Structure-Acidity-IR Spectra Correlations for p-Substituted N-Phenylsulfonylbenzamides†

Abstract
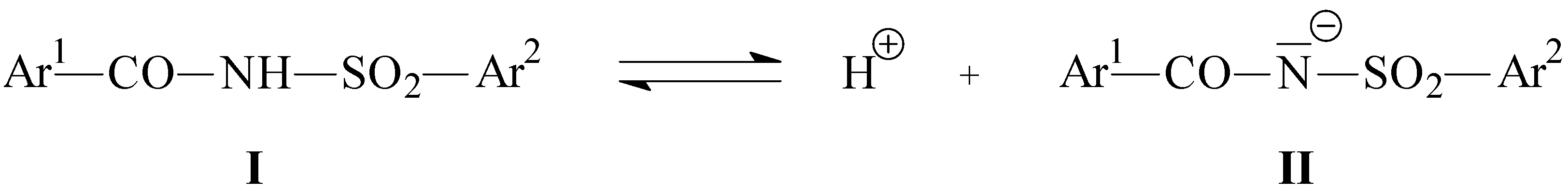
:Introduction
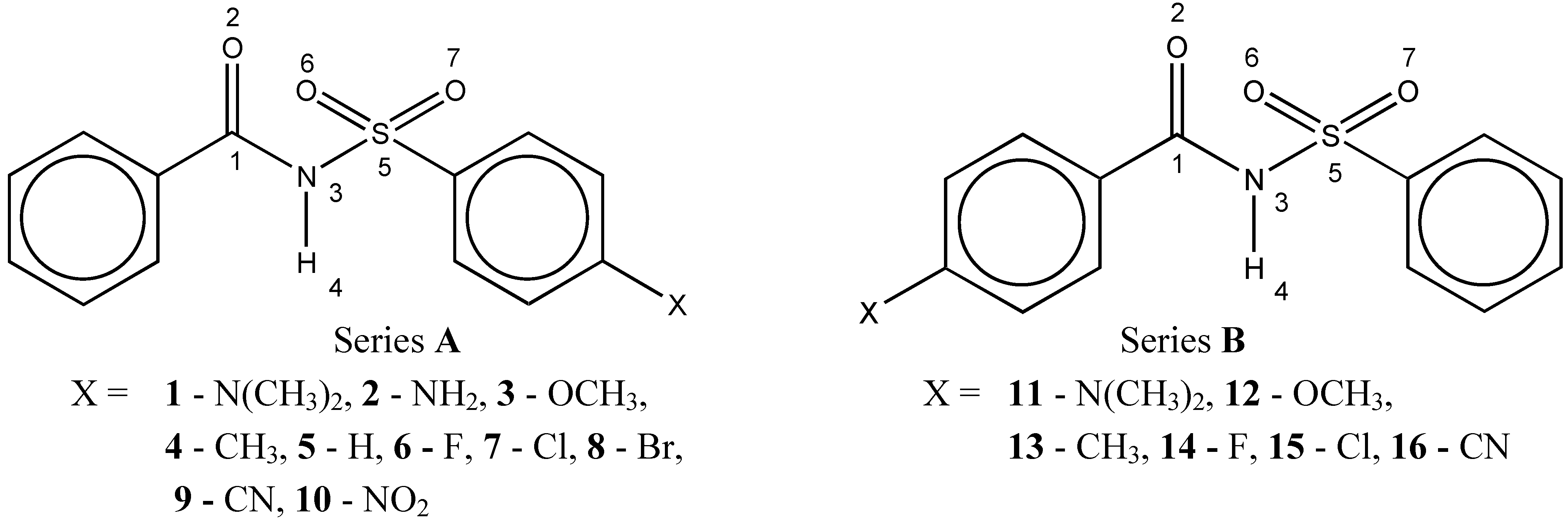
Results and Discussion

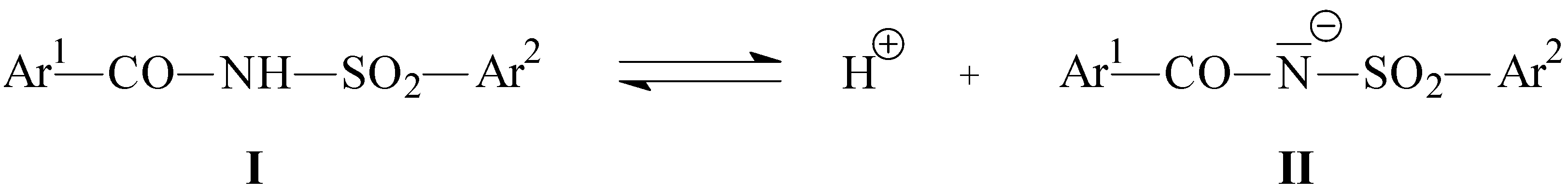
{kind=link}
{kind=link}
{kind=link}
| Compound | / cm-1 | |||
| νs(SO2) | νas(SO2) | ν (C=O) | ν (N-H) | |
| 1 | 1156.4 | 1344.4 | 1697.2 | -a |
| 2 | 1161.2 | 1346.0 | 1699.2 | -a |
| 3 | 1162.4 | 1350.4 | 1700.0 | 3278.4 |
| 4 | 1168.0 | 1350.8 | 1701.6 | 3280.0 |
| 5 | 1171.2 | 1351.5 | 1702.4 | 3279.2 |
| 6 | 1171.6 | 1353.6 | 1702.4 | 3281.6 |
| 7 | 1169.2 | 1354.0 | 1702.4 | 3273.6 |
| 8 | 1170.8 | 1354.4 | 1702.4 | 3274.0 |
| 9 | 1172.0 | 1357.6 | 1704.4 | -a |
| 10 | 1173.2 | 1357.8 | 1704.8 | -a |
| 11 | 1165.2 | 1345.6 | 1686.4 | 3288.2 |
| 12 | 1168.0 | 1347.6 | 1698.8 | 3281.6 |
| 13 | 1170.4 | 1348.8 | 1702.4 | 3280.0 |
| 14 | 1171.2 | 1349.6 | 1705.6 | 3276.8 |
| 15 | 1171.2 | 1350.4 | 1706.0 | 3273.5 |
| 16 | 1173.0 | 1352.8 | 1710.4 | 3267.2 |
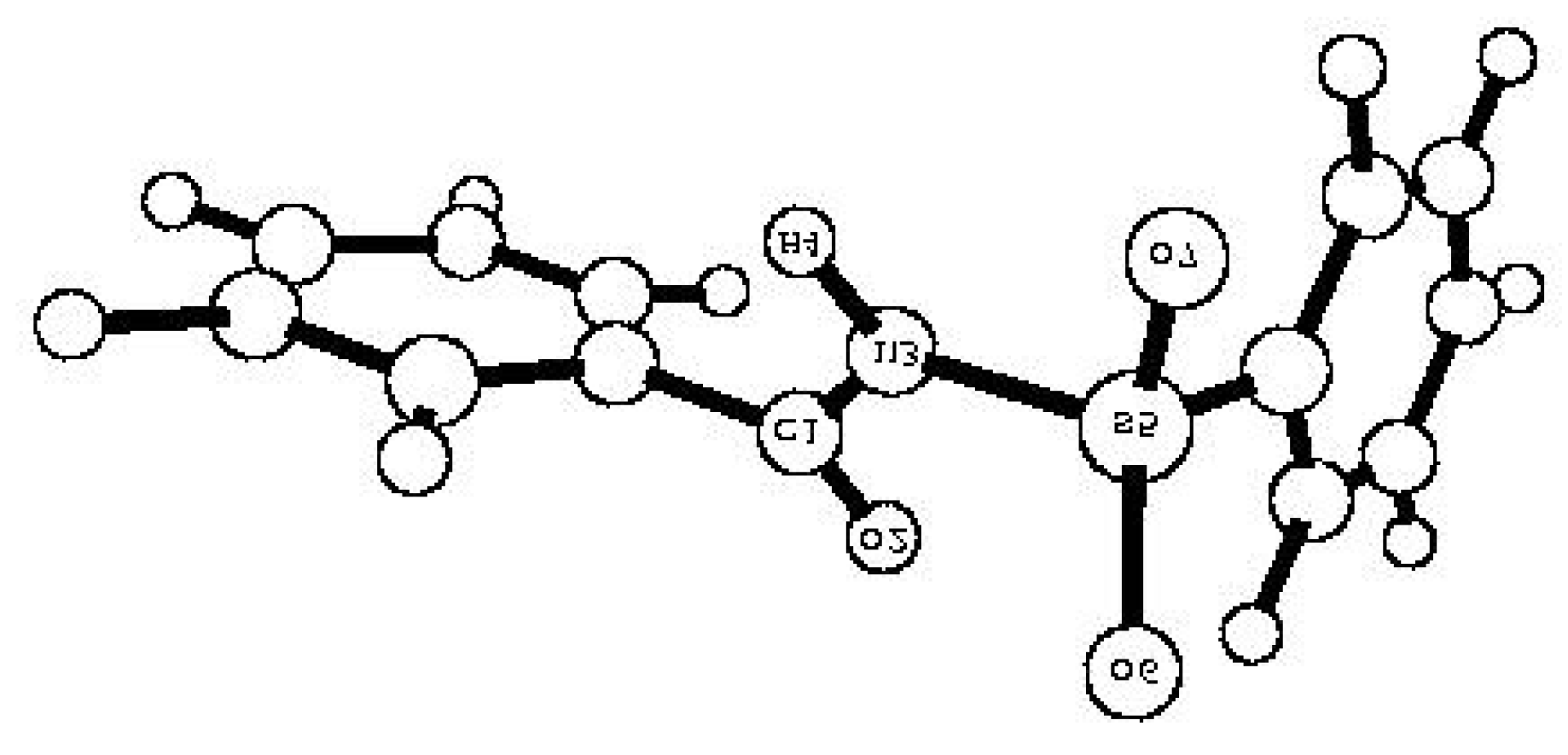
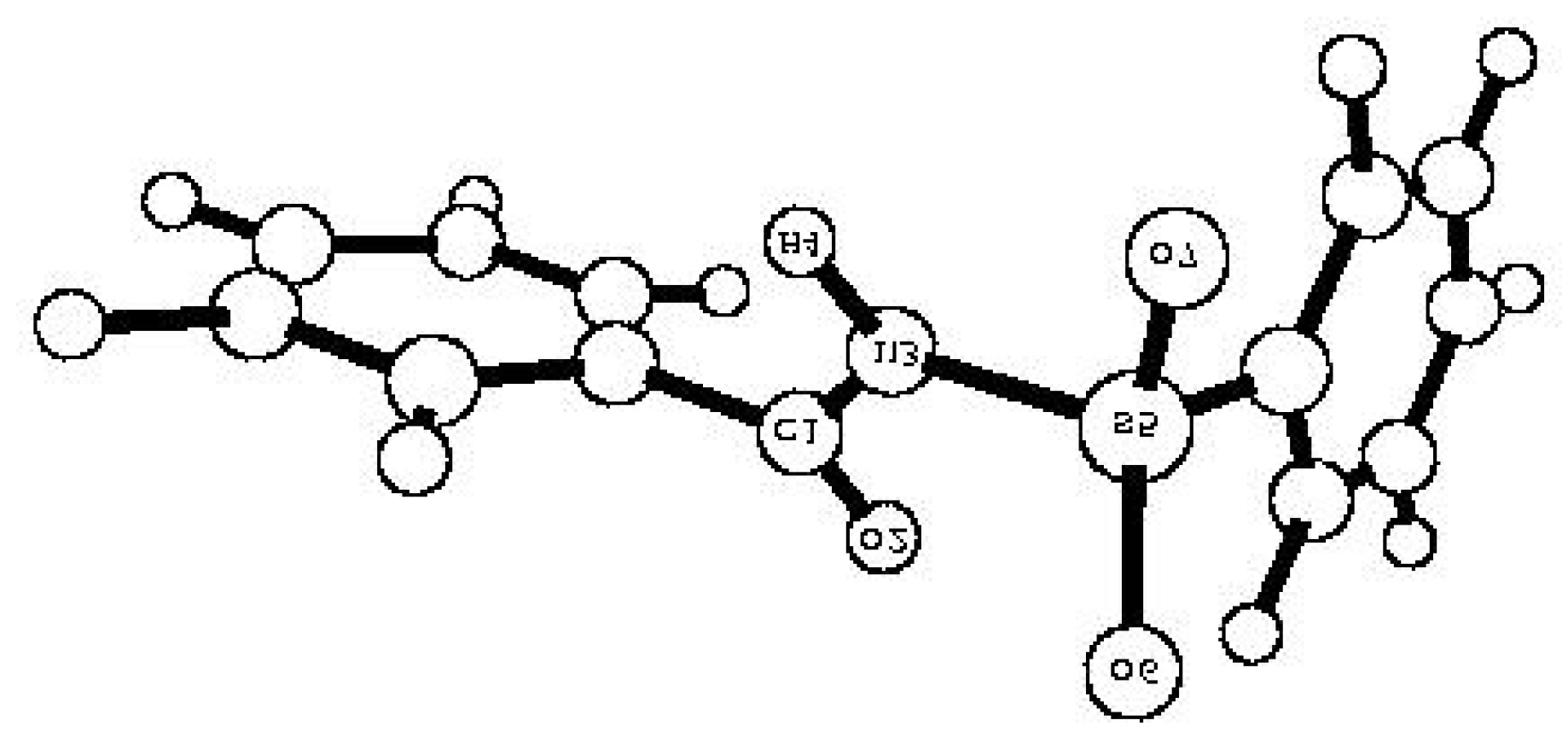
| Comp. | qM(C1) | -qM(O2) | -qM(N3) | qM(H4) | qM(S5) | -qM(O6) | -qM(O7) | p(S5=O6) | p(S5=O7) | p(N3-H4) | p(C1=O2) |
| 1 | 0.421 | 0.385 | 0.635 | 0.197 | 2.400 | 0.819 | 0.841 | 1.2023 | 1.2298 | 0.9311 | 1.8024 |
| 2 | 0.421 | 0.385 | 0.636 | 0.197 | 2.400 | 0.818 | 0.841 | 1.2025 | 1.2303 | 0.9309 | 1.8020 |
| 3 | 0.422 | 0.387 | 0.637 | 0.198 | 2.402 | 0.819 | 0.839 | 1.2051 | 1.2307 | 0.9308 | 1.8009 |
| 4 | 0.421 | 0.385 | 0.638 | 0.198 | 2.397 | 0.817 | 0.839 | 1.2049 | 1.2320 | 0.9307 | 1.8035 |
| 5 | 0.422 | 0.384 | 0.639 | 0.199 | 2.396 | 0.817 | 0.838 | 1.2057 | 1.2325 | 0.9306 | 1.8043 |
| 6 | 0.423 | 0.386 | 0.640 | 0.200 | 2.402 | 0.815 | 0.837 | 1.2077 | 1.2348 | 0.9302 | 1.8037 |
| 7 | 0.423 | 0.385 | 0.640 | 0.199 | 2.399 | 0.815 | 0.837 | 1.2061 | 1.2342 | 0.9307 | 1.8039 |
| 8 | 0.422 | 0.385 | 0.639 | 0.199 | 2.399 | 0.815 | 0.837 | 1.2074 | 1.2345 | 0.9308 | 1.8045 |
| 9 | 0.425 | 0.384 | 0.644 | 0.202 | 2.402 | 0.813 | 0.835 | 1.2103 | 1.2370 | 0.9298 | 1.8067 |
| 10 | 0.427 | 0.383 | 0.648 | 0.204 | 2.403 | 0.811 | 0.832 | 1.214 | 1.2405 | 0.9293 | 1.8093 |
| 11 | 0.433 | 0.390 | 0.641 | 0.199 | 2.394 | 0.817 | 0.840 | 1.2038 | 1.2318 | 0.9345 | 1.7997 |
| 12 | 0.433 | 0.389 | 0.640 | 0.198 | 2.396 | 0.817 | 0.8390 | 1.2050 | 1.2318 | 0.9304 | 1.8000 |
| 13 | 0.425 | 0.386 | 0.641 | 0.199 | 2.397 | 0.816 | 0.839 | 1.2054 | 1.2327 | 0.9304 | 1.8037 |
| 14 | 0.423 | 0.383 | 0.637 | 0.197 | 2.399 | 0.815 | 0.837 | 1.2075 | 1.2346 | 0.9309 | 1.8036 |
| 15 | 0.421 | 0.383 | 0.637 | 0.198 | 2.398 | 0.816 | 0.837 | 1.2069 | 1.2341 | 0.9307 | 1.8037 |
| 16 | 0.416 | 0.377 | 0.636 | 0.197 | 2.402 | 0.815 | 0.836 | 1.2096 | 1.2359 | 0.9313 | 1.8070 |
| Compound | -ΔHf (I)a/kJ.mol-1 | -ΔHf (II)b/kJ.mol-1 |
| 1 | 243.61 | 424.92 |
| 2 | 237.80 | 421.18 |
| 3 | 383.98 | 572.48 |
| 4 | 263.21 | 449.88 |
| 5 | 222.47 | 411.31 |
| 6 | 403.44 | 604.03 |
| 7 | 248.79 | 446.70 |
| 8 | 186.55 | 388.34 |
| 9 | 68.93 | 282.88 |
| 10 | 245.96 | 476.03 |
| 11 | 200.01 | 386.47 |
| 12 | 381.25 | 574.27 |
| 13 | 261.86 | 449.48 |
| 14 | 401.22 | 602.18 |
| 15 | 248.10 | 445.87 |
| 16 | 70.72 | 283.07 |
| No.b | y | x | Series | nc | rd | se | Ff | ρ | q |
| 1 | ν(C=O) | σ+ | A | 10 | 0.992 | 0.31 | 493 | 2.86±0.13 | 1702.4 |
| 2 | B | 7 | 0.979 | 1.71 | 116 | 9.94±0.92 | 1704.7 | ||
| 3 | ν(SO2) | σ+ | A | 9g | 0.995 | 0.52 | 742 | 5.89±0.22 | 1261.0 |
| 4 | B | 7 | 0.991 | 0.35 | 287 | 3.24±0.19 | 1260.7 | ||
| 5 | ν(N-H) | σ | A | poor | correlation | ||||
| 6 | B | 7 | 0.989 | 1.05 | 231 | -14.19±0.93 | 3277.4 | ||
| 7 | ν(C=O) | p(C=O) | A | 8h | 0.967 | 0.42 | 85 | 590.45±64.00 | 637.0 |
| 8 | B | 6i | 0.883 | 2.09 | 14 | 1578.91±418.81 | -1143.6 | ||
| 9 | ν(SO2) | p(S=O) | A + B | 15g | 0.887 | 1.99 | 48 | 1253.49±180.70 | -269.7 |
| 10 | ν(N-H) | p(N-H) | A + B | 11j | 0.841 | 2.53 | 22 | -12558.56±2697.25 | 14965.1 |
| 11 | ν(C=O) | pKHA(MeOH) | A | 10 | 0.964 | 0.65 | 105 | -2.82±0.27 | 1724.8 |
| 12 | B | 6k | 0.966 | 2.12 | 55 | -12.72±1.71 | 1807.6 | ||
| 13 | ν(C=O) | pKHA(CH3CN) | A | 10 | 0.962 | 0.67 | 99 | -1.95±0.20 | 1735.7 |
| 14 | B | 6k | 0.968 | 2.03 | 60 | -8.62±1.11 | 1853.8 | ||
| 15 | ν(C=O) | pKHA(DMF) | A | 10 | 0.950 | 0.77 | 74 | -2.04±0.24 | 1718.5 |
| 16 | B | 6k | 0.970 | 1.98 | 64 | -9.85±1.23 | 1784.4 | ||
| 17 | ν(C=O) | pKHA(DMSO) | A | 10 | 0.923 | 0.95 | 46 | -1.42±0.21 | 1710.7 |
| 18 | B | 6k | 0.941 | 2.75 | 31 | -8.22±1.47 | 1755.6 | ||
| 19 | ν(C=O) | pKHA(Py) | A | 10 | 0.931 | 0.90 | 52 | -2.09±0.29 | 1711.9 |
| 20 | B | 6k | 0.956 | 2.38 | 43 | -9.13±1.40 | 1746.7 | ||
| 21 | ν(SO2) | pKHA(MeOH) | A | 10 | 0.962 | 1.43 | 99 | -5.97±0.60 | 1308.8 |
| 22 | B | 7 | 0.962 | 0.74 | 61 | -3.66±0.47 | 1290.1 | ||
| 23 | ν(SO2) | pKHA(CH3CN) | A | 10 | 0.958 | 1.49 | 90 | -4.12±0.43 | 1331.8 |
| 24 | B | 7 | 0.941 | 0.91 | 39 | -2.41±0.39 | 1302.2 | ||
| 25 | ν(SO2) | pKHA(DMF) | A | 10 | 0.954 | 1.56 | 82 | -4.35±0.48 | 1295.8 |
| 26 | B | 7 | 0.944 | 0.90 | 41 | -2.76±0.43 | 1282.8 | ||
| 27 | ν(SO2) | pKHA(DMSO) | A | 10 | 0.922 | 2.02 | 45 | -3.01±0.45 | 1278.9 |
| 28 | B | 7 | 0.956 | 0.80 | 53 | -2.40±0.33 | 1275.3 | ||
| 29 | ν(SO2) | pKHA(Py) | A | 10 | 0.957 | 1.52 | 86 | -4.55±0.49 | 1282.1 |
| 30 | B | 7 | 0.951 | 0.84 | 47 | -2.63±0.38 | 1272.6 | ||
| 31 | ν(N-H) | pKHA(MeOH) | A + B | 11l | 0.962 | 1.57 | 111 | 9.33±0.89 | 3200.5 |
| 32 | ν(N-H) | pKHA(CH3CN) | A + B | 11l | 0.976 | 1.26 | 179 | 6.43±0.48 | 3164.9 |
| 33 | ν(N-H) | pKHA(DMF) | A + B | 11l | 0.970 | 1.41 | 141 | 7.27±0.61 | 3217.1 |
| 34 | ν(N-H) | pKHA(DMSO) | A + B | 11l | 0.938 | 1.99 | 66 | 5.75±0.71 | 3240.6 |
| 35 | ν(N-H) | pKHA(Py) | A + B | 11l | 0.963 | 1.56 | 114 | 6.77±0.64 | 3244.8 |
| 36 | pKHA(MeOH) | ΔEm | A + B | 15m | 0.904 | 0.31 | 58 | 0.0631±0.0083 | 20.61 |
| 37 | pKHA(CH3CN) | ΔEm | A + B | 15n | 0.912 | 0.43 | 64 | 0.0914±0.0114 | 35.61 |
| 38 | pKHA(DMF) | ΔEm | A + B | 15n | 0.913 | 0.39 | 65 | 0.0852±0.0105 | 24.99 |
| 39 | pKHA(DMSO) | ΔEm | A + B | 15n | 0.878 | 0.61 | 44 | 0.1083±0.0169 | 27.64 |
| 40 | pKHA(Py) | ΔEm | A + B | 15n | 0.894 | 0.45 | 52 | 0.0868±0.0121 | 21.86 |
| 41 | pKHA(MeOH) | qM(H4) | A | 8o | 0.950 | 0.25 | 56 | -447.59±59.81 | 97.31 |
| 42 | B | 5p | 0.883 | 0.45 | 11 | 1017.23±312.46 | -192.75 | ||
| 43 | pKHA(CH3CN) | qM(H4) | A | 8o | 0.948 | 0.36 | 53 | -624.68±85.87 | 141.83 |
| 44 | B | 5p | 0.880 | 0.65 | 10 | 1443.32±449.37 | -267.68 | ||
| 45 | pKHA(DMF) | qM(H4) | A | 8o | 0.945 | 0.36 | 50 | -601.61±85.18 | 128.03 |
| 46 | B | 5p | 0.898 | 0.55 | 13 | 1333.49±376.78 | -255.22 | ||
| 47 | pKHA(DMSO) | qM (H4) | A | 8o | 0.927 | 0.58 | 36 | -827.36±136.77 | 171.06 |
| 48 | B | 5p | 0.850 | 0.87 | 8 | 1676.08±599.21 | -324.82 | ||
| 49 | pKHA(Py) | qM(H4) | A | 8o | 0.910 | 0.47 | 29 | -595.98±110.61 | 123.50 |
| 50 | B | 5p | 0.912 | 0.50 | 15 | 1314.15±342.19 | -254.81 | ||
| Dependence | Series | na | Rb | sc | Fd | a | b | |
| ν(C=O) vs. pKHAe | A | 5 | 0.940 | 0.20 | 23 | 2.14±.0.45 | -3.92 | |
| ν(C=O) vs. pKHAf | B | 5 | 0.928 | 0.77 | 19 | 7.57±1.75 | -16.27 | |
| ν(N-H) vs. pKHAg | A + B | 5 | 0.940 | 0.54 | 23 | -5.82±1.22 | 12.15 |
| No.b | y | x1 | x2 | Series | nc | Rd | se | Ff | ρ1 | ρ2 | q | ||||
| 51 | ν (C=O) | qM(C1) | qM(O2) | A | 10 | 0.836 | 1.4 | 8 | 819.8 | 417.5 | 1515.9 | ||||
| 52 | B | 7 | 0.883 | 4.4 | 7 | -664.3 | 760.6 | 2275.9 | |||||||
| 53 | ν (N-H) | qM(N3) | qM(H4) | A | poor correlation | ||||||||||
| 54 | B | 7 | 0.931 | 3.0 | 13 | -4174.0 | -3082.4 | 1222.6 | |||||||
| 55 | ν(SO2) | qM(S5) | qM(O6,O7) | A | 10 | 0.925 2.1 | 24 | -576.5 | 2008.0 | 4303.5 | |||||
| 56 | B | 7 | 0.830 | 1.8 | 4 | 727.3 | 179.5 | -425.1 | |||||||
Experimental
General
Computational Details
Acknowledgements
References and Notes
- Matsumoto, K.; Kurihara, K.; Suzuki, Y.; Koyama, M.; Kai, F.; Watanabe, T.; Inoue, S. Jpn. Kokai Tokkyo Koho JP 60152401 (80152401); [C. A. 1986, 104, 125043p.], 1985.
- Pallos, F. M. U.S. Pat. 4266078; [C. A. 1981, 95, 80481g], 1981.
- Donaruma, L.G.; Dombroski, J. R.; Razzano, J. J. Med. Chem. 1971, 14, 993.
- Donaruma, L.G.; Razzano, J. J. Med. Chem. 1971, 14, 244.
- Donaruma, L.G.; Dombroski, J. R. J. Med. Chem. 1971, 14, 460.
- DEHYDAG Deutsche Hydrierwerke G.m.b.H. Brit. Pat. 879707; [C. A. 1963, 58, 5272b], 1961.
- Strauss, W.; Michael, G.; Willmund, W. D. U.S. Pat. 3023151; [C. A. 1963, 58, 6456c], 1962.
- Doetzer, R.; Todt, E.; Hauschildt, H.G. Ger. Offen. 2453830; [C. A. 1976, 85, 132849y], 1976.
- Ludwig, M.; Parik, P.; Kulhanek, J. Collect. Czech. Chem. Commun. 1995, 60, 841.
- Ludwig, M.; Parik, P. Org. Reactiv. 1995, 28, 75.
- Perjessy, A.; Kolehmainen, E.; Fabian, W.M.F.; Ludwig, M.; Laihia, K.; Kulhanek, J.; Sustekova, Z. Sulfur Letters 2002, 25, 71.
- Perjessy, A.; Jones, R.G.; McClair, S.L.; Wilkins, J.M. J. Org. Chem. 1983, 48, 1266.
- Hansch, C.; Leo, A. Substituent Constants for Correlation Analysis in Chemistry and Biology; Wiley: New York, 1979. [Google Scholar]
- Marcus, Y. Chem. Soc. Rev. 1993, 22, 409.
- Castan, P.; Dagnac, P. J. Chim. Phys. 1972, 69, 545.
- Stewart, J. J. P. J. Comput. Chem. 1989, 10, 209 and 221.
- Clark, T. VAMP Erlangen Vectorized Molecular Orbital Package Version 4.40. 1992; In Computer-Chemie-Centrum; University Erlangen, Nürnberg, Germany. [Google Scholar]
- AMPAC 6.55. 1999; Semichem, 7128 Summit, KS 66216, USA.
- Baker, J. J. Comput. Chem. 1986, 7, 385.
- SYBYL 6.8. Tripos Associates, St. Louis, Mo., USA.
- Sample Availability: The samples of compounds 1 - 16 investigated in this work are available from Dr. Patrik Parik, Department of Organic Chemistry, Faculty of Chemical Technology, University of Pardubice, nam. Cs. legii 565, CZ - 532 10 Pardubice, Czech Republic, Phone: +420 466 037 075, Fax: +420 466 037 067, E-mail: Patrik.Parik@upce.cz.
© 2004 by MDPI (http:www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Perjessy, A.; Fabian, W.M.F.; Parik, P.; Ludwig, M.; Loos, D.; Sustekova, Z. Structure-Acidity-IR Spectra Correlations for p-Substituted N-Phenylsulfonylbenzamides†. Molecules 2004, 9, 213-222. https://doi.org/10.3390/90400213
Perjessy A, Fabian WMF, Parik P, Ludwig M, Loos D, Sustekova Z. Structure-Acidity-IR Spectra Correlations for p-Substituted N-Phenylsulfonylbenzamides†. Molecules. 2004; 9(4):213-222. https://doi.org/10.3390/90400213
Chicago/Turabian StylePerjessy, Alexander, Walter M.F. Fabian, Patrik Parik, Miroslav Ludwig, Dusan Loos, and Zora Sustekova. 2004. "Structure-Acidity-IR Spectra Correlations for p-Substituted N-Phenylsulfonylbenzamides†" Molecules 9, no. 4: 213-222. https://doi.org/10.3390/90400213
APA StylePerjessy, A., Fabian, W. M. F., Parik, P., Ludwig, M., Loos, D., & Sustekova, Z. (2004). Structure-Acidity-IR Spectra Correlations for p-Substituted N-Phenylsulfonylbenzamides†. Molecules, 9(4), 213-222. https://doi.org/10.3390/90400213