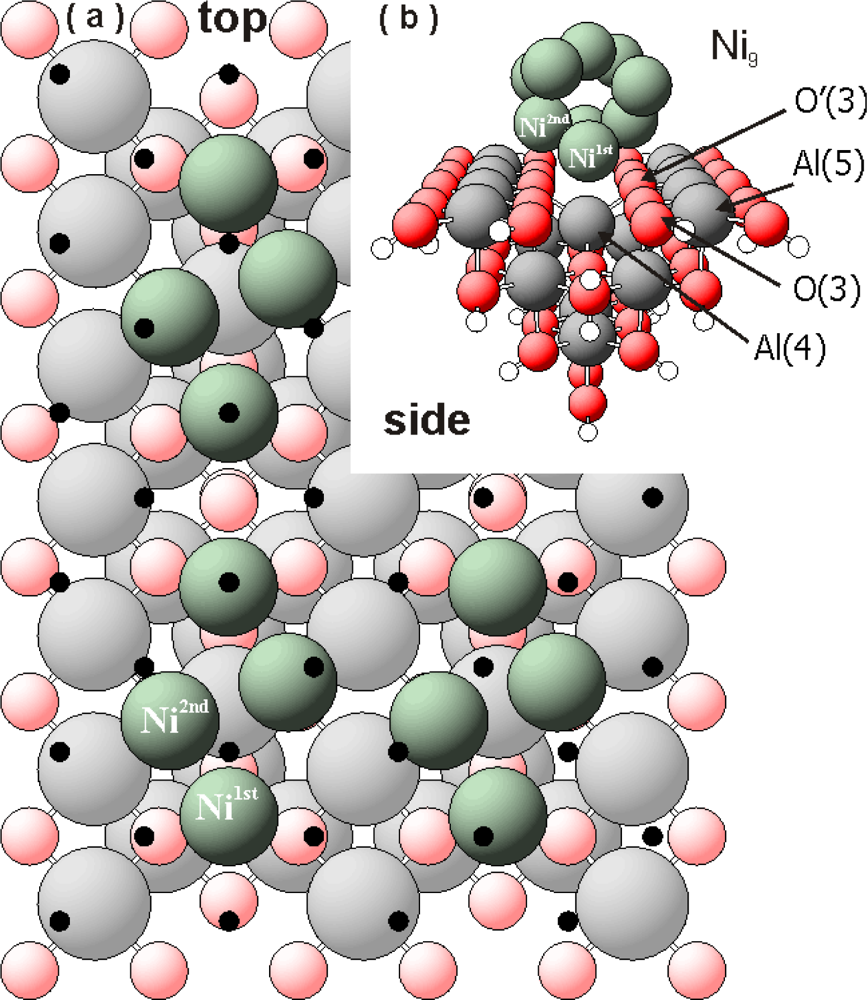
Figure 1a shows only the interface atoms (green spheres) of the Ni
9 cluster after relaxation. The interfacing Ni atoms correspond to a coverage θ
Ni∼0.40 mL. The figure was created by multiplying the calculated Ni/Al
2O
3 cluster in x and y direction (see
Figure 1b). Black dots symbolize the mismatch of an “ideal” Ni(100) monolayer with the γ-Al
2O
3 surface. Strong vertical and lateral re-arrangements of the interface Ni atoms in the Ni
9 clusters with respect to the position on the Ni(100) surface are indicated by our DFT results. Part of the interfacing nickel atoms (Ni
1st) seem to prefer the “valley regions” between the AlO
5 rows, close to the O(3) centres. These O(3) centres are mostly influenced creating strong bonds with the first (interfacing) nickel atoms.
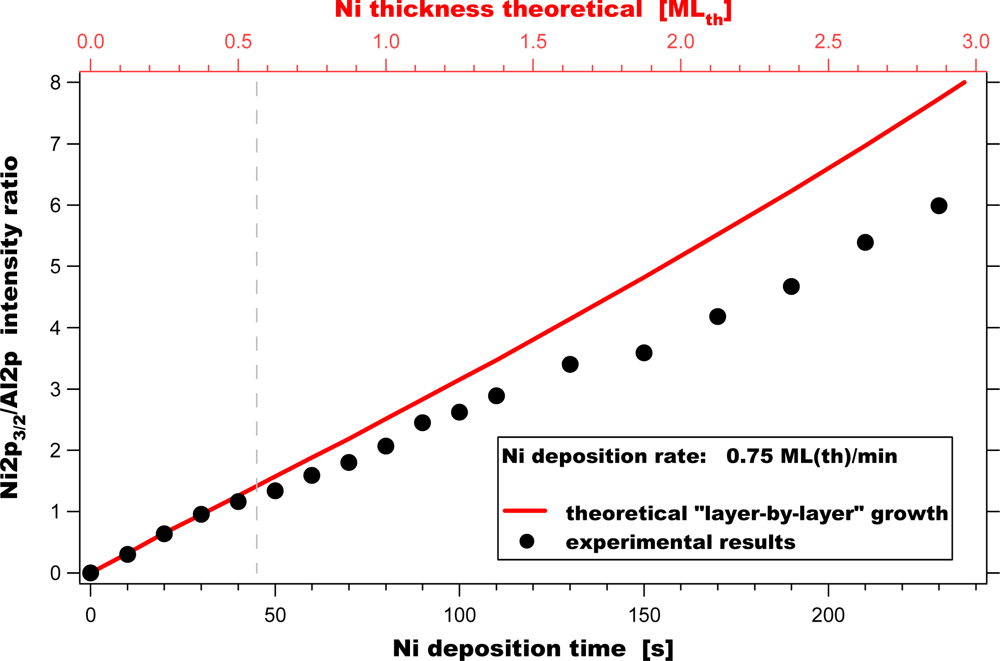
This finding is in good accordance with our experimental results (see
Figure 2), where we observed the existence of electronically strongly altered Ni adsorbed on γ-Al
2O
3 in the first stage of the deposition. Our experimental studies suggest that Ni does not form clusters immediately, but one-dimensional (or possibly small two-dimensional) agglomerates, initially. This seems to be valid for nickel coverages up to Θ
Ni∼0.5 mL. Further growth of these initial agglomerates following a layer-by-layer growth mode (see the red line) was not found. Our data clearly show a deviation from the expected behaviour exhibiting smaller Ni 2p
3/2/Al 2p values. This indicates that in the second stage three-dimensional Ni clusters are formed on the surface. We conclude that Ni deposition on γ-Al
2O
3 follows a “modified” Stranski-Krastanov growth mode under the applied experimental conditions, which is in accordance with the findings of Jacobs
et al. [
21].
Summarizing, we conclude that nickel is stabilized on the γ-Al2O3 surface influencing the electronic properties of the newly formed surface. Our DFT data suggest that at low coverages (≤0.2 mL) Ni prefers being localized in AlO4 tetrahedra between rows of AlO5. The DFT results are in good agreement with the experimentally obtained results from the initial stage of Ni deposition, where the formation of a “partial Ni monolayer” is suggested. Further Ni deposition first leads to three-dimensional agglomerates, which are finally transferred to Ni clusters on the surface by continued Ni deposition, as was derived from the slow approach of the XPS binding energy to the value of bulk Ni. For Ni7 and Ni9 clusters, the initially deposited Ni atoms, which represent the interface nickel atoms (“Ni1st”), are bound strongly to the oxygen of the support and are located in positions closer to O(3) centres as well as between rows of AlO5 with adsorption energy, which varies with the size of the cluster. The astonishingly good agreement between the experimental data and our theoretical studies shows the good accuracy of cluster model calculations for investigating metal-support structures.
3.2. Water Adsorption on Metal Oxide Surfaces: TiO2 and Al2O3
The effects of water adsorption on different catalysts are of high importance, because water is present in many processes, such as the methanation of syngas or the SCR process with humid exhaust gas. TiO2 and Al2O3 are interesting materials for both processes, which are discussed as urea decomposition catalysts or as support for methanation catalysts.
Molecular and dissociative adsorption of water is possible on both substrates [
22–
28].
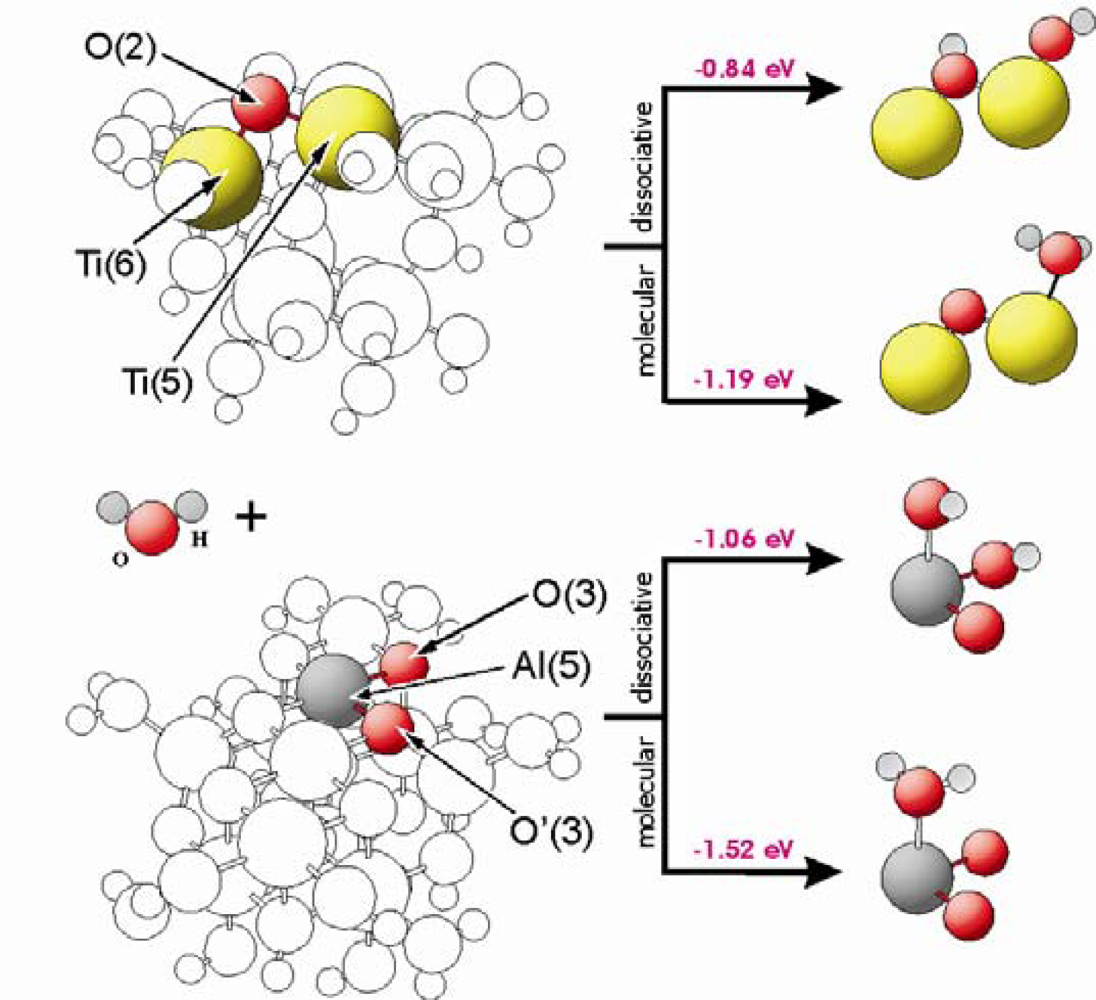
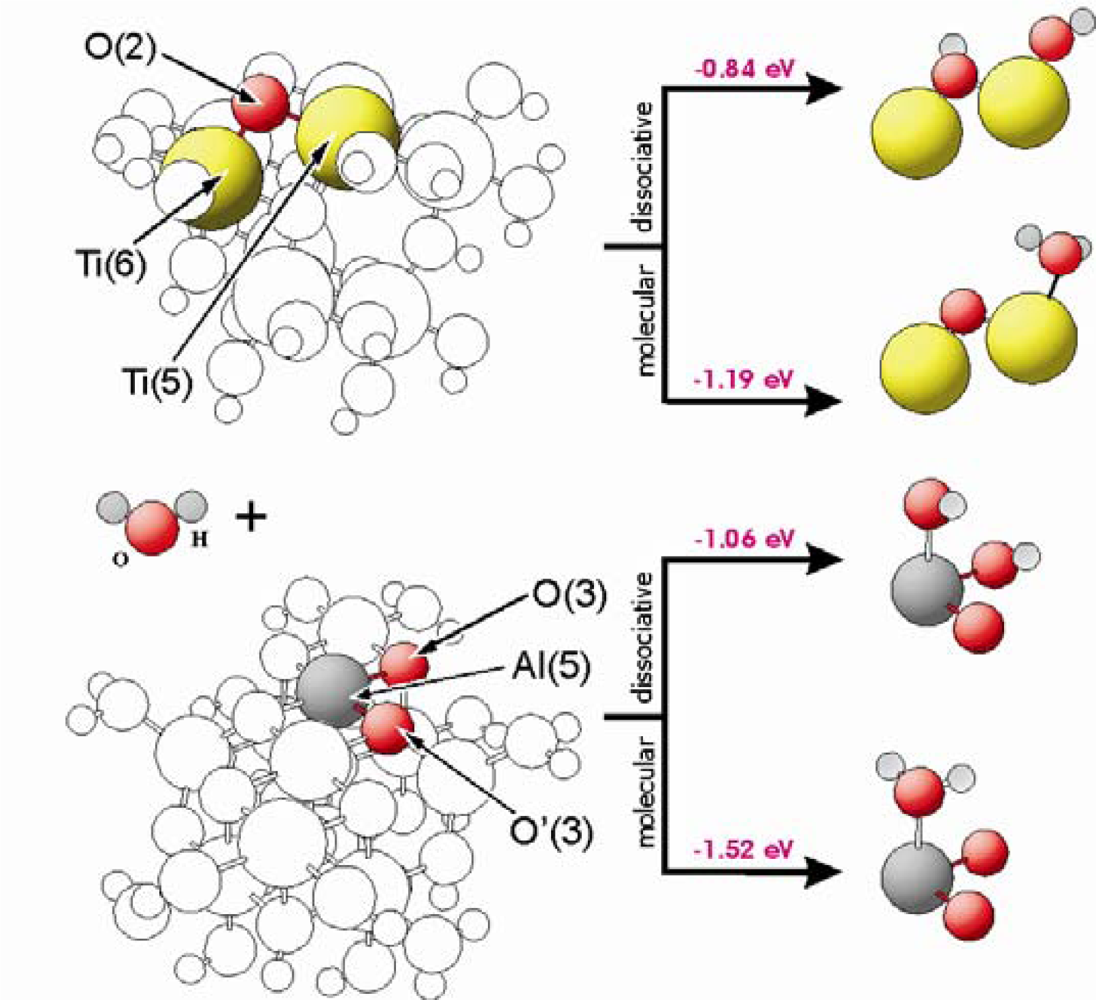
Figure 3 shows the corresponding geometric and the electronic structures of the surfaces for both types of catalysts. The calculations of the water adsorption showed that the M(5) centres (M = Ti, Al) are involved on the TiO
2 as well as the Al
2O
3 surface. Concerning dissociative adsorption, one hydrogen atom of the water molecule is transferred to and stabilized at a surface oxygen centre. In case of TiO
2, O(2) centres are able to bind the hydrogen atom. For Al
2O
3, the situation is more complicated, because at the surface exist two types of 3-fold coordinated oxygen centres, O(3) and O’(3). The O(3) centres are bound to Al(5) and Al(6) centres, while O’(3) are linked to Al(5) and Al(4) centres. As visible in
Table 1, the oxygen in O(3) has a lower charge and it is stronger bound than in case of the O’(3) centres. Our results show that hydrogen prefers to be adsorbed on the O(3) oxygen sides. In case of hydrogen bound to O’(3) centres, our optimisation results indicate an immediate hydrogen transfer to the O(3) centres. Water can be stabilized molecularly at a M(5) centre with an adsorption energy of −1.19 eV and −1.52 eV for TiO
2 and Al
2O
3, respectively, whereas water adsorbs dissociatively with an adsorption energy of E
ad = −0.84 eV and −1.06 eV on both materials at a M(5) centers.
Table 2 shows the calculated characteristic vibrational frequencies of water being bound to TiO
2 and Al
2O
3. Although molecular adsorption of water should be energetically even more favoured, dissociative adsorption of water is observed over the whole temperature range and even at low temperatures (50 °C/323 K) [
29].
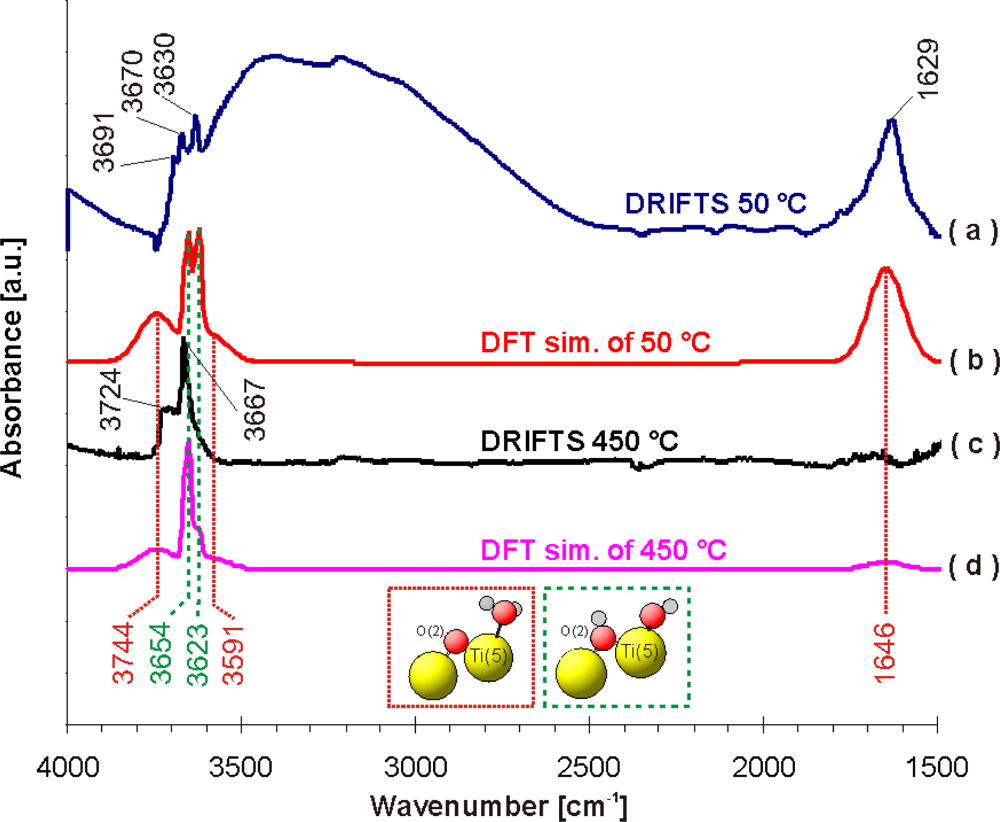
Figure 4 shows a comparison between simulated and measured IR spectra of water adsorbed on TiO
2. Only bands deriving from water adsorption are visible in case of the theoretical spectrum. The band at 1,629 cm
−1 found in the experimental DRIFT spectrum at 50 °C is in good agreement with the vibrational frequency of 1,646 cm
−1 [δ(H-O-H)] obtained from DFT calculations for molecularly adsorbed water with respect to the used approximations. There is also a fair match between the experimental band at 3,691 cm
−1 in the DRIFT spectrum and the theoretical band at 3,744 cm
−1 [ν(O(2)H)] in the “DFT” spectrum. On first view, it seems that the experimental band at 3,630 cm
−1 is also caused by molecularly adsorbed water. However, we believe that this band can rather be attributed to dissociatively adsorbed water, which was found to have a theoretical frequency of 3,623 cm
−1 [ν(O(1)H)]. The experimental bands at 3,670/3,667 cm
−1 in the DRIFT spectra at 50 °C and 450 °C, respectively, are also compatible with dissociatively adsorbed water with a calculated band at 3,654 cm
−1 [ν(O
s(2)H)] (see green dotted line in
Figure 4). The stretching vibration at 3,623 cm-1 [ν(O(1)H)] is connected with hydroxyl groups adsorbed at the Ti(5) centres and the stretching vibration at 3,654 cm
−1 [ν(O
s(2)H)] results from hydroxyl groups formed with the O
s(2) surface oxygen atoms [O
s(2)-H]. However, a detailed analysis of the experimental spectra at 450 °C (see
Figure 4c) shows that the absorbance of the ν[O(1)H] vibration is much weaker than that of ν[O
s(2)-H] one, which suggests a higher population of O
s(2)-H sites (with theoretical frequency 3,654 cm-1, see
Figure 4d). A lower population of hydroxyl groups at Ti(5) centres is a very important feature for the adsorption of HNCO on these sites as a prerequisite for the hydrolysis reaction. It should be noted that the DRIFT spectrum of TiO
2 at 50 °C shows also a broad band in the range 2,500–3,740 cm
−1, which is typical for the presence of liquid water due to the formation of hydrogen bridging bonds [
30].
The comparison of the calculated vibration frequencies with the DRIFT experiments suggests that at lower temperatures (50 °C) both molecular (bands at 1,629 and 3,691 cm
−1) and dissociative (bands at 3,630 and 3,670 cm
−1) adsorption of water occurs with higher amounts of molecularly adsorbed water. At higher temperatures (450 °C) mainly dissociative adsorption (bands at 3,667 and 3,724 cm
−1) was observed (see
Figures 4c and
d).
The combination of theoretical and experimental vibrational spectroscopic studies allows the identification of surface species and how they are adsorbed on the surface and show in detail which species remain on the surface even after drying of the catalyst. We have demonstrated such a comparison for water adsorption, which always occur under humid process conditions on heterogeneous catalysts, but it is also useful for studying other adsorbates, for example isocyanic acid, which is hydrolyzed over TiO2 in the urea-SCR process.
3.3. Isocyanic Acid Behaviour at Different Catalysts
Isocyanic acid (HNCO) is formed from the thermolysis of urea, which is used as an ammonia precursor compound in the selective catalytic reduction of nitrogen oxides in diesel engines. HNCO itself hydrolyses to ammonia and carbon dioxide. The hydrolysis of isocyanic acid is possible over a variety of metal oxides, among which TiO2 and Al2O3 were selected to study the mechanism both theoretically and with in situ DRIFT investigations.
The first calculation has been made for the isocyanic acid molecule in the gas phase in order to validate the electronic parameters’ accuracy. A comparison between results from DFT codes (StoBe and Gaussian98) and experimentally derived vibrational frequencies of pure HNCO are shown in
Table 3. Very good agreement of theoretical frequencies and experimental data of Ranier
et al. [
31] has been found for the calculations with the StoBe code.
As a next step the adsorption of isocyanic acid TiO
2 and Al
2O
3 has been investigated. The calculations revealed that dissociative as well as molecular adsorption of HNCO is possible and energetically feasible on both the TiO
2(101) [
6,
7] and the Al
2O
3(100) surface [
8,
32,
33].
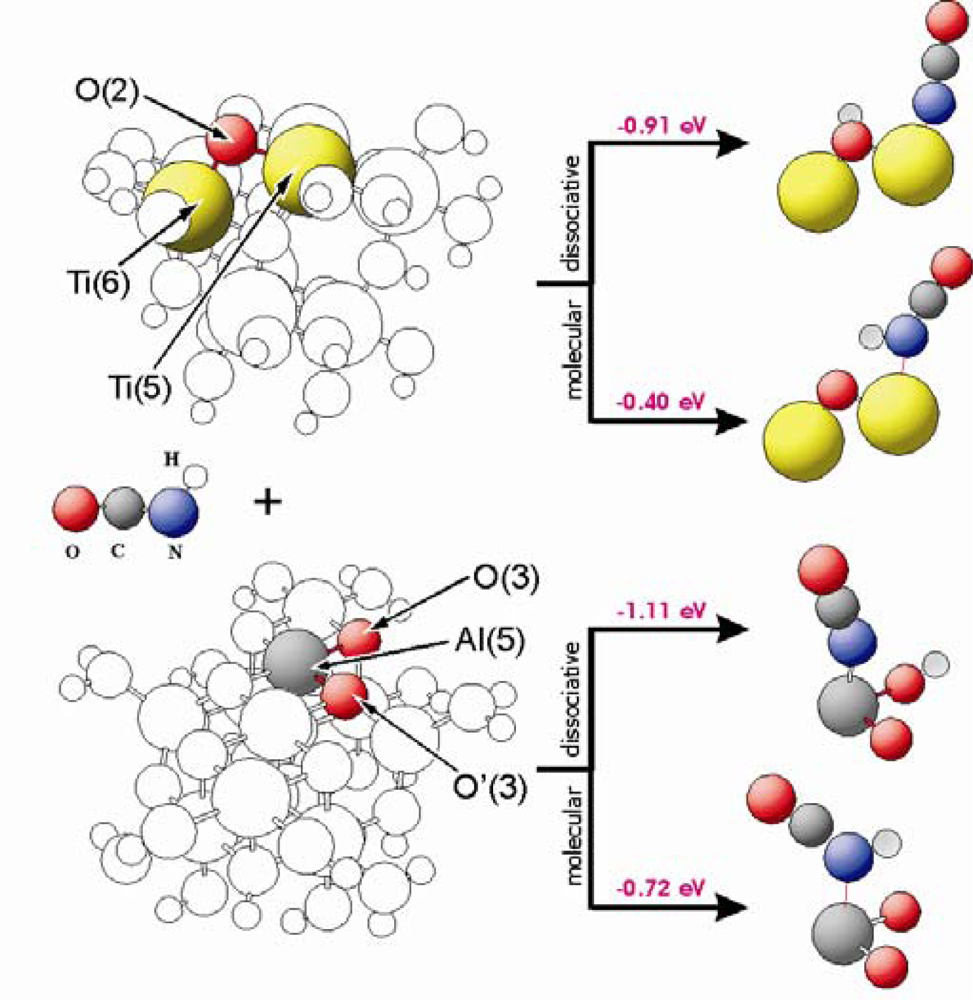
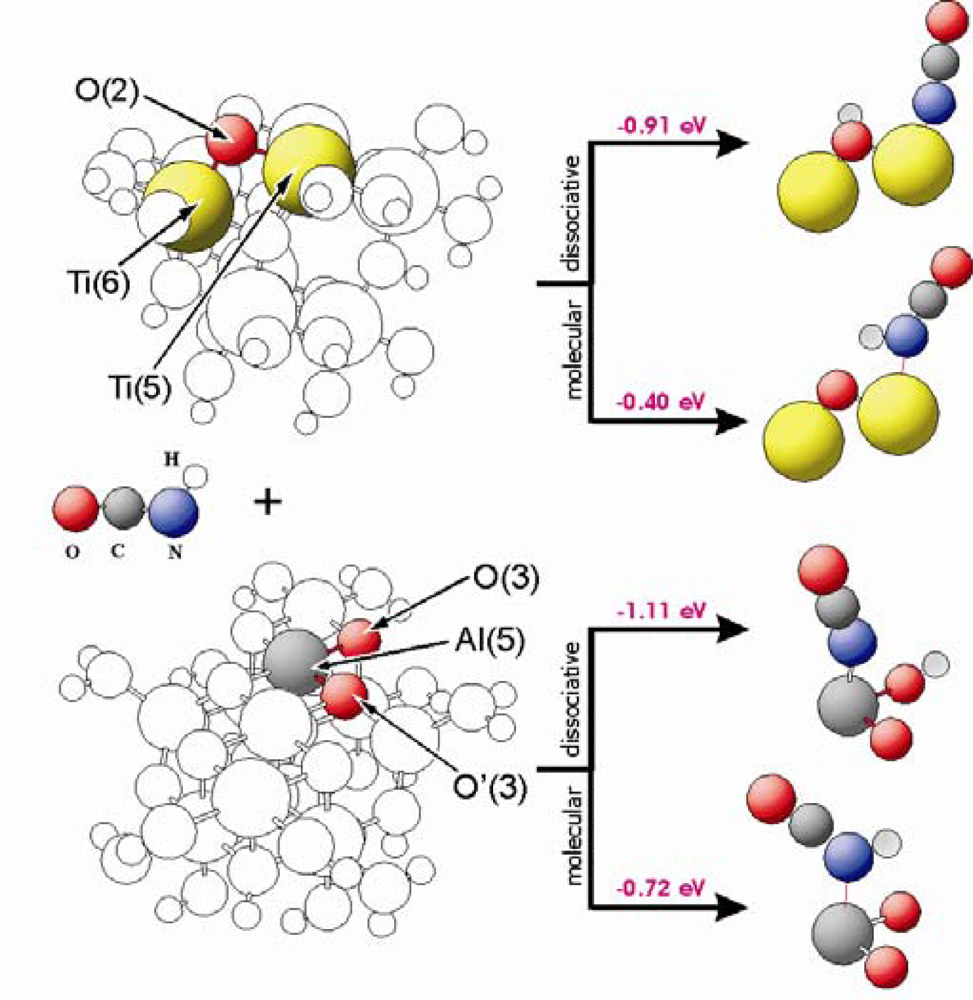
Figure 5 shows adsorption energies as well as geometric structures of HNCO interacting with different sites of TiO
2(101) and Al
2O
3(100). These surfaces are represented by Ti
8O
28H
24 and Al
11O
30H
27 clusters, respectively. The HNCO or –NCO groups are stabilized at the M(5) centres of these clusters. In case of dissociative adsorption on Al
2O
3, hydrogen is bound to the O(3) oxygen side in parallel to the adsorption of water (see paragraph 3.2). The HNCO molecule is bound to the M(5) centres with stabilization energies of −0.40 eV and −0.72 eV for TiO
2 and Al
2O
3, respectively. The energies for dissociative adsorption are −0.91 eV and −1.11 eV for TiO
2 and Al
2O
3, respectively. As shown in
Figure 3, water adsorbs dissociatively with an adsorption energy of E
ad = −0.84 eV (TiO
2) and −1.06 eV (Al
2O
3). This means that dissociative adsorption of isocyanic acid is favourable, and competitive with respect to water adsorption.
Due to the fact that the catalysts are working under humid conditions, any investigation of the reaction mechanism of the HNCO hydrolysis has to consider co-adsorption of HNCO and H2O on the catalyst surface. This approach comprises the competitive adsorption of water and isocyanic acid, as well as the interaction of water with isocyanic acid adsorbates. It also requires the presence of free neighboured five-fold coordinated metal sites.
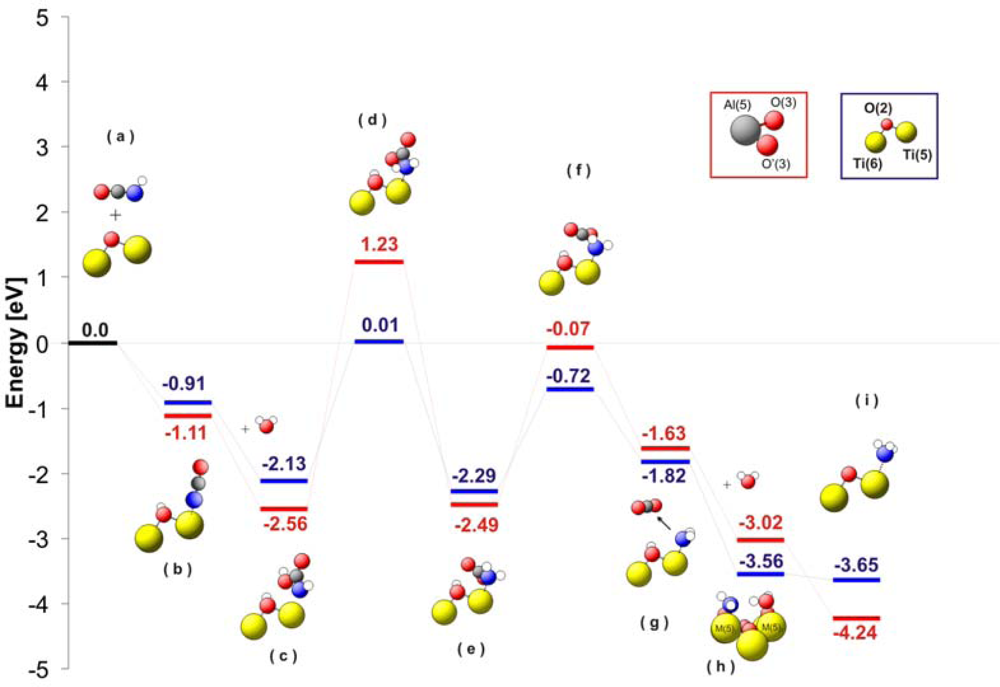
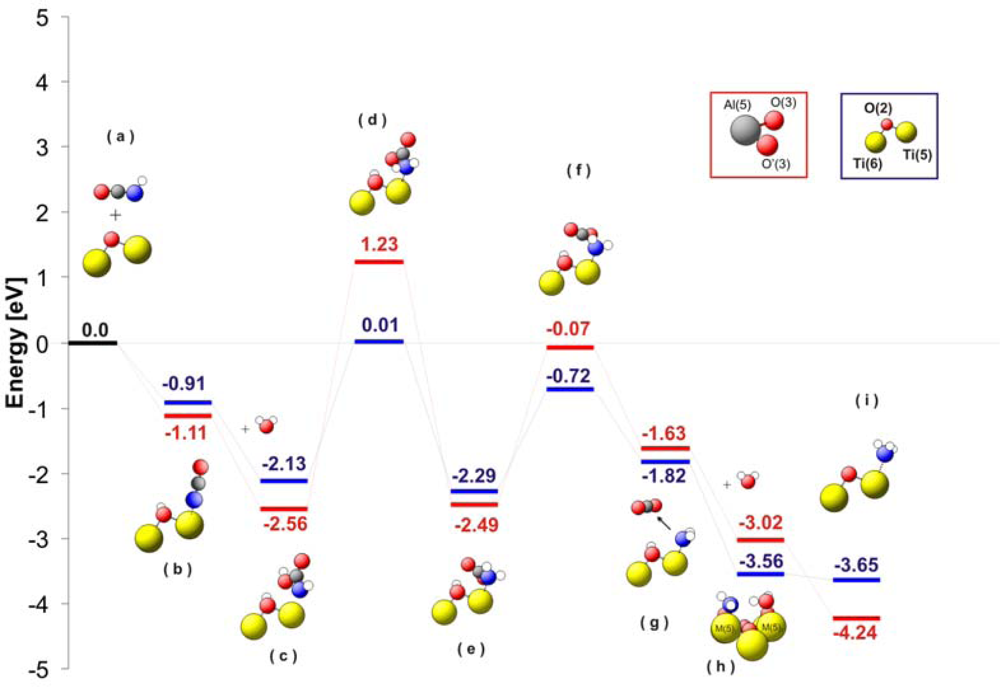
Figure 6 shows a comparison of the energy levels for the different intermediates observed during hydrolysis of isocyanic acid. The first important step of the reaction mechanism is dissociative adsorption of HNCO (see
Figure 6b). After stabilization at the surface, –NCO groups are attacked by a water molecule. As consequence of this water attack, a carbamic acid complex is formed (-NHCOOH) without energy barrier. The carbamic acid is strongly adsorbed on the surface with an energy of −2.13 eV for TiO
2 and −2.56eV for Al
2O
3. Thereupon, the carbamic acid changes its conformation in a highly endothermic process transferring hydrogen from carbonylic oxygen to the nitrogen atom (see
Figure 6d). Consequently, a carbamate complex (–NH
2CO
2) is formed at the surface (
Figure 6e). This carbamate complex decarboxylates (
Figure 6g) and the NH
2 group remains at the M(5) site. The formation of ammonia requires an additional hydrogen atom, which can be obtained from a water molecule adsorbed on a neighbouring M(5) site. This hydrogen transfer from a second water molecule is facilitated by the very low energy level of the system after the adsorption of water (−3.56 eV for TiO
2 and −3.02 eV for Al
2O
3;
Figure 6h). The NH
3 is finally released from the catalyst surface enriching the catalyst surface with OH groups. This is in agreement with the
in situ DRIFTS experiments described below, since strong OH/water vibrations are visible after reaction always. As an additional proof for the accuracy of our reaction mechanism a comparison of the theoretical vibrational spectra of the reaction intermediates of the HNCO adsorption on TiO
2 and Al
2O
3 with the
in situ DRIFT spectrum for TiO
2 has been performed. An example of these spectra is shown in
Figure 7 for the TiO
2 catalyst.
In the following, we give a brief introduction how to obtain theoretical IR spectra. The calculations of the vibrational frequencies were performed using harmonic approximations with additional anharmonicity fit as discussed in Chapter 2 [
18]. The vibrational spectrum of the individual adsorbates includes all vibrations and their characteristic intensities (see
Table 4). The frequency of a vibration is based on mechanical properties (anharmonically oscillating atomic masses) whereas its intensity is a function of the change in dipole moment [
34]. The amplitude of a peak in a vibrational spectrum is proportional to the square of the first derivative of the dipole momentum of the molecule with respect to one of the normal-mode vibrational coordinates (a combination of nuclear displacement coordinates of the equilibrium geometry).
Therefore in our case the intensity of a certain vibrational mode of an adsorbate is a function of the square of its dipole momentum. The final spectrum of an individual adsorbate has been described by Gaussian line shapes function with the same line width (50 cm−1) in order to obtain realistic peak shapes.
A complete theoretical vibrational spectrum was obtained by convolution of the vibrational spectra of all individual adsorbates determined to be present in the considered reaction path under the reaction condition applied (
e.g., water or hydroxyl groups adsorbed at neighboured metal centres), applying Gaussian line-shapes. The wavenumbers are reported as obtained from the calculations. The adsorption of isocyanic species with different surroundings has been considered with presence or absence of water, for example NCO and NCO
aq, see
Table 4. Due to the fact that in DRIFT spectroscopy the measured intensity of a certain vibration is a function of the number of adsorbed molecules, the theoretical spectrum of the individual adsorbates/intermediates have to be scaled separately to simulate the different populations of the individual adsorbates. The aim is to obtain the best possible fit to the experimental data.
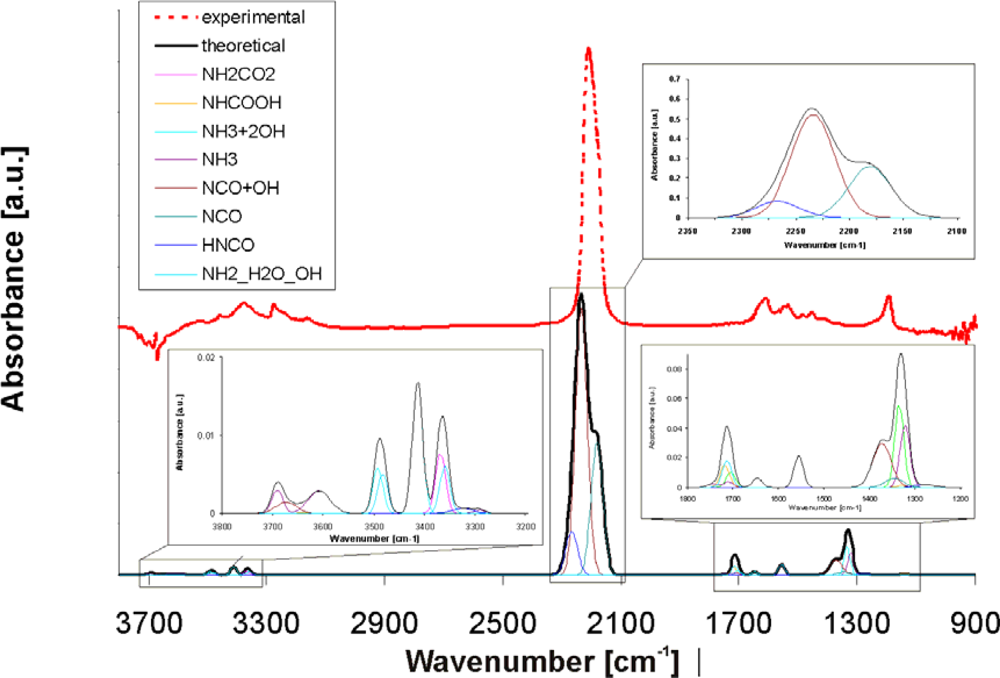
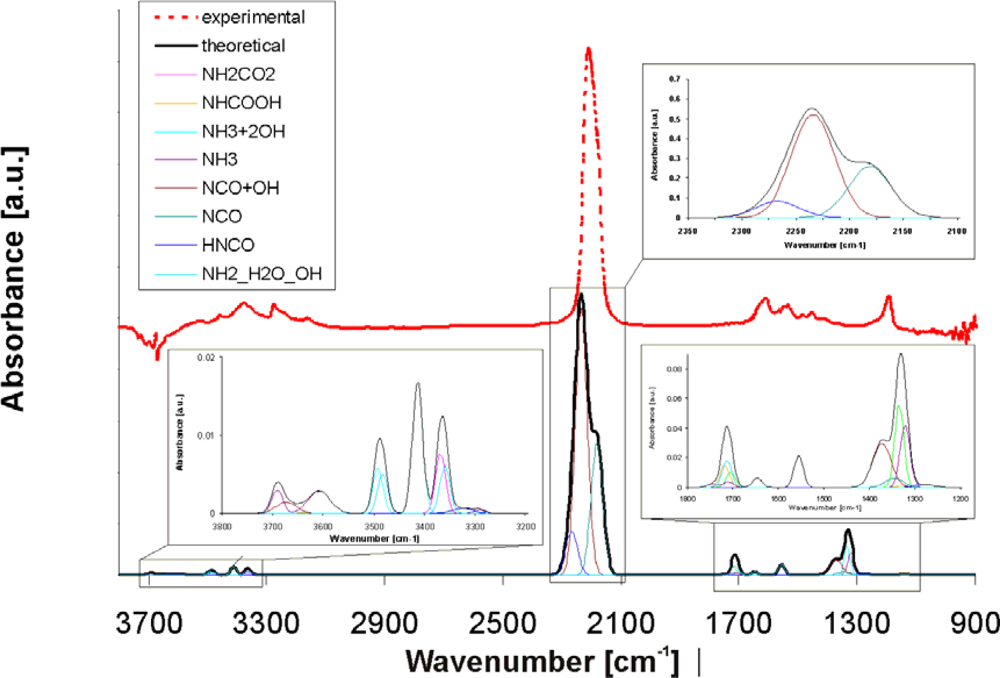
The detailed theoretical spectrum comprising all individual adsorbates/intermediates, which are considered for the hydrolysis of isocyanic acid, is shown in
Figure 7. The red curve shows the experimental DRIFT spectrum of the system HNCO/TiO
2 without water at 150 °C. A strong band at 2,209 cm
−1 together with less intense bands in the range 3,524–3,163 and 1,619–1,190 cm
−1 are visible. The strong band at 2,209 cm
−1 is assigned to the asymmetric stretching vibrations of –NCO groups adsorbed on the surface. From our theoretical studies we found that the main contribution to this band comes from dissociatively adsorbed isocyanic acid, stabilized on M(5) centres, evoking the vibration at 2,234 cm
−1 in the neighbourhood of strong OH groups (see brown individual curve) and the vibration at 2,181 cm
−1 in the absence of OH groups (light blue individual curve). However, small amounts of HNCO adsorbed in molecular form can be found at 2,266 cm
−1 (light blue individual curve), which is very close to the vibration of gaseous HNCO (2,259 cm
−1).
In the case of the Al
2O
3 catalyst the theoretically predicted vibrations of the -NCO groups are at 2,274 cm
−1 in case of dissociative adsorption and at 2,264 cm
−1 in case of molecularly adsorbed HNCO. These vibrations are also in good agreement with experimental data presented recently by Ozensoy
et al. [
22], who recorded TD-FTIR spectra at 120 °C and who found a band at 2,254 cm
−1. The calculated vibrations of NCO
aq and HNCO are very close to each other suggesting that on Al
2O
3 rather isocyanic acid in molecular form can be found. This is in good agreement with the higher adsorption energy of molecular HNCO on Al
2O
3 (−0.72 eV) compared to TiO
2 (−0.40 eV) (see
Figure 5).
The theoretical bands below 1,800 cm
−1 and above 3,000 cm
−1 in
Figure 7 have a similar shape as in the experimental TiO
2 spectrum, but they are shifted to higher frequencies. This could be due to differences in mass, bond strength or other effects. The low frequency bands in the 1,200–1,800 cm
−1 range in the calculated spectrum correspond to the bending vibrations of NH
2 or NH
3 as well as NH
2CO
2 and NHCOOH and the symmetric stretching vibrations of NCO influenced by water. The high frequency bands in the 3,000–3,500 cm
−1 range are the sum of the N-H stretching vibrations of the individual adsorbates influenced by water.
The intermediate complexes, such as –NCO or carbamic acid, have higher stabilisation energies at the Al2O3 surface, which simply means that it will be more difficult to convert them into the following surface complexes. This finding is in agreement with our experimental result that Al2O3 has a lower activity than TiO2 for the HNCO hydrolysis.
3.4. Theoretical Modelling of Binding Energies of Different Elements
As the last example we have selected the investigation of the deactivation of V
2O
5/WO
3-TiO
2 SCR catalysts by lubrication oil additives, which is of high significance for the NO
x reduction in diesel vehicles. In this project the influence of dopant metals on the binding energies of oxygen in a V-W-Ti-O catalyst was investigated theoretically by modelling the doping of VO
x [
9]. In this type of catalyst the active face are two dimensionally spreaded VO
x surface polymers and consequently, we limited our study on the influence of the metals on the active face only.
The two dimensional VO
x surface species were described by clusters, which consist of six vanadium atoms, V
6O
20H
10, as shown as inset in
Figure 8. The V
6O
20H
10 cluster represents all possible single-, double- and three-fold coordinated oxygen centres and five-fold coordinated vanadium centres on a orthorhombic V
2O
5(010) surface as well as the particular hole between the vanadyl-oxygen centres, where dopant metals are especially stabilized. Two dopant metals were tested, K and Ca, which were positioned in the energetically favourable hole position between vanadyl oxygen centres, following theoretical studies of Witko
et al. [
36].
XPS analyses of the individual components of the V-W-Ti-O catalyst, mainly V
2O
5, TiO
2, WO
3 and TiO
2-WO
3, was carried out to obtain information about binding energies and full-width-at-half-maximum data for the oxygen O1s region. The spectra were measured in our VG ESCALAB 220i XL set-up using the Mg X-ray source applying 15 kV and 30 mA in large area geometry. For a more detailed description see ref. [
9]. Sample charging was in the order of 10eV. The binding energy (BE) scale was adjusted by setting the main C1s peak to 284.5 eV. The spectra were deconvoluted applying Gaussian-Lorentzian line-shapes and a Shirley-type background.
As background for calculations of theoretical binding energies the Koopmans theorem can be taken. If the ground state of the system is considered and assuming the remaining electrons as being inert, then the binding energy of a particular electron is equal to the negative energy of the orbital eigenvalue: E
b = −ɛ
b. However, especially valence electrons respond due to the removal of a photoelectron, and also their relaxation and correlation energy must be included in the binding energy calculations. The relaxation and correlation energy can be included by calculating the binding energy as difference of the total energy between the ground state system (E
totGround) and the final state after emission of a particular electron, and the electron hole in the system (E
totFinal):
This definition is more accurate then Koopmans theorem and guarantees that relaxation of the remaining electrons is included.
XPS binding energies of the O 1s region of the V
6O
20H
10 cluster were calculated as the total energy difference between ground state and the core ionized state [
37]. The theoretically obtained binding energies were shifted by i) subtracting the sample work-function, and by ii) adding a relativistic correction (+0.33 eV for O) [
38]. The effective core potentials (ECP) were used for all other oxygen atoms to localize the core hole on the particular oxygen centres, O(1), O(2) and O(3).
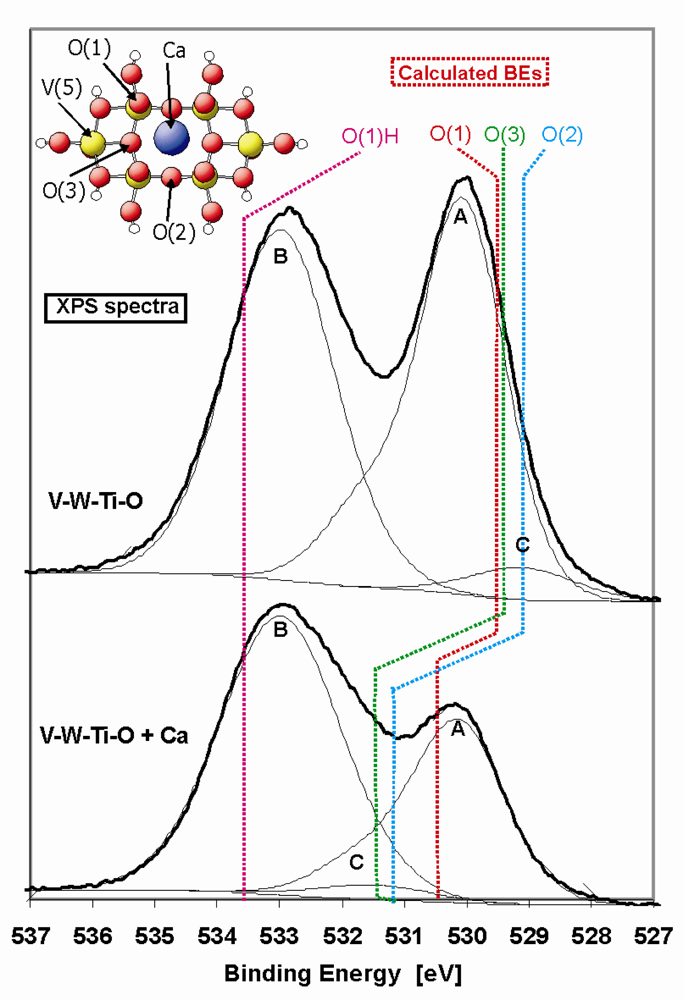
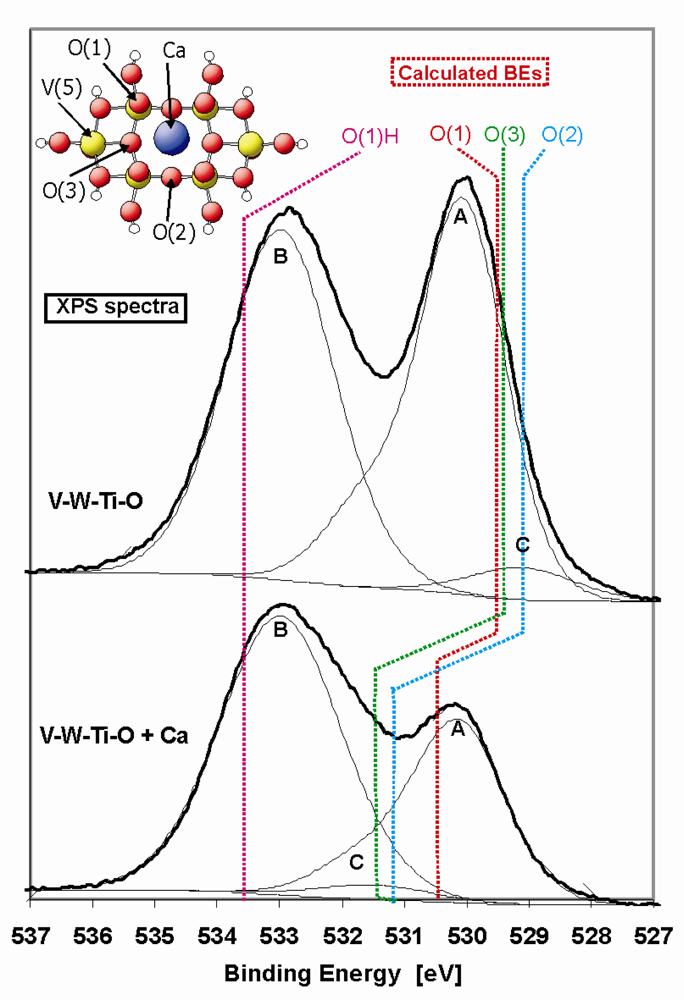
In
Figure 8 the O 1s XP spectrum of the pure and metal-doped V-W-Ti-O catalyst is presented. The spectra exhibit a two-peak structure at about 530 eV and 533 eV, which can be correlated with a TiO
2-WO
3 (peak A) compound and hydroxyl groups (peak B), respectively. For the deconvolution of the measured spectra, reference samples of single and mixed metal oxides were used (TiO
2, WO
3, V
2O
5 and TiO
2-WO
3, see
Table 5). Peak A (529.8 eV) and peak B (533.0 eV) were kept fixed during the fitting. The derived binding energies are listed in
Table 5. The asymmetry in peak A is caused by oxygen in WO
3 that has a higher binding energy (531.7 eV) and therefore TiO
2-WO
3 has been chosen as most suitable reference sample for this peak. Additionally, a small peak at about 529 eV can be distinguished (peak C, see
Figure 8, V-W-Ti-O sample), which is likely connected with V
2O
5. During deconvolution the ratio between the content of V
2O
5 and TiO
2 in the catalyst has been taken into account. In the upper spectrum of the V-W-Ti-O catalyst, the peak at lower BE corresponds quite well with our theoretical predictions of the binding energies of the different surface oxygen groups (see
Table 5 and
Figure 8) of pure V
2O
5. The binding energies of oxygen on a pure V
2O
5 (010) surface are localized at about 529.3 eV, which is exactly the position of peak C in case of the V-W-Ti-O sample.
The terminal oxygen, O(1), is most important for the SCR reaction [
36], for which the possible formation of OH group was considered in the calculation of the oxygen binding energy in O(1). The binding energy of terminal oxygen of Brønsted acidic V-OH site is equal 533.5 eV, which corresponds well with the second prominent peak (BE about 533 eV). However, it has to be noted that Brønsted acidic sites could also come from other components of the catalyst, mainly TiO
2.
Doping of the V-W-Ti-O with Ca leads to a decrease of the O1s signal located at the lower binding energy. Additionally, the peak labelled as “B” starts to be more asymmetric towards the lower binding energy range. We assign this asymmetry to the appearance of a third peak “C” at an energy position between 531–532 eV (see
Figure 8, V-W-Ti-O + Ca), which is in agreement with a theoretically determined binding energy shift after Ca-doping (see
Table 5). The theoretically determined binding energies of oxygen at the metal doped V
2O
5(010) surface are also shifted to higher energies (centred at 530.9 eV). However, the predicted shift of the oxygen binding energies was somewhat lower than the experimental value. A reason for this may be the additional shift of O binding energies in real V-W-Ti-O system compared to pure V
2O
5. However, the trend of the O1s peak shift is similar and offers a proper explanation of the role of the metal-dopant. Changes in the O1s signal after doping is connected with the formation of strong bonds between surface oxygen centres and the metal-dopant as well as an electron transfer from the dopant to the oxygen centre. Consequently, the oxygen becomes increasingly negatively charged and therefore exhibits a higher binding energy. Due to the charge transfer and a coupling with additional elements, the surface oxygen is supposed to be less reactive than in pure V-W-Ti-O catalyst.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}