From Labdanes to Drimanes. Degradation of the Side Chain of Dihydrozamoranic Acid.

1
Departamento de Química, Universidade da Beira Interior, 6201-001 Covilhã, Portugal
2
Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad de Salamanca, 37008 Salamanca, Spain
*
Author to whom correspondence should be addressed.
Molecules 2004, 9(5), 300-322; https://doi.org/10.3390/90500300
Submission received: 23 February 2004
/
Revised: 4 March 2004
/
Accepted: 5 March 2004
/
Published: 30 April 2004
(This article belongs to the Special Issue Carbanion chemistry from Carboxylic acids in honor of Prof. Ramón Mestres on his 65th anniversary)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:A new route for the degradation of the saturated side chain of dihydrozamoranic acid has been devised, giving an advanced intermediate, compound 14, useful for the synthesis of insect antifeedants such as warburganal and polygodial.
Introduction
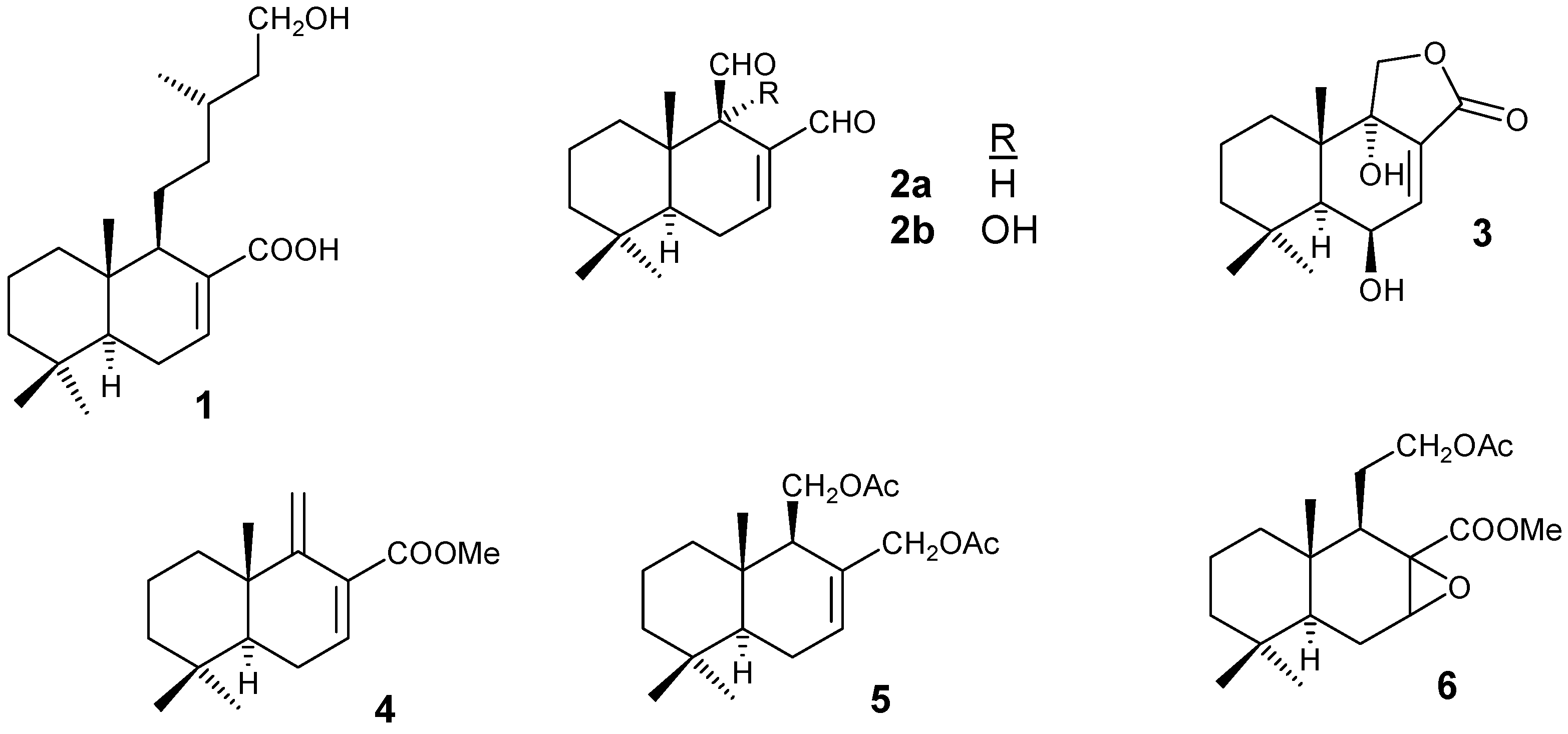
One of the main problems of our civilization is the shortage of food, especially in Third World countries. The intensity of the problem will increase as the global caloric demand is likely to double over the next ten years. This may be addressed in two ways; by better distribution of existing resources and by increasing total food supplies. With a rising productive area the number of pests has also increased considerably and thus the use of herbicides and insecticides [1]. Several insect species have developed resistance against agrochemicals causing not only a significant increase in the amount of chemicals used but also in the environmental pollutant level. The strategy used by plants to protect themselves from insect attack is the biosynthesis of secondary metabolites with antifeedant activity. These natural products are highly specific to some insect species and are completely inactive against other species useful to human beings [2]. Moreover, these compounds are biodegradable and there is no danger of accumulation or environmental pollution [3]. Among the natural antifeedants, azadirachtin isolated from Azadirachta indica [4], some clerodane diterpenoids such as jodrellin A and B isolated from Schutellaria woronowii Juss [5] and somedrimanes such as warburganal (2b) isolated from Warburgia ugandensis [6] and polygodial (2a) isolated from Polygonum hydropiper [7] should be highlighted because their specific and high antifeedant activity against Spodoptera species [8], which causes more than 30% crop losses in India [9]. In our lab several bioactive drimanes (Figure 1) have been synthetised starting from zamoranic acid [10].
Figure 1.

Results and Discussion
Dihydrozamoranic [11] acid (1), is a diterpene isolated from Halimium verticillatum and Halimium viscosum, being a main component in the first case. This compound’s only difference with zamoranic acid is the saturation of the side chain. The transformation of this compound into an intermediate readily transformed in turn into natural compounds with biological activity would be of interest. Some of these intermediates are diene 4, the diacetylderivative 5 [12] and epoxide 6. The last compound has been transformed by us into poligodial (2a), warburganal (2b) and pereniporin A (3).
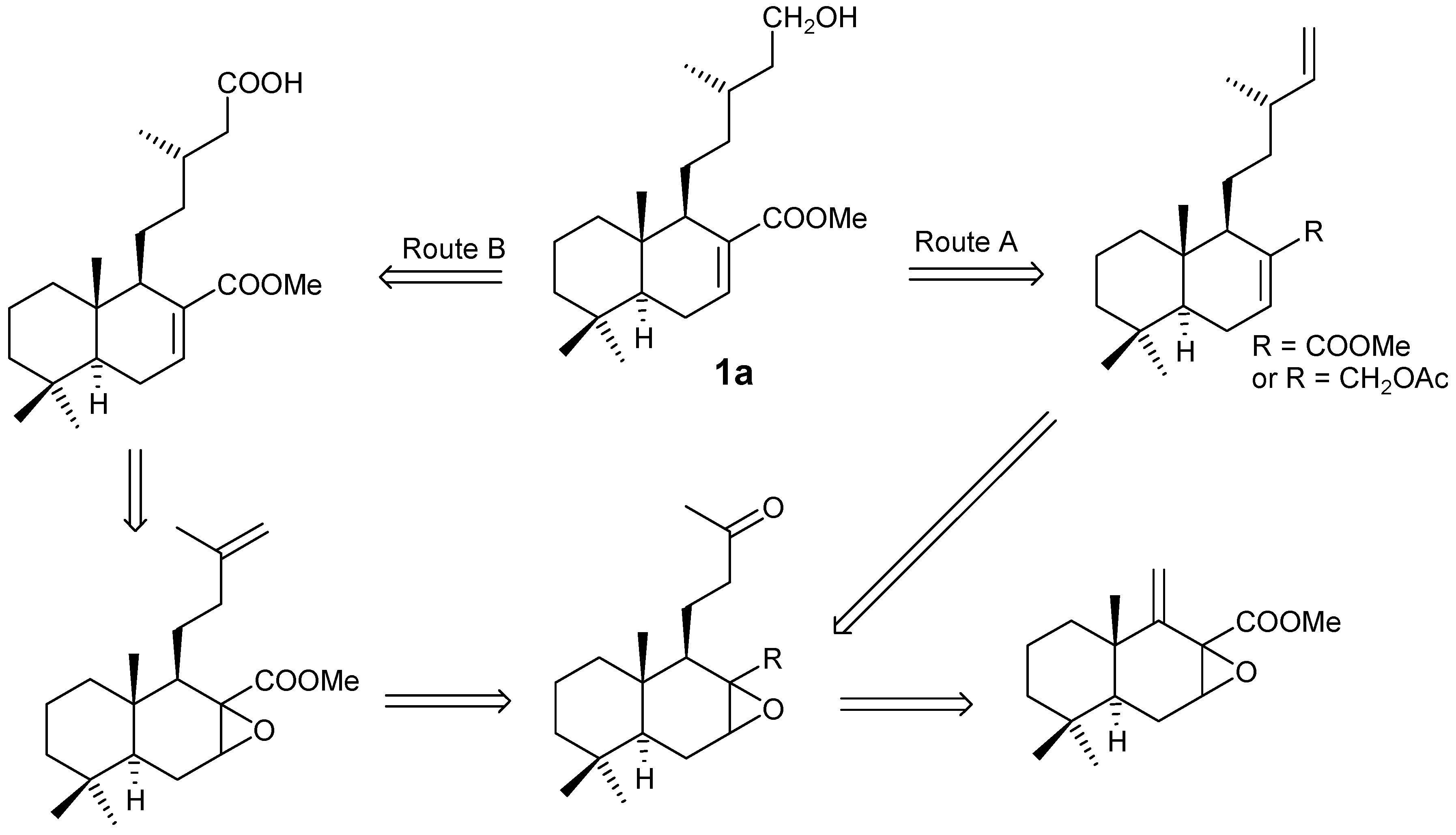
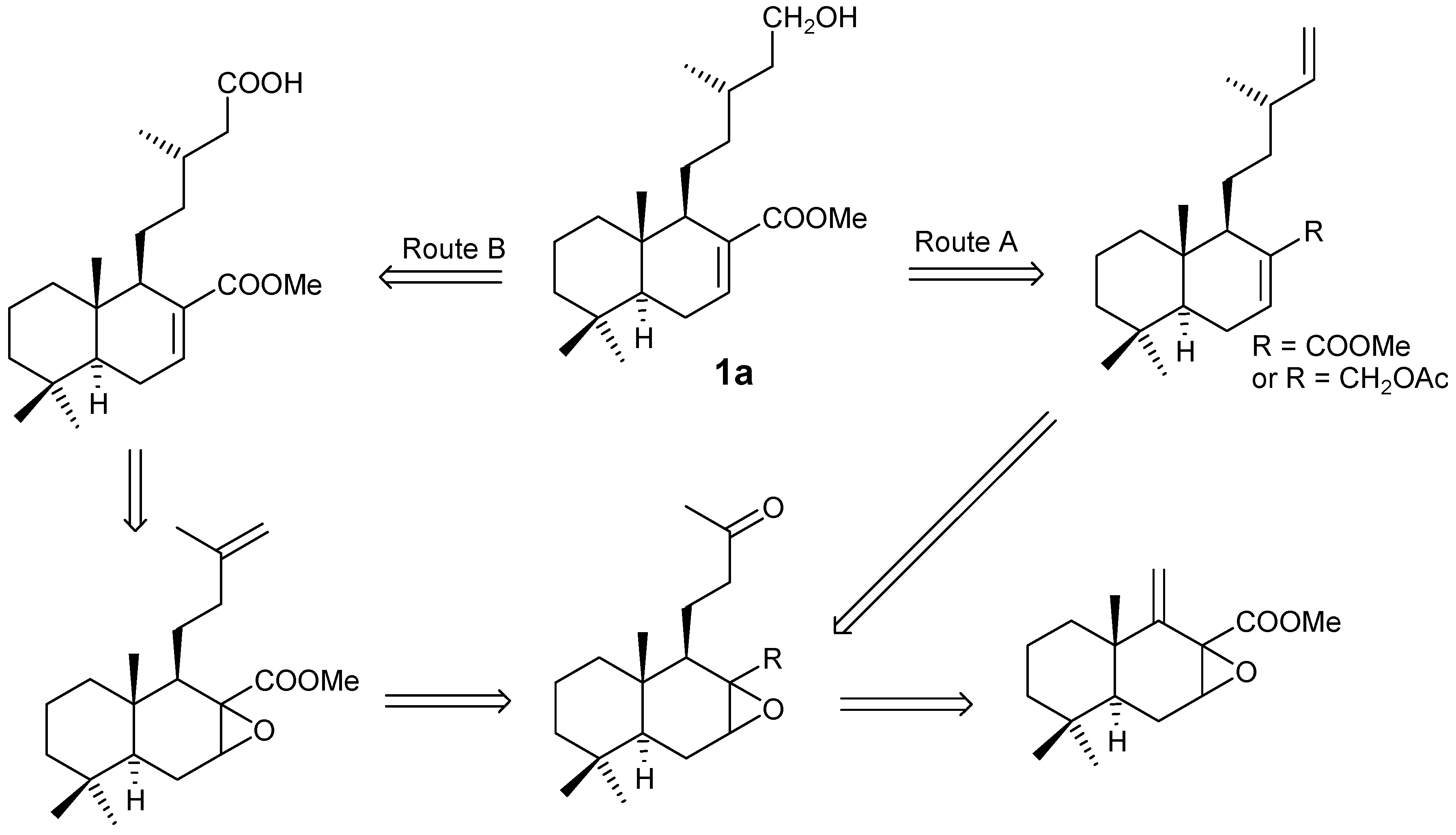
Starting from dihydrozamoranic acid methyl ester (1a), the retrosynthetic analysis as indicated below (Scheme 1), could be accomplished by two routes. In both cases the main degradation reaction is an elimination of the terminal carbon of the side chain, either by transformation into a double bond or by oxidative decarboxylation of an acid.
Scheme 1.
Route A
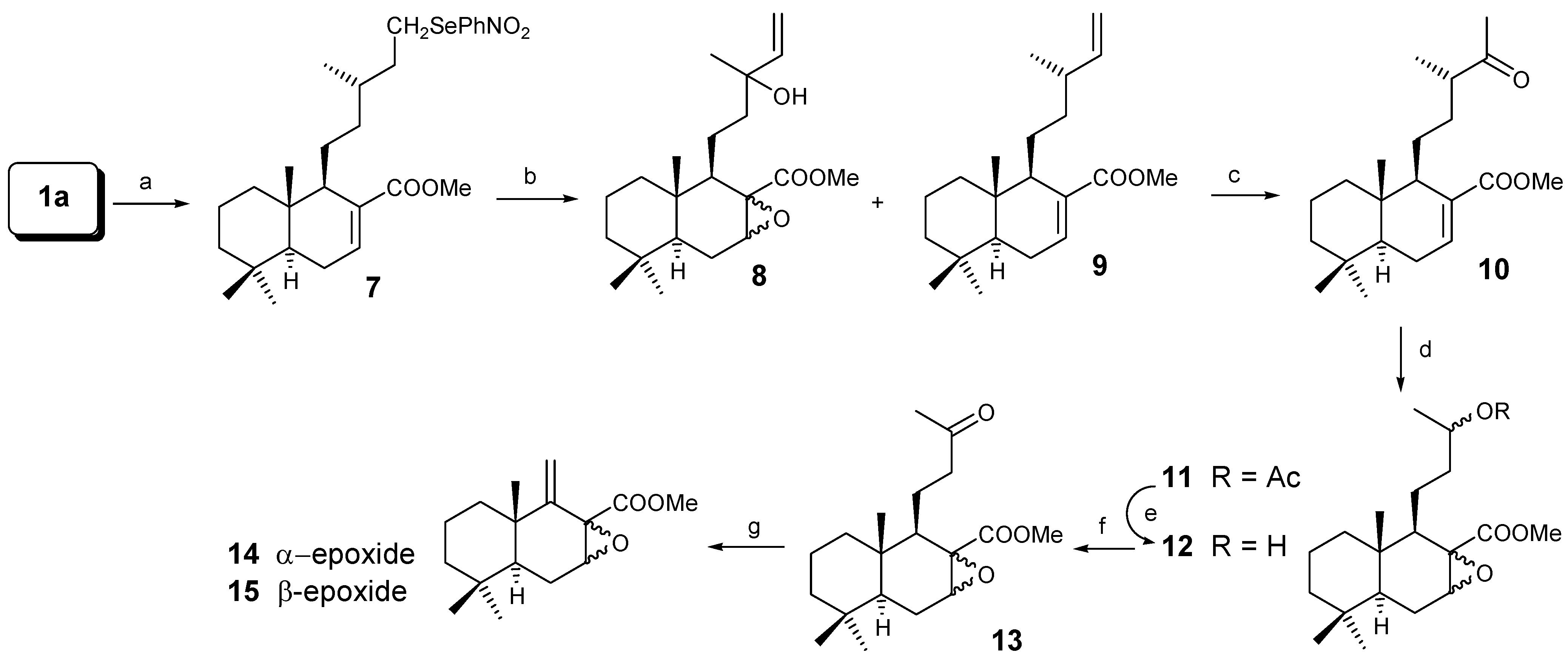
The first route followed for the degradation of the side chain is shown in Scheme 2. Treatment of 1a with o-nitrophenylselenocyanate (nBu3P/THF) [13] gave an 87% yield of the nitrophenylselenyl derivative 7, which with H2O2 [14], produced a minor derivative 8 (4%) and compound 9 in 92% yield. Treatment of 9 under Wacker conditions [15] with CuCl/PdCl2/O2, lead to ketone 10 in 80% yield. Baeyer-Villiger oxidation [16] of 10 with m-CPBA gives 11, a 1:2 mixture of α and β epoxides, in 95% yield. This compound shows a degradation of the side chain by two carbons.
The hydrolysis of 11 with K2CO3/MeOH gives 12 (85% yield), a mixture of epoxy alcohols that by oxidation with CrO3/Py [17] gave the epoxyketones 13 (82%). As in previous studies, the degradation of the nor-derivatives to the drimane skeleton was done by Norrish II type reaction [18]. Photochemical treatment (hυ; λ = 366 nm) of 13 lead to olefins 14 (9.5%) and 15 (22.5%), while a (39%) of 13 was recovered, so the global yield is quite good when the recovery of the starting material is taken into account.
Scheme 2.
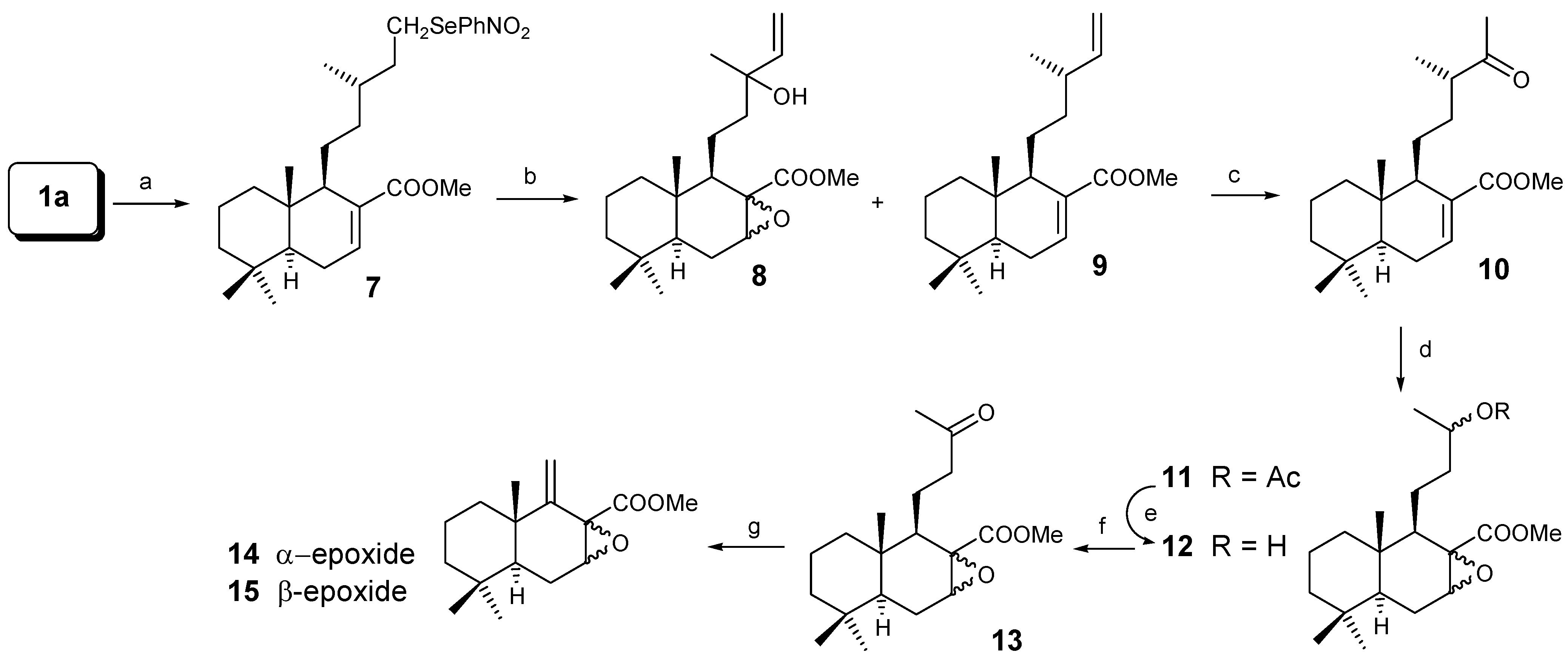
Reagents: (a) o-NO2PhSeCN/nBu3P/THF; (b) H2O2/TFH; (c) CuCl/PdCl2/O2/DMF;
(d) m-CPBA/CH2Cl2; (e) K2CO3/MeOH; (f) CrO3/Py; (g) Norrish II
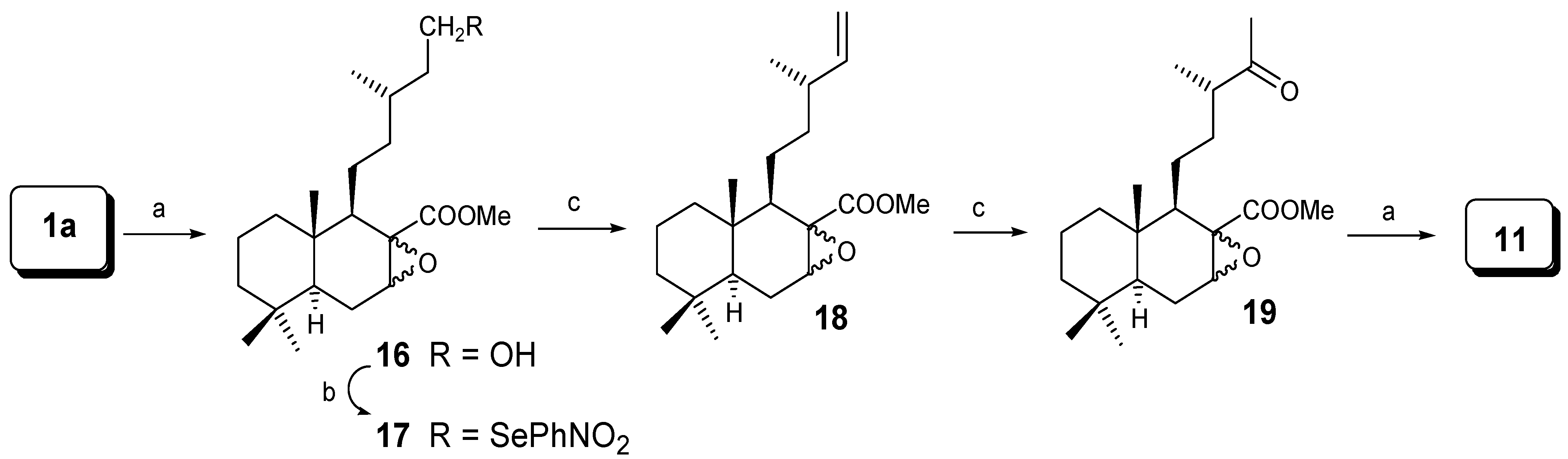
The synthesis of compound 11 could be achieved by epoxidation and then degradation, as shown in Scheme 3. Treatment of dihydrozamoranic acid methyl ester (1a) with m-CPBA led to a 67% yield of the mixture of epoxides 16 in a ratio 3:2 (α:β), which by treatment with o-nitrophenylselenocyanate gives the mixture 17 in 75% yield. Oxidation of 17 with H2O2 lead to the Δ14 olefin 18, in 86% yield. Wacker oxidation with CuCl/PdCl2 and O2 gave the corresponding ketone, 19 (80%). Baeyer-Villiger oxidation with m-CPBA gave 11 (94% yield).
Scheme 3.

Reagents: (a) m-CPBA/CH2Cl2; (b) o-NO2PhSeCN/nBu3P/THF; (c) H2O2/THF; (d) CuCl/PdCl2/O2/DMF.
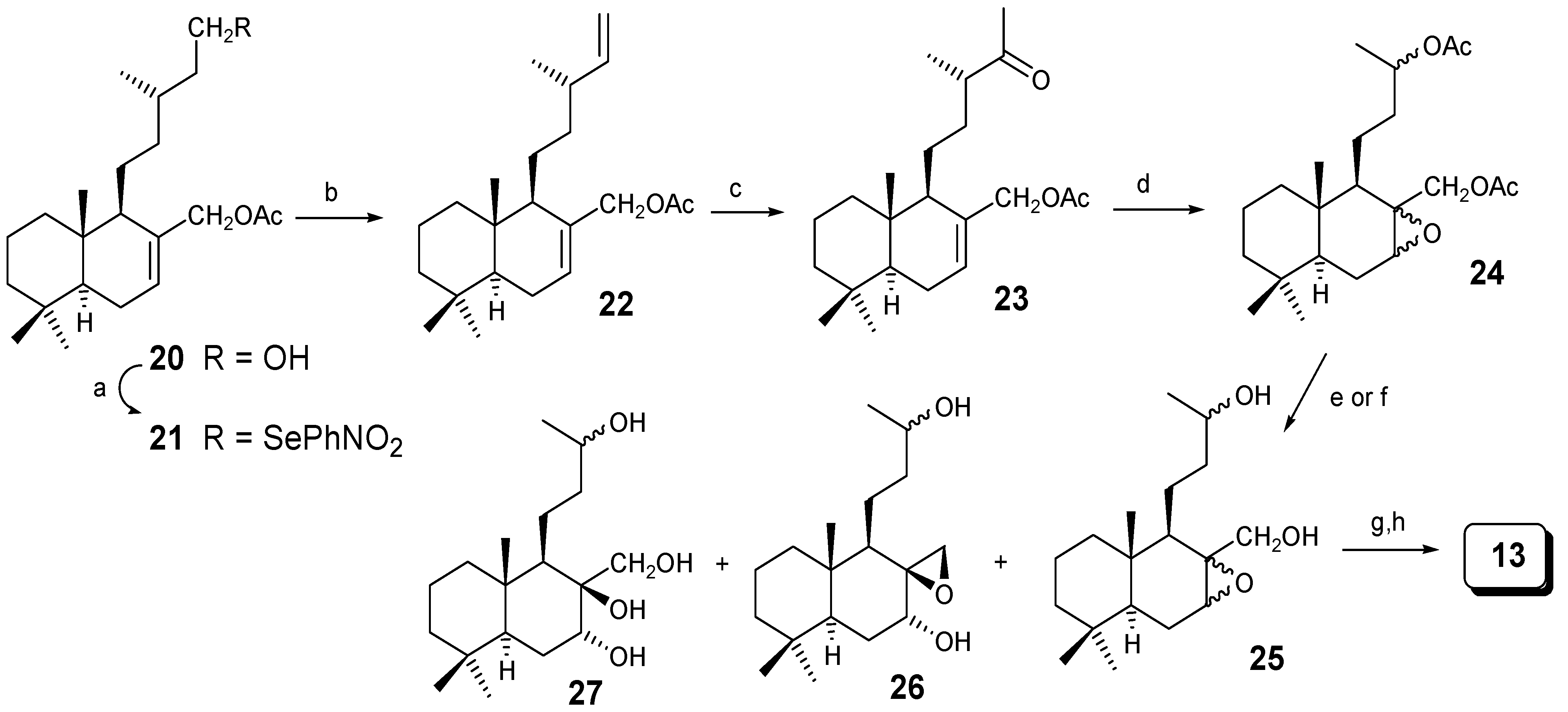
Compound 13 can also be obtained in a different manner, starting from 20, a natural compound isolated from H. verticillatum and H. viscosum [19] (Scheme 4), which in a similar manner as described before was transformed into compound 24. Hydrolysis of 24 under basic conditions gave 25, the epoxide of Payne rearrangement [20] 26 and 27 that were separated by CC. Compound 25 was oxidized to give 13, previously transformed into drimanes 14/15 (Scheme 2).
Scheme 4.

Reagents: (a) o-NO2PhSeCN/nBu3P/THF; (b) H2O2/THF; (c) CuCl/PdCl2/O2/ DMF; (d) m-CPBA/CH2Cl2; (e) K2CO3/MeOH; (f) NaOH/MeOH; (g) CrO3/AcOH; (h) CH2N2/ether.
Route B
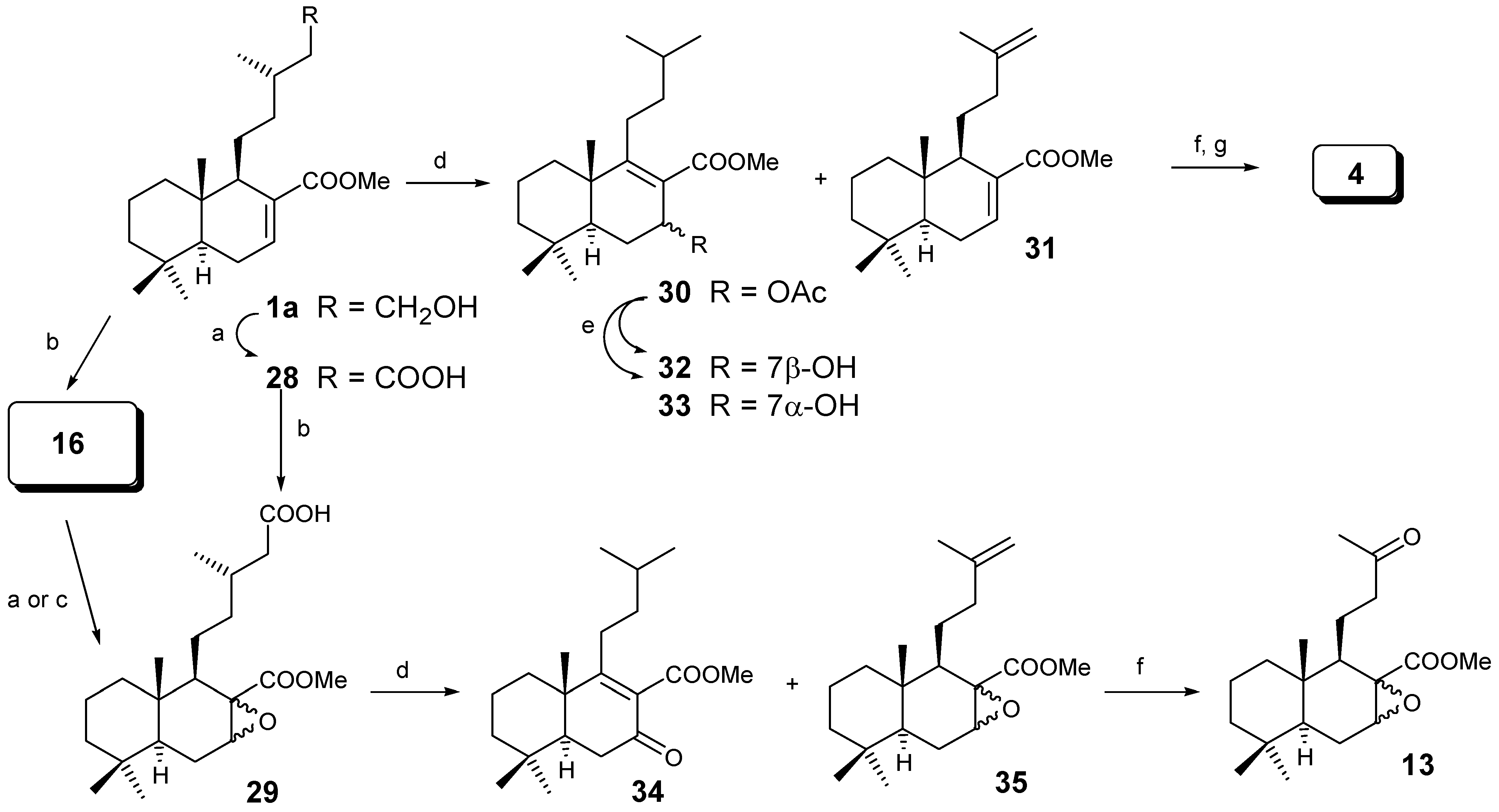
A second approach is based on the degradation of the side chain by an oxidative decarboxylation process (Scheme 5). The synthesis of the acid group on C-15 and epoxide C-7 can be acheived in two different ways. Oxidation of 1a with CrO3/AcOH gives acid 28 in 60% yield, which by treatment with m-CPBA lead to the 3:2 mixture of α and β epoxides 29 in 60% yield. Alternatively, treatment of 1a with m-CPBA gives the 3:2 mixture of α and β epoxides 16, then oxidation with PDC/DMF [21] or CrO3/AcOH lead to the same 3:2 mixture of α and β epoxides 29, (in 74% and 56% yields) respectively.
Scheme 5.
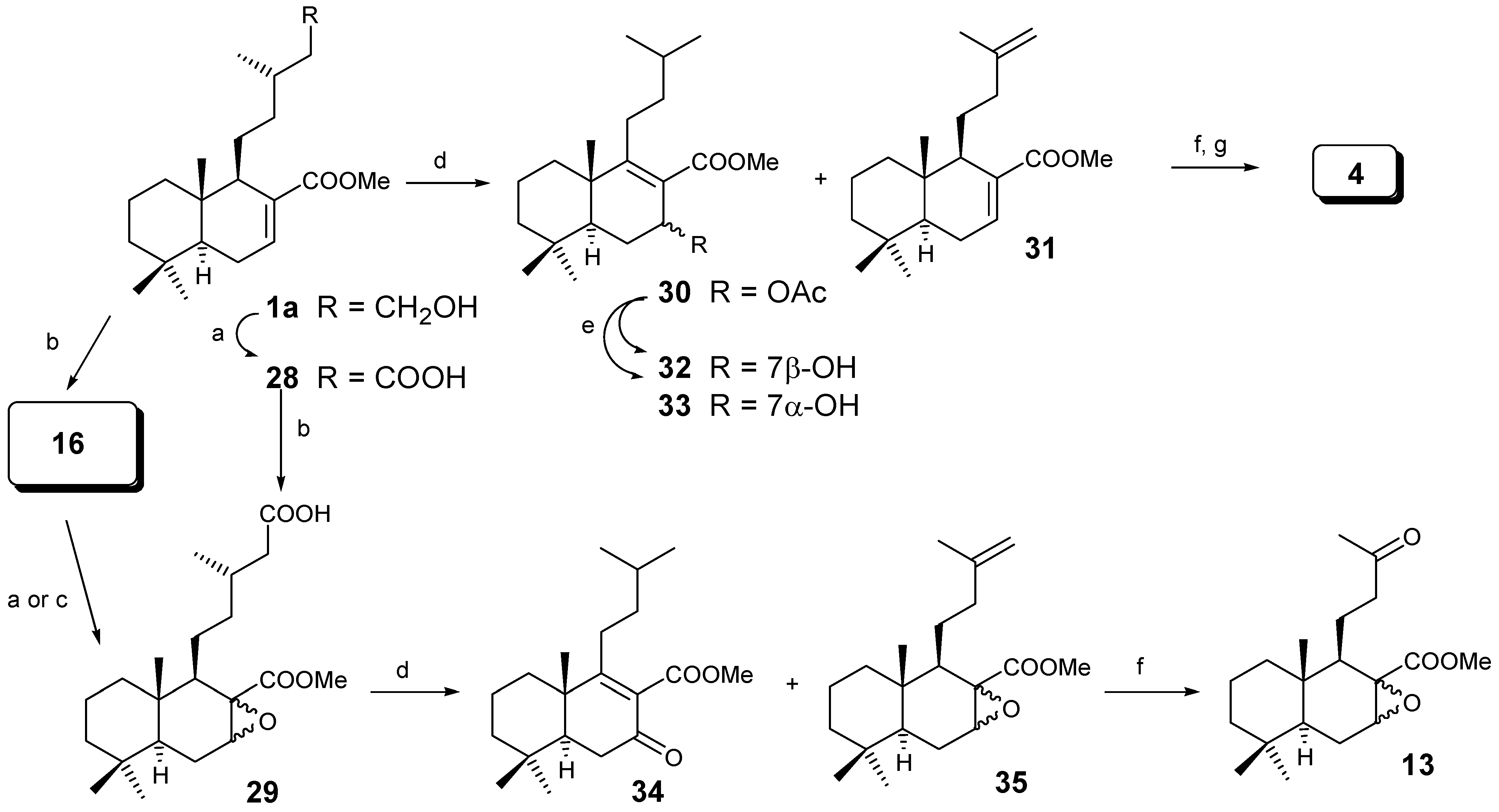
Reagents: (a) CrO3/AcOH; (b) m-CPBA/CH2Cl2; (c) PDC/DMF; (d) (AcO)4Pb/ (AcO)2Cu/ Py/C6H6; (e) K2CO3/MeOH; (f) O3/CH2Cl2; (g) Norrish II
Oxidative decarboxylation of 28 gave 30 and 31 in 40% and 8% yield, respectively. The major component was hydrolysed to give alcohols 32 and 33. It is very interesting to note that the decarboxylation takes place giving mainly an isobutyl group in the parent compound 30.
The minor compound 31 was transformed by ozonolysis and Norrish II type reaction into diene 4, already transformed into active drimanes [12]. As the yield was very poor it was decided to do the same reaction with compound 29, leading again, to a major component 34 (62%) and a terminal olefin 35 (12%) which was transformed into epoxides 13.
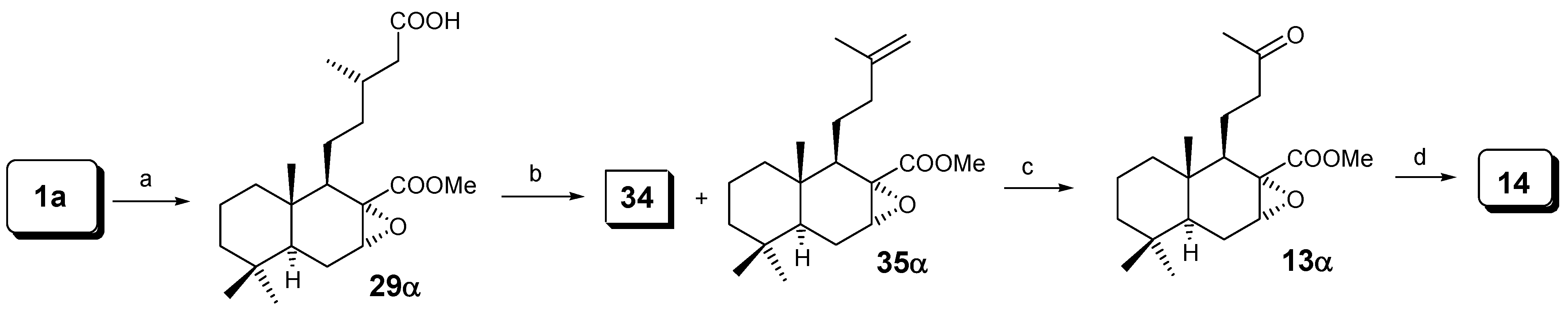
As we have seen with m-CPBA, a mixture of epoxides at C-7 was always obtained. In order to obtain only one epoxide, dihydrozamoranic acid methyl ester (1a) was treated with dimethyldioxirane [23], giving selectively only compound 29α in 45% yield and producing the oxidation of the primary alcohol into the acid at C-15; following the same sequence as in Scheme 6 only compound 14 was obtained.
Scheme 6.
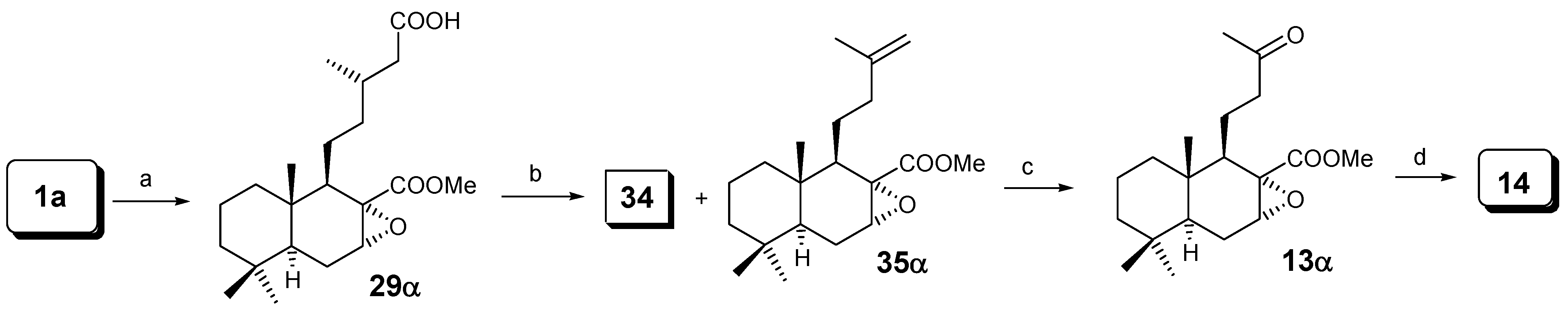
Reagents: (a) dimethyldioxirane / acetone; (b) (AcO)4Pb/(AcO)2Cu/Py/C6H6;
(c) O3/ CH2Cl2; (d) Norrish II
Conclusions
We have developed a new procedure to obtain drimane 14 from dihydrozamoranic acid, making use of methodology developed in our laboratory [24] that could be useful for further transformations.
Experimental
General
Unless otherwise stated, all chemicals were purchased as the highest purity commercially available and were used without further purification. IR spectra (thin film) were recorded on a MATTSON-GENESIS II FT-IR spectrophotometer. 1H- and 13C-NMR spectra were recorded in deuterochloroform and referenced to the residual peak of CHCl3 at δ 7.26 ppm and δ 77.0 ppm, for 1H- and 13C-, respectively, on a Bruker WP-200 SY or a Bruker DRX 400 MHz instrument. Chemical shifts are reported in δ ppm and coupling constants (J) are given in Hz. MS were performed at a VG-TS 250 spectrometer at 70 eV ionising voltage. Mass spectra are represented at m/z (% rel. int.). HRMS were recorded on a VG Platform (Fisons) spectrometer using chemical ionisation (ammonia as gas). Optical rotations were determined at a digital ADP 220 polarimeter in 1 dm cells. Diethyl ether, THF and benzene were distilled from sodium, and pyridine and dichloromethane were distilled from calcium hydride under an Ar atmosphere. The raw material 1 was isolated from a hexane extract of Halimium verticillatum as reported in reference [11].
Reaction of 1a: Synthesis of methyl 15-o-nitrophenylseleno-7-labden-17-oate (7).
To a solution of 1a (750 mg, 2.2 mmol) in dry THF (3.3 mL) was added o-nitrophenyl-selenocyanate (635.8 mg, 2.8 mmol) and the mixture was stirred under an inert atmosphere for 20 min. at 48ºC. nBu3P (504.5 mg, 2.5 mmol) was added to the reaction mixture, that was stirred for 48 min., then the solvent was removed and the residue chromatographed (95:5 hexane/EtOAc) yielding 652.5 mg (87%) of 7; 1H-NMR δ: 8.30 (d, 1H, J=8.2, H-3’), 7.47 and 7.27 (both m, 3H, H-4’, H-5’ and H-6’), 6.66 (m, 1H, H-7), 3.70 (s, 3H, MeOOC), 2.95 (m, 2H, H-15), 2.15 – 1.30 (m, 11H), 1.22 – 0.90 (m, 6H), 0.98 (d, 3H, J=5.8 Hz, Me-16), 0.92 (s, 3H, Me-19), 0.89 (s, 3H, Me-18) and 0.84 (s, 3H, Me-20); 13C-NMR δ: 39.4 (C-1), 18.4 (c-2), 41.9 (C-3), 32.6 (C-4), 49.3 (C-5), 23.8 (C-6), 137.0 (C-7), 135.2 (C-8), 50.9 (C-9), 36.8 (C-10), 25.6 (C-11), 37.8 (C-12), 34.3 (C-13), 34.7 (C-14), 23.8 (C-15), 19.3 (C-16), 169.6 (C-17), 33.0 (C-18), 21.8 (C-19), 14.3 (C-20), 51.2 (COOMe), 134.0 (C-1’), 146.3 (C-2’), 126.3 (C-3’), 128.9 (C-4’), 133.4 (C-5’), 125.1 (C-6’); IR cm-1: 2925, 1714, 1644, 1590, 1564, 1515, 1332, 1247,1068, 730.
Oxidation of 7 with H2O2: Synthesis ofmethyl 13-hydroxy-7,14-labdadien-17-oate (8) andmethyl 7,14-labdadien-17-oate (9).
To a solution of 7 (600 mg, 1.15 mmol) in THF (6 mL) was added 30% H2O2 (0.30 mL, 130 mg, 2.0 mmol) and the mixture was stirred for 12 h. The solvent was evaporated at reduced pressure and the crude product chromatographed eluting with a hexane/EtOAc gradient yielding 24 mg (4%) of 8 (95:5 hexane/EtOAc) and 552 mg (92%) of 9 (98:2 hexane/EtOAc).
Compound 8: 1H-NMR δ: 6.69 (m, 1H, H-7), 5.86 (dd, 1H, J=17.4, 10.7, H-14), 5.19 (d, 1H, J=17.4, H-15a), 5.02 (d, 1H, J=17.4, H-15b), 3.69 (s, 3H, COOMe), 2.25 – 1.35 (m, 8H), 1.25 – 0.90 (m, 6H), 1.22 (s, 3H, Me-16), 0.89 (s, 3H, Me-19), 0.85 (s, 3H, Me-18) and 0.80 (s, 3H, Me-20); 13C-NMR δ: 39.3 (C-1), 18.4 (C-2), 41.9 (C-3), 32.6 (C-4), 49.3 (C-5), 22.0 (C-6), 137.4 and 137.6 (C-7, epimers), 135.5 and 135.7 (C-8, epimers), 50.7 and 50.8 (C-9, epimers), 36.9 (C-10), 23.9 (C-11), 42.6 and 42.8 (C-12, epimers), 73.1 (C-13), 144.9 and 145.4 (C-14, epimers), 111.3 and 111.6 (C-15, epimers), 169.5 (C-17), 33.0 (C-18), 21.9 (C-19), 14.2 (C-20), 51.3 (COOMe); IR cm-1: 3446, 3086, 2924, 1717, 1646, 1245,1071, 917; MS m/z (EI+): 334 (M+, <1%), 316 (4), 284 (19), 249 (15), 248 (79), 233 (28), 203 (15), 192 (29) 175 (25), 124 (15), 109 (56), 105 (16), 91 (24), 81 (33), 79 (34), 77 (15), 71 (100), 69 (28), 67 (20), 59 (15), 55 (45); HRMS (CI) for C21H34O3 (MH+): Calc 334.2508; Found 334.2519.
Compound 9: 1H-NMR δ: 6.51 (m, 1H, H-7), 5.56 (ddd, 1H, J=17.3, 10.1, 7.3, H-14), 4.82 (dd, 1H, J=17.3 Hz, 2.1 Hz, H-15a), 4.78 (dd, 1H, J=10.1, 2.1 Hz, H-15b), 3.59 (s, 3H, COOMe), 2.15 – 1.65 (m, 4H), 1.60 – 1.25 (m, 6H), 1.20 – 0.90 (m, 5H), 0.87 (d, 3H, J=6.7, Me-16), 0.81 (s, 3H, Me-19), 0.77 (s, 3H, Me-18) and 0.72 (s, 3H, Me-20); 13C-NMR δ: 39.4 (C-1), 18.3 (C-2), 41.9 (C-3), 32.5 (C-4), 49.2 (C-5), 23.6 (C-6), 136.2 (C-7), 135.4 (C-8), 50.8 (C-9), 36.6 (C-10), 25.7 (C-11), 37.8 (C-12), 38.5 (C-13), 144.4 (C-14), 112.2 (C-15), 20.1 (C-16), 169.3 (C-17), 33.0 (C-18), 21.8 (C-19), 14.2 (C-20), 50.9 (COOMe); [α]D20 - 16.7 (c 0.9 in CHCl3); IR cm-1: 3076, 2951, 1722, 1643, 1434, 1246, 1066, 909; MS m/z (EI+) 318 (M+, 1.8%), 248 (10), 195 (3), 194 (9), 165 (3), 162 (13), 137 (8), 135 (12), 124 (79), 109 (100), 95 (36), 81 (41), 69 (50), 67 (34), 55 (83).
Wacker oxidation of 9: Synthesis of methyl 14-oxo-7-labden-17-oate (10).
A solution of PdCl2 (128 mg, 0.72 mmol) and CuCl (3.5 g, 3.6 mmol) in DMF (10 mL) and H2O (1 mL) was activated for 30 min with O2. A solution of 9 (1.1 g, 3.6 mmol) in DMF (8 mL) was added and stirred with O2 atmosphere at room temperature for 36 h. Then to this mixture was added a cooled solution of 3N HCl and the mixture was extracted with CH2Cl2 (3 x 30 mL) and washed successively with 10% NaHCO3 and water, dried, filtered, evaporated and chromatographed (9:1 hexane/EtOAc) to give 910 mg (80%) of 10; 1H-NMR δ: 6.45 (m, 1H, H-7), 3.49 (s, 3H, COOMe), 2.24 (sex, 1H, J=6.8, H-13), 2.00 – 1.60 (m, 4H), 1.91 (s, 3H, Me-15), 1.40 – 0.90 (m, 10H), 0.85 (d, 3H, J=6.8, Me-16), 0.69 (s, 3H, Me-19), 0.65 (s, 3H, Me-18) and 0.60 (s, 3H, Me-20); 13C-NMR δ: 39.0 (C-1), 18.1 (C-2), 41.5 (C-3), 32.3 (C-4), 48.9 (C-5), 23.5 (C-6), 136.8 (C-7), 134.6 (C-8), 50.5 (C-9), 36.4 (C-10), 25.4 (C-11), 34.1 (C-12), 47.6 (C-13), 211.8 (C-14), 27.1 (C-15), 15.6 (C-16), 168.7 (C-17), 32.7 (C-18), 21.5 (C-19), 13.9 (C-20), 50.8 (COOMe); [α]D20 -11.1 (c 0.2 in CHCl3); IR cm-1: 2927, 1713, 1645, 1461, 1365, 1254, 1065; MS m/z (EI+) 334 (M+, <0.8%), 302 (2), 263 (5), 211 (8), 179 (37), 124 (32), 109 (100), 105 (20), 95 (16), 91 (32), 81 (26), 79 (32), 77 (17), 69 (26), 67 (17), 59 (12), 55 (30); HRMS (CI) for C21H34O3 (MH+): Calcd 334.2508; Found 334.2517.
Baeyer-Villiger reaction and epoxidation of 10 with m-CPBA: Synthesis of methyl 13-acetoxy-7,8-epoxy-14,15-dinor-labdan-17-oate (11).
To a solution of 10 (334.5 mg, 1.0 mmol) in dry CH2Cl2 (5 mL), m-CPBA (345.0 mg, 2.0 mmol) was added and the mixture stirred at room temperature. After 12 h, the solvent was removed and ether was added. The organic phase was washed with 40% Na2S2O3, 10% Na2CO3 and water until neutrality, dried over Na2SO4, filtered and evaporated to give 11 (317.8 mg, 95%). 1H-NMR δ: 4.60 (sex, 1H, J=6.1, H-13), 3.53 and 3.51 (two s, 2 x 3H, 2 x COOMe), 3.10 (d, 1H, J=6.2, H-7 β-epoxide), 3.00 (sb, 1H, H-7 α-epoxide), 2.00 – 1.78 (m, 1H), 1.81 and 1.79 (each s, 2 x H, 2 x OOCMe), 1.60 – 1.05 (m, 13H), 0.97 (d, 3H, J=6.1, Me-16), 0.66 and 0.62 (each s, 3 x 3H, Me-18, Me-19 and Me-20); 13C-NMR δ: 38.2 and 40.1 (C-1, isomers), 18.3 and 17.8 (C-2), 41.6 (C-3), 32.6 and 32.7 (C-4 isomers), 45.6 (α-isomer) and 47.1 (β-isomer) (C-5), 20.7 (C-6), 57.5 (α-isomer) and 59.4 (β-isomer) (C-7), 58.8 (α-isomer) and 60.2 (β-isomer) (C-8), 49.1 (β-isomer) and 53.5 (α-isomer) (C-9), 34.7 and 35.6 (C-10 isomers), 22.6 (C-11), 34.9 (C-12), 70.4 (C-13), 21.1 (C-16), 170.2 and 170.7 (C-17 isomers), 32.4 and 33.0 (C-18 isomers), 21.7 (C-19), 14.2 and 14.9 (C-20 isomers), 51.9 and 52.1 (2XCOOMe), 170.2 and 170.7 (2XMeCOO), 19.8 (MeCOO); IR cm-1: 2930, 1731, 1715, 1461, 1251, 1142.
Hydrolysis of 11: Synthesis of methyl 13-hydroxy-7α,8α-epoxy-14,15-dinor-labdan-17-oate (12α) and methyl 13-hydroxy-7β,8β-epoxy 14,15-dinor-labdan-17-oate (12β).
To a solution of 11 (250 mg, 0.68 mmol) in methanol (12 mL) was added K2CO3 (150 mg). The reaction mixture was stirred at room temperature for 1 h, water was added and the mixture extracted with ether, washed with 2N HCl and H2O, dried, filtered and evaporated yielding 212 mg (85%) of 12.
Compound 12α: 1H-NMR δ: 3.72 (s, 3H, COOMe), 3.71 (m, 1H, H-13), 3.22 (sb, 1H, H-7), 2.14 (dd, 1H, J=4.3, 14.4, H-6), 1.80 – 1.00 (m, 13H), 1.14 (d, 3H, J=6.1, Me-16), 0.87 (s, 3H,Me-19), 0.86 (s, 3H, 18) and 0.83 (s, 3H, Me-20); 13C-NMR δ: 38.5 (C-1), 18.6 (C-2), 41.8 (C-3), 33.0 (C-4), 45.9 (C-5), 20.1 (C-6), 57.8 (C-7), 59.1 (C-8), 53.8 (C-9), 35.0 (C-10), 21.9 (C-11), 38.3 (C-12), 67.7 (C-13), 23.6 (C-16), 171.1 (C-17), 32.6 (C-18), 21.8 (C-19) 14.6 (C-20), 52.2 (COOMe); [α]D20 + 6.9 (c 0.4 in CHCl3); IR cm-1: 3491, 2981, 1732, 1450, 1375, 1167, 1026.
Compound 12β: 1H-NMR δ: 3.72 (m, 1H, H-13), 3.71 (s, 3H, COOMe), 3.30 (d, 1H, J=6.3, H-7), 2.10 – 0.90 (m, 14H), 1.16 (d, 3H, J=6.2, Me-16); 0.87 (s, 3H, Me-19); 0.84 (s, 3H, Me-18) and 0.82 (s, 3H, Me-20); 13C-NMR δ: 38.5 (C-1), 18.0 (C-2), 41.9 (C-3), 32.9 (C-4), 47.7 (C-5), 21.0 (C-6), 59.7 (C-7), 60.5 (C-8), 49.4 (C-9), 35.9 (C-10), 23.2 (C-11), 40.6 (C-12), 68.1 (C-13), 23.4 (C-16), 172.7 (C-17), 33.2 (C-18), 21.9 (C-19), 15.2 (C-20), 52.3 (COOMe); IR cm-1: 3486, 2985, 1736, 1455, 1371, 1164, 1026.
Oxidation of 12 with CrO3/Py: Synthesis of methyl 7α,8α-epoxy-13-oxo-14,15-dinor-labdan-17-oate (13α) and methyl 7β,8β-epoxy-13-oxo-14,15-dinor-labdan-17-oate (13β).
Pyridine (1 mL) and dry CH2Cl2 (3 mL) were placed in a 50 mL Erlenmeyer flask externally cooled with ice. CrO3 (660 mg, 0.65 mmol) was added in small portions with stirring until a think yellow paste was obtained. This was allowed to reach room temperature and was stirred for 15 min under N2. Following this 12 (324.0 mg, 1.0 mmol) dissolved in dry CH2Cl2 (3 mL) was added and the mixture was stirred vigorously for 1.5 hours. The mixture was filtered and chromatographed on silica-gel, yielding 265.7 mg (82%) of 13. In some fractions the α isomer is the major compound and in other fractions, it is the β isomer.
Compound 13α: 1H-NMR δ: 3.62 (s, 3H, COOMe), 3.14 (sb, 1H, H-7), 2.35 – 2.20 (m, 1H, H-6), 2.01 (s, 3H, Me-16), 1.90 – 0.85 (m, 13H), 0.78 (s, 3H, Me-19), 0.77 (s, 3H, Me-18) and 0.73 (s, 3H, Me-20); 13C-NMR δ: 38.1 (C-1), 17.9 (C-2), 41.6 (C-3), 32.7 (C-4), 45.7 (C-5), 18.3 (C-6), 57.5 (C-7), 58.7 (C-8), 53.1 (C-9), 34.8 (C-10), 21.6 (C-11), 42.2 (C-12), 207.6 (C-13), 29.8 (C-16), 170.5 (C-17), 32.4 (C-18), 21.6 (C-19), 14.2 (C-20), 52.2 (COOMe); IR cm-1: 2926, 1732, 1712, 1461, 1282, 1158, 1062, 897.
Compound 13β: 1H-NMR δ: 3.73 (s, 3H, COOMe), 3.31 (d, 1H, J=6.4, H-7), 2.55 – 2.40 (m, 1H, H-6), 2.11 (s, 3H, Me-16), 2.10 – 0.90 (m, 13H), 0.88 (s, 3H, Me-19), 0.85 (s, 3H, Me-18) and 0.82 (s, 3H, Me-20); 13C-NMR δ: 40.5 (C-1), 18.0 (C-2), 41.9 (C-3), 32.9 (C-4), 46.9 (C-5), 20.9 (C-6), 59.9 (C-7), 60.1 (C-8), 49.4 (C-9), 36.0 (C-10), 20.9 (C-11), 42.6 (C-12), 208.2 (C-13), 29.7 (C-16), 172.7 (C-17), 33.3 (C-18), 21.9 (C-19), 15.2 (C-20), 53.5 (COOMe); IR cm-1: 2950, 1733, 1714, 1275, 1163, 1053.
Norrish type II reaction of 13: Synthesis of methyl 7α,8α-epoxy-9-drimen-12-oate (14) and methyl 7β,8β-epoxy-9-drimen-12-oate (15).
A solution of 13 (250.0 mg, 0.77 mmol) in dry hexane (250 mL) was placed in a quartz flask and a stream of dry N2 was bubbled through. The solution was irradiated with UV light (Hanau TQ-150, high pressure) for 90 min. Removal of solvent afforded a yellow oil which was purified by chromatography on silica-gel eluting with 98:2 hexane-EtOAc to yield 23.3 mg (9.3%) of 14, 56.3 mg (22.5%) of 15 and 97.5 mg (39%) of the starting material 13.
Compound 14: 1H-NMR δ: 5.25 (s, 1H, H-11a), 5.13 (s, 1H, H-11b), 3.76 (s, 3H, COOMe), 3.50 (t, 1H, J=1.9, H-7), 2.20 (ddd, 1H, J=1.9, 4.3, 15.1 Hz, H-6a), 1.80 (m, 2H, H-6b and H-1), 1.53 (m, 1H), 1.44 – 1.40 (m, 3H), 1.20 (dd, 1H, J=4.3, 13.1 Hz, H-5), 1.12 (m, 1H, H-3), 1.05 (s, 3H, Me-15), 0.90 (s, 3H, Me-14), and 0.85 (s, 3H, Me-13); 13C-NMR δ: 36.4 (C-1), 18.4 (C-2), 41.8 (C-3), 33.0 (C-4), 41.4 (C-5), 22.2 (C-6), 58.5 (C-7), 57.7 (C-8), 150.8 (C-9), 37.0 (C-10), 114.9 (C-11), 170.0 (C-12), 32.8 (C-13), 22.4 (C-14), 20.2 (C-15), 52.6 (COOMe); [α]D20 + 97.8 (c 0.5 in CHCl3); IR cm-1: 3098, 1743, 1633, 1373, 1242, 1159, 1047, 902; MS m/z (EI+) 264 (M+, 8%), 263 (3), 249 (49), 232 (26), 217 (35), 205 (22), 189 (46), 175 (29), 161 (74), 154 (30), 147 (59), 135 (88), 121 (69), 107 (87), 105 (92), 91 (100), 79 (80); HRMS (CI) for C16H24O3 (MH+): Calcd 264.1725; Found 264.1736.
Compound 15: 1H-NMR δ: 5.33 (s, 1H, H-11a), 5.19 (s, 1H, H-11b), 3.79 (s, 3H, COOMe), 3.57 (d, 1H, J=6.2, H-7), 2.13 (ddd, 1H, J=5.0, 6.2, 15.1 Hz, H-6a), 1.87 (m, 2H, H-6b and H-1), 1.52 – 1.38 (m, 4H), 1.22 (dd, 1H, J=5.0, 13.2, H-5), 1.13 (m, 1H), 1.09 (s, 3H, Me-15), 0.89 (s, 3H, Me-13) and 0.86 (s, 3H, Me-14); 13C-NMR δ: 38.5 (C-1), 18.5 (C-2), 41.8 (C-3), 33.4 (C-4), 48.2 (C-5), 21.3 (C-6), 60.6 (C-7), 60.2 (C-8), 151.1 (C-9), 37.6 (C-10), 115.0 (C-11), 170.0 (C-12), 32.8 (C-13), 21.7 (C-14), 22.7 (C-15), 52.4 (COOMe); [α]D20 + 73.5 (c 0.5 in CHCl3); IR cm-1: 3096, 2923, 1744, 1635, 1275, 1244, 1151, 1043, 902; MS m/z (EI+) 264 (M+, 3%), 263 (3), 249 (49), 232 (26), 217 (35), 205 (22), 189 (46), 175 (29), 161 (74), 154 (30), 147 (59), 135 (88), 121 (69), 107 (87), 105 (92), 91 (100), 79 (80); HRMS (CI) for C16H24O3 (MH+): Calcd 264.1725; Found 264.1737.
Treatment of 1a with m-CPBA: Synthesis of methyl 7,8(α+β)epoxy-15-hydroxylabdan-17-oate (16).
Compound 1a (490 mg, 1.5 mmol) was dissolved in CH2Cl2 (10 mL) and m-CPBA (380 mg, 2.2 mmol) was added. The mixture was stirred at 40º C and monitored by TLC. After 4 h the reaction was complete and the solvent evaporated. Work-up afforded 480 mg of crude product that after chromatography on silica-gel gave 323 mg (67.3%) of 16 in the 7:3 hexane/EtOAc fractions. 1H-NMR δ: 3.68 (s, 3H, COOMe), 3.64 (m, 2H, H-15), 3.26 (d, 1H, J=6.3, H-7 β-epoxide), 3.18 (sb, 1H, H-7 α-epoxide), 2.09 – 1.88 (m, 3H), 1.82 – 0.90 (m, 14H), 0.89 (d, 3H, J=6.7, Me-16), 0.84 (s each, 2 x 3H, Me-18 and Me-19) and 0.80 (s, 3H, Me-20); 13C-NMR δ (α-epoxide): 38.4 (C-1), 18.0 (C-2), 41.8 (C-3), 32.8 (C-4), 45.7 (C-5), 20.9 (C-6), 57.7 (C-7), 59.0 (C-8), 54.2 (C-9), 35.0 (C-10), 24.6 (C-11), 36.2 (C-12), 30.1 (C-13), 40.5 (C-14), 60.9 (C-15), 19.6 (C-16), 171.4 (C-17), 32.6 (C-18), 21.9 (C-19), 15.2 (C-20), 52.1 (COOMe); 13C-NMR δ (β-epoxide): 39.2 (C-1), 18.6 (C-2), 41.9 (C-3), 33.2 (C-4), 48.4 (C-5), 21.5 (C-6), 59.6 (C-7), 60.7 (C-8), 49.4 (C-9), 35.9 (C-10), 24.6 (C-11), 36.6 (C-12), 29.9 (C-13), 40.5 (C-14), 60.9 (C-15), 19.7 (C-16), 172.7 (C-17), 32.8 (C-18), 21.8 (C-19), 14.6 (C-20), 52.1 (COOMe); IR cm-1: 3445, 1732, 1457, 1283, 1063, 758; HRMS (CI) for C21H36O4 (MH+): Calcd 352.2614; Found 352.2625.
Reaction of 16: Synthesis of methyl 15-o-nitrophenylseleno-7α,8α-epoxylabdan-17-oate (17α) and methyl 15-o-nitrophenylseleno-7β,8β-epoxylabdan-17-oate (17β)
To a solution of 16 (450 mg, 1.3 mmol) in dry THF (3.0 mL) was added o-nitrophenylselenocyanate (363.3 mg, 1.6 mmol) and stirred under inert atmosphere for 20 min at 48º C. n-Bu3P (322.9 mg, 1.6 mmol) was added to reactional mixture, stirred for 48 min, the solvent was removed at reduced pressure and the residue chromatographed yielding with hexane/EtOAc 95:5, 338.0 mg (75%) of 17.
Compound 17α: 1H-NMR δ: 8.27 (d, 1H, J=8.1, H-3’), 7.60 – 7.18 (m, 3H, H-4’, H-5’ and H-6’), 3.67 (s, 3H, COOMe), 3.21 (sb, 1H, H-7), 2.89 (m, 2H, H-15), 2.24 – 1.90 (m, 3H), 1.85 – 0.90 (m, 14H), 0.94 (d, 3H, J=6.1, Me-16), 0.86 (s each, 2 x 3H, Me-19 and Me-18) and 0.83 (s, 3H, Me-20); 13C-NMR δ: 38.7 (C-1), 18.7 (C-2), 42.0 (C-3), 33.1 (C-4), 46.2 (C-5), 21.7 (C-6), 57.9 (C-7), 59.2 (C-8), 54.7 (C-9), 35.2 (C-10), 22.1 (C-11), 35.0 (C-12), 34.1 (C-13), 35.9 (C-14), 23.9 (C-15), 19.6 (C-16), 171.0 (C-17), 32.7 (C-18), 21.9 (C-19), 14.6 (C-20), 52.2 (COOMe), 134.8 (C-1’), 146.9 (C-2’), 126.5 (C-3’), 129.1 (C-4’), 133.6 (C-5’), 125.3 (C-6’); IR cm-1: 2930, 1740, 1596, 1514, 1331, 715, 711.
Compound 17β: 1H-NMR δ: 8.29 (d, 1H, J=8.2, H-3’), 7.40 – 7.30 (m, 3H,H-4’, H-5’ and H-6’), 3.71 (s, 3H, COOMe), 3.32 (d, 1H, J=6.4, H-7), 2.90 (m, 2H, H-15), 2.10 – 1.90 (m, 2H), 1.85 – 0.90 (m, 15H), 1.06 (d, 3H, J=6.1, Me-16), 0.90 (s, 3H, Me-19), 0.85 (s, 3H, Me-18) and 0.85 (s, 3H, Me-20); 13C-NMR (CDCl3) δ: 40.7 (C-1), 18.1 (C-2), 42.0 (C-3), 33.3 (C-4), 48.4 (C-5), 21.1 (C-6), 59.8 (C-7), 60.7 (C-8), 49.5 (C-9), 35.9 (C-10), 24.8 (C-11), 34.9 (C-12), 34.1 (C-13), 36.1 (C-14), 23.9 (C-15), 19.4 (C-16), 172.7 (C-17), 32.9 (C-18), 22.0 (C-19), 15.4 (C-20), 52.3 (COOMe), 134.4 (C-1’), 146.5 (C-2’), 126.5 (C-3’), 129.0 (C-4’), 133.6 (C-5’), 125.3 (C-6’); IR cm-1: 3062, 2929, 1733, 1590, 1513, 1332, 1037, 754.
Oxidation of 17 with H2O2: Synthesis of methyl 7,8(α+β)-epoxy-14-labden-17-oate (18).
To a solution of 17 (330.0 mg, 0.61 mmol) in THF (5 mL) was added 30% H2O2 (0.07 mL, 42 mg, 1.2 mmol) and the mixture was stirred for 12 h. The solvent was evaporated at reduced pressure and the crude product chromatographed yielding with 98:2 hexane/EtOAc, 284 mg (86%) of 18 as a mixture of epoxides. Data is given for a fraction in which the β-isomer predominates.
Compound 18β: 1H-NMR δ: 5.63 (ddd, 1H, J=7.7, 10.2, 17.5, H-14), 4.93 (dd, 1H, J=3.1, 17.5, H-15a), 4.91 (dd, 1H, J=3.1, 10.2, H-15b), 3.72 (s, 3H, COOMe), 3.29 (d, 1H, J=6.3, H-7), 2.15 – 1.90 (m, 2H, H-6), 1.85 – 0.98 (m, 13H), 0.96 (d, 3H, J=6.7, Me-16), 0.88 (s, 3H, Me-19) and 0.83 (s each, 2X3H, Me-18 and Me-20); 13C-NMR δ: 40.7 (C-1), 18.1 (C-2), 42.0 (C-3), 32.9 (C-4), 48.0 (C-5), 21.0 (C-6), 59.7 (C-7), 60.6 (C-8), 49.5 (C-9), 35.9 (C-10), 24.9 (C-11), 36.0 (C-12), 38.2 (C-13), 144.3 (C-14), 112.9 (C-15), 20.4 (C-16), 172.7 (C-17), 33.3 (C-18), 22.0 (C-19), 15.3 (C-20), 52.3 (COOMe); IR cm-1: 3074, 2926, 1733, 1649,1460, 1241.
Wacker oxidation of 18: Synthesis of methyl 7,8(α+β)-epoxy-14-oxolabdan-17-oate (19).
To a solution of PdCl2 (64 mg, 0.36 mmol) and CuCl (1.7 g, 1.8 mmol) in DMF (8 ml) and H2O (1 mL) was activated for 30 min with O2. A solution of 18 (550 mg, 1.8 mmol) in DMF (7 mL) was added and stirred with O2 atmosphere at room temperature for 36 h. Then the reaction mixture was added a cooled solution of 3N HCl, extracted with CH2Cl2 (3 x 30 mL) and washed successively with 10% NaHCO3 and water, dried, filtered, evaporated and chromatographed (eluting with 9:1 hexane/EtOAc) to give 440 mg (80%) of 19. A fraction collected during this CC was purified to give 4% of the α-isomer of 19, which was used for characterization purposes.
Compound 19α: 1H-NMR δ: 3.76 (s, 3H,COOMe), 3.23 (sb, 1H, H-7), 2.47 (sex, 1H, J=6.6, H-13), 2.11 (s, 3H, Me-15), 1.84 – 0.90 (m, 14H), 1.06 (d, 3H, J=6.6, Me-16), 0.86 (s each, 2X3H, Me-19 and Me-18) and 0.84 (s, 3H, Me-20); 13C-NMR δ: 38.5 (C-1), 18.6 (C-2), 41.9 (C-3), 33.0 (C-4), 46.0 (C-5), 21.9 (C-6), 57.8 (C-7), 59.0 (C-8), 54.4 (C-9), 35.1 (C-10), 21.9 (C-11), 32.3 (C-12), 47.4 (C-13), 212.1 (C-14), 28.0 (C-15), 16.4 (C-16), 170.8 (C-17), 32.6 (C-18), 21.9 (C-19), 14.5 (C-20), 52.3 (COOMe); m/z (EI+) 350 (M+, 10%), 318 (24), 300 (55), 285 (55), 261 (73), 257 (100), 233 (59), 229 (27), 219 (23), 201 (67), 177 (15), 163 (27), 151 (15), 141 (16), 123 (34), 109 (43), 95 (19), 79 (22), 69 (23), 55 (36); [α]D20 + 11.7 (c 0.5 in CHCl3); IR cm-1: 2926, 1731, 1718, 1461, 1282, 1158, 769; HRMS (CI) for C21H34O4 (MH+): Calcd 350.2457; Found 350.2468.
Reaction of 19 with m-CPBA: Synthesis of 11.
Compound 19 (300 mg, 0.86 mmol) was dissolved in anhydrous CH2Cl2 (10 mL) and m-CPBA (276 mg, 1.6 mmol) was added. The mixture was stirred at 30º C and monitored by TLC. After 5 h the reaction was complete and the solvent evaporated. Work-up afforded 290 mg of crude product that after chromatography on silica-gel gave in the 9:1 hexane/EtOAc fractions 264 mg (88%) of 11 as an epoxide mixture.
Reaction of 20: Synthesis of 17-acetoxy-15-o-nitrophenylseleno-7-labdene (21).
To a solution of 20 (450 mg, 1.3 mmol) in dry THF (3.0 mL) was added o-nitrophenylselenocyanate (363.3 mg, 1.6 mmol) and the mixture was stirred under an inert atmosphere for 20 min at 48º C. Then n-Bu3P (322.9 mg, 1.6 mmol) was added to the mixture, which ws stirred for 48 min, then the solvent was removed under reduced pressure and the residue chromatographed (95:5 hexane/EtOAc) yielding 382.5 mg (85%) of 21. 1H-NMR δ: 8.29 (d, 1H, J=8.2, H-3’), 7.55 – 7.26 (m, 3H, H-4’, H-5’ and H-6’), 4.57 (d, 1H, J=12.1, H-17a), 4.38 (d, 1H, J=12.1, H-17b), 2.85 (m, 2H, H-15), 2.03 (s, 3H, OOCMe), 2.00 – 0.95 (m, 17H), 0.97 (d, 3H, J=5.7, Me-16), 0.88 (s, 3H, Me-19), 0.85 (s, 3H, Me-18) and 0.75 (s, 3H, Me-20); 13C-NMR δ: 39.0 (C-1), 18.7 (C-2), 42.1 (C-3), 32.9 (C-4), 49.6 (C-5), 23.8 (C-6), 129.1 (C-7), 134.0 (C-8), 52.5 (C-9), 36.7 (C-10), 23.9 (C-11), 38.3 (C-12), 34.4 (C-13), 34.8 (C-14), 23.9 (C-15), 19.4 (C-16), 67.8 (C-17), 33.0 (C-18), 21.8 (C-19), 13.6 (C-20), 170.6 (MeCOO), 21.2 (Me-COO), 134.7 (C-1’), 146.7 (C-2’), 126.4 (C-3’), 129.1 (C-4’), 133.6 (C-5’), 125.2 (C-6’); IR cm-1: 2930, 1733, 1591, 1515, 1332, 1247, 737.
Oxidation of 21 with H2O2: Synthesis of 17-acetoxy-7,14-labdadiene (22).
To a solution of 21 (330 mg, 0.61 mmol) in THF (5 mL) was added 30% H2O2 (0.07 mL, 42 mg, 1.2 mmol) and this mixure was stirred for 12 h. The solvent was evaporated under reduced pressure and the crude product chromatographed (95:5 hexane/EtOAc) yielding 297 mg (90%) of 22. 1H-NMR δ: 5.71 (m, 1H, H-7), 5.58 (ddd, 1H, J=7.7, 10.3, 17.1, H-14), 4.89 (dd, 1H, J=2.1, 17.1, H-15a), 4.86 (dd, 1H, J=2.1, 17.1, H-15b), 4.50 (d, 1H, J=12.1, H-17a), 4.30 (d, 1H, J=12.1, H-17b), 1.99 (s, 3H, OOCMe), 1.95 – 0.80 (m, 15H), 0.92 (d, 3H, J=6.7, Me-16), 0.83 (s, 3H, Me-19), 0.80 (s, 3H, Me-18) and 0.69 (s, 3H, Me-20); 13C-NMR δ: 38.9 (C-1), 18.6 (C-2), 42.0 (C-3), 32.8 (C-4), 49.5 (C-5), 23.6 (C-6), 128.5 (C-7), 134.1 (C-8), 52.3 (C-9), 36.6 (C-10), 24.0 (C-11), 38.3 (C-12), 38.5 (C-13), 144.1 (C-14), 112.8 (C-15), 20.4 (C-16), 67.7 (C-17), 32.9 (C-18), 21.7 (C-19), 13.4 (C-20), 170.5 (MeCOO), 21.0 (Me-COO); [α]D20 - 3.9 (c 0.5 in CHCl3); IR cm-1: 3076, 1742, 1640, 1240, 911.
Wacker oxidation of 22: Synthesis of 23.
A solution of PdCl2 (64 mg, 0.34 mmol) and CuCl (1.6 g, 1.6 mmol) in DMF (8 mL) and H2O (1 mL) was activated for 30 min with O2. A solution of 22 (520 mg, 1.6 mmol) in DMF (7 mL) was then added and the mixture stirred with an O2 atmosphere at room temperature for 36 h, then it was added to a cooled solution of 3N HCl, extracted with CH2Cl2 (3 x 30 mL) and washed successively with 10% NaHCO3 and water, dried, filtered, evaporated and chromatographed (9:1 hexane/EtOAc) to give 416 mg (80%) of 23.
Treatment of 23 with m-CPBA: Synthesis of 13,17-diacetoxy-7,8-epoxy-14,15-dinor-labdane (24).
Compound 23 (300 mg, 0.86 mmol) was dissolved in anhydrous CH2Cl2 (10 mL) and m-CPBA (276 mg, 1.6 mmol) was added. The mixture was stirred at 40º C and monitored by TLC. After 4 h the reaction was complete and the solvent evaporated. Work-up afforded 290 mg of crude product that after chromatography on silica-gel giving 264 mg (88%) of 24 (as a mixture of epoxides) in the 9:1 hexane/EtOAc fractions. 1H-NMR δ: 4.95 – 4.71 (m, 1H, H-13), 4.20 (d, 1H, J=12.1, H-17a), 4.03 (d, 1H, J=12.1, H-17b), 3.22 (d, 1H, J=6.1, H-7 β-isomer), 3.14 (sb, 1H, H-7 α-isomer), 2.04 and 1.99 (s each, 2X3H, 2X-OOCMe), 1.85 – 0.75 (m, 14H), 1.19 (d, 3H, J=6.2, Me-16), 0.83 (s, 3H, Me-19), 0.82 (s, 3H, Me-18) and 0.72 (s, 3H, Me-20); 13C-NMR δ: 38.0 and 38.4 (C-1, isomers), 18.5 (C-2), 41.9 (C-3), 32.9 (C-4), 45.6 (C-5), 20.3 (C-6), 57.7 (C-7), 58.6 (C-8), 54.1 (C-9), 35.6 (C-10), 22.1 (C-11), 38.0 and 38.4 (C-12, isomers), 70.8 (C-13), 20.8 (C-16), 67.1 (C-17), 32.5 (C-18), 21.8 (C-19), 14.0 (C-20), 170.3 and 170.9 (2 x MeCOO), 21.2 (2 x Me-COO); IR cm-1: 2925, 1724, 1246, 1024.
Hydrolysis of 24: Synthesis of 13,17-dihydroxy-7,8-epoxy-14,15-dinor-labdane (25), 7α,13-dihydroxy-7,17-epoxy-14,15-dinor-labdane (26) and 7α,8β,13,17-tetrahydroxy-14,15-dinor-labdane (27).
Method A: To a solution of 24 (98.5 mg, 0.26 mmol) in methanol (4 mL) was added K2CO3 (120 mg). The reaction mixture was stirred at room temperature for 10 h, water was added and the mixture extracted with ether washed with 2N HCl and H2O, dried, filtered, evaporated and chromatographed yielding 59.1 mg of 25+26 (60%), and 29.6 mg of 27 (30%).
Method B: To a solution of 24 (110 mg, 0.29 mmol) was added a solution of 4% NaOH in methanol (6 mL). The reaction mixture was stirred at room temperature for 5 h, water was added and the mixture extracted with ether, washed with 2N HCl and H2O, dried, filtered, evaporated and chromatographed yielding 33 mg (30%) of 25, 16.5 mg (15%) of 26 and 36.3 mg (33%) of 27.
Compound 25: 1H-NMR δ: 3.83 (d, 1H, J= 12.2, H-17a), 3.70 (m, 1H, H-13), 3.65 (d, 1H, J=12.2 Hz, H-17b), 3.30 (m, 1H, H-7, α and β isomer), 1.90 – 0.90 (m, 14H), 1.42 (d, 3H, J=6.8, Me-16), 0.87 (s, 3H, Me-19) and 0.85 (s each, 2 x 3H, Me-18 and Me-20); 13C-NMR δ: 38.5 and 40.9 (C-1, isomers), 18.0 and 18.4 (C-2, isomers), 41.6 and 41.9 (C-3, isomers), 32.9 (C-4), 45.6 (C-5), 20.4 (C-6), 57.0 (C-7), 58.6 (C-8), 54.3 (C-9), 35.8 (C-10), 22.8 (C-11), 38.5 and 40.9 (C-12 isomer), 67.8 and 67.9 (C-13 isomers), 23.5 and 23.9 (C-16 isomers), 65.4 (C-17), 32.6 and 33.1 (C-18 isomers), 21.6 and 21.9 (C-19 isomers), 13.9 and 14.2 (C-20 isomers); IR cm-1: 3420, 2924, 1474, 1253, 1090, 1070.
Compound 26: 1H-NMR δ: 3.70 (m, 1H, H-13), 3.34 (t, 1H, J=2.9, H-7), 2.68 (d, 1H, J=4.3, H-17a), 2.44 (d, 1H, J=4.3, H-17b), 1.89 – 0.95 (m, 14H), 1.16 (d, 3H, J=6.7, Me-16), 0.87 (s, 3H, Me-19) and 0.85 (s each, 2X3H, Me-18 and Me-20); 13C-NMR δ: 38.5 and 40.9 (C-1 isomers), 18.0 and 18.4 (C-2 isomers), 41.6 and 41.9 (C-3 isomers), 32.9 (C-4), 46.8 (C-5), 27.7 (C-6), 73.5 (C-7), 61.2 (C-8), 46.9 (C-9), 35.8 (C-10), 22.8 (C-11), 38.5 and 40.9 (C-12 isomers), 67.8 and 67.9 (C-13 isomers), 23.5 and 23.9 (C-16 isomers), 48.6 (C-17), 32.6 and 33.1 (C-18 isomers), 21.6 and 21.9 (C-19 isomers),13.9 and 14.2 (C-20 isomers); IR cm-1: 3420, 2924, 1472, 1252, 1090, 1070.
Compound 27: 1H-NMR δ: 3.78 – 3.65 (m, 1H, H-13), 3.75 (d, 1H, J=12.0, H-17a), 3.53 (d, 1H, J=12.0, H-17b), 3.17 (sb, 1H, H-7), 1.90 – 1.00 (m, 14H), 1.15 (d, 3H, J=6.4, Me-16), 0.85 (s, 3H, Me-19), 0.83 (s, 3H, Me-19) and 0.80 (s, 3H, Me-20); 13C-NMR δ: 38.7 (C-1), 18.2 (C-2), 42.0 (C-3), 33.1 (C-4), 46.6 (C-5), 25.6 (C-6), 70.7 (C-7), 76.5 (C-8), 50.2 (C-9), 35.8 (C-10), 20.9 (C-11), 42.0 (C-12), 68.6 (C-13), 23.7 (C-16), 66.3 (C-17), 33.1 (C-18), 21.7 (C-19), 15.1 (C-20); IR cm-1: 3420, 1080.
Oxidation of 1a: Synthesis of 7-labden-17-methoxycarbonyl-15-oic acid (28).
CrO3 (620 mg, 6.2 mmol) was added to 90% acetic acid (10.0 mL). After stirring for 15 min, 1a (1.4 g, 4.17 mmol) in CH2Cl2 (10 mL) were added and stirring was continued at 40º C for 40 h. MeOH was added and stirred for 30 min, the solvent removed in vacuo. The reaction product was extracted with ether, the ether solution was washed with a solution of 4% NaOH and the pH of the aqueous solution was adjusted to a value of 2 with HCl and the acidic products were recovered by extraction with ether. The solution of the acidic products was dried with anhydrous Na2SO4. Solvent removal and chromatography on silica-gel gave 860 mg (60%) of 28 in the 6:4 hexane/EtOAc fractions. 1H-NMR δ: 9.70 (sb, 1H, COOH), 6.62 (m, 1H, H-7), 3.62 (s, 3H, COOMe), 2.42 – 1.70 (m, 4H), 1.68 – 0.90 (m, 13H), 0.94 (d, 3H, J=6.9, Me-16), 0.87 (s, 3H, Me-19), 0.84 (s, 3H, Me-18) and 0.79 (s, 3H, Me-20); 13C-NMR δ: 39.5 (C-1), 18.5 (C-2), 42.0 (C-3), 32.7 (C-4), 49.4 (C-5), 23.9 (C-6), 137.2 (C-7), 135.3 (C-8), 50.9 (C-9), 36.9 (C-10), 25.6 (C-11), 38.1 (C-12), 31.0 (C-13), 41.3 (C-14), 179.7 (C-15), 19.7 (C-16), 170.7 (C-17), 33.1 (C-18), 21.9 (C-19), 14.3 (C-20), 51.3 (COOMe); IR cm-1: 3500 – 2500, 2929, 1710, 1645, 1254.
Epoxidation of 28: Synthesis of 29.
To a solution of 28 (846 mg, 2.41 mmol) in dry CH2Cl2 (5 mL), m-CPBA (800 mg, 4.6 mmol) was added and the mixture stirred at 40º C. After 30 h, the solvent was removed and ether was added, the organic phase was washed with 40% Na2S2O3, 10% Na2CO3 and water to neutrality, dried over Na2SO4, filtered and evaporated to give 29 (508 mg, 60%).
Oxidation of 16: Synthesis of 7,8(α+β)epoxylabdan-17-methoxycarbonyl-15-oic acid (29).
Method A: CrO3 (235 mg, 2.3 mmol) was added to 90% acetic acid (5.0 mL). After stirring for 15 min, 16 (330 mg, 0.9 mmol) in CH2Cl2 (5 mL) and glacial acetic acid (2 mL) were added and stirring was continued at room temperature for 8 h. MeOH was added and the solvent removed in vacuo. Work-up afforded 320 mg of crude product that after chromatography on silica-gel gave 181.5 mg (55%) of 29 in the EtOAc fractions.
Method B: To a stirred solution of PDC (1.2 mg, 3.3 mmol) in DMF (12.0 mL) was added 16 (340 mg, 1.0 mmol). After 40 h at room temperature the solvent is evaporated, extracted with ether, washed with 7-10 vol of water and dried over Na2SO4. The residue was chromatographed on silica-gel affording 889 mg (74%) of 29 in the EtOAc fractions.
Compound 29: 1H-NMR δ: 3.70 (s, 3H, COOMe), 3.28 (d, 1H, J=6.3, H-7 β), 3.26 (sb, 1H, H-7α), 2.40 – 2.00 (m, 3H), 1.95 – 1.00 (m, 14H), 0.96 (d, 3H, J=6.7, Me-16), 0.88 (s each, 2X3H, Me-18 and Me-19) and 0.85 (s, 3H, Me-20); 13C-NMR δ: 38.6 (C-1), 18.6 (C-2), 42.0 (C-3), 33.0 (C-4), 46.0 (C-5), 21.6 (C-6), 57.8 (C-7), 59.0 (C-8), 54.5 (C-9), 35.1 (C-10), 21.9 (C-11), 36.0 (C-12), 30.6 (C-13), 41.2 (C-14), 178.4 (C-15), 19.8 (C-16), 170.8 (C-17), 32.6 (C-18), 21.8 (C-19), 14.5 (C-20), 52.1 (COOMe); IR cm-1: 3550 – 2500, 1730, 1700, 1089, 758; HRMS (CI) for C21H34O5 (MH+): Calcd 366.4917; Found 366.4928.
Reaction of 28: Synthesis of methyl 15-nor-7(α+β)-acetoxy-8-labden-17-oate (30) and methyl 15-nor-7,13-labdadien-17-oate (31).
To a solution of 28 (343 mg, 0.98 mmol) in dry C6H6 (10 mL) was added dry pyridine (0.1 mL) under a N2 atmosphere and the mixture was stirred at room temperature. After 15 min, dry (AcO)2Cu (60 mg, 0.33 mmol) was added and the reaction mixture was heated at 80º C. (AcO)4Pb (1.280 g, 2.9 mmol) was added in six portions over the next 6 h. The solvent was removed and extracted with ether. The ethereal solution of neutral products was washed with water and dried over Na2SO4, filtered and evaporated to give 273 mg of crude mixture that by CC afforded 103.2 mg of 30 and 21.8 mg of 31.
Compound 30: 1H-NMR δ: 5.69 (t, 1H, J=8.7, H-7β), 5.68 (m, 1H, H-7α), 3.66 (s, 2 x 3H, 2 x COOMe), 2.60 – 2.00 (m, 4H), 1.98 (s, 2 x 3H, 2 x OOCMe), 1.97 – 1.10 (m, 10H), 1.09 (s, 3H, Me-20, β), 0.97 (s, 3H, Me-20, α), 0.87 (d, 2 x 3H, J=6.9, Me-16), 0.84 (d, 2 x 3H, J=6.9, Me-14) and 0.83 (s, 4 x 3H, Me-18 and Me-19); IR cm-1: 2956, 1733, 1651, 1238.
Compound 31: 1H-NMR δ: 6.61 (m, 1H, H-7), 4.63 (s, 2H, H-14), 3.69 (s, 3H, COOMe), 2.40 – 1.71 (m, 6H), 1.68 (s, 3H, Me-16), 1.62 – 0.98 (m, 8H), 0.88 (s, 3H, Me-19), 0.85 (s, 3H, Me-18) and 0.81 (s, 3H, Me-20); 13C-NMR (CDCl3) δ: 39.3 (C-1), 18.5 (C-2), 42.0 (C-3), 32.8 (C-4), 49.4 (C-5), 23.8 (C-6), 136.9 (C-7), 135.4 (C-8), 50.7 (C-9), 36.9 (C-10), 26.8 (C-11), 39.4 (C-12), 146.8 (C-13), 109.3 (C-14), 22.5 (C-16), 169.8 (C-17), 33.1 (C-18), 21.9 (C-19), 14.3 (C-20), 51.3 (COOMe); [α]D20 - 19.0 (c 0.2 in CHCl3); IR cm-1: 3079, 2960, 1720, 1652, 798.
Reaction of 31 with O3 and Norrish type II reaction: Synthesis of 5.
A solution of 31 (100 mg, 0.3 mmol), in CH2Cl2 (4 mL) was cooled to –78º C with acetone/Dry Ice. Ozone (about 5.2 g of O3/h) was bubbled through this solution for 1.5 min. To the cooled reaction mixture Ph3P (164.5 mg, 0.6 mmol) in CH2Cl2 (3 mL) was added and then it was gradually allowed to reach room temperature. The solvent was removed under reduced pressure and the residue chromatographed on silica-gel to give 75 mg (75%) of product. A solution of the reaction product (75 mg, 0.24 mmol) in dry hexane (250 mL) was placed in a quartz flask and a stream of dry N2 was bubbled through it. The solution was irradiated with UV light (Hanau TQ-150, high pressure) for 2 h. Removal of solvent afforded a yellow oil which was purified by chromatography on silica-gel eluting with 95:5 hexane/EtOAc to yield 37.5 mg (50%) of 5 and 34.2 mg (45%) of the starting material.
Hydrolysis of 30: Synthesis of methyl 15-nor-7β-hydroxy-8-labden-17-oate (32) and methyl 15-nor-7α-hydroxy-8-labden-17-oate (33).
To a solution of 30 (41.4 mg, 0.12 mmol) in methanol (4 mL) was added K2CO3 (60 mg). The reaction mixture was stirred at room temperature for 10 h, water was added and the mixture extracted with ether, washed with 2N HCl and H2O, dried, filtered, evaporated and chromatographed yielding 15.5 mg of 32 and 10.4 mg of 33.
Compound 32: 1H-NMR δ: 4.68 (t, 1H, J=8.7, H-7), 3.76 (s, 3H, COOMe), 2.62 (m, 1H), 2.20 – 1.10 (m, 14H), 1.10 (s, 3H, Me-20), 0.90 (s, 3H, Me-19), 0.89 (d, 3H, J=6.8, Me-16), 0.88 (d, 3H, J=6.8, Me-14) and 0.86 (s, 3H, Me-18); 13C-NMR δ: 40.1 (C-1), 18.7 (C-2), 41.4 (C-3), 33.0 (C-4), 49.5 (C-5), 28.4 (C-6), 69.7 (C-7), 128.6 (C-8), 158.5 (C-9), 40.6 (C-10), 26.6 (C-11), 35.9 (C-12), 29.1 (C-13), 22.3 (C-14), 22.3 (C-16), 170.4 (C-17), 33.0 (C-18), 21.7 (C-19), 20.1 (C-20), 51.3 (COOMe); [α]D20 + 38.4 (c 0.2 in CHCl3); IR cm-1: 3400, 2940, 1720, 1630, 1460, 1250, 1060; MS m/z (EI+) 322 (M+, 3%), 304 (17), 290 (4), 273 (4), 263 (17), 248 (19), 230 (7), 219 (20), 201 (19), 191 (10), 177 (14), 173 (13), 166 (30), 163 (63), 159 (12), 151 (19), 149 (13), 147 (11), 145 (16), 142 (32), 139 (15), 133 (19), 131 (17), 129 (11), 123 (26), 119 (37), 117 (17), 109 (60), 105 (40), 91 (40), 83 (21), 81 (27), 79 (27), 77 (22), 59 (20), 55 (70), 43 (79), 41 (100); HRMS (CI) for C20H34O3 (MH+): Calcd 322.2508; Found 322.2520.
Compound 33: 1H-NMR δ: 4.44 (m, 1H, H-7), 3.76 (s, 3H, COOMe), 2.61 – 2.41 (m, 2H), 1.99 – 1.10 (m, 11H), 0.98 (s, 3H, Me-20), 0.96 (d, 3H, J=6.8, Me-16), 0.88 (d, 3H, J=6.8, Me-14) and 0.86 (s, 2X3H, Me-18 and Me-19); 13C-NMR δ: 40.1 (C-1), 18.9 (C-2), 41.4 (C-3), 33.0 (C-4), 44.9 (C-5), 27.1 (C-6), 65.8 (C-7), 126.3 (C-8), 161.7 (C-9), 41.3 (C-10), 27.2 (C-11), 35.6 (C-12), 29.2 (C-13), 22.3 (C-14), 22.4 (C-16), 170.6 (C-17), 33.1 (C-18), 21.8 (C-19), 18.5 (C-20), 51.4 (COOMe); [α]D20 + 41.4 (c 0.3 in CHCl3); IR cm-1: 3450, 2950, 1715, 1620, 1460, 1230, 1100; MS m/z (EI+) 322 (M+, 1%), 304 (17), 263 (17), 248 (16), 234 (17), 233 (100), 219 (25), 201 (28), 191 (15), 177 (16), 167 (10), 164 (18), 163 (98), 159 (15), 149 (11), 147 (11), 145 (18), 133 (20), 121 (19), 119 (48), 117 (17), 105 (40), 95 (23), 91 (46), 83 (24), 81 (22), 79 (25), 77 (23), 69 (50), 67 (22), 55 (55), 43 (73), 41 (92).
Reaction of 29: Synthesis of methyl 7-oxo-15-nor-8-labden-17-oate (34) and methyl 7β,8β-epoxy-15-nor-13-labden-17-oate (35β).
To a solution of 29 (250 mg, 0.68 mmol) in dry C6H6 (10 mL) was added dry pyridine (0.1 mL) under a N2 atmosphere and the mixture was stirred at room temperature. After 15 min, dry (AcO)2Cu (50 mg, 0.27 mmol) was added and the reaction mixture is heated to 80º C. Over the next 6 h (AcO)4Pb (774 mg, 1.74 mmol) was added in six portions. The solvent was removed and the residue extracted with ether. The ethereal solution was washed with a solution of 4% NaOH for extraction of the unreacted acid 29 (44.2 mg). The solution of neutral products was washed with water and dried over Na2SO4, filtered and evaporated to give 195 mg of a mixture that by CC afforded 155 mg (62%) of 34 and 30 mg (12%) of 35.
Compound 34: 1H-NMR δ: 3.76 (s, 3H, COOMe), 2.60 – 2.00 (m, 4H), 1.90 – 1.12 (m, 13H), 1.12 (s, 3H, Me-20), 0.87 (d, 3H, J=7.2, Me-16), 0.86 (s each, 2X3H, Me-18 and Me-19) and 0.85 (d, 3H, J=7.2, Me-14); 13C-NMR δ: 38.9 (C-1), 18.3 (C-2), 41.0 (C-3), 33.2 (C-4), 49.8 (C-5), 34.9 (C-6), 196.4 (C-7), 132.1 (C-8), 167.9 (C-9), 40.5 (C-10), 28.1 (C-11), 34.9 (C-12), 29.0 (C-13), 22.0 (C-14), 22.1 (C-16), 172.7 (C-17), 32.4 (C-18), 21.3 (C-19), 18.3 (C-20), 52.1 (COOMe); [α]D20 + 22.3 (c 0.4 in CHCl3); IR cm-1: 2956, 1750, 1667, 1583, 1462, 1348, 1242, 1144, 1095, 1022, 801; MS m/z (EI+) 320 (M+, 0.8%), 288 (25), 273 (14), 245 (100), 217 (41), 189 (27), 175 (29), 161 (25), 149 (32), 135 (23), 121 (22), 109 (39), 91 (31), 79 (22), 77 (17); HRMS (CI) for C20H32O3 (MH+): Calcd 320.2351; Found 320.2363.
Compound 35β: 1H-NMR δ: 4.65 (s, 1H, H-14a), 4.63 (s, 1H, H-14b), 3.70 (s, 3H, COOMe), 3.28 (d, 1H, J=6.4, H-7), 2.22 – 0.99 (m, 14H), 1.67 (s, 3H, Me-16), 0.86 (s, 3H, Me-19), 0.83 (s, 3H, Me-19) and 0.81 (s, 3H, Me-20); 13C-NMR δ: 40.5 (C-1), 18.1 (C-2), 42.0 (C-3), 32.9 (C-4), 47.9 (C-5), 25.8 (C-6), 59.7 (C-7), 60.6 (C-8), 49.4 (C-9), 35.9 (C-10), 21.0 (C-11), 37.3 (C-12), 146.0 (C-13), 109.8 (C-14), 22.5 (C-16), 172.5 (C-17), 33.2 (C-18), 22.0 (C-19), 15.3 (C-20), 52.3 (COOMe); IR cm-1: 3070, 2931, 1740, 1650, 1454, 1381, 1283, 1161, 1054, 891, 769; MS m/z (EI+) 320 (M+, 0.8%), 281 (8), 243 (7), 234 (9), 219 (49), 207 (23), 193 (20), 135 (26), 123 (44), 109 (100), 95 (65), 93 (43), 81 (83), 79 (45); HRMS (CI) for C20H32O3 (MH+): Calcd 320.2351; Found 320.2362.
Reaction of 35 with O3: Synthesis of 13.
A solution of 35 (220 mg, 0.69 mmol) in CH2Cl2 (6 mL), was cooled to –78º C with acetone/dry ice. Ozone (about 5.2 g of O3/h) was bubbled through this solution for 8 min. To the cooled reaction mixture Ph3P (230 mg, 0.90 mmol) in CH2Cl2 (6 mL) was added and the mixture was gradually allowed to reach room temperature. The solvent was then removed under reduced pressure and the residue chromatographed on silica-gel affording 16.7 mg (7.6%) of 35 and 158.4 mg (72%) of 13.
Reaction of 1a with dimethyldioxirane: Synthesis of 7α,8α-epoxylabdan-17-methoxycarbonyl-15-oic acid (29α).
To a solution of 1a (196 mg, 0.58 mmol) in acetone (5 mL) was added dimethyldioxirane (8 mL, 0.1 M). The reaction is carried out at room temperature. After 20 min, the solvent was removed and the residue chromatographed on silica-gel (elution with 7:3 hexane/EtOAc) affording 88.2 mg (45%) of 29α. 1H-NMR δ: 3.72 (s, 3H, COOMe), 3.22 (sb, 1H, H-7), 2.40 – 1.00 (m, 17H), 0.96 (d, 3H, J=6.7, Me-16), 0.89 (s, 3H, Me-19), 0.88 (s, 3H, Me-18) and 0.85 (s, 3H, Me-20); 13C-NMR δ: 38.6 (C-1), 18.6 (C-2), 42.0 (C-3), 33.0 (C-4), 46.0 (C-5), 21.6 (C-6), 57.8 (C-7), 59.0 (C-8), 54.5 (C-9), 35.1 (C-10), 21.9 (C-11), 36.0 (C-12), 30.6 (C-13), 41.2 (C-14), 178.4 (C-15), 19.8 (C-16), 170.8 (C-17), 32.6 (C-18), 21.8 (C-19), 14.5 (C-20), 52.1 (COOMe); IR cm-1: 3380 – 2600, 1736, 1712, 1437, 1279, 1161, 1054, 755.
Reaction of 29α: Synthesis of 34 and methyl 7α,8α-epoxy-15-nor-13-labden-17-oate (35α).
To a solution of 29α (250 mg, 0.68 mmol) in dry C6H6 (10 mL) was added dry pyridine (0.1 mL) under a N2 atmosphere and the mixture was stirred at room temperature. After 15 min, dry (AcO)2Cu (42 mg, 0.23 mmol) was added and the reaction mixture was heated to 80º C. Over 6 h additional (AcO)4Pb (882.8 mg, 2.0 mmol) was added in six portions. The solvent was removed and the residue extracted with ether. The ethereal solution was washed with a solution of 4% NaOH to extract the unreacted acid 29α (25 mg). The solution of neutral products was washed with water and dried over Na2SO4, filtered and evaporated to give 220 mg of a mixture that after CC afforded 149 mg (60%) of 34 and 30.1 mg (12%) of 35α. 1H-NMR δ: 4.71 (s, 1H, H-14a), 4.68 (s, 1H, H-14b), 3.73 (s, 3H, COOMe), 3.23 (sb, 1H, H-7), 2.20 – 1.00 (m, 14H), 1.63 (s, 3H, Me-16), 0.89 (s, 3H, Me-19), 0.87 (s, 3H, Me-18) and 0.85 (s, 3H, Me-20); 13C-NMR δ: 38.5 (C-1), 18.6 (C-2), 42.0 (C-3), 33.0 (C-4), 46.0 (C-5), 22.0 (C-6), 57.8 (C-7), 59.0 (C-8), 53.3 (C-9), 35.3 (C-10), 22.3 (C-11), 36.9 (C-12), 145.2 (C-13), 110.7 (C-14), 22.2 (C-16), 171.0 (C-17), 32.6 (C-18), 21.9 (C-19), 14.6 (C-20), 52.2 (COOMe); IR cm-1: 3073, 2927, 1733, 1649, 1437, 1277, 1053, 886, 768.
Reaction of 35α with O3: Synthesis of 13α.
A solution of 35α (100 mg, 0.31 mmol) in CH2Cl2 (5 mL), was cooled to –78º C. Ozone (about 5.2 g of O3/h) was bubbled through this solution for 8 min. To the cooled reaction mixture Ph3P (158.4 mg, 0.62 mmol) in CH2Cl2 (6 mL) was added and the mixture was gradually allowed to reach room temperature. The solvent was then removed under reduced pressure and the residue chromatographed on silica-gel affording 6.0 mg (6%) of 35α and 72.0 mg (72%) of 13α.
Norrish type II reaction of 13α: Synthesis of 14.
A solution of 13α (100 mg, 0.31 mmol) in dry hexane (250 mL) was placed in a quartz flask and a stream of dry N2 was bubbled through. The solution was irradiated with UV light (Hanau TQ-150, high pressure) for 90 min. Removal of the solvent afforded a yellow oil which was purified by chromatography on silica-gel eluting with 98:2 hexane/EtOAc, to yield 28.5 mg (28%) of 14 and 40.0 mg (40%) of the starting material 13α.
Acknowledgments
The authors are grateful to A. Lithgow, Servicio General de Resonancia Magnética Nuclear, Facultad de Ciencias Químicas, Universidad de Salamanca for the NMR spectra.
References
- Ware, W. G. “Pesticides, Theory and Application”; W. H. Freeman and Company: San Francisco, 1983. [Google Scholar]
- Van Beek, T. A.; De Groot, A. Recl. Trav. Chim. Pays Bas 1986, 105, 513, and references cited therein.
- Knusli, E. “Industrial aspects of the practical use of natural products or derivatives in the protection of crops”. In “Natural Products and the Protection of Plants”; Marini-Bettolo, G. B., Ed.; Pontificia Accademia Scientiarium Scripta Varia, 1977; Volume 41, p. 755. [Google Scholar]
- Butterworth, J. H.; Morgan, E. D. J. Chem. Soc., Chem. Commum. 1968, 23.
- Anderson, J. C.; Blaney, W. M.; Cole, M. D.; Fellons, L. L.; Ley, S. V.; Shephard, R. N.; Simmonds, M. S. J. Tetrahedron Lett. 1989, 30, 4737.
- Kubo, I.; Lee, Y. W.; Pettei, M. J.; Pilkiewicz, F.; Nakanishi, K. J. J. Chem. Soc., Chem. Commum. 1976, 1013.
- Barnes, C. S.; Loder, J. W. Aust. J. Chem. 1962, 15, 32.
- Nakanishi, K.; Rube, I. Israel J. Chemistry 1977, 16, 28.
- Singhal, S.; Mathur, S. C. Chem. Ind. 1993, 112.
- Urones, J. G.; Marcos, I. S.; Pérez, B. G.; Díez, D.; Lithgow, A. M.; Gómez, P. M.; Basabe, P.; Garrido, N. M. Tetrahedron 1994, 50, 10995.
- Urones, J. G.; Marcos, I. S.; Martín, D. D.; Brito Palma, F. M.; Rodilla, J. M. Phytochemistry 1987, 26, 3037.
- Urones, J. G.; Marcos, I. S.; Martín, D. D. Tetrahedron 1988, 44, 112.
- Grieco, P. A.; Jaw, J. Y. J. Org. Chem. 1981, 46, 1215.
- Mizuno, M.; Cava, M. P.; Gabrito, A. F. J. Org. Chem. 1976, 41, 1485. [CrossRef]
- Tsuji, J.; Shimizu, I.; Yamamoto, K. Tetrahedron Lett. 1976, 34, 2975.
- Lamers, Y. M.; Rusu, G.; Wijnberg, J. B.; de Groot, A. Tetrahedron 2003, 59, 9361. [CrossRef]
- Kubo, I.; Mura, I.; Pettei, M. J.; Lee, Y. W.; Pikiewicz, F.; Nakanishi, K. Tetrahedron Lett. 1997, 52, 4553.
- Nakamo, T.; Maillo, M. A. Synth. Commun. 1981, 11, 463.
- Urones, J. G.; Marcos, I. S.; Gómez Pérez, B.; Lithgow, A. M.; Díez, D.; Gómez, P. M.; Basabe, P.; Garrido, N. M. Tetrahedron 1995, 51, 1845.
- Payne, J. J. Org. Chem. 1962, 27, 3819. [CrossRef]
- Corey, E. J.; Schmidt, G. Tetrahedron Lett. 1979, 37, 399.
- Bacha, J. D.; Cochi, J. K. Tetrahedron 1968, 24, 2215.
- Adam, W.; Chan, Y. Y.; Cremer, D.; Gauss, J.; Scheutzow, D.; Schindler, M. J. Org. Chem. 1987, 52, 2800. [CrossRef]
- Urones, J. G.; Marcos, I. S.; Pérez, B. G.; Lithgow, A. M.; Díez, D.; Basabe, P.; Gómez, P. M. Tetrahedron Lett. 1994, 35, 3781.
- Sample availability: Small amounts (mgs) of the final compounds are available from the authors.
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Rodilla, J.M.L.; Díez, D.; Urones, J.G.; Rocha, P.M. From Labdanes to Drimanes. Degradation of the Side Chain of Dihydrozamoranic Acid. Molecules 2004, 9, 300-322. https://doi.org/10.3390/90500300
AMA Style
Rodilla JML, Díez D, Urones JG, Rocha PM. From Labdanes to Drimanes. Degradation of the Side Chain of Dihydrozamoranic Acid. Molecules. 2004; 9(5):300-322. https://doi.org/10.3390/90500300
Chicago/Turabian StyleRodilla, Jesús M.L., D. Díez, J. G. Urones, and Pedro M. Rocha. 2004. "From Labdanes to Drimanes. Degradation of the Side Chain of Dihydrozamoranic Acid." Molecules 9, no. 5: 300-322. https://doi.org/10.3390/90500300