Results and Discussion
The structure of dermolactone
3, the major orange-red pigment in the fruit bodies of the Australian toadstool
Dermocybe kula [
1] was determined by spectroscopic methods but, due to the scarcity of material, the absolute configuration of
3 was not accessible by chemical methods. To solve this problem the synthetic approach to (
S)-dermolactone (
3) summarized in
Scheme 1was developed [
2].
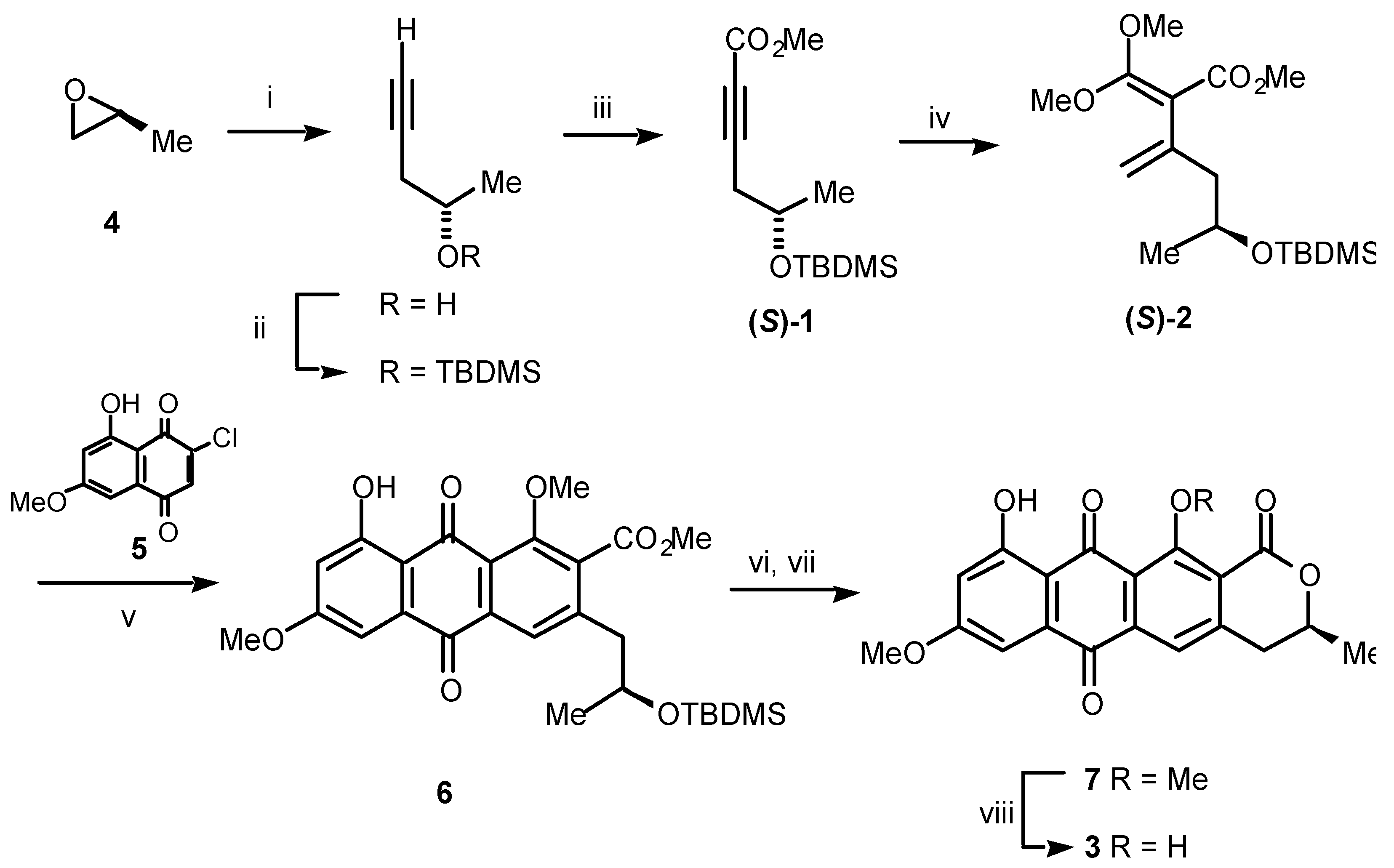
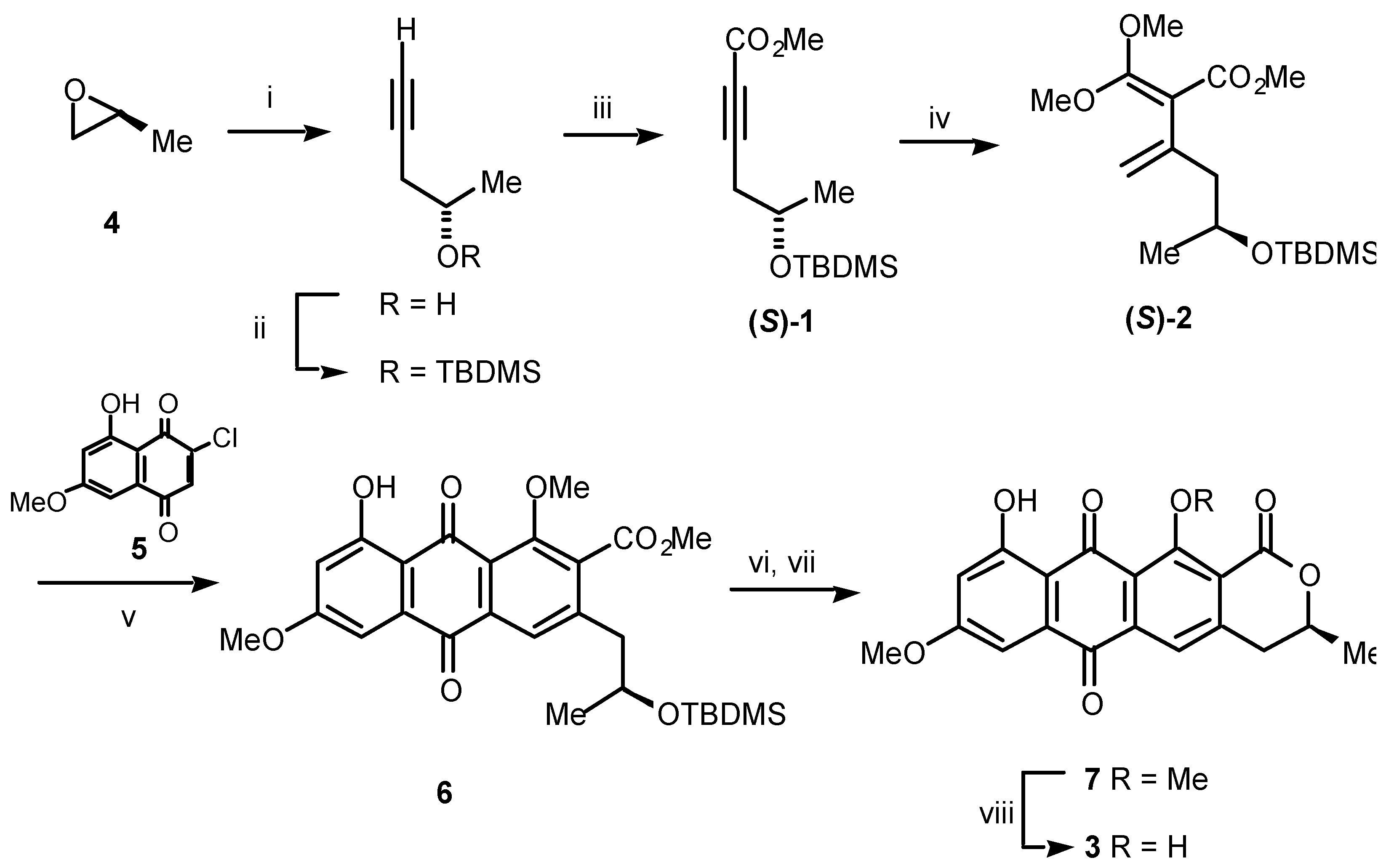
Scheme 1.
Reagents and conditions: (i) LiC≡CH:H2N(CH2)2NH2 complex, DMSO, 0 °C, 67%; (ii) TBDMSCl, imidazole, DMF, rt, 93%; (iii) a) n-BuLi, ether, –78 °C; b) ClCO2Me, –78 °C, 82%; (iv) (MeO)2C=CH2, sealed tube, 165 °C, 24 h, 28%; (v) 5, sealed tube, 160 °C, 4 h, 69%; (vi) 1 M H2SO4, THF, rt; (vii) p-TsOH, CH2Cl2, rt, 89% (2 steps); (viii) BCl3, CH2Cl2, 0 °C, 100%.
It begins with (
S)-propylene oxide (
4)
, which was converted straightforwardly over three steps to the acetylene (
S)-
1. Heating (
S)-
1 with ketene dimethyl acetal gave the important diene (
S)-
2. This diene reacted regiospecifically with the 2-chloro-1,4-naphthoquinone
5 to deliver, after aromatisation, the anthraquinone
6. Exposure of
6 to dilute sulfuric acid both removed the silyl protecting group and effected cyclisation to give the lactone
7. Finally, selective cleavage of the sterically encumbered
peri-methyl ether in
7 by using boron trichloride at low temperature delivered the enantiomerically pure (
S)-dermolactone (
3). The synthetic material was identical spectroscopically to the natural product with the important exception that the specific rotation of the synthetic ([
α]
D +169.3) and natural materials ([
α]
D +45.9) differed significantly. Further work on natural
3 by using chiral shift reagents and chiral HPLC analysis established unequivocally that natural dermolactone is an anisochiral mixture (exists as an unequal mixture of enantiomers) in which the (
S)-enantiomer predominates over the (
R) to the extent of 64 to 36% (28% e.e.) [
2].
It appeared to us that the chemistry developed for the synthesis of (
S)-dermolactone (
3) had the potential for application to the synthesis of other, mainly quinonoid natural products. In particular, we recognised that chiral dienes such as (
S)-
2 (and their enantiomers) could, in principle, react with a wide range of quinonoid dienophiles leading to a host of polycyclic systems The first target we chose to illustrate this potential was the mould metabolite xanthomegnin (
8). (3
R,3′
R)-Xanthomegnin (
8), first isolated from the mould
Trichophyton megnini [
3], has since been isolated from various
Penicillium and
Aspergillus species. It shows antibiotic and antifungal activity and is one of a small group of metabolites used as taxonomic markers in various
Penicillium species. Zeeck and co-workers [
4] synthesised (3
R,3′
R)-xanthomegnin (
8) from the corresponding 'monomer', (
R)-semixanthomegnin (
9), a cometabolite of
8 in
T. megnini, however, neither xanthomegnin (
8) nor semixanthomegnin (
9) had been the subjects of total synthesis prior to our work.
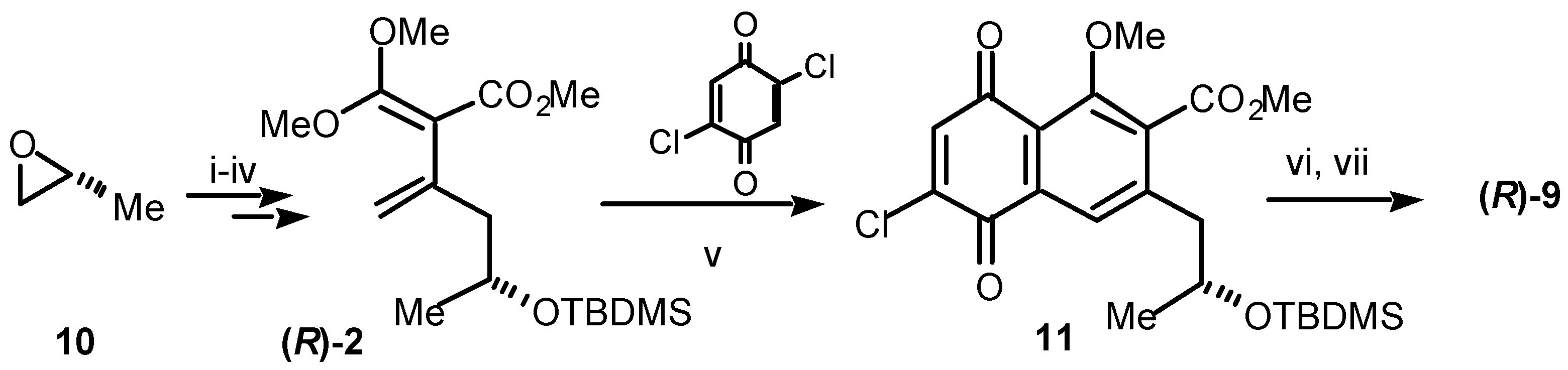
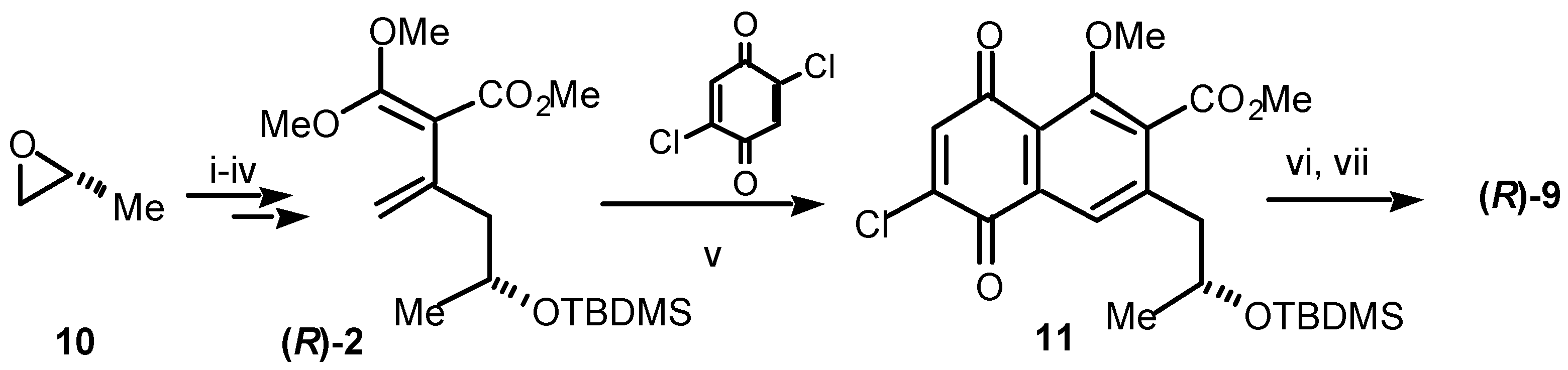
Our synthesis of (
R)-semixanthomegnin (
9), is shown in
Scheme 2. In the knowledge that the stereochemistry of the natural product is (
R), our approach began from (
R)-propylene oxide (
10). Thereafter, the path followed a parallel sequence to that shown in
Scheme 1, at least as far as the second Diels-Alder step. Thus, (
R)-propylene oxide (
10) was converted over four steps to the diene (
R)-
2. The chiral diene
2 underwent smooth Diels-Alder cycloaddition with 2,5-dichlorobenzoquinone yielding, after aromatisation, the chloronaphthoquinone
11. Finally, conversion to (
R)-semixanthomegnin (
9) involved consecutive replacement of chloride by methoxide, deprotection of the alcohol, lactonisation and selective demethylation to give the naturally derived product
9 [5]. The spectroscopic data for synthetic
9 proved identical to those of the naturally derived material. This is the first total synthesis of (
R)-semixanthomegnin (
9) in enantiopure form and still represents the only formal total synthesis of (3
R,3′
R)-xanthomegnin (
8).
Scheme 2.
Reagents and conditions: (i) LiC≡CH:H2N(CH2)2NH2 complex, DMSO, 0 °C, 45%; (ii) TBDMSCl, imidazole, DMF, rt, 86%; (iii) a) n-BuLi, THF, –78 °C; b) ClCO2Me, –78 °C, 72%; (iv) (MeO)2C=CH2, sealed tube, 165 °C, 24 h, 43%; (v) 2,5-dichloro-1,4-benzoquinone, benzene, sealed tube, 145 °C, 3 h, 16%; (vi) NaOMe, MeOH, 0 °C, 64%; (vii) BCl3, CH2Cl2, 0 °C, 73%.
To this point we have seen that the novel (
R)- and (
S)-dienes
2 can be used to produce tetracyclic and tricyclic quinones that incorporate a peripheral lactone ring. We next sought to adapt this chemistry to bicyclic lactone systems. Such systems are widespread in nature and are generally referred to as isochromanones, and this is where our attention was drawn next.
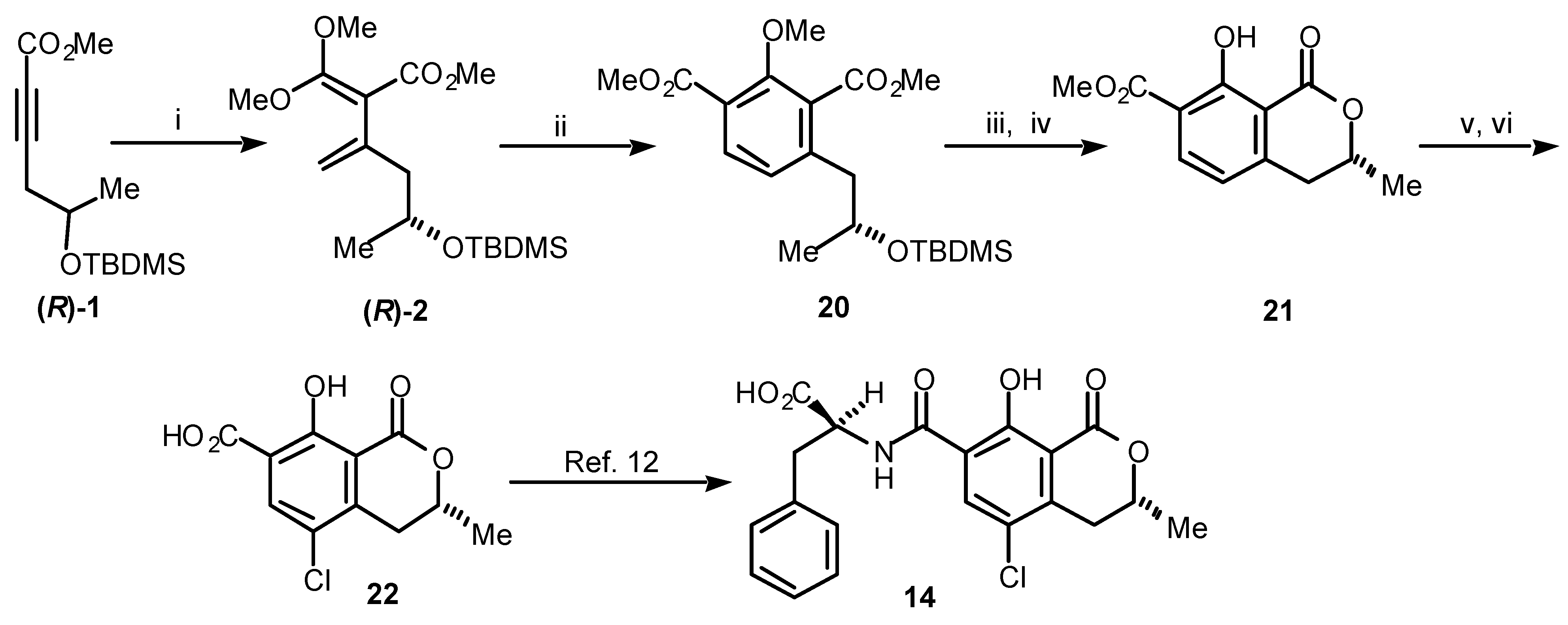
Natural isochromanones have been found in a diverse range of natural organisms that includes,
inter alia, the fungi, plants and insects and they display a variety of biological activities [
6]. The enantiomeric melleins
12 and
13 are the structural parents of the family and occur themselves in both stereochemical modifications [
7,
8]. (
R)-Ochratoxin A (
14), first isolated by Steyn and co-workers [
9], is arguably the most dangerous member of the group and has, consequently, been the subject of numerous publications [
10]. It occurs in several
Penicillium and
Aspergillus species and is a powerful nephrotoxin, immunosuppressant, teratogen and carcinogen. Its implication in human diseases and its occurrence at extremely low levels in wheat and other agricultural products has led to continuing development in analytical methods for its detection and quantification. Excessive levels of ochratoxin A (
14) continue to threaten Australia’s wheat exports to Japan and the USA. Despite its significance, before our work [
11] there had been no reported total synthesis of (
R)-ochratoxin A (
14), although it had been produced [
12] from the less toxic (
R)-ochratoxin α (
22), a cometabolite of
14 in several moulds.
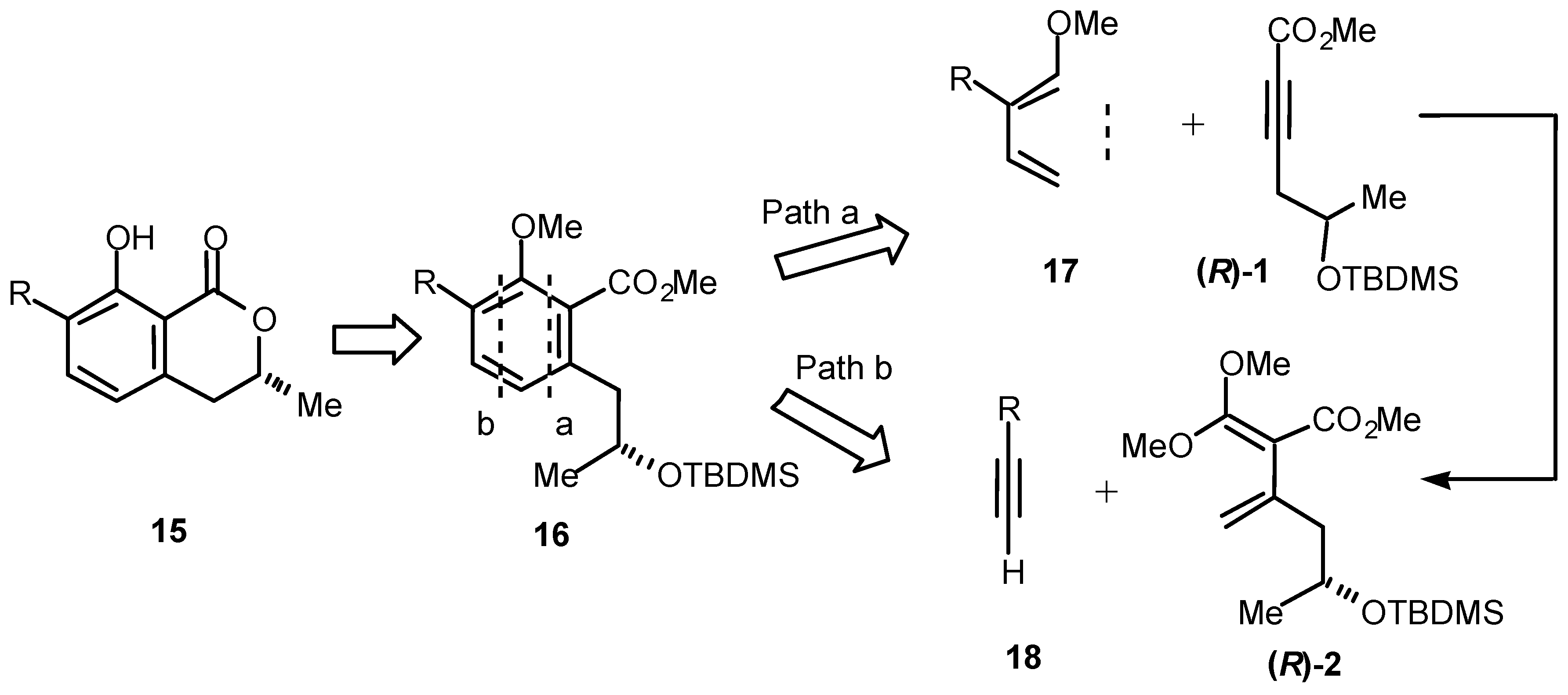
Our approach to chiral 7-substituted 3-methyl-8-hydroxyisochromanones such as
15 is shown in
Scheme 3 and, as may be seen, the pathway can proceed from either a diene of the type
17 or an acetylene of the type
18, by cycloaddition with (
R)-
1 or (
R)-
2, respectively, in the (
R)-series. Bearing in mind that (
R)-
2 is derived from (
R)-
1,
Scheme 3 depicts an extremely versatile route to isochromanones. We have successfully applied both of these strategies to the synthesis of members of the isochromanone family.
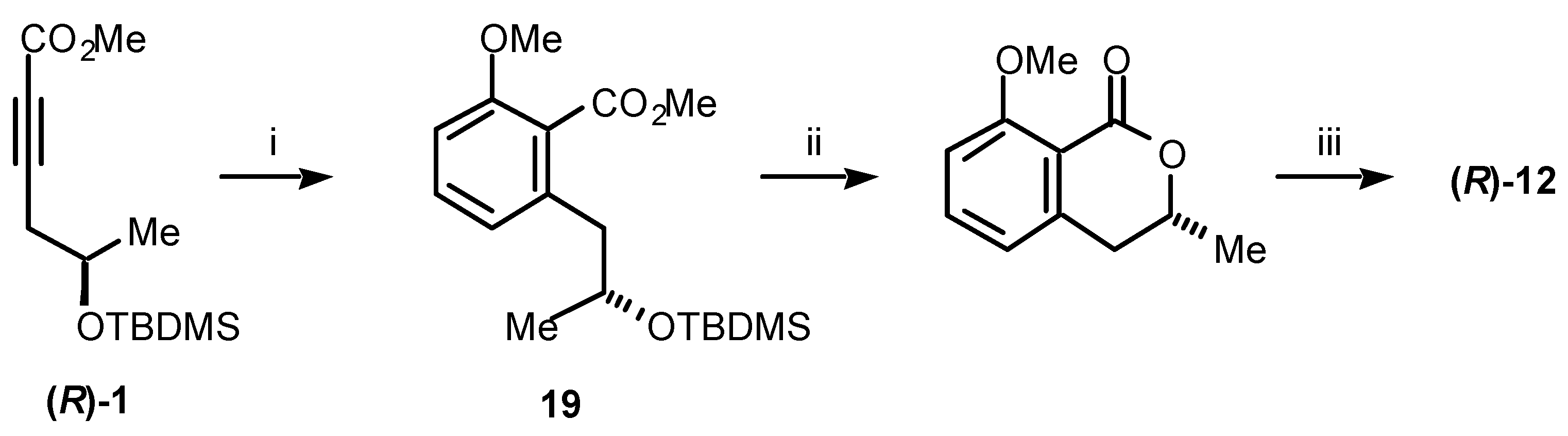
For the synthesis of (
R)-mellein (
12) we were able to employ
Path a (
Scheme 3) [
13]. Thus, the acetylenic ester (
R)-
1, prepared as shown in
Scheme 1, undergoes effective cycloaddition with 1-methoxy-1,3-cyclohexadiene to give, after expulsion of ethylene and aromatisation, the chiral benzoate
19 in high yield (
Scheme 4). Removal of the silyl group with concomitant cyclisation and, finally, cleavage of the methyl ether gave (
R)-mellein (
12) in an overall yield of 30% from (
R)-propylene oxide. (
S)-Mellein (
13) was prepared in parallel fashion, in 29% yield, from (
S)-propylene oxide [
13]. This economical synthesis has further allowed the (
R)- and (
S)-melleins,
12 and
13, respectively, to serve as precursors to some other important natural products (
vide infra).
Scheme 4.
Reagents and conditions: (i) 1-Methoxy-1,3-cyclohexadiene, sealed tube, 185 °C, 26 h, 79%; (ii) p-TsOH, CH2Cl2, rt, 84%; (iii) HBr, AcOH, reflux, 97%.
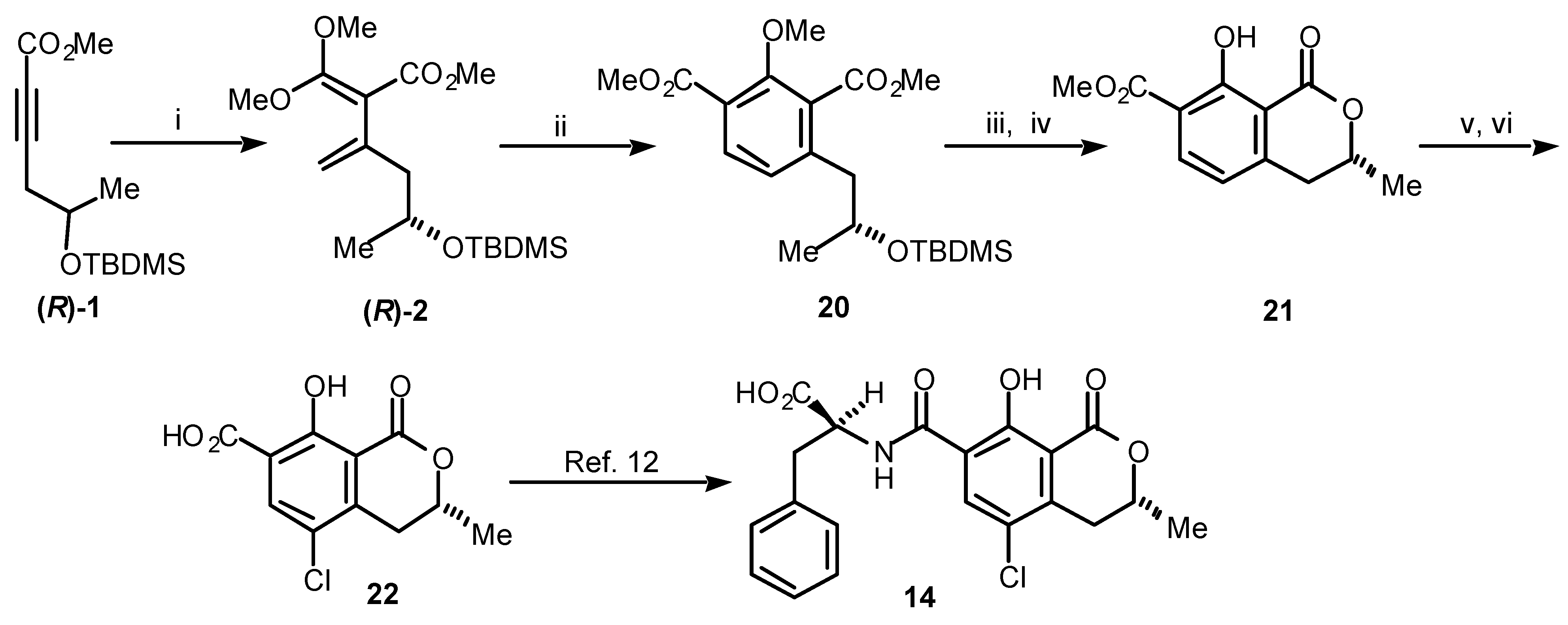
Turning now to our total synthesis of (
R)-ochratoxin α [
11], which still remains the only synthesis of (
R)-
22 published to date, we followed
Path b in
Scheme 3. Thus, the acetylenic ester (
R)-
1 was converted to the diene (
R)-
2 (
Scheme 5). The diene (
R)-
2was reacted with methyl propiolate (
18, R = CO
2Me) to give the chiral benzoate
20. Treatment of
20 with
p-toluenesulfonic acid and cleavage of the phenolic methyl ether yielded the 7-carboxymethylmellein
21. Finally, chlorination of
21 at C 5 and subsequent saponification gave (
R)-ochratoxin α (
22), which has been converted to (
R)-ochratoxin A by others [
12].
Scheme 5.
(i) (MeO)2C=CH2, sealed tube, 165 °C, 23 h; (ii) methyl propiolate (18, R = CO2Me), sealed tube, 145 °C, 22 h, 69% (2 steps); (iii) p-TsOH, CH2Cl2, rt, 82%; (iv) BCl3, CH2Cl2, 0 °C, 92%; (v) SO2Cl2, CH2Cl2, rt; (vi) LiOH.H2O, MeOH, reflux, 68% (2 steps).
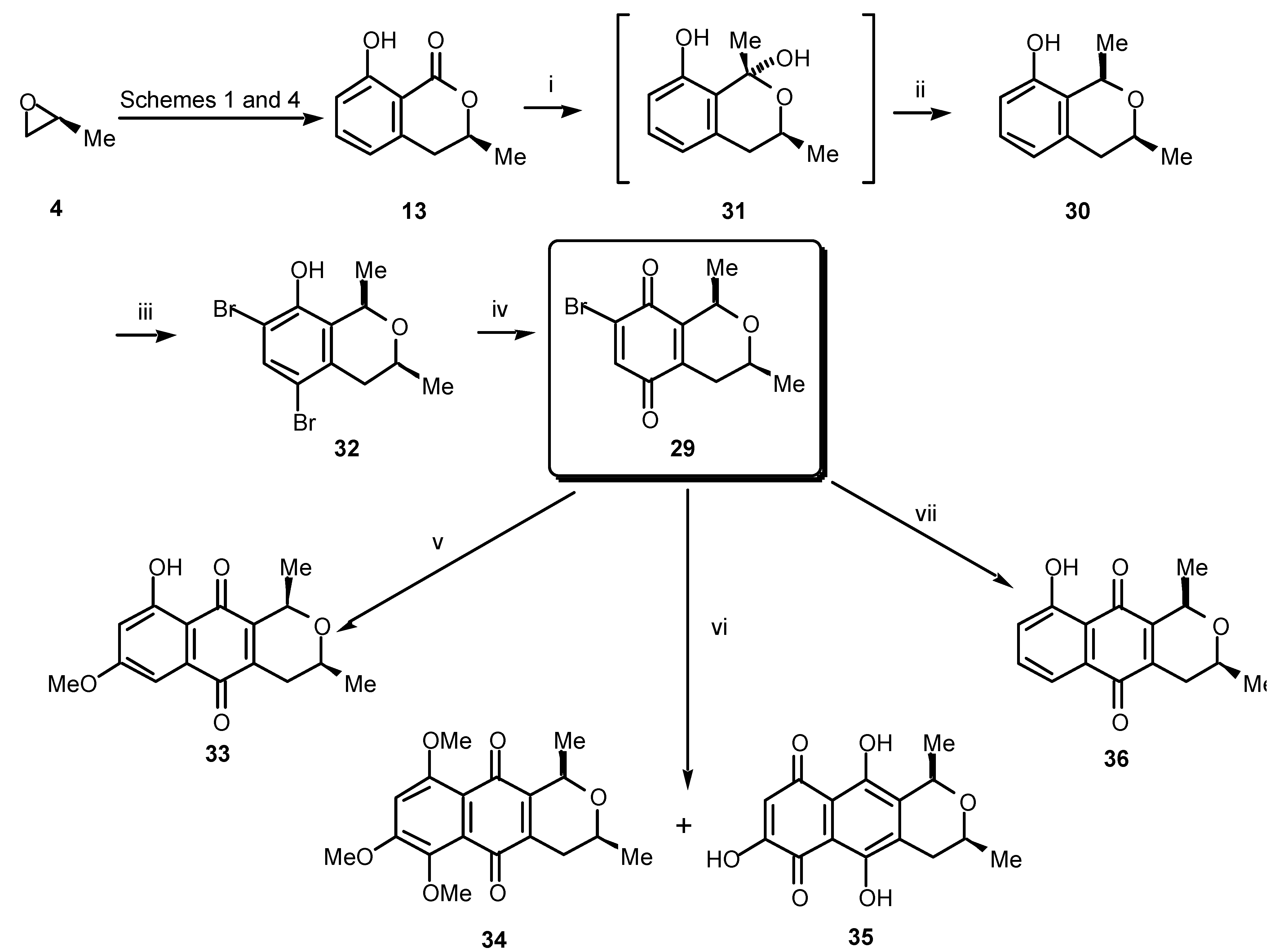
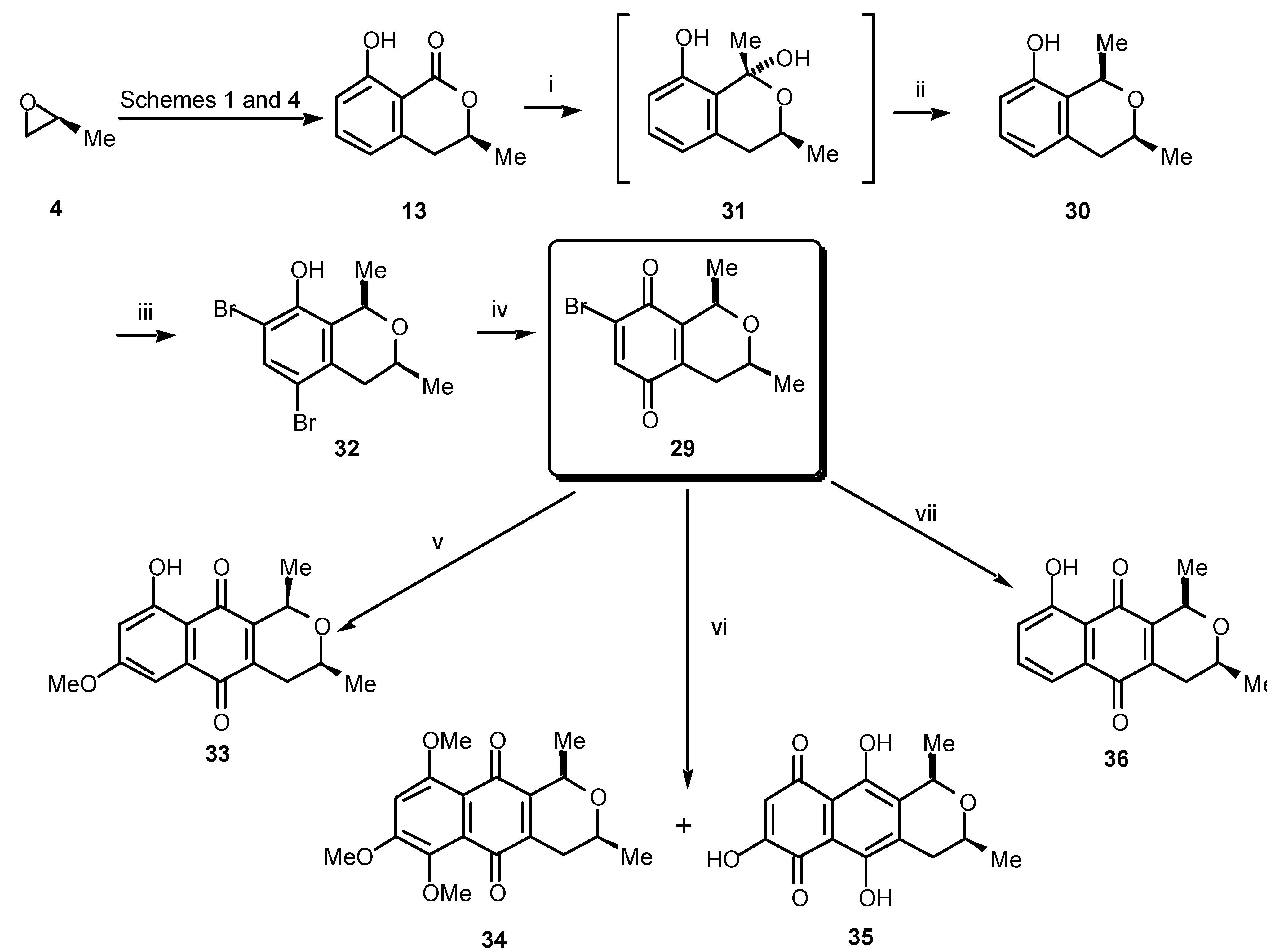
The chemistry described to this point has made possible the synthesis of tetracyclic, tricyclic and bicyclic lactones using the chiral diene
2 and/or the acetylene
1. The stimulation to enter the next phase of our work came from our isolation from the fungus
Dermocybe cardinalis of a new family of 1,3-dimethylbenzopyranoquinone natural products that we have called the cardinalins [
14]. The compounds
23–
26 are representative of the group that now numbers fifteen members. Cardinalin 1 (
23), is stereochemically the most complex member (9 stereogenic centres and an axis of chirality) while cardinalin 3 (
24), is the simplest.
Although we know the chirality at the axis in these molecules only the relative stereochemistry of the various chiral centres can be deduced from the spectroscopic data. The cardinalins 4 (25) and 5 (26) are biologically the most active members with IC50 values of 0.40 μg/mL against P388 murine leukaemia cell lines.
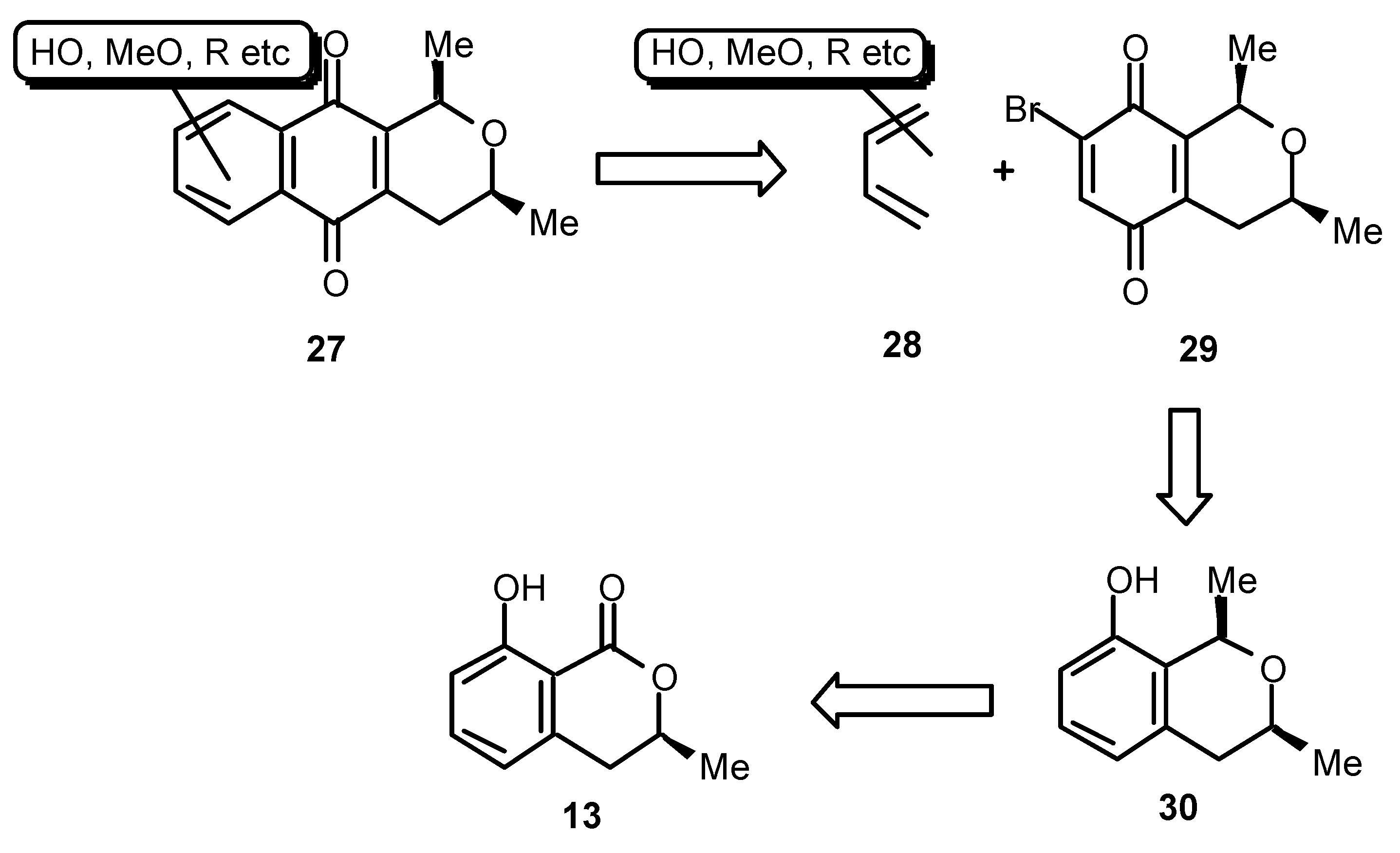
Our isolation of the cardinalins drew our attention to the synthesis of 1,3-dialkylbenzoisochromanquinones in general. These compounds have been known as plant, insect and bacterial metabolites for many years [
15] and a considerable effort has been devoted to their total synthesis [
16]. Ventiloquinones L (
33), E (
34) and G (
35) are plant constituents [
17], the absolute configuration of which was not, hitherto, known with certainty. They have yet to be the subjects of total synthesis. Notably, the structure of ventiloquinone L (
33) corresponds to one half of the cardinalin 3 system
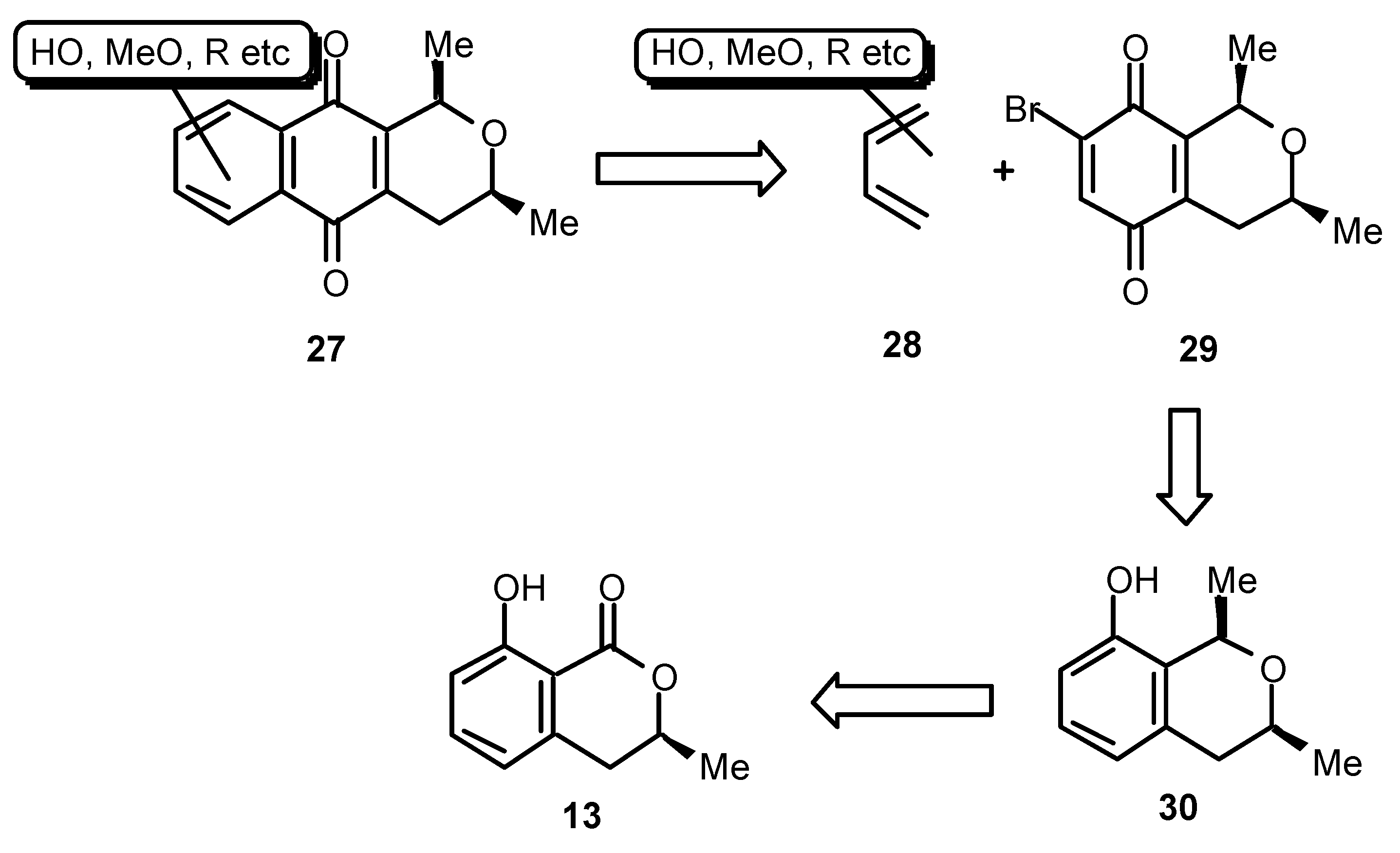
24. We sought therefore to develop a versatile method for the synthesis of 1,3-dialkylbenzoisochromanquinones in enantiomerically predetermined form. Our concept is shown in a retrosynthetic sense in
Scheme 6. Thus, a naphthoquinone such as
27 may be made available by cycloaddition between a suitably substituted butadiene of the type
28 and the chiral benzoisochromanquinone
29. The quinone
29 should, in turn, be available from the phenol
30 derived from (
S)-mellein (
13), the synthesis of which we have already covered. The most important step in this sequence then becomes the stereochemical integrity of the C 1 alkylation, which must inherit itsconfiguration from the chirality of the pre-existing C 3 methyl group. Fortunately, just such a conversion has been reported by Kraus [
18].
In practice (
Scheme 7), treatment of (
S)-mellein (
13) with methylmagnesium bromide (or methyllithium) gave the (1
R,3
S)-1,3-dimethylbenzopyran
30 via the lactol
31. The lactol
31 could be characterised spectroscopically, then reduced diastereospecifically to
30 in excellent yield by using triethylsilane and trifluoroacetic acid. The benzopyran
30 was subsequently treated with
N-bromosuccinimide to give
32, which, upon oxidation, afforded the chiral isochromanquinone
29 in high yield. Regioselective cycloaddition between the quinone
29 and 1,3-dimethoxy-1-trimethylsilyloxy-1,3-butadiene gave ventiloquinone L (
33), which proved identical in all respects, including specific rotation, with the natural material. This is the first total synthesis of ventiloquinone L (
33) in enantiomerically pure form [
19].
To illustrate the versatility of our method we have also produced several other members of the ventiloquinone and other related groups. Thus, merely by ringing changes in the diene employed in the cycloaddition reaction with the pivotal bromoquinone
29 will lead to a different substitution pattern in the left-hand peripheral ring in the quinonoid product. For example, when we used 1,3,4-trimethoxy-1-trimethylsilyloxy-1,3-butadiene and
29, a complex mixture of products was obtained from which the ventiloquinones E (
34) and G (
35) could be isolated (
Scheme 7). By cycloaddition with 1-methoxy-1-trimethylsilyloxy-1,3-butadiene, bromoquinone
29 gave the 8-
O-desmethyl ether
36 of the plant root constituent eleutherin [
20].
Scheme 7.
Reagents and conditions: (i) MeMgBr (3.5 equiv.), ether, 0 °C; (ii) Et3SiH, TFA, CH2Cl2, –80 °C, 92% (2 steps); (iii) NBS (2 equiv.), DMF, rt, 82%; (iv) CAN, MeCN, H2O, rt, 86%; (v) 1,3-dimethoxy-1-trimethylsilyloxy-1,3-butadiene, benzene, 60 °C, 41%; (vi) 1,3,4-trimethoxy-1-trimethylsilyloxy-1,3-butadiene, benzene, rt; (vii) 1-methoxy-1-trimethylsilyloxy-1,3-butadiene, benzene, rt, 10%.
To summarize, we have envisioned, developed and demonstrated a versatile new route to 1,3-dialkylbenzoisochromanquinone natural products. Even more potential in the method becomes apparent once it is recognized that the C 3 residue is dependent on the choice of the chiral oxirane starting material, while the C 1 substituent is derived directly from the organometallic reagent used, and that both can, in principle, be adapted in innumerable ways to yield 1,3-disubstituted benzoisochromanquinones incorporating innumerable functional groups at C 1 and C 3. We will report on the realization of some of this potential in later papers of this series.
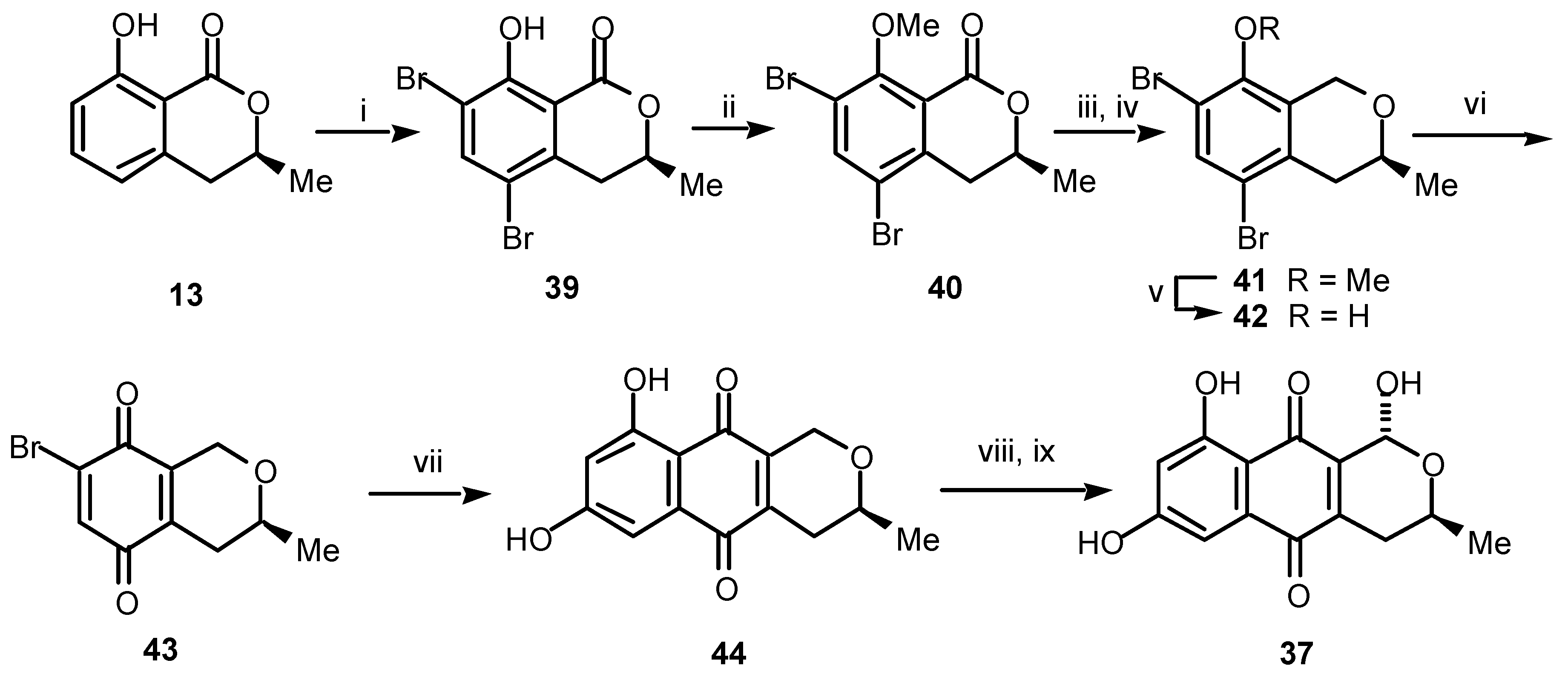
Thysanone (
37) (no absolute stereochemistry is yet implied) was isolated by chemists at Merck, Sharp and Dohme during a screening program looking for drugs active against the common cold [
21]: thysanone shows significant activity against human rhinovirus 3C protease (IC50 of 13 μg/mL). The structure and relative stereochemistry of thysanone, as shown, was deduced from the spectroscopic data and a single crystal X-ray analysis of the methyl acetal derivative
38. We sought to establish the absolute stereochemistry of thysanone unequivocally and for the first time by way of its total synthesis in enantiomerically pure form.
Scheme 8.
Reagents and conditions: (i) NBS (2 equiv.), DMF, rt, 82%; (ii) Me2SO4, K2CO3, acetone, reflux, 98%; (iii) DIBAL-H, toluene, –78 °C; (iv) Et3SiH, TFA, CH2Cl2, 0 °C, 93% (2 steps); (v) Bn2Se2, NaBH4, DMF, reflux, 86%; (vi) CAN, MeCN, H2O, rt, 93%; (vii) 1-methoxy-1,3-bis(trimethylsilyloxy)-1,3-butadiene, toluene, reflux, 73%; (viii) Br2, CCl4, hν; (ix) H2O, THF, rt, 85% (over 2 steps).
For the synthesis of (1
R,3
S)-thysanone (
37) we began from (
S)-mellein (
13), which was firstly dibrominated (
Scheme 8) and the resulting dibromoisochromanone
39 was protected as the 8-
O-methyl ether
40. To avoid difficulties encountered when the lactone carbonyl group was retained, it was temporarily removed over two steps. Firstly, exposure of
40 to di-isobutylaluminium hydride gave the corresponding lactol which, on subsequent exposure to triethylsilane afforded the pyran
41. Demethylation of
41 gave the phenol
42 which, on oxidation, gave the isochromanquinone
43. Cycloaddition between the new chiral bromoquinone
43 and 1-methoxy-1,3-bis(trimethylsilyloxy)-1,3-butadiene gave the pyranonaphthoquinone
44 in good yield. Finally, efficient benzylic bromination of
44 followed by hydrolysis afforded (1
R,3
S)-thysanone (
37) [
22]. Direct comparison of the spectroscopic data recorded for our synthetic (1
R,3
S)-thysanone (
37) and the corresponding data for the natural product (kindly provided by Dr S. B. Singh, Merck, Sharp and Dohme, Rahway, New Jersey, USA), including the respective CD spectra, confirmed the identity of the natural and synthetic materials [
22].
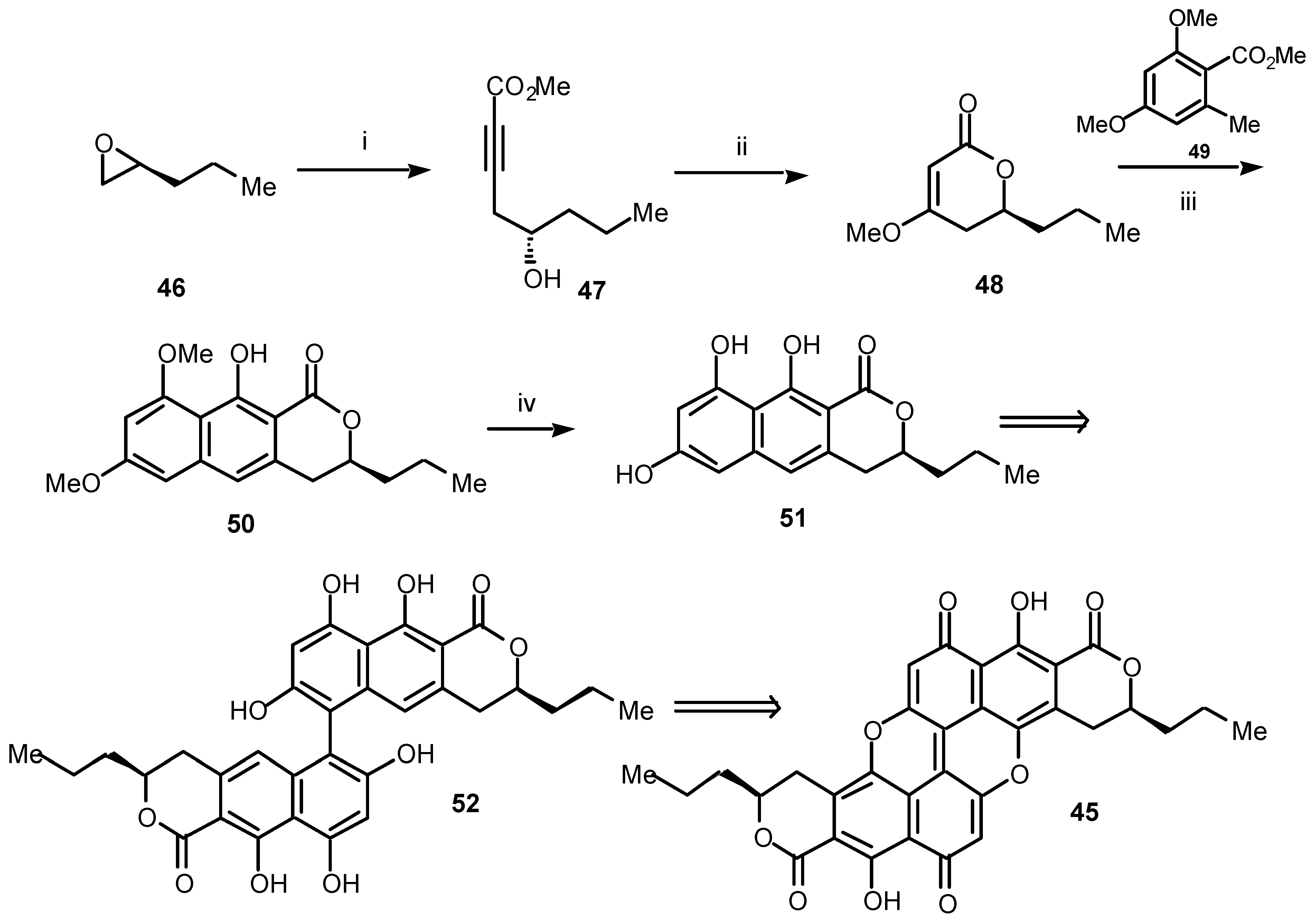
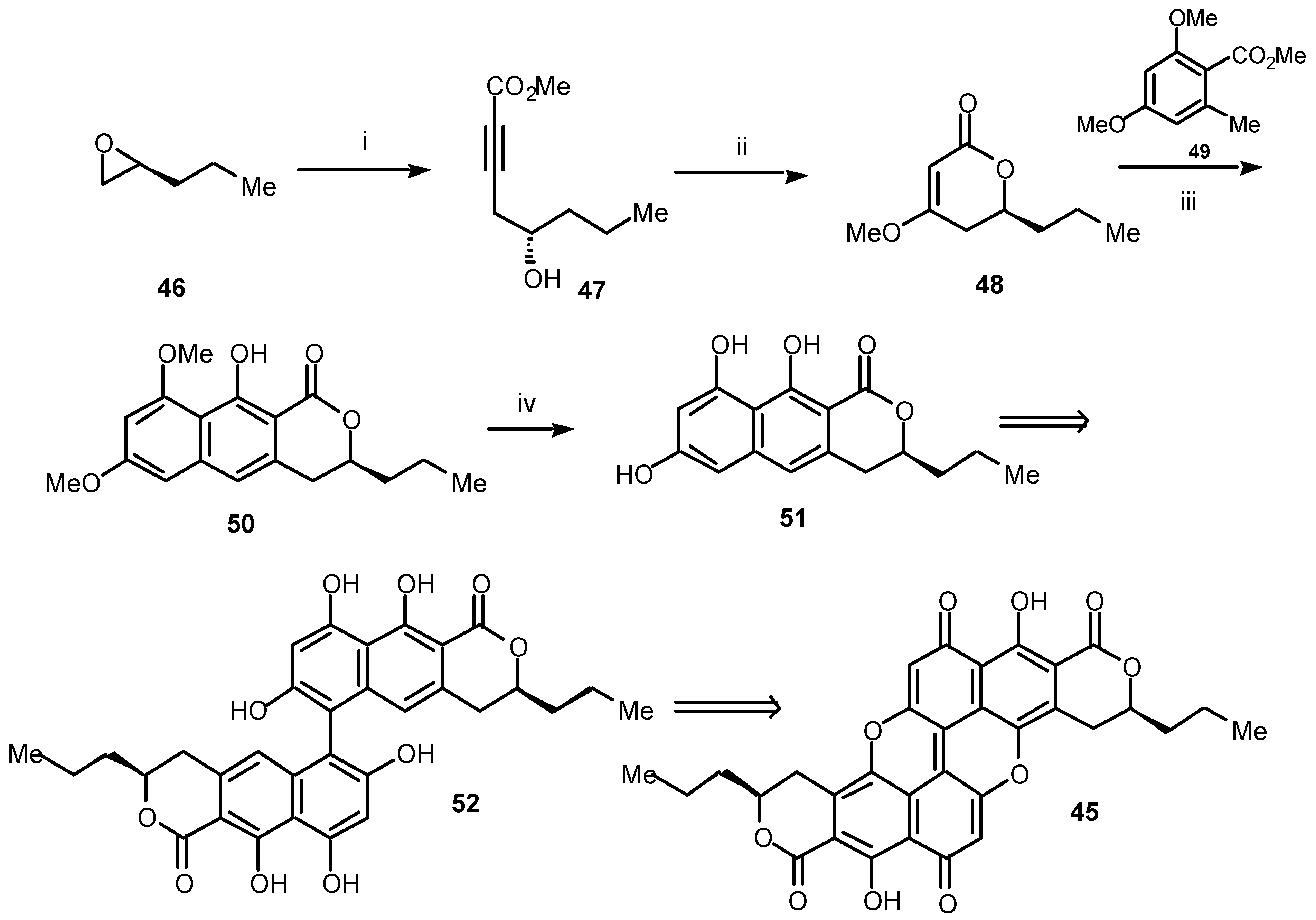
During the syntheses discussed so far we have used cycloaddition chemistry employing either acetylenic esters of the type
1 or dienes of the type
2 to assemble the appropriate aromatic ring system. In looking towards future progress, we are currently developing a somewhat different strategy beginning, as before, from a chiral acetylenic ester, in this case
47 (
Scheme 9) for the assembly of polycyclic benzopyranones such as the unique blue-green, extended quinone pigment, xylindein (
45) and some of its rare, as yet unexplored, analogues and relatives [
15].
The attractive blue-green colour of wood infected by the fungus
Chlorociboria aeruginosa which used to be sold commercially as ‘Tunbridge ware’, is due to the production by the fungus of the extended quinone xylindein
45. The pigment
45 was first obtained over 100 years ago [
23], and its structure was determined independently by Todd and co-workers [
24] and by Edwards and Kale [
25] at the Universities of Cambridge and Bradford, respectively. It is only recently, and after the start of our own research, that Saikawa
et al. established the (3
S,3′
S)-stereochemistry of xylindein (
45) by obtaining an X-ray crystal structure of a solvate of a derivative of the natural product [
26].
Our approach to the first synthesis of (3
S,3′
S)-xylindein (
45) is shown in
Scheme 9. It began with an assumption: that extended quinones such as
45 should be formed by oxidative coupling between two benzoisochromanones such as
51 which, in turn, should become available by a tandem Michael-Dieckmann condensation [
27] between the benzylic carbanion, generated from an orsellinate ester such as
49, and a chiral pyranone such as
48. The chiral Michael receptor
48 should itself be the product of conjugate addition of methoxide to the key acetylene
47 [28]. To date, this strategy has been successful (
Scheme 9), at least, as far as the benzoisochromanone
51.
Scheme 9.
Reagents and conditions: (i) methyl propiolate (18, R = CO2Me), n-BuLi, BF3.Et2O, THF, –78 °C, 68%; (ii) NaOMe, MeOH, rt, 86%; (iii) 49, LDA, THF, –78 °C, 31%; (iv) BBr3, CH2Cl2, rt, 92%.
Thus, opening the (S)-propyl oxirane (46) by using the acetylide anion of methyl propiolate gave the acetylenic alcohol 47. Treatment of 47 with sodium methoxide in methanol afforded the new, chiral pyranone 48 in high yield. Initial conjugate addition to the pyranone 48 of the carbanion 49 followed, in situ, by intramolecular Dieckmann cyclization gave the benzoisochromanone 50, albeit in moderate yield. Finally, the benzoisochromanone 50 was smoothly demethylated to afford the important chiral benzoisochromanone 51. We are currently engaged in assessing methods for the oxidative coupling of 51 to give the dehydro-dimer 52, which we hope will ultimately lead to (3S,3′S)-xylindein (45). Pleasingly, at this stage it appears that this protocol will provide a very short and versatile entry to benzoisochromanones, such as 51, from readily available chiral epoxides. This opens the way for the synthesis of a large group of benzoisochromanones and provides the first route to previously inaccessible extended quinones.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}