Total and Local Quadratic Indices of the Molecular Pseudograph's Atom Adjacency Matrix: Applications to the Prediction of Physical Properties of Organic Compounds

Abstract
:Introduction
- Research on drugs, toxics or generally any organic molecules with a common skeleton, which is responsible for the activity or property under study.
- Study of the reactivity of specific sites of a series of molecules, which can undergo a chemical reaction or enzymatic metabolism.
- In the study of molecular properties such as spectroscopic measurements, which are calculated experimentally in a local fashion
- In any general case where it is necessary to study not the molecule as a whole, but rather some local properties of certain fragments, then the definition of local descriptors could be necessary.
Results and Discussion
Computational methods. Mathematical definition of the molecular descriptor
Molecular vector space
Total quadratic indices; [qk(x)].
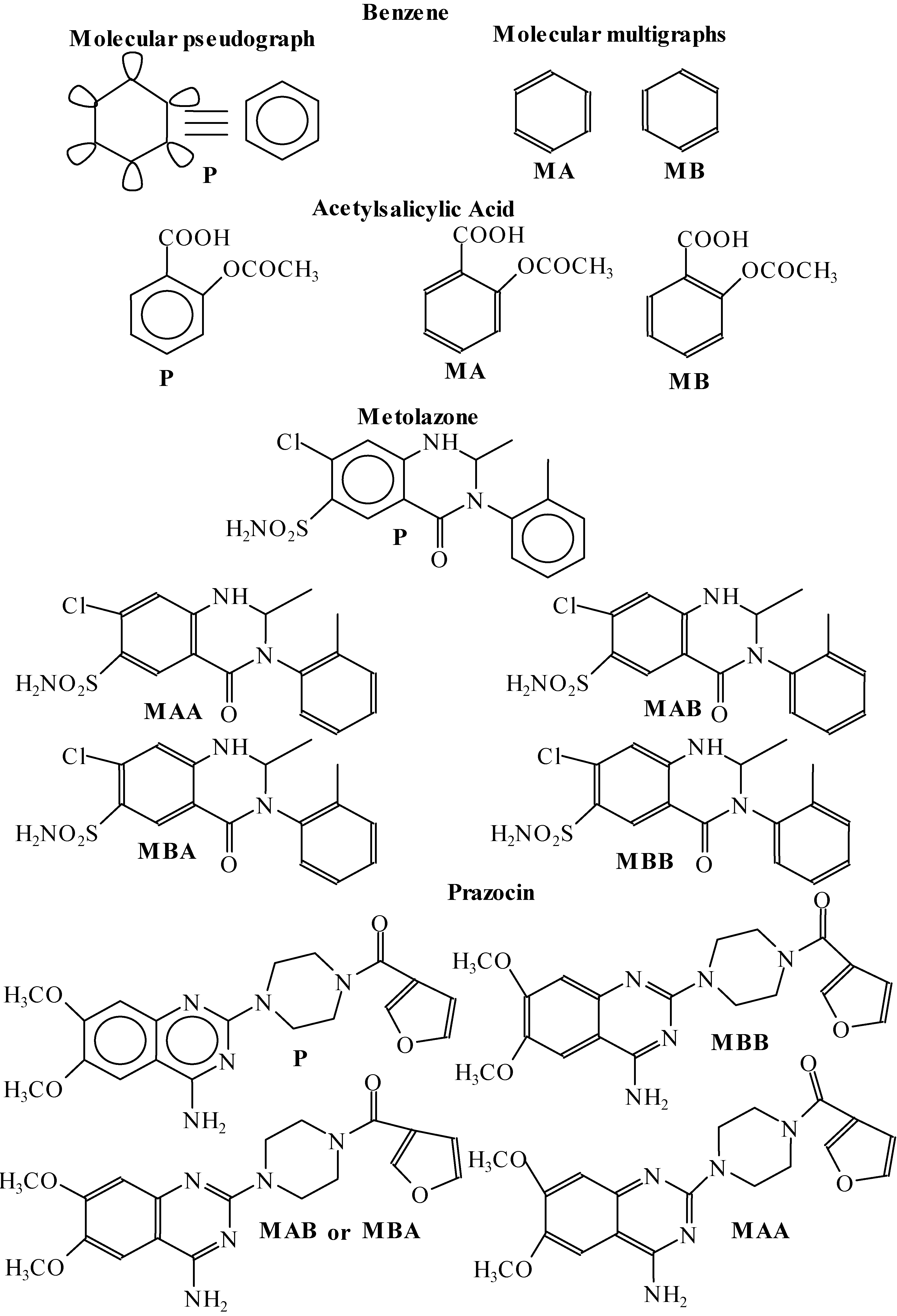

{kind=link}
| Benzene | ||||||||
|---|---|---|---|---|---|---|---|---|
| q0(x) | q1(x) | q2(x) | q3(x) | q4(x) | q5(x) | q6(x) | q7(x) | |
| P | 41.5014 | 124.5042 | 373.5126 | 1120.5378 | 3361.6134 | 10084.8402 | 30254.5206 | 90763.5618 |
| MKA | 41.5014 | 124.5042 | 373.5126 | 1120.5378 | 3361.6134 | 10084.8402 | 30254.5206 | 90763.5618 |
| MKB | 41.5014 | 124.5042 | 373.5126 | 1120.5378 | 3361.6134 | 10084.8402 | 30254.5206 | 90763.5618 |
| Acetylsalicylic acid | ||||||||
| q0(x) | q1(x) | q2(x) | q3(x) | q4(x) | q5(x) | q6(x) | q7(x) | |
| P | 102.4477 | 268.8912 | 873.5982 | 2566.8034 | 8381.4114 | 25593.6122 | 83330.7872 | 260026.931 |
| MKA | 102.4477 | 268.8912 | 873.5982 | 2549.8376 | 8284.7898 | 25063.374 | 81351.7828 | 250745.988 |
| MKB | 102.4477 | 268.8912 | 873.5982 | 2566.5118 | 8389.425 | 25513.2092 | 83389.772 | 258104.308 |
| Eq0(x) | Eq1(x) | Eq2(x) | Eq3(x) | Eq4(x) | Eq5(x) | Eq6(x) | Eq7(x) | |
| P | 40.1956 | 58.3597 | 265.963 | 510.2749 | 2171.4817 | 4947.1654 | 19328.9482 | 49869.8377 |
| MKA | 40.1956 | 58.3597 | 265.963 | 500.226 | 2133.2198 | 4618.7534 | 18773.2472 | 44486.7656 |
| MKB | 40.1956 | 58.3597 | 265.963 | 508.5631 | 2201.8503 | 4802.1696 | 19870.6695 | 47162.9747 |
| Hq0(x) | Hq1(x) | Hq2(x) | Hq3(x) | Hq4(x) | Hq5(x) | Hq6(x) | Hq7(x) | |
| P | 4.84 | 6.974 | 10.626 | 33.682 | 67.54 | 270.578 | 670.604 | 2600.972 |
| MKA | 4.84 | 6.974 | 10.626 | 33.682 | 67.54 | 269.632 | 647.306 | 2589.686 |
| MKB | 4.84 | 6.974 | 10.626 | 33.682 | 67.54 | 271.766 | 653.092 | 2639.868 |
| Metolazone | ||||||||
| q0(x) | q1(x) | q2(x) | q3(x) | q4(x) | q5(x) | q6(x) | q7(x) | |
| P | 171.9119 | 485.942 | 1711.0469 | 5439.1693 | 19235.232 | 62338.8312 | 220106.56 | 721470.089 |
| MKAA | 171.9119 | 485.942 | 1711.0469 | 5424.1812 | 19161.672 | 61839.7906 | 218582.941 | 710431.996 |
| MKAB | 171.9119 | 485.942 | 1711.0469 | 5411.9254 | 19107.9148 | 61560.958 | 217543.348 | 706114.062 |
| MKBA | 171.9119 | 485.942 | 1711.0469 | 5426.3854 | 19199.863 | 61837.827 | 219141.462 | 710613.352 |
| MKBB | 171.9119 | 485.942 | 1711.0469 | 5414.1296 | 19146.1058 | 61558.9944 | 218101.869 | 706307.674 |
| Eq0(x) | Eq1(x) | Eq2(x) | Eq3(x) | Eq4(x) | Eq5(x) | Eq6(x) | Eq7(x) | |
| P | 61.2415 | 133.8902 | 554.1099 | 1558.9199 | 6272.0672 | 18784.7951 | 73539.8425 | 228597.096 |
| MKAA | 61.2415 | 133.8902 | 554.1099 | 1545.5098 | 6202.9256 | 18310.0294 | 72577.097 | 218343.795 |
| MKAB | 61.2415 | 133.8902 | 554.1099 | 1539.3819 | 6196.7977 | 18225.9483 | 72439.9618 | 217339.95 |
| MKBA | 61.2415 | 133.8902 | 554.1099 | 1553.8419 | 6260.6838 | 18444.8521 | 73549.9487 | 220551.513 |
| MKBB | 61.2415 | 133.8902 | 554.1099 | 1547.714 | 6254.5559 | 18360.771 | 73412.8135 | 219553.796 |
| Hq0(x) | Hq1(x) | Hq2(x) | Hq3(x) | Hq4(x) | Hq5(x) | Hq6(x) | Hq7(x) | |
| P | 14.52 | 15.378 | 46.376 | 146.608 | 380.556 | 1654.686 | 4353.734 | 19526.76 |
| MKAA | 14.52 | 15.378 | 46.376 | 146.608 | 381.216 | 1662.65 | 4285.534 | 19850.446 |
| MKAB | 14.52 | 15.378 | 46.376 | 146.608 | 381.216 | 1662.65 | 4284.588 | 19835.926 |
| MKBA | 14.52 | 15.378 | 46.376 | 146.608 | 380.27 | 1647.096 | 4238.41 | 19605.3 |
| MKBB | 14.52 | 15.378 | 46.376 | 146.608 | 380.27 | 1647.096 | 4237.464 | 19590.78 |
| Prazocin | ||||||||
| q0(x) | q1(x) | q2(x) | q3(x) | q4(x) | q5(x) | q6(x) | q7(x) | |
| P | 198.7612 | 541.9074 | 1696.6156 | 5358.4782 | 17314.5582 | 56186.8214 | 183864.863 | 603661.363 |
| MKAA | 198.7612 | 541.7274 | 1694.1796 | 5323.0646 | 17197.7804 | 55637.9444 | 181811.302 | 595116.828 |
| MKAB | 198.7612 | 541.7274 | 1694.3596 | 5327.7986 | 17244.174 | 55914.3384 | 183221.047 | 601548.719 |
| MKBB | 198.7612 | 541.7274 | 1694.3596 | 5335.6406 | 17224.5402 | 55735.215 | 181942.392 | 595274.105 |
| Eq0(x) | Eq1(x) | Eq2(x) | Eq3(x) | Eq4(x) | Eq5(x) | Eq6(x) | Eq7(x) | |
| P | 67.3401 | 144.9615 | 468.8527 | 1384.3378 | 4526.6829 | 14281.5586 | 46761.2533 | 151360.249 |
| MKAA | 67.3401 | 146.3595 | 475.5165 | 1381.8781 | 4632.9291 | 14424.8713 | 48134.0569 | 153961.075 |
| MKAB | 67.3401 | 146.3595 | 474.1185 | 1363.4944 | 4559.3158 | 14146.1775 | 47209.3348 | 151083.318 |
| MKBB | 67.3401 | 146.3595 | 474.1185 | 1377.4643 | 4553.9629 | 14140.7919 | 46743.0601 | 149152.807 |
| Hq0(x) | Hq1(x) | Hq2(x) | Hq3(x) | Hq4(x) | Hq5(x) | Hq6(x) | Hq7(x) | |
| P | 9.68 | 10.252 | 30.932 | 64.152 | 216.128 | 645.392 | 2236.476 | 7512.296 |
| MKAA | 9.68 | 10.252 | 30.932 | 64.152 | 220.088 | 668.8 | 2359.72 | 7965.76 |
| MKAB | 9.68 | 10.252 | 30.932 | 62.832 | 208.516 | 616.484 | 2135.1 | 7120.168 |
| MKBB | 9.68 | 10.252 | 30.932 | 62.832 | 208.516 | 615.912 | 2111.956 | 7031.288 |
 Isonicotinic acid Molecular Structure | 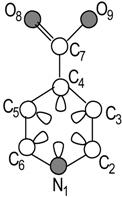 Molecular Pseudograph (G) (Hydrogen Suppressed-pseudograph) | X=[N1 C2 C3 C4 C5 C6 C7 O8 O9] Molecular Vector: X∊ℜ9 and ℜ9∊E; E: Molecular Vector Space In the definition of the X, as molecular vector, the chemical symbol of the element is used to indicate the corresponding electronegativity value. That is: if we write O it means χ(O), oxygen Mulliken electronegativity or some atomic property, which characterizes each atom in the molecule. Therefore, if we use the canonical bases of R9, the coordinates of any vector X coincide with the components of that molecular vector Xt =[233 263 263 263 263 263 263 3.17 3.17] Xt = transposed of X and it means the vector of the coordinates of X in the Canonical basis of R9 (a row vector) X: vector of coordinates of X in the Canonical basis of R9 (a column vector) | |
| = XtM0X=67.0281 | |||
| = XtM1X=183.7166 | |||
| = XtM2X=589.963 | M(G): Adjacency Matrix Among Vertices of the Molecular Pseudograph (G) | ||
| = XtM3X=1784.6905 | |||
| = XtM4X=5707.7232 | |||
Local quadratic indices; [qkL(x)]
=1/2 kaij either vi or vj is contained in the specific fragment but not both
at the same time
=0 otherwise
Calculation of total and local quadratic indices
- 1)
- Total and Local indices of zero order [q0(x) and q0L(x)]. These indices are obtained when the matrix M is raised to the power 0 (k=0). A matrix raised to the power 0 is the identity matrix (I); which is constituted by the elements aii=1 [M0(i, i)=1]. Since the zero order matrix is diagonal, its quadratic form contains only the terms with the squares of the coordinates (an atomic property) of the X vector in canonical bases. Generally, we can establish that.andwhere n and m are the number of atoms in the molecule or in the fragment FR under study, respectively.
- 2)
- Total and local quadratic indices of first order [q1(x) and q1L(x)]. These indices are obtained when the matrix M is raised to the unit power (M1= M) and multiplied by the matrices Xt and X. We can write the expression for q1(x) and q1L(x) in the forms:and
- 3)
- Total and local quadratic indices of second order [q2(x) and q2L(x)]. In general, these indices are calculated as:and
q2(x, F1)=1.(XC2.XO4)+1.(XO4.XC5)+1.(XO4)2=1.(2.63.3.17) +1.(3.17.2.63)
+1.(3.17)2=26.7231;
q2(x, F2)=2.(XC1.XC3) +1.(XC2.XC5) +1.(XC4.XC5) +3.(XC3)2 +1.(XC5)2=2.(2.63.2.63)
+1.(2.63.2.63) +1.(3.17.2.63) +3.(2.63)2+ 1.(2.63)2=56.7554, and
q2(x, F3)=2.(XC1.XC3) +1.(XC2.XC4) +1.(XC2.XC5) +4.(XC1)2 +5.(XC2)2=2.(2.63.2.63)
+1.(2.63.3.17) +1.(2.63.2.63) +4.(2.63)2 +5.(2.63)2=91.3399.
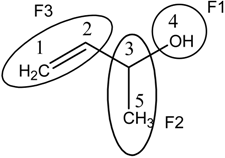 Molecular Structure of 1-methylallyl alchohol (But-3-en-2-ol) | X=[C1 C2 C3 O4 C5] Molecular Vector: X∊ℜ5 and 5∊ℜE; E: Molecular Vector Space In the definition of the X, as molecular vector, the chemical symbol of the element is used to indicate the corresponding electronegativity value. That is: if we write O it means χ(O), oxygen Mulliken electronegativity or some atomic property, which characterizes each atom in the molecule. Therefore, if we use the canonical bases of ℜ5, the coordinates of any molecular vector X coincide with the components of that molecular vector. Xt = [2.63 2.63 2.63 3.17 2.63] Xt = transposed of X and it means the vector of the coordinates of X in the Canonical basis of ℜ5 (a row vector) X: vector of coordinates of X in the Canonical basis of ℜ5 (a column vector) | ||||
| The zero, first and second powers of the molecular “pseudograph’s” total atom adjacency matrix. | |||||
| The zero, first and second powers of the molecular “pseudograph’s” local atom adjacency matrix of each one of 3 fragments shown in the molecule of 1-methylallyl alcohol | |||||
The TOMO-COMD software
- (1)
- qk(x) and qkH(x) are the k-th total quadratic indices calculated using the k-th power of the matrices [Mk(G) or Mk(GH)] of the molecular pseudograph (G) considering and not considering hydrogen atoms, respectively.
- (2)
- EqkL(x) [or EqkLH(x)] and H qkL(x) are the k-th local quadratic indices calculated using a k-th power of the local matrices [MkL(G, FR)] of the molecular pseudograph (G) not considering (or considering) hydrogen atoms for heteroatoms (S,N,O) and hydrogen bonding heteroatoms, respectively.
Physical properties data sets for QSPR studies
- a)
- b)
- c)
- d)
| no. | Alkane | q0H(x) | q1H(x) | q2H(x) | q3H(x) | q4H(x) | q0(x) | q2(x) | q3(x) | q5(x) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 42.8738 | 83.2658 | 211.8872 | 461.6846 | 1097.3462 | 13.8338 | 13.8338 | 13.8338 | 13.8338 |
| 2 | 3 | 59.4707 | 120.2436 | 319.0366 | 749.5692 | 1876.432 | 20.7507 | 41.5014 | 55.3352 | 110.6704 |
| 3 | 4 | 76.0676 | 157.2214 | 426.186 | 1037.8236 | 2666.8698 | 27.6676 | 69.169 | 110.6704 | 290.5098 |
| 4 | 2M3 | 76.0676 | 157.2214 | 426.5558 | 1048.8058 | 2757.6878 | 27.6676 | 83.0028 | 124.5042 | 373.5126 |
| 5 | 5 | 92.6645 | 194.1992 | 533.3354 | 1326.078 | 3457.6774 | 34.5845 | 96.8366 | 166.0056 | 498.0168 |
| 6 | 2M4 | 92.6645 | 194.1992 | 533.7052 | 1337.43 | 3559.4776 | 34.5845 | 110.6704 | 193.6732 | 664.0224 |
| 7 | 22MM3 | 92.6645 | 194.1992 | 534.4448 | 1359.3944 | 3741.1136 | 34.5845 | 138.338 | 221.3408 | 885.3632 |
| 8 | 6 | 109.2614 | 231.177 | 640.4848 | 1614.3324 | 4248.485 | 41.5014 | 124.5042 | 221.3408 | 719.3576 |
| 9 | 2M5 | 109.2614 | 231.177 | 640.8546 | 1625.6844 | 4350.655 | 41.5014 | 138.338 | 249.0084 | 899.197 |
| 10 | 3M5 | 109.2614 | 231.177 | 640.8546 | 1626.0542 | 4361.6372 | 41.5014 | 138.338 | 262.8422 | 982.1998 |
| 11 | 22MM4 | 109.2614 | 231.177 | 641.5942 | 1648.3884 | 4554.2554 | 41.5014 | 166.0056 | 304.3436 | 1314.211 |
| 12 | 23MM4 | 109.2614 | 231.177 | 641.2244 | 1637.4062 | 4463.4374 | 41.5014 | 152.1718 | 290.5098 | 1175.873 |
| 13 | 7 | 125.8583 | 268.1548 | 747.6342 | 1902.5868 | 5039.2926 | 48.4183 | 152.1718 | 276.676 | 940.6984 |
| 14 | 2M6 | 125.8583 | 268.1548 | 748.004 | 1913.9388 | 5141.4626 | 48.4183 | 166.0056 | 304.3436 | 1134.3716 |
| 15 | 3M6 | 125.8583 | 268.1548 | 748.004 | 1914.3086 | 5152.8146 | 48.4183 | 166.0056 | 318.1774 | 1231.2082 |
| 16 | 3E.5 | 125.8583 | 268.1548 | 748.004 | 1914.6784 | 5164.1666 | 48.4183 | 166.0056 | 332.0112 | 1328.0448 |
| 17 | 22MM5 | 125.8583 | 268.1548 | 748.7436 | 1936.6428 | 5345.8026 | 48.4183 | 193.6732 | 359.6788 | 1577.0532 |
| 18 | 23MM5 | 125.8583 | 268.1548 | 748.3738 | 1926.0304 | 5265.9668 | 48.4183 | 179.8394 | 359.6788 | 1521.718 |
| 19 | 24MM5 | 125.8583 | 268.1548 | 748.3738 | 1925.2908 | 5244.0024 | 48.4183 | 179.8394 | 332.0112 | 1328.0448 |
| 20 | 33MM5 | 125.8583 | 268.1548 | 748.7436 | 1937.3824 | 5367.767 | 48.4183 | 193.6732 | 387.3464 | 1770.7264 |
| 21 | 223MMM4 | 125.8583 | 268.1548 | 749.1134 | 1948.7344 | 5469.5672 | 48.4183 | 207.507 | 415.014 | 1992.0672 |
| 22 | 8 | 142.4552 | 305.1326 | 854.7836 | 2190.8412 | 5830.1002 | 55.3352 | 179.8394 | 332.0112 | 1162.0392 |
| 23 | 2M7 | 142.4552 | 305.1326 | 855.1534 | 2202.1932 | 5932.2702 | 55.3352 | 193.6732 | 359.6788 | 1355.7124 |
| 24 | 3M7 | 142.4552 | 305.1326 | 855.1534 | 2202.563 | 5943.6222 | 55.3352 | 193.6732 | 373.5126 | 1466.3828 |
| 25 | 4M7 | 142.4552 | 305.1326 | 855.1534 | 2202.563 | 5943.992 | 55.3352 | 193.6732 | 373.5126 | 1480.2166 |
| 26 | 3E.6 | 142.4552 | 305.1326 | 855.1534 | 2202.9328 | 5955.344 | 55.3352 | 193.6732 | 387.3464 | 1590.887 |
| 27 | 22MM6 | 142.4552 | 305.1326 | 855.893 | 2224.8972 | 6136.6102 | 55.3352 | 221.3408 | 415.014 | 1826.0616 |
| 28 | 23MM6 | 142.4552 | 305.1326 | 855.5232 | 2214.2848 | 6057.1442 | 55.3352 | 207.507 | 415.014 | 1784.5602 |
| 29 | 24MM6 | 142.4552 | 305.1326 | 855.5232 | 2213.915 | 6046.162 | 55.3352 | 207.507 | 401.1802 | 1673.8898 |
| 30 | 25MM6 | 142.4552 | 305.1326 | 855.5232 | 2213.5452 | 6034.4402 | 55.3352 | 207.507 | 387.3464 | 1563.2194 |
| 31 | 33MM6 | 142.4552 | 305.1326 | 855.893 | 2225.6368 | 6159.3142 | 55.3352 | 221.3408 | 442.6816 | 2047.4024 |
| 32 | 34MM6 | 142.4552 | 305.1326 | 855.5232 | 2214.6546 | 6068.4962 | 55.3352 | 207.507 | 428.8478 | 1881.3968 |
| 33 | 23ME5 | 142.4552 | 305.1326 | 855.5232 | 2214.6546 | 6068.866 | 55.3352 | 207.507 | 428.8478 | 1895.2306 |
| 34 | 33ME5 | 142.4552 | 305.1326 | 855.893 | 2226.3764 | 6181.6484 | 55.3352 | 221.3408 | 470.3492 | 2254.9094 |
| 35 | 223MMM5 | 142.4552 | 305.1326 | 856.2628 | 2237.3586 | 6272.4664 | 55.3352 | 235.1746 | 484.183 | 2365.5798 |
| 36 | 224MMM5 | 142.4552 | 305.1326 | 856.2628 | 2236.2492 | 6239.5198 | 55.3352 | 235.1746 | 442.6816 | 2033.5686 |
| 37 | 233MMM5 | 142.4552 | 305.1326 | 856.2628 | 2237.7284 | 6283.4486 | 55.3352 | 235.1746 | 498.0168 | 2476.2502 |
| 38 | 234MMM5 | 142.4552 | 305.1326 | 855.893 | 2226.0066 | 6170.6662 | 55.3352 | 221.3408 | 456.5154 | 2088.9038 |
| 39 | 2233MMMM4 | 147.2952 | 305.1326 | 857.0024 | 2260.4324 | 6487.049 | 55.3352 | 262.8422 | 553.352 | 3001.9346 |
| 40 | 9 | 159.0521 | 342.1104 | 961.933 | 2479.0956 | 6620.9078 | 62.2521 | 207.507 | 387.3464 | 1383.38 |
| 41 | 2M8 | 159.0521 | 342.1104 | 962.3028 | 2490.4476 | 6723.0778 | 62.2521 | 221.3408 | 415.014 | 1577.0532 |
| 42 | 3M8 | 159.0521 | 342.1104 | 962.3028 | 2490.8174 | 6734.4298 | 62.2521 | 221.3408 | 428.8478 | 1687.7236 |
| 43 | 4M8 | 159.0521 | 342.1104 | 962.3028 | 2490.8174 | 6734.7996 | 62.2521 | 221.3408 | 428.8478 | 1715.3912 |
| 44 | 3E.7 | 159.0521 | 342.1104 | 962.3028 | 2491.1872 | 6746.1516 | 62.2521 | 221.3408 | 442.6816 | 1826.0616 |
| 45 | 4E.7 | 159.0521 | 342.1104 | 962.3028 | 2491.1872 | 6746.5214 | 62.2521 | 221.3408 | 442.6816 | 1853.7292 |
| 46 | 22MM7 | 159.0521 | 342.1104 | 963.0424 | 2513.1516 | 6927.4178 | 62.2521 | 249.0084 | 470.3492 | 2047.4024 |
| 47 | 23MM7 | 159.0521 | 342.1104 | 962.6726 | 2502.5392 | 6847.9518 | 62.2521 | 235.1746 | 470.3492 | 2019.7348 |
| 48 | 24MM7 | 159.0521 | 342.1104 | 962.6726 | 2502.1694 | 6837.3394 | 62.2521 | 235.1746 | 456.5154 | 1922.8982 |
| 49 | 25MM7 | 159.0521 | 342.1104 | 962.6726 | 2502.1694 | 6836.5998 | 62.2521 | 235.1746 | 456.5154 | 1895.2306 |
| 50 | 26MM7 | 159.0521 | 342.1104 | 962.6726 | 2501.7996 | 6825.2478 | 62.2521 | 235.1746 | 442.6816 | 1770.7264 |
| 51 | 33MM7 | 159.0521 | 342.1104 | 963.0424 | 2513.8912 | 6950.1218 | 62.2521 | 249.0084 | 498.0168 | 2296.4108 |
| 52 | 34MM7 | 159.0521 | 342.1104 | 962.6726 | 2502.909 | 6859.6736 | 62.2521 | 235.1746 | 484.183 | 2144.239 |
| 53 | 35MM7 | 159.0521 | 342.1104 | 962.6726 | 2502.5392 | 6848.3216 | 62.2521 | 235.1746 | 470.3492 | 2019.7348 |
| 54 | 44MM7 | 159.0521 | 342.1104 | 963.0424 | 2513.8912 | 6950.8614 | 62.2521 | 249.0084 | 498.0168 | 2324.0784 |
| 55 | 23ME6 | 159.0521 | 342.1104 | 962.6726 | 2502.909 | 6860.0434 | 62.2521 | 235.1746 | 484.183 | 2171.9066 |
| 56 | 24ME6 | 159.0521 | 342.1104 | 962.6726 | 2502.5392 | 6848.6914 | 62.2521 | 235.1746 | 470.3492 | 2047.4024 |
| 57 | 33ME6 | 159.0521 | 342.1104 | 963.0424 | 2514.6308 | 6973.1956 | 62.2521 | 249.0084 | 525.6844 | 2545.4192 |
| 58 | 34ME6 | 159.0521 | 342.1104 | 962.6726 | 2503.2788 | 6871.3954 | 62.2521 | 235.1746 | 498.0168 | 2268.7432 |
| 59 | 223MMM6 | 159.0521 | 342.1104 | 963.4122 | 2525.613 | 7063.6438 | 62.2521 | 262.8422 | 539.5182 | 2642.2558 |
| 60 | 224MMM6 | 159.0521 | 342.1104 | 963.4122 | 2524.8734 | 7041.6794 | 62.2521 | 262.8422 | 511.8506 | 2393.2474 |
| 61 | 225MMM6 | 159.0521 | 342.1104 | 963.4122 | 2524.5036 | 7029.5878 | 62.2521 | 262.8422 | 498.0168 | 2268.7432 |
| 62 | 233MMM6 | 159.0521 | 342.1104 | 963.4122 | 2525.9828 | 7074.9958 | 62.2521 | 262.8422 | 553.352 | 2766.76 |
| 63 | 234MMM6 | 159.0521 | 342.1104 | 963.0424 | 2514.6308 | 6973.1956 | 62.2521 | 249.0084 | 525.6844 | 2462.4164 |
| 64 | 235MMM6 | 159.0521 | 342.1104 | 963.0424 | 2513.8912 | 6950.4916 | 62.2521 | 249.0084 | 498.0168 | 2241.0756 |
| 65 | 244MMM6 | 159.0521 | 342.1104 | 963.4122 | 2525.2432 | 7053.0314 | 62.2521 | 262.8422 | 525.6844 | 2517.7516 |
| 66 | 334MMM6 | 159.0521 | 342.1104 | 963.4122 | 2526.3526 | 7086.3478 | 62.2521 | 262.8422 | 567.1858 | 2863.5966 |
| 67 | 33EE5 | 159.0521 | 342.1104 | 963.0424 | 2515.3704 | 6995.8996 | 62.2521 | 249.0084 | 553.352 | 2766.76 |
| 68 | 223MME5 | 159.0521 | 342.1104 | 963.4122 | 2525.9828 | 7075.7354 | 62.2521 | 262.8422 | 553.352 | 2766.76 |
| 69 | 233MME5 | 159.0521 | 342.1104 | 963.4122 | 2526.7224 | 7097.6998 | 62.2521 | 262.8422 | 581.0196 | 2988.1008 |
| 70 | 234MEM5 | 159.0521 | 342.1104 | 963.0424 | 2514.6308 | 6973.9352 | 62.2521 | 249.0084 | 525.6844 | 2490.084 |
| 71 | 2233(M)5 | 159.0521 | 342.1104 | 964.1518 | 2549.4264 | 7301.3002 | 62.2521 | 290.5098 | 636.3548 | 3513.7852 |
| 72 | 2234(M)5 | 159.0521 | 342.1104 | 963.782 | 2537.3348 | 7177.5356 | 62.2521 | 276.676 | 581.0196 | 2960.4332 |
| 73 | 2244(M)5 | 159.0521 | 342.1104 | 964.1518 | 2547.2076 | 7235.407 | 62.2521 | 290.5098 | 553.352 | 2766.76 |
| 74 | 2334(M)5 | 159.0521 | 342.1104 | 963.782 | 2538.0744 | 7199.5 | 62.2521 | 276.676 | 608.6872 | 3209.4416 |
| no. | q7(x) | q11(x) | q13(x) | q15(x) | no. | q7(x) | q11(x) | q13(x) | q15(x) | |
| 1 | 13.8338 | 13.8338 | 13.8338 | 13.8338 | 38 | 9531.4882 | 198335.19 | 904716.69 | 4126913 | |
| 2 | 221.3408 | 885.3632 | 1770.7264 | 3541.4528 | 39 | 16033.374 | 452213.09 | 2398545.7 | 12719902 | |
| 3 | 760.859 | 5215.3426 | 13653.9606 | 35746.539 | 40 | 4980.168 | 65018.86 | 235174.6 | 850778.7 | |
| 4 | 1120.5378 | 10084.84 | 30254.5206 | 90763.562 | 41 | 6031.5368 | 88812.996 | 341252.18 | 1311776.3 | |
| 5 | 1494.0504 | 13446.454 | 40339.3608 | 121018.08 | 42 | 6695.5592 | 106326.59 | 424531.65 | 1696120.7 | |
| 6 | 2268.7432 | 26450.226 | 90307.0464 | 308327.73 | 43 | 6930.7338 | 114032.01 | 463003.45 | 1880234.8 | |
| 7 | 3541.4528 | 56663.245 | 226652.979 | 906611.92 | 44 | 7580.9224 | 131365.76 | 547431.13 | 2281968.3 | |
| 8 | 2337.9122 | 24665.665 | 80097.702 | 260089.27 | 45 | 7802.2632 | 138504.01 | 583675.69 | 2459760.3 | |
| 9 | 3250.943 | 42538.935 | 153901.025 | 556810.45 | 46 | 9019.6376 | 178179.34 | 795498.84 | 3556836 | |
| 10 | 3665.957 | 51060.556 | 190560.595 | 711181.82 | 47 | 8715.294 | 163432.51 | 708926.91 | 3076720.1 | |
| 11 | 5658.0242 | 104763.37 | 450774.373 | 1939581.8 | 48 | 8148.1082 | 146859.62 | 623696.87 | 2648840.7 | |
| 12 | 4717.3258 | 75546.382 | 302199.361 | 1208811.3 | 49 | 7871.4322 | 135571.24 | 562274.8 | 2331382.6 | |
| 13 | 3209.4416 | 37406.595 | 127713.642 | 436041.38 | 50 | 7082.9056 | 113326.49 | 453305.96 | 1813223.8 | |
| 14 | 4233.1428 | 58959.656 | 220040.423 | 821202.04 | 51 | 10679.694 | 233182.53 | 1091597.5 | 5112419.1 | |
| 15 | 4772.661 | 71797.422 | 278515.895 | 1080447.4 | 52 | 9545.322 | 189772.07 | 846421.05 | 3775354.7 | |
| 16 | 5312.1792 | 84994.867 | 339979.469 | 1359917.9 | 53 | 8687.6264 | 160831.76 | 692022.01 | 2977614.8 | |
| 17 | 6944.5676 | 135156.23 | 596541.124 | 2633153.2 | 54 | 10956.37 | 245024.27 | 1159383.1 | 5486153.1 | |
| 18 | 6418.8832 | 114045.85 | 480641.547 | 2025600.3 | 55 | 9752.829 | 196688.97 | 883301.96 | 3966786.8 | |
| 19 | 5312.1792 | 84994.867 | 339979.469 | 1359917.9 | 56 | 8908.9672 | 168329.68 | 731310 | 3176683.2 | |
| 20 | 8078.9392 | 168108.34 | 766835.202 | 3497959.3 | 57 | 12367.417 | 292640.21 | 1423940.7 | 6928990.7 | |
| 21 | 9462.3192 | 212155.16 | 1004001.87 | 4751080.3 | 58 | 10347.682 | 215309.26 | 982144.46 | 4480103.8 | |
| 22 | 4094.8048 | 51060.556 | 180365.084 | 637115.66 | 59 | 12962.271 | 312505.54 | 1534901.6 | 7539338 | |
| 23 | 5132.3398 | 73886.326 | 280632.467 | 1066267.8 | 60 | 11219.212 | 247237.67 | 1161361.3 | 5456410.4 | |
| 24 | 5782.5284 | 90279.379 | 356995.043 | 1411988.3 | 61 | 10347.682 | 215309.26 | 982144.46 | 4480103.8 | |
| 25 | 5907.0326 | 94443.353 | 377759.577 | 1511024.5 | 62 | 13833.8 | 345845 | 1729225 | 8646125 | |
| 26 | 6543.3874 | 110767.24 | 455782.209 | 1875475.9 | 63 | 11537.389 | 253324.55 | 1186995.4 | 5561796.3 | |
| 27 | 8092.773 | 160236.91 | 714156.091 | 3184194.9 | 64 | 10071.006 | 202803.51 | 909378.68 | 4076654.9 | |
| 28 | 7677.759 | 142142.3 | 611606.132 | 2631603.8 | 65 | 12090.741 | 279636.43 | 1345613.7 | 6476127.5 | |
| 29 | 6986.069 | 121585.27 | 507105.607 | 2114869.8 | 66 | 14456.321 | 368504.76 | 1860549.3 | 9393758.9 | |
| 30 | 6294.379 | 101443.26 | 406533.881 | 1628127.6 | 67 | 13833.8 | 345845 | 1729225 | 8646125 | |
| 31 | 9503.8206 | 205390.43 | 955196.222 | 4442545 | 68 | 13833.8 | 345845 | 1729225 | 8646125 | |
| 32 | 8258.7786 | 159185.54 | 698869.742 | 3068226.2 | 69 | 15327.85 | 402729.59 | 2064003 | 10577877 | |
| 33 | 8369.449 | 163114.34 | 720035.456 | 3178412.4 | 70 | 11786.398 | 263948.9 | 1249026.1 | 5910463.4 | |
| 34 | 10804.198 | 248026.2 | 1188364.92 | 5693798.4 | 71 | 19228.982 | 572193.64 | 3118774.9 | 16996954 | |
| 35 | 11523.555 | 273010.04 | 1328584.32 | 6465267.9 | 72 | 15051.174 | 389172.46 | 1979229.4 | 10066248 | |
| 36 | 9365.4826 | 199303.56 | 920044.537 | 4247972.6 | 73 | 13833.8 | 345845 | 1729225 | 8646125 | |
| 37 | 12242.913 | 298408.9 | 1472843.18 | 7269219.2 | 74 | 16821.901 | 461274.23 | 2415270.8 | 12646528 |
Data analysis
QSPR applications
B.p. (oC)=-204.184(±3.262) +1.44048(±0.026).q1H(x) -9.29x10-3(±0.427x10-3).q0(x).q2(x) +2.91x10-7 (±1.75x10-8).q0(x).q13(x) -0.11678(±0.028).q2(x)
N=74 R=0.9988 q2=0.9970 F(4.69)=7068.1 s=2.35 MAE=2.11 p<0.0000
MV (cm3)=39.72(±2.441) +0.7651(±0.031).q0H(x) -4.4x10-7(±1.08x10-7).q15(x) +4.634x10-3(±0.214 x10-3).q0(x).q2(x) -1.74x10-3(±0.132x10-3).q0(x).q3(x)
N=69 R=0.9991 q2=0.9973 F(4.69)=8916.5 s= 0.75 MAE=0.53 p<0.0000
MR (cm3)=3.2327(±0.048) +1.734x10-2(±4.71x10-5). q3H(x) -0.01012(±0.302x10-3).q3(x) +7.486x10-3 (±0.836x10-3).q2(x)
N= 69 R=0.9999 q2=0.9999 F(3.65)= 2.52x105 s= 0.049 MAE=0.0322 p<0.00
HV (KJ/mol)=-1.35607(±0.327) +0.07648(±0.001).q2H(x) -0.1309(±0.004).q2(x) +1.19x10-5(±9.3x10-7) .q11(x)
N=69 R=0.998 q2= 0.9955 F(3.65)=5469.5 s= 0.34 MAE=0.32 p<0.0000
TC (oC)=-71.6809(±6.373) +0.2399(±0.007).q3H(x) -0.02165(±0.001).q0(x).q2(x) +0.83x10-3(±6.01x10-5) .q0(x). q5(x)
N=74 R=0.9953 q2= 0.9892 F(3.70)=2460.1 s=5.66 MAE=5.34 p<0.0000
PC (atm)=54.7074(±0.786) -6.998x10-3(±0.265x10-3).q4H(x)+5.95x10-4(±3.72x10-5).q0(x).q3(x)
N=74 R=0.9803 q2=0.9575 F(2.71)= 878.64 s= 0.86 MAE=0.64 p<0.0000
ST (dyn/cm)=-3.49402(±1.097) +0.04848(±0.001).q2H(x)
-0.00163(±0.122x10-3).q0(x).q2(x) +1.21x10-5(±5.15x10-7).q0(x).q7(x) -0.01617(±0.006).q2(x) N=68 R=0.9892 q2= 0.9734 F(4.63)=722.14 s= 0.29 MAE=0.23 p<0.0000 |
| Connectivity Indices | ad hoc Descriptors | Moments of E Matrix | Quadratic Indices of M Matrix | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Prop. | na | R | s | na | R | s | na | R | s | na | R | s |
| Bp | 5 | 0.9995 | 1.86 | 5 | 0.9989 | 2.0 | 4 | 0.9984 | 2.48 | 4 | 0.9988 | 2.35 |
| MV | 5 | 0.9995 | 0.5 | 5 | 0.9995 | 0.4 | 5 | 0.9993 | 0.6 | 4 | 0.9991 | 0.75 |
| MR | 5 | 0.9999 | 0.05 | 5 | 0.9999 | 0.05 | 4 | 0.9999 | 0.05 | 3 | 0.9999 | 0.05 |
| HV | 5 | 0.9989 | 0.2 | 5 | 0.9969 | 0.4 | 3 | 0.9988 | 0.2 | 3 | 0.9980 | 0.34 |
| TC | 5 | 0.9975 | 4.1 | 5 | 0.9970 | 4.8 | 5 | 0.9944 | 5.4 | 3 | 0.9953 | 5.66 |
| PC | 5 | 0.9904 | 0.6 | 5 | 0.9889 | 0.7 | 5 | 0.9854 | 0.6 | 2 | 0.9803 | 0.86 |
| ST | 5 | 0.9929 | 0.2 | 5 | 0.9945 | 0.2 | 6 | 0.9869 | 0.3 | 4 | 0.9892 | 0.29 |
-78.0818x10 –5(±9.932x10-5).Eq9LH(x)
-10.266162(±0.707).Eq2LH(x) +10.956280x10-5(±1.32x10-5).E q14L(x)
| Equation | Set | Correlation Coefficient (R) | Standard Error (S) | Fischer ratio (F) | Average Deviation |
|---|---|---|---|---|---|
| Eq. 26 | Complete | 0.9938 | 4.006 | 1446.9 | 2.82 |
| Randić and Basak /48/ | Complete | 0.9938 | 4.039 | 2193 | 2.90 |
| Eq. 27 | Training Test | 0.9979 0.9938 | 2.97 3.17 | 1390.7 2177.9 | 2.13 2.15 |
| Eq. 11 /44/ | Training Test | 0.9953 0.9948 | 2.903 3.025 | 5733 2529 | 2.20 2.50 |
| Eq. 12 /44/ | Training Test | 0.9953 0.9948 | 3.008 2.833 | 2764 1296 | 2.20 2.48 |
| Eq. 13 /44/ | Training Test | 0.9954 0.9949 | 2.874 2.871 | 2018 841 | 2.03 2.63 |
| Alkyl alcohol | Bp exp (oC) | Bp calc. (Eq.26) | ∆* | % ∆ | Bp cal. Ref./48/ |
|---|---|---|---|---|---|
| 1. methanol | 64.70 | 65.50 | -0.80 | -1.24 | 65.24 (-0.54) |
| 2. ethanol | 78.30 | 78.43 | -0.13 | -0.17 | 77.69 (0.61) |
| 3. 1-propanol | 97.20 | 95.63 | 1.57 | 1.62 | 96.42 (0.77) |
| 4. 2. propanol | 82.30 | 85.83 | -3.53 | -4.28 | 84.11 (-1.81) |
| 5. 1-butanol | 117.70 | 113.40 | 4.30 | 3.65 | 115.67 (2.03) |
| 6. 2-butanol | 99.60 | 102.87 | -3.27 | -3.28 | 102.43 (-2.83) |
| 7. 2-methyl-1-propanol | 107.90 | 108.66 | -0.76 | -0.71 | 109.15 (-1.25) |
| 8. 2-methyl-2-propanol | 82.40 | 87.68 | -5.28 | -6.41 | 84.52 (-2.12) |
| 9. 1-pentanol | 137.80 | 133.16 | 4.64 | 3.36 | 134.92 (2.88) |
| 10. 2-pentanol | 119.00 | 120.59 | -1.59 | -1.34 | 121.68 (-2.68) |
| 11. 3-pentanol | 115.30 | 119.90 | -4.60 | -3.99 | 120.75 (-5.45) |
| 12. 2-methyl-1-butanol | 128.70 | 126.39 | 2.31 | 1.80 | 127.97 (0.73) |
| 13. 3-methyl-1-butanol | 131.20 | 127.13 | 4.07 | 3.10 | 128.90 (2.30) |
| 14. 2.methyl-2-butanol | 102.00 | 104.57 | -2.57 | -2.52 | 102.41 (-0.41) |
| 15. 3-methyl-2-butanol | 111.50 | 115.75 | -4.25 | -3.81 | 114.72 (-3.22) |
| 16. 2,2-dimethyl-1-propanol | 113.10 | 117.54 | -4.44 | -3.93 | 115.84 (-2.74) |
| 17. 1-hexanol | 157.13 | 153.12 | 4.01 | 2.55 | 154.17 (2.83) |
| 18. 2-hexanol | 139.90 | 140.35 | -0.45 | -0.32 | 140.92 (-1.02) |
| 19. 3-hexanol | 135.40 | 137.63 | -2.23 | -1.64 | 139.99 (-4.59) |
| 20. 2-methyl-1-pentanol | 148.00 | 146.14 | 1.86 | 1.25 | 147.22 (0.78) |
| 21.3-methyl-1-pentanol | 152.40 | 146.89 | 5.51 | 3.61 | 147.72 (4.8) |
| 22. 4-methyl-1-pentanol | 151.80 | 148.97 | 2.83 | 1.86 | 148.15 (3.65) |
| 23. 2-methyl-2-pentanol | 121.40 | 122.25 | -0.85 | -0.70 | 121.66 (-0.25) |
| 24. 3-methyl-2-pentanol | 134.20 | 133.42 | 0.78 | 0.58 | 133.55 (0.65) |
| 25. 4-methyl-2-pentanol | 131.70 | 134.27 | -2.57 | -1.95 | 134.90 (-3.20) |
| 26. 2-methyl-3-pentanol | 126.50 | 132.77 | -6.27 | -4.96 | 134.31 (-7.81) |
| 27. 3-methyl-3-pentanol | 122.40 | 121.45 | 0.95 | 0.78 | 120.30 (2.10) |
| 28. 2-ethyl-1-butanol | 146.50 | 144.11 | 2.39 | 1.63 | 146.79 (-0.29) |
| 29. 2,2-dimethyl-1-butanol | 136.80 | 135.21 | 1.59 | 1.16 | 134.37 (2.43) |
| 30. 2,3-dimethyl-1-butanol | 149.00 | 140.07 | 8.93 | 6.00 | 140.77 (8.23) |
| 31. 3.3-dimethyl-1-butanol | 143.00 | 136.82 | 6.18 | 4.32 | 136.11 (6.89) |
| 32. 2,3-dimethyl-2-butanol | 118.60 | 117.30 | 1.30 | 1.10 | 114.28 (4.32) |
| 33. 3,3-dimethyl-2-butanol | 120.00 | 124.47 | -4.47 | -3.72 | 121.00 (-1.00) |
| 34. 1-heptanol | 176.30 | 173.38 | 2.92 | 1.66 | 173.41 (2.87) |
| 35. 3-heptanol | 156.80 | 157.38 | -0.58 | -0.37 | 159.24 (-2.44) |
| 36. 4-heptanol | 155.00 | 155.35 | -0.35 | -0.23 | 159.24 (-4.24) |
| 37. 2-methyl-2-hexanol | 142.50 | 142.00 | 0.50 | 0.35 | 140.90 (1.60) |
| 38. 3-methyl-3-hexanol | 142.40 | 139.13 | 3.27 | 2.30 | 139.55 (2.85) |
| 39. 3-ethyl-3-pentanol | 142.50 | 138.32 | 4.18 | 2.93 | 138.37 (4.13) |
| 40. 2,3-dimethyl-2-pentanol | 139.70 | 134.92 | 4.78 | 3.42 | 133.11 (6.59) |
| 41.3,3-dimethyl-2-pentanol | 133.00 | 142.09 | -9.09 | -6.83 | 139.67 (-6.57) |
| 42. 2.2-dimethyl-3-pentanol | 136.00 | 141.49 | -5.49 | -4.04 | 139.32 (-3.32) |
| 43. 2,3-dimethyl-3-pentanol | 139.00 | 134.17 | 4.83 | 3.48 | 132.18 (6.82) |
| 44. 2,4-dimethyl-3-pentanol | 138.80 | 145.64 | -6.84 | -4.93 | 145.34 (-6.54) |
| 45. 1-octanol | 195.20 | 193.67 | 1.53 | 0.78 | 192.58 (2.62) |
| 46. 2-octanol | 179.80 | 180.57 | -0.77 | -0.43 | 179.33 (0.47) |
| 47. 2-ethyl-1-hexanol | 184.60 | 183.82 | 0.78 | 0.42 | 185.29 (-0.69) |
| 48. 2,2,3trimethyl-3-pentanol | 152.20 | 142.73 | 9.47 | 6.22 | 152.78 (-0.57) |
| 49. 1-nonanol | 213.10 | 213.97 | -0.87 | -0.41 | 211.91 (1.19) |
| 50. 2-nonanol | 198.50 | 200.85 | -2.35 | -1.19 | 198.66 (-0.16) |
| 51. 3-nonanol | 194.70 | 197.60 | -2.90 | -1.49 | 197.73 (-3.03) |
| 52. 4-nonanol | 193.00 | 195.07 | -2.07 | -1.07 | 197.73 (-4.73) |
| 53. 5-nonanol | 195.10 | 194.87 | 0.23 | 0.12 | 197.73 (-2.63) |
| 54. 7-methyl-1-octanol | 206.00 | 210.01 | -4.01 | -1.95 | 205.46 (0.54) |
| 55. 2,6-dimethyl-4-heptanol | 178.00 | 182.72 | -4.72 | -2.65 | 185.69 (-7.69) |
| 56. 3,5-dimethyl-4-hexanol | 187.00 | 180.99 | 6.01 | 3.21 | 183.83 (3.17) |
| 57. 3,3,5-trimethyl-1-hexanol | 193.00 | 192.54 | 0.46 | 0.24 | 186.98 (6.02) |
| 58. 1-decanol | 230.20 | 234.27 | -4.07 | -1.77 | 231.15 (-0.95) |
| Alkyl alcohol | Bp exp (oC) | Bp calc. (Eq. 27) | ∆* | % ∆ | Bp calc. (Eq. 11) |
|---|---|---|---|---|---|
| 1. methanol | 64.70 | 66.03 | -1.33 | -2.06 | 64.68 (0.02) |
| 2. ethanol | 78.30 | 75.96 | 2.34 | 2.99 | 77.36 (0.94) |
| 3. 1-propanol | 97.20 | 97.44 | -0.24 | -0.24 | 96.80 (0.40) |
| 4. 2. propanol | 82.30 | 80.69 | 1.61 | 1.96 | 78.24 (4.06) |
| 6.2-butanol | 99.60 | 100.08 | -0.48 | -0.48 | 97.68 (1.92) |
| 8. 2-methyl-2-propanol | 82.40 | 81.63 | 0.77 | 0.93 | 84.97 (-2.57) |
| 9. 1-pentanol | 137.80 | 137.06 | 0.74 | 0.54 | 135.69 (2.11) |
| 11. 3-pentanol | 115.30 | 118.40 | -3.10 | -2.69 | 117.13 (-1.83) |
| 14. 2.methyl-2-butanol | 102.00 | 101.74 | 0.26 | 0.26 | 104.41 (-2.41) |
| 16. 2,2-dimethyl-1-propanol | 113.10 | 116.94 | -3.84 | -3.40 | 117.11 (4.01) |
| 18. 2-hexanol | 139.90 | 138.73 | 1.17 | 0.83 | 136.57 (3.33) |
| 20. 2-methyl-1-pentanol | 148.00 | 147.82 | 0.18 | 0.12 | 148.68 (-0.68) |
| 22. 4-methyl-1-pentanol | 151.80 | 149.11 | 2.69 | 1.77 | 148.68 (3.12) |
| 26. 2-methyl-3-pentanol | 126.50 | 131.41 | -4.91 | -3.88 | 130.11 (-3.61) |
| 27. 3-methyl-3-pentanol | 122.40 | 121.41 | 0.99 | 0.81 | 123.86 (-1.46) |
| 29. 2,2-dimethyl-1-butanol | 136.80 | 132.03 | 4.77 | 3.49 | 136.55 (0.25) |
| 34. 1-heptanol | 176.30 | 175.42 | 0.88 | 0.50 | 174.57 (1.73) |
| 35. 3-heptanol | 156.80 | 156.88 | -0.08 | -0.05 | 156.01 (0.79) |
| 37. 2-methyl-2-hexanol | 142.50 | 140.89 | 1.61 | 1.13 | 143.30 (-0.80) |
| 39. 3-ethyl-3-pentanol | 142.50 | 140.75 | 1.75 | 1.23 | 143.30 (-0.80) |
| 41.3,3-dimethyl-2-pentanol | 133.00 | 136.16 | -3.16 | -2.37 | 137.43 (-4.43) |
| 44. 2,4-dimethyl-3-pentanol | 138.80 | 143.48 | -4.68 | -3.37 | 143.10 (-4.30) |
| 45. 1-octanol | 195.20 | 194.46 | 0.74 | 0.38 | 194.01 (1.19) |
| 48. 2,2,3trimethyl-3-pentanol | 152.20 | 154.18 | -1.98 | -1.30 | 144.16 (8.04) |
| 49. 1-nonanol | 213.10 | 213.32 | -0.22 | -0.10 | 213.45 (-0.35) |
| 52. 4-nonanol | 193.00 | 195.49 | -2.49 | -1.29 | 194.89 (-1.89) |
| 53. 5-nonanol | 195.10 | 195.34 | -0.24 | -0.12 | 194.89 (0.21) |
| 56. 3,5-dimethyl-4-hexanol | 187.00 | 178.80 | 8.20 | 4.39 | 181.99 (5.01) |
| 58. 1-decanol | 230.20 | 232.18 | -1.98 | -0.86 | 232.86 (-2.66) |
| Alkyl alcohol | Bp exp. (oC) | Bp calc. (Eq. 27) | ∆* | % ∆ | Bp calc.(Eq. 11) |
|---|---|---|---|---|---|
| 5. 1-butanol | 117.70 | 117.50 | 0.20 | 0.17 | 116.25 (1.45) |
| 7. 2-methyl-1-propanol | 107.90 | 112.68 | -4.78 | -4.43 | 109.79 (-1.89) |
| 10. 2-pentanol | 119.00 | 119.23 | -0.23 | -0.20 | 117.13 (1.87) |
| 12. 2-methyl-1-butanol | 128.70 | 130.00 | -1.30 | -1.01 | 129.34 (-0.64) |
| 13. 3-methyl-1-butanol | 131.20 | 131.11 | 0.09 | 0.07 | 129.23 (1.97) |
| 15. 3-methyl-2-butanol | 111.50 | 114.17 | -2.67 | -2.39 | 110.67 (0.83) |
| 17. 1-hexanol | 157.13 | 156.38 | 0.75 | 0.48 | 155.13 (1.87) |
| 19. 3-hexanol | 135.40 | 137.52 | -2.12 | -1.57 | 136.57 (-1.17) |
| 21.3-methyl-1-pentanol | 152.40 | 147.35 | 5.05 | 3.31 | 148.68 (3.72) |
| 23. 2-methyl-2-pentanol | 121.40 | 121.16 | 0.24 | 0.20 | 123.86 (-2.46) |
| 24. 3-methyl-2-pentanol | 134.20 | 131.27 | 2.93 | 2.18 | 130.11 (4.09) |
| 25. 4-methyl-2-pentanol | 131.70 | 132.55 | -0.85 | -0.65 | 130.11 (1.59) |
| 28. 2-ethyl-1-butanol | 146.50 | 146.12 | 0.38 | 0.26 | 148.68 (-2.18) |
| 30. 2,3-dimethyl-1-butanol | 149.00 | 141.00 | 8.00 | 5.37 | 142.22 (6.78) |
| 31. 3.3-dimethyl-1-butanol | 143.00 | 133.59 | 9.41 | 6.58 | 136.55 (6.45) |
| 32. 2,3-dimethyl-2-butanol | 118.60 | 119.44 | -0.84 | -0.71 | 117.40 (1.20) |
| 33. 3,3-dimethyl-2-butanol | 120.00 | 120.08 | -0.08 | -0.06 | 117.99 (2.01) |
| 36. 4-heptanol | 155.00 | 156.58 | -1.58 | -1.02 | 156.01 (-1.01) |
| 38. 3-methyl-3-hexanol | 142.40 | 141.12 | 1.28 | 0.90 | 143.30 (-0.90) |
| 40. 2,3-dimethyl-2-pentanol | 139.70 | 138.02 | 1.68 | 1.20 | 136.84 (2.86) |
| 42. 2.2-dimethyl-3-pentanol | 136.00 | 136.45 | -0.45 | -0.33 | 137.43 (-1.43) |
| 43. 2,3-dimethyl-3-pentanol | 139.00 | 138.90 | 0.10 | 0.07 | 136.84 (2.16) |
| 46. 2-octanol | 179.80 | 177.28 | 2.52 | 1.40 | 175.45 (4.35) |
| 47. 2-ethyl-1-hexanol | 184.60 | 182.69 | 1.91 | 1.03 | 187.56 (-2.96) |
| 50. 2-nonanol | 198.50 | 196.41 | 2.09 | 1.05 | 194.89 (3.61) |
| 51. 3-nonanol | 194.70 | 195.53 | -0.83 | -0.43 | 194.89 (-0.19) |
| 54. 7-methyl-1-octanol | 206.00 | 205.50 | 0.50 | 0.24 | 207.00 (1.00) |
| 55. 2,6-dimethyl-4-heptanol | 178.00 | 183.63 | -5.63 | -3.16 | 181.99 (-3.99) |
| 57. 3,3,5-trimethyl-1-hexanol | 193.00 | 190.45 | 2.55 | 1.32 | 188.43 (4.57) |
-0.3016(±4.718).q2(x) -1.75x10-5(±3.75x10-6).q14(x)
+6.42x10-6(±1.34x10-6) .q15(x)
| Equation | Set | Correlation Coefficient (R) | Standard Error (S) | Fischer ratio (F) |
|---|---|---|---|---|
| Eq. (28) two descriptors | Training Test | 0.9823 0.9726 | 7.8211 10.245 | 1058.2 421.21 |
| Eq. (29) Five descriptors | Training Test | 0.9927 0.9938 | 5.0145 4.7865 | 5257.9 2025.4 |
| Eq. (1)/(25). Six descriptors | Training Test | 0.9937 0.9943 | 4.800 4.696 | 960 2094.8 |
| no | Cycloalkane | Obsd (oC) | Cald [Eq. 28] | Res. | Cald [Eq. 29] | Res. | Cald [Eq. 1 /25 ] | Res. |
|---|---|---|---|---|---|---|---|---|
| 1 | cyclopropane | -32.8 | -14.82 | -17.98 | -16.07 | -16.73 | -36.99 | 4.19 |
| 2 | cyclobutane | 12.51 | 15.29 | -2.78 | 14.64 | -2.13 | 1.77 | 10.74 |
| 3 | spiropentane | 40.6 | 48.20 | -7.60 | 43.20 | -2.60 | 49.42 | -8.82 |
| 4 | methylcyclobutane | 36.3 | 38.57 | -2.27 | 38.83 | -2.53 | 33.49 | 2.81 |
| 5 | cyclopentane | 49.262 | 48.20 | 1.06 | 43.20 | 6.06 | 52.5 | -3.24 |
| 6 | 1,1-dimethylcyclopropane | 20.63 | 24.92 | -4.29 | 26.19 | -5.56 | 23.95 | -3.32 |
| 7 | cis-1,2-dimethylcyclopropane | 37.03 | 31.74 | 5.29 | 31.66 | 5.37 | 30.15 | 6.88 |
| 8 | ethylcyclopropane | 36 | 38.57 | -2.57 | 37.95 | -1.95 | 37.46 | -1.46 |
| 9 | bicyclo[3.1.0]hexane | 79.2 | 85.14 | -5.94 | 73.57 | 5.63 | 85.82 | -6.62 |
| 10 | 1,1-dimethylcyclobutane | 56 | 55.03 | 0.97 | 53.43 | 2.57 | 54.31 | 1.69 |
| 11 | cis-1,2-dimethylcyclobutane | 68 | 61.85 | 6.15 | 62.67 | 5.33 | 62.41 | 5.59 |
| 12 | tras-1,2-dimethylcyclobutane | 60 | 61.85 | -1.85 | 62.67 | -2.67 | 62.41 | -2.41 |
| 13 | cis-1,3-dimethylcyclobutane | 60.5 | 61.85 | -1.35 | 61.01 | -0.51 | 59.56 | 0.94 |
| 14 | tras-1,3-dimethylcyclobutane | 57.5 | 61.85 | -4.35 | 61.01 | -3.51 | 59.56 | -2.06 |
| 15 | cyclohexane | 80.738 | 75.50 | 5.24 | 76.05 | 4.68 | 84.36 | -3.62 |
| 16 | methylcyclopentane | 71.812 | 68.68 | 3.14 | 69.84 | 1.98 | 75.98 | -4.17 |
| 17 | 1,1,2-trimethylcyclopropane | 52.48 | 48.20 | 4.28 | 50.35 | 2.13 | 54.66 | -2.18 |
| 18 | cis,cis-1,2,3,-trimethylcyclopropane | 71 | 55.03 | 15.97 | 71.01 | -0.01 | 61.37 | 9.63 |
| 19 | cis,trans-1,2,3,-trimethylcyclopropane | 66 | 55.03 | 10.97 | 55.10 | 10.90 | 61.37 | 4.63 |
| 20 | cis-1-ethyl-2-ethylcyclopropane | 70 | 91.96 | -21.96 | 70.495 | -0.495 | 64.86 | 5.14 |
| 21 | propylcyclopropane | 68.5 | 68.68 | -0.18 | 68.11 | 0.39 | 72.82 | -4.32 |
| 22 | isopropylcyclopropane | 58.34 | 61.85 | -3.51 | 62.15 | -3.81 | 63.18 | -4.84 |
| 23 | bicyclo[3.2.0]heptane | 109.3 | 115.24 | -5.94 | 103.51 | 5.79 | 112.2 | -2.9 |
| 24 | bicyclo[4.1.0]heptane | 111.5 | 115.24 | -3.74 | 103.60 | 7.90 | 111.69 | -0.19 |
| 25 | 2-cyclopropylbutane | 90.98 | 91.96 | -0.98 | 91.89 | -0.91 | 94.75 | -3.77 |
| 26 | propylcyclobutane | 100.6 | 115.24 | -14.64 | 103.52 | -2.92 | 100.42 | 0.18 |
| 27 | isopropylcyclobutane | 92.7 | 91.96 | 0.74 | 93.02 | -0.32 | 91.13 | 1.57 |
| 28 | methylcyclohexane | 100.93 | 98.78 | 2.15 | 100.42 | 0.52 | 104.36 | -3.43 |
| 29 | 1,1-dimethylcyclopentane | 87.846 | 85.14 | 2.71 | 86.44 | 1.40 | 90.62 | -2.77 |
| 30 | trans-1,2-dimethylcyclopentane | 91.869 | 91.96 | -0.09 | 93.48 | -1.61 | 98.15 | -6.28 |
| 31 | cis-1,3-dimethylcyclopentane | 91.725 | 91.96 | -0.24 | 93.68 | -1.95 | 95.52 | -3.79 |
| 32 | trans-1,3-dimethylcyclopentane | 90.773 | 91.96 | -1.19 | 93.68 | -2.90 | 95.52 | -4.75 |
| 33 | 1,1,2,2-tetramethylcyclopropane | 75.6 | 64.66 | 10.94 | 75.64 | -0.04 | 74.28 | 1.32 |
| 34 | 1,1,2,3-tetramethylcyclopropane | 78.5 | 71.49 | 7.01 | 78.08 | 0.42 | 84.01 | -5.51 |
| 35 | 1-methyl-1-isopropylcyclopropane | 82.1 | 78.31 | 3.79 | 80.28 | 1.82 | 84.83 | -2.73 |
| 36 | 1,1-dimethylcyclopropane | 88.67 | 85.14 | 3.53 | 84.92 | 3.75 | 92.95 | -4.28 |
| 37 | 2-methylbicyclo[2.2.1]heptane | 125.8 | 138.53 | -12.73 | 127.90 | -2.10 | 130.33 | -4.53 |
| 38 | 3,3-dimethylbicyclo[3.1.0]hexane | 115.3 | 124.88 | -9.58 | 119.06 | -3.76 | 110.49 | 4.81 |
| 39 | 1,1,3,3-tetramethylcyclobutane | 78.2 | 94.77 | -16.57 | 75.30 | 2.90 | 86.57 | -8.37 |
| 40 | trans-1,2-diethylcyclobutane | 115.5 | 122.07 | -6.57 | 121.40 | -5.90 | 122.24 | -6.74 |
| 41 | methylcycloheptane | 134 | 128.89 | 5.11 | 131.20 | 2.80 | 133.38 | 0.62 |
| 42 | 1,1-dimethylcyclohexane | 119.54 | 115.24 | 4.30 | 116.01 | 3.53 | 116.49 | 3.05 |
| 43 | trans-1,2-imethylcyclohexane | 123.42 | 122.07 | 1.35 | 124.23 | -0.81 | 123.9 | -0.48 |
| 44 | cis-1,3-dimethylcyclohexane | 120.09 | 122.07 | -1.98 | 123.67 | -3.59 | 121.28 | -1.19 |
| 45 | trans-1,3-dimethylcyclohexane | 124.45 | 122.07 | 2.38 | 123.67 | 0.78 | 121.28 | 3.17 |
| 46 | cis-1,4-dimethylcyclohexane | 124.32 | 122.07 | 2.25 | 124.90 | -0.58 | 121.51 | 2.81 |
| 47 | ethylcyclohexane | 131.78 | 128.89 | 2.89 | 130.24 | 1.54 | 133.19 | -1.41 |
| 48 | cyclooctane | 151.14 | 135.72 | 15.42 | 137.47 | 13.67 | 145.2 | 5.89 |
| 49 | 1,1,2-trimethylcyclopentane | 113.73 | 108.42 | 5.31 | 110.08 | 3.65 | 112.39 | 1.34 |
| 50 | cis,cis-1,1,3-trimethylcyclopentane | 123 | 115.24 | 7.76 | 116.71 | 6.29 | 117 | 6 |
| 51 | cis,trans-1,1,3-trimethylcyclopentane | 117.5 | 115.24 | 2.26 | 116.71 | 0.79 | 117 | 0.5 |
| 52 | trans,cis-1,1,3-trimethylcyclopentane | 110.2 | 115.24 | -5.04 | 116.71 | -6.51 | 117 | -6.8 |
| 53 | 1-ethyl-1-methylcyclopentane | 121.52 | 115.24 | 6.28 | 115.75 | 5.77 | 121.05 | 0.47 |
| 54 | isopropylcyclopentane | 126.42 | 122.07 | 4.35 | 123.75 | 2.67 | 127.4 | -0.98 |
| 55 | 1,1,2-trimethyl-2-ethylcyclopropane | 104 | 94.77 | 9.23 | 108.34 | -4.34 | 103.22 | 0.78 |
| 56 | 1-methyl-1,2-diethylcyclopropane | 108.5 | 108.42 | 0.08 | 110.79 | -2.29 | 114.83 | -6.83 |
| 57 | 7,7-bicycloylbicyclo[2.2.1]heptane | 143.5 | 124.88 | 18.62 | 141.78 | 1.72 | 143.2 | 0.3 |
| 58 | 2-ethylbicyclo[2.2.1]heptane | 146.5 | 168.64 | -22.14 | 157.75 | -11.25 | 154.66 | -8.16 |
| 59 | 4-methylspiro[5.2]octane | 149 | 161.81 | -12.81 | 155.20 | -6.20 | 151.49 | -2.49 |
| 60 | 1,2-dimethylcycloheptane | 153 | 152.18 | 0.82 | 154.91 | -1.91 | 150.71 | 2.29 |
| 61 | 1,1,2-trimethylcyclohexane | 145.2 | 138.53 | 6.67 | 140.19 | 5.01 | 136.28 | 8.92 |
| 62 | 1,1,3-trimethylcyclohexane | 136.63 | 138.53 | -1.90 | 137.22 | -0.59 | 130.74 | 5.88 |
| 63 | 1,1,4-trimethylcyclohexane | 135 | 138.53 | -3.53 | 141.47 | -6.47 | 131.32 | 3.68 |
| 64 | 1-ethyl-1-methylcyclohexane | 152.16 | 145.35 | 6.81 | 145.61 | 6.55 | 144.59 | 7.57 |
| 65 | propylcyclohexane | 156.72 | 159.00 | -2.28 | 160.30 | -3.58 | 159.77 | -3.06 |
| 66 | isopropylcyclohexane | 154.76 | 152.18 | 2.59 | 154.45 | 0.31 | 150.6 | 4.16 |
| 67 | cyclononane | 178.4 | 165.82 | 12.58 | 168.18 | 10.22 | 171.95 | 6.45 |
| 68 | 1,1,2,2-tetramethylcyclopentane | 135 | 124.88 | 10.12 | 129.67 | 5.33 | 124.67 | 10.36 |
| 69 | 1,1,3,3--tetramethylcyclopentane | 117.96 | 124.88 | -6.92 | 125.09 | -7.13 | 115.29 | 2.67 |
| 70 | cis-1,2-dimethyl-1-ethylcyclopentane | 143 | 138.53 | 4.47 | 139.53 | 3.47 | 140.15 | 3.15 |
| 71 | trans-1,2-dimethyl-1-ethylcyclopentane | 142 | 138.53 | 3.47 | 139.53 | 2.47 | 140.15 | 2.15 |
| 72 | 1-methyl-1-propylcyclopentane | 146 | 145.35 | 0.65 | 145.04 | 0.96 | 147.4 | -1.4 |
| 73 | 1,1-diethylcyclopentane | 151 | 145.35 | 5.65 | 145.05 | 5.95 | 148.92 | 2.08 |
| 74 | trans-1,3-dietjhylcyclopentane | 150 | 152.18 | -2.18 | 152.91 | -2.91 | 150.87 | -0.87 |
| 75 | cis-1-methyl-3-isopropylcyclopentane | 142 | 145.35 | -3.35 | 147.58 | -5.58 | 141.76 | 1.76 |
| 76 | trans-1-methyl-3-isopropylcyclopentane | 143 | 145.35 | -2.35 | 147.58 | -4.58 | 141.76 | 2.76 |
| 77 | isobutylcyclopentane | 147.95 | 152.18 | -4.23 | 154.29 | -6.34 | 151.47 | -3.52 |
| 78 | sec-butylcyclopentane | 154.35 | 152.18 | 2.17 | 153.42 | 0.93 | 153.79 | 0.56 |
| 79 | 2-cyclopropylhexane | 142.95 | 152.18 | -9.23 | 152.67 | -9.72 | 150.35 | -7.4 |
| 80 | 3-cyclobutylpentane | 151.5 | 152.18 | -0.68 | 152.06 | -0.56 | 146.12 | 5.38 |
| no | Cycloalkane | Obsd (oC) | Cald [Eq. 28] | Res. | Cald [Eq. 29] | Res. | Cald [Eq. 1 /25 ] | Res. |
|---|---|---|---|---|---|---|---|---|
| 1 | methylcyclopropane | 0.73 | 8.46 | -7.73 | 8.35 | -7.62 | -2.34 | 3.07 |
| 2 | trans-1,2-dimethylcyclopropane | 28.21 | 31.74 | -3.53 | 31.66 | -3.45 | 30.15 | -1.94 |
| 3 | bicyclo[2.2.0]hexane | 80.2 | 85.14 | -4.94 | 73.41 | 6.79 | 78.97 | 1.23 |
| 4 | ethylcyclobutane | 70.6 | 68.68 | 1.92 | 68.71 | 1.89 | 68.66 | 1.94 |
| 5 | 1-ethyl-1-methylcyclopropane | 56.77 | 55.03 | 1.74 | 55.46 | 1.31 | 60.36 | -3.59 |
| 6 | trans-1,2-diethylcyclopropane | 65 | 91.96 | -26.96 | 64.80 | 0.2 | 64.86 | 0.14 |
| 7 | cycloheptane | 118.79 | 105.61 | 13.18 | 106.76 | 12.03 | 116.11 | 2.68 |
| 8 | cis-1,2-dymethylcyclopentane | 99.532 | 91.96 | 7.57 | 93.48 | 6.05 | 98.15 | 1.382 |
| 9 | ethylcyclopentane | 103.46 | 98.78 | 4.68 | 99.56 | 3.90 | 107.67 | -4.204 |
| 10 | spiro[5.2]octane | 125.5 | 138.53 | -13.03 | 128.38 | -2.88 | 135.02 | -9.52 |
| 11 | cis-1,2-dimethylcyclohexane | 129.72 | 122.07 | 7.65 | 124.23 | 5.49 | 123.9 | 5.828 |
| 12 | trans-1,4-dimethylcyclohexane | 119.35 | 122.07 | -2.72 | 124.90 | -5.55 | 121.51 | -2.159 |
| 13 | 1,1,2-trimethylcyclopentane | 104.89 | 108.42 | -3.53 | 110.08 | -5.18 | 106.86 | -1.967 |
| 14 | propylcyclopentane | 130.95 | 128.89 | 2.06 | 129.68 | 1.27 | 136.57 | -5.621 |
| 15 | 2-cyclopropylpentane | 117.74 | 122.07 | -4.33 | 122.09 | -4.35 | 123.66 | -5.92 |
| 16 | cis-bicyclo[4.3.0]nonane | 166 | 175.46 | -9.46 | 164.38 | 1.62 | 164.59 | 1.41 |
| 17 | 1,1-dimethyl-2-ethylcyclopentane | 138 | 138.53 | -0.53 | 138.78 | -0.78 | 138.33 | -0.33 |
| 18 | 1,1-dimethylcyclopentane | 133 | 138.53 | -5.53 | 139.46 | -6.46 | 133.37 | -0.37 |
| 19 | cis-1,3-diethylcyclopentane | 150 | 152.18 | -2.18 | 152.91 | -2.91 | 150.87 | -0.87 |
| 20 | butylcyclopentane | 156.6 | 159.00 | -2.40 | 160.22 | -3.62 | 163.27 | -6.67 |
| 21 | tert-butylcyclopentane | 144.85 | 138.53 | 6.32 | 140.05 | 4.80 | 138.18 | 6.67 |
| 22 | dicyclobutylmethane | 161.8 | 175.46 | -13.66 | 164.47 | -2.67 | 152.11 | 9.69 |
| 23 | 1,5-dimethylspiro[3.3]heptane | 132.2 | 154.99 | -22.79 | 135.25 | -3.05 | 142.44 | -10.24 |
| 24 | 4-methylspiro[5.2]octane | 149 | 161.81 | -12.81 | 155.20 | -6.20 | 151.49 | -2.49 |
| 25 | 2,6-dimethylbicyclo[3.2.1]octane | 164.5 | 191.92 | -27.42 | 165.4 | -0.90 | 165.41 | -0.91 |
| 26 | 3,7-dimethylbicyclo[3.3.0]octane | 166 | 191.92 | -25.92 | 166.03 | -0.03 | 165.6 | 0.4 |
| Compound | Obs. (oC) | Calc. | Res. | R-CV | Compound | Obs. (oC) | Calc. | Res. | R-CV |
|---|---|---|---|---|---|---|---|---|---|
| Chlorobenzene | 132.00 | 130.79 | 1.21 | 1.34 | Mesitylene | 165.00 | 169.99 | -4.99 | -5.24 |
| m-Nitrochlorobenzene | 236.00 | 235.11 | 0.89 | 1.25 | Prehnitene | 205.00 | 191.08 | 13.92 | 15.14 |
| p-Nitrochlorobenzene | 239.00 | 237.21 | 1.79 | 2.48 | Isodurene | 197.00 | 191.08 | 5.92 | 6.44 |
| Aniline | 184.00 | 187.35 | -3.35 | -3.57 | Durene | 195.00 | 191.08 | 3.92 | 4.26 |
| Phenol | 181.00 | 174.56 | 6.44 | 6.78 | Pentamethylbenzene | 231.00 | 212.18 | 18.82 | 21.97 |
| o-Cresol | 191.00 | 193.84 | -2.84 | -2.95 | Ethylbenzene | 136.00 | 141.26 | -5.26 | -5.54 |
| m-Cresol | 201.00 | 194.85 | 6.15 | 6.34 | n-Propylbenzene | 152.00 | 158.55 | -6.55 | -7.01 |
| p-Cresol | 201.00 | 195.22 | 5.78 | 5.95 | tert-Butylbenzene | 169.00 | 179.64 | -10.64 | -11.92 |
| o-Toluic Acid | 259.00 | 265.28 | -6.28 | -6.68 | p-Cymene | 177.00 | 179.64 | -2.64 | -2.96 |
| m- Toluic Acid | 263.00 | 266.40 | -3.40 | -3.63 | Biphenyl | 255.00 | 257.78 | -2.78 | -3.20 |
| p- Toluic Acid | 275.00 | 267.05 | 7.95 | 8.52 | Diphenylmethane | 263.00 | 271.25 | -8.25 | -9.32 |
| o-Tolualdehyde | 196.00 | 197.50 | -1.50 | -1.56 | Styrene | 145.00 | 153.11 | -8.11 | -8.65 |
| m-Tolualdehyde | 199.00 | 198.25 | 0.75 | 0.78 | Phenylacetaldehyde | 193.00 | 200.62 | -7.62 | -8.65 |
| p-Tolualdehyde | 205.00 | 198.68 | 6.32 | 6.61 | Diphenylether | 259.00 | 281.11 | -22.11 | -24.23 |
| o-Bromophenol | 194.00 | 191.36 | 2.64 | 2.82 | Benzyl Alcohol | 205.00 | 194.72 | 10.28 | 10.72 |
| p-Fluorophenol | 185.00 | 189.05 | -4.05 | -6.64 | α-Phenylethyl Alcohol | 205.00 | 212.19 | -7.19 | -7.54 |
| o-Phenylenediamine | 252.00 | 265.08 | -13.08 | -15.96 | β-Phenylethyl Alcohol | 221.00 | 211.43 | 9.57 | 10.37 |
| p-Phenylenediamine | 267.00 | 267.44 | -0.44 | -0.53 | α-Picoline | 128.00 | 136.75 | -8.75 | -9.50 |
| o-Toluidine | 200.00 | 207.11 | -7.11 | -7.48 | β-Picoline | 143.00 | 139.17 | 3.83 | 4.10 |
| m-Toluidine | 203.00 | 207.85 | -4.85 | -5.08 | γ-Picoline | 144.00 | 139.75 | 4.25 | 4.53 |
| p-Toluidine | 200.00 | 208.13 | -8.13 | -8.51 | Phthalyc Anhydride | 284.00 | 280.66 | 3.34 | 4.85 |
| Benzoic Acid | 250.00 | 245.95 | 4.05 | 4.28 | Naphthalene | 218.00 | 215.18 | 2.82 | 3.23 |
| Benzaldehyde | 178.00 | 177.58 | 0.42 | 0.45 | 1-Methylnaphthalene | 241.00 | 236.28 | 4.72 | 5.23 |
| m-Anisidine | 251.00 | 244.98 | 6.02 | 6.52 | 2-Methylnaphthalene | 240.00 | 236.28 | 3.72 | 4.12 |
| p-Anisidine | 244.00 | 245.78 | -1.78 | -1.93 | 1-Naphtylamine | 301.00 | 292.10 | 8.90 | 9.90 |
| o-Nitroaniline | 284.00 | 287.79 | -3.79 | -5.32 | 2-Naphtylamine | 294.00 | 294.61 | -0.61 | -0.69 |
| N-Methylaniline | 196.00 | 184.00 | 12.00 | 12.38 | 1-Naphthol | 280.00 | 277.96 | 2.04 | 2.20 |
| Acetophenone | 202.00 | 196.57 | 5.43 | 5.65 | 2-Naphthol | 286.00 | 281.38 | 4.62 | 5.04 |
| Benzophenone | 308.00 | 310.04 | -2.04 | -2.33 | Phenylthiol | 169.50 | 157.48 | 12.02 | 12.85 |
| Benzoyl Chloride | 197.00 | 200.84 | -3.84 | -4.08 | 9,10-Anthraquinone | 380.00 | 374.99 | 5.01 | 9.29 |
| o-Xylene | 144.00 | 148.89 | -4.89 | -5.12 | Pyrrole | 130.00 | 120.91 | 9.09 | 10.14 |
| m-Xylene | 139.00 | 148.89 | -9.89 | -10.35 | Pyridine | 115.00 | 120.15 | -5.15 | -5.75 |
| p-Xylene | 138.00 | 148.89 | -10.89 | -11.40 | Furfuryl Alcohol | 171.00 | 175.81 | -4.81 | -5.42 |
| 1, 2, 3-Trimethyl benzene | 176.00 | 169.99 | 6.01 | 6.32 | Phenylacetic Acid | 266.00 | 275.84 | -9.84 | -12.57 |
| Pseudocumene | 169.00 | 169.99 | -0.99 | -1.04 | Cathechol | 245.00 | 237.21 | 7.79 | 8.70 |
Colinearity between variables and redundancy of information
| Compound | Obs. (oC) | Cal. | Res. | Compound | Obs. (oC) | Cal. | Res. |
|---|---|---|---|---|---|---|---|
| o-Chlorotoluene | 159.00 | 150.17 | 8.83 | sec-butylbenzene | 173.50 | 172.02 | 1.48 |
| m-Chlorotoluene | 162.00 | 151.12 | 10.88 | tert-butylbenzene | 284.00 | 284.72 | -0.72 |
| p-Chlorotoluene | 162.00 | 151.48 | 10.52 | Cinnamylic Alcohol | 257.50 | 239.34 | 18.16 |
| o-Nitrobenzene | 245.00 | 229.54 | 15.46 | 1,4-Dihidronaphthalene | 212.00 | 199.52 | 12.48 |
| m-Chlorophenol | 214.00 | 196.98 | 17.02 | Isoquinoline | 243.00 | 222.61 | 20.39 |
| m-Phenylendiamine | 287.00 | 266.86 | 20.14 | Phenanthrene | 340.00 | 323.67 | 16.33 |
| o-Chloroaniline | 209.00 | 207.48 | 1.52 | Thiophene | 84.00 | 90.31 | -6.31 |
| m-Nitroaniline | 307.00 | 292.21 | 14.79 | m-Bromophenol* | 236.00 | 194.79 | 41.21 |
| N,N-Dimethylaniline | 194.00 | 182.57 | 11.43 | o-Anisidine* | 225.00 | 241.99 | -16.99 |
| Diphenylaniline | 302.00 | 301.00 | 1.00 | p-Nitroaniline* | 232.00 | 293.93 | -61.93 |
| n-Propylbenzene | 159.00 | 154.73 | 4.27 | Hexamethylbenzene* | 264.00 | 233.28 | 30.72 |
| n-Butylbenzene | 183.00 | 168.20 | 14.80 | Furan* | 32.00 | 105.28 | -73.28 |
| Isobutylbenzene | 171.00 | 173.72 | -2.72 |
| eq2(x) | Heq1L(x) | Eeq1L(x) | Eeq5L(x) | eq0H(x) | Eeq4LH(x) |
|---|---|---|---|---|---|
| 1.0000 | 0.1824 | 0.4142 | -0.3593 | -0.8106 | -0.1738 |
| 1.0000 | 0.3980 | 0.1503 | -0.0116 | -0.4667 | |
| 1.0000 | -0.2225 | -0.2098 | -0.6433 | ||
| 1.0000 | 0.1378 | -0.5776 | |||
| 1.0000 | 0.1826 | ||||
| 1.0000 |
| Descriptors | Multiple R | Multiple R-square | R-square change | Partial Correlation. | Tolernce | R-square |
|---|---|---|---|---|---|---|
| eq2(x) | 0.8063 | 0.6501 | 0.6501 | 0.9421 | 0.2060 | 0.7940 |
| Heq1L(x) | 0.9653 | 0.9317 | 0.2817 | 0.9527 | 0.6936 | 0.3064 |
| Eeq1L(x) | 0.9775 | 0.9555 | 0.0238 | 0.5129 | 0.0366 | 0.9634 |
| Eeq5L(x) | 0.9865 | 0.9732 | 0.0176 | -0.6647 | 0.0346 | 0.9654 |
| eq0H(x) | 0.9885 | 0.9772 | 0.0040 | 0.4687 | 0.2657 | 0.7343 |
| Eeq4LH(x) | 0.9904 | 0.9809 | 0.0037 | 0.4046 | 0.0221 | 0.9779 |
Interpretation of QSPR models
Molecular Branching)
 | |||||||
| Atom (f) | q0L(x, f) | q1L(x, f) | q2L(x, f) | q14L(x, f) | q15L(x, f) | BpA [0C; (Eq. 28)] | BpB [oC; (Eq. 29) |
| a | 6.9169 | 27.6676 | 55.3352 | 3605884 | 9077470 | 47.07 | 34.90 |
| b | 6.9169 | 20.7507 | 55.3352 | 3153885 | 7816879 | 25.19 | 20.34 |
| c | 6.9169 | 13.8338 | 48.4183 | 2717007 | 6759769 | 6.73 | 8.92 |
| e | 6.9169 | 13.8338 | 34.5845 | 1744048 | 4293673 | 13.55 | 13.68 |
| f | 6.9169 | 6.9169 | 13.8338 | 687788.9 | 1744048 | 1.91 | 7.30 |
| d | 6.9169 | 6.9169 | 27.6676 | 1462530 | 3605884 | -4.91 | 2.01 |
| g | 6.9169 | 13.8338 | 27.6676 | 1493988 | 3759467 | 16.96 | 16.56 |
| h | 6.9169 | 6.9169 | 13.8338 | 605581.5 | 1493988 | 1.91 | 7.09 |
| Total | 55.3352 | 110.6704 | 276.676 | 15470712 | 38551176 | 108.42 | 110.79 |
BpB (a)=(-108.197/8) +1.6358.q0L(x, a) +2.038.q1L(x, a)–0.3016.q2L(x, a)
-1.75x10-5.q14L(x, a) +6.42x10-6. q15L(x, a)=34.90 oC
BpA (b)= (-105.146/8) +3.1629.q1L(x, b)–0.4933.q2L(x, b)=25.19 oC
BpB (b)= (-108.197/8) +1.6358.q0L(x, b)+2.038.q1L(x, b)–0.3016.q2L(x, b)
-1.75x10-5.q14L(x, b) +6.42x10-6. q15L(x, b)=20.34 oC
BpA (c)= (-105.146/8) +3.1629.q1L(x, c)–0.4933.q2L(x, c)=6.73 oC
BpB (c)= (-108.197/8) +1.6358.q0L(x, c)+2.038.q1L(x, c)–0.3016.q2L(x, c)
-1.75x10-5.q14L(x, c) +6.42x10-6. q15L(x, c)=8.92 oC
BpA (d)= (-105.146/8) +3.1629.q1L(x, d)–0.4933.q2L(x, d)=-4.91 oC
BpB (d)= (-108.197/8) +1.6358.q0L(x, d)+2.038.q1L(x, d)–0.3016.q2L(x, d)
-1.75x10-5.q14L(x, d) +6.42x10-6. q15L(x, d)=13.68 oC
BpA (e)= (-105.146/8) +3.1629.q1L(x, e)–0.4933.q2L(x, e)=13.55 oC
BpB (e)= (-108.197/8) +1.6358.q0L(x, e) +2.038.q1L(x, e)–0.3016.q2L(x, e)
-1.75x10-5.q14L(x, e) +6.42x10-6. q15L(x, e)=13.68 oC
BpA (f)= (-105.146/8) +3.1629.q1L(x, f)–0.4933.q2L(x, f)=1.91 oC
BpB (f)= (-108.197/8) +1.6358.q0L(x, f)+2.038.q1L(x, f)–0.3016.q2L(x, f)
-1.75x10-5.q14L(x, f) +6.42x10-6. q15L(x, f)=7.30 oC
BpA (g)= (-105.146/8) +3.1629.q1L(x, g)–0.4933.q2L(x, g)=16.96 oC
BpB (g)= (-108.197/8) +1.6358.q0L(x, g)+2.038.q1L(x, g)–0.3016.q2L(x, g)
-1.75x10-5.q14L(x, g) +6.42x10-6. q15L(x, g)=16.56 oC
BpA (h)= (-105.146/8) +3.1629.q1L(x, h)–0.4933.q2L(x, h)=1.91 oC
BpB (h)= (-108.197/8) +1.6358.q0L(x, h)+2.038.q1L(x, h)–0.3016.q2L(x, h)
-1.75x10-5.q14L(x, h) +6.42x10-6. q15L(x, h)=7.09 oC
BpB (Molecule)=-108.197+1.6358.q0(x)+2.038.q1(x)–0.3016.q2(x)-1.75x10-5.q14(x)
+6.42x10-6. q15(x)=110.79 oC
Conclusions
Acknowledgements
References
- Devlin, J. P. (Ed.) High Throughput Screening; Marcel Dekker: New York, 2000.
- Broach, J. R.; Thorner, J. High-Throughput Screening for Drug Discovery. Nature 1996, 384 Suppl., 14–16. [Google Scholar]
- Walters, W. P.; Stahl, M. T.; Murcko, M. A. Virtual Screening-an Overview. Drug Disc Today. 1998, 3, 160–178. [Google Scholar]
- Drie, J. H. V.; Lajinees, M. S. Approaches to Virtual Library Design. Drug Disc Today. 1998, 3, 274–283. [Google Scholar]
- de Julián-Ortiz, J. V.; Gálvez, J.; Muñoz-Collado, C.; García- Domenech, R.; Gimeno-Cardona, C. Virtual Combinatorial Synthesis and Computational Screening of New Potential Anti-Herpes Compounds. J Med Chem. 1999, 42, 3308–3314. [Google Scholar]
- Van de Waterbeemd, H.; Carter, R. E.; Grassy, G.; Kubinyi, H.; Martin, Y. C.; Tute, M. S.; Willett, P. Annu. Rep. Med. Chem. 1998, 33, 397.
- Karelson, M. Molecular Descriptors in QSAR/ QSPR; John Wiley & Sons: New York, 2000. [Google Scholar]
- Katritzky, A. R.; Gordeeva, E. V. Traditional Topological Indexes vs Electronic, Geometrical, and Combined Molecular Descriptors in QSAR/QSPR Research. J. Chem. Inf. Comput. Sci. 1993, 33, 835. [Google Scholar]
- Kier, L. B.; Hall, L. H. Molecular Structure Description. The Electrotopological State; Academic Press: New York, 1999. [Google Scholar]
- Balaban, A. Topological and Stereochemical Molecular Descriptors for Databases Useful in QSAR, Similarity/Dissimilarity and Drug Design. SAR QSAR Environ. Res. 1998, 8, 1–21. [Google Scholar]
- Estrada, E. On the Topological Sub-Structural Molecular Design (TOSS-MODE) in QSPR/QSAR and Drug Design Research. SAR QSAR Environ. Res. 2000, 11, 55–73. [Google Scholar]
- Randić, M. Encyclopedia of Computational Chemistry; Schleyer, P. V. R., Ed.; John Wiley & Sons: New York, 1998; Vol. 5, pp. 3018–3032. [Google Scholar]
- Rouvray, D. H. Mathematical and Computational Concepts in Chemistry; Trinajstic, N., Ed.; Ellis Horwood: Chichester, 1986; pp. 295–306. [Google Scholar]
- Balaban, A. T. (Ed.) From Chemical Graphs to Three-Dimensional Geometry; Plenum Press: New York, 1997.
- Todeschini, R.; Consoni, V. Handbook of molecular descriptors; Wiley VCH, Weinheim: Germany, 2000. [Google Scholar]
- Topological Indices and Related Descriptors in QSAR and QSPR; Devillers, J.; Balaban, A. T. (Eds.) Gordon and Breach: Amsterdam, the Netherlands, 1999.
- Estrada, E.; Uriarte, E. Recent Advances on the Role of Topological Indices in Drug Discovery Research. Curr. Med. Chem. 2001, 8, 1699–1714. [Google Scholar]
- Wiener, H. Structural Determination of Paraffin Boiling Point. J. Am. Chem. Soc. 1947, 69, 17–20. [Google Scholar]
- Balaban, A. T. Highly Discriminant Distance-Based Topological Index. Chem. Phys. Lett. 1982, 89, 399–404. [Google Scholar]
- Randić, M. Characterization of Molecular Branching. J. Am. Chem. Soc. 1975, 69, 6609–6615. [Google Scholar]
- Kier, L. B.; Hall, L. H. Molecular Structure Description. The Electrotopological State; Academic Press: New York, 1999. [Google Scholar]
- Plavšić, D.; Nikolić, S.; Trinajstić, N.; Mihalić, Z. On the Harary Index for the Characterization of Chemical Graphs. J. Math. Chem. 1993, 12, 235–250. [Google Scholar]
- Estrada, E. Spectral Moment of Edge Adjacency Matrix in Molecular Graphs.1. Definition and Application to the Prediction of Physical Properties of Alkanes. J. Chem. Inf. Comp. Sci. 1996, 36, 846–849. [Google Scholar]
- Estrada, E. Spectral Moment of Edge Adjacency Matrix in Molecular Graphs. 2. Molecules Containing Heteroatom and QSAR Applications. J. Chem. Inf. Comp. Sci. 1997, 37, 320–328. [Google Scholar]
- Estrada, E. Spectral Moment of Edge Adjacency Matrix in Molecular Graphs 3. Molecules Containing Cycles. J. Chem. Inf. Comp. Sci. 1998, 38, 123–27. [Google Scholar]
- Randić, M. Generalized Molecular Descriptors. J. Math. Chem. 1991, 7, 155–168. [Google Scholar]
- Mihalic, Z.; Trinajstić, N. A Graph-Theoretical Approach to Structure-Property Relationships. J. Chem. Educ. 1992, 69, 701–712. [Google Scholar]
- Diudea, M. V. (Ed.) QSPR/QSAR Studies by Molecular Descriptors; Nova Science, Huntington: New York, 2001.
- Ivanciuc, O.; Ivanciuc, T.; Cabrol–Bass, D.; Balaban, A. T. Evaluation in Quantitative Structure–Property Relationship Models of Structural Descriptors Derived from Information–Theory Operators. J. Chem. Inf. Comput. Sci. 2000, 40, 631–643. [Google Scholar]
- Balaban, T.; Mills, D.; Ivanciuc, O.; Basak, S. C. Reverse Wiener Indices. Croat. Chem. Acta. 2000, 73, 923. [Google Scholar]
- Ivanciuc, O.; Ivanciuc, T.; Klein, D. J.; Seitz, W. A.; Balaban, A. T. Wiener Index Extension by Counting Even/Odd Graph Distances. J. Chem. Inf. Comput. Sci. 2001, 41, 536–549. [Google Scholar]
- Torrens, F. Valence Topological Charge-Transfer Indices for Dipole Moments. Molecules 2003, 8, 169–185. [Google Scholar]
- Rios–Santamarina, I.; García–Domenech, R.; Cortijo, J.; Santamaria, P.; Morcillo, E. J.; Gálvez, J. Natural Compounds with Bronchodilator Activity Selected by Molecular Topology. Internet Electron. J. Mol. Des. 2002, 1, 70–79. http://www.biochempress.com. [Google Scholar]
- Marino, D. J. G.; Peruzzo, P. J.; Castro, E. A.; Toropov, A. A. QSAR Carcinogenic Study of Methylated Polycyclic Aromatic Hydrocarbons Based on Topological Descriptors Derived from Distance Matrices and Correlation Weights of Local Graph Invariants. Internet Electron. J. Mol. Des. 2002, 1, 115–133. http://www.biochempress.com. [Google Scholar]
- Ivanciuc, O. QSAR Comparative Study of Wiener Descriptors for Weighted Molecular Graphs. J. Chem. Inf. Comput. Sci. 2000, 40, 1412–1422. [Google Scholar]
- Marrero, Y.; Romero, V. TOMO-COMD software. Central University of Las Villas, 2002; TOMO-COMD (TOpological MOlecular COMputer Design) for Windows, version 1.0 is a preliminary experimental version; in the future a professional version may be obtained upon request to Y. Marrero: yovanimp@qf.uclv.edu.cu; ymarrero77@yahoo.es. [Google Scholar]
- Cotton, F. A. Advanced Inorganic Chemistry; Revolucionaria: Havana; p. 103.
- Ross, K. A.; Wright, C.R.B. Matemáticas Discretas; Prentice Hall Hispanoamericana: México, 1990. [Google Scholar]
- Noriega, T. Álgebra; Revolucionaria: Havana, Cuba, 1990; pp. 2-10, 43-49. [Google Scholar]
- Maltsev, A. I. Fundamentos del álgebra lineal; Mir: Moscow, 1976; pp. 68, 262. [Google Scholar]
- Garrido, L. G. Introduccion a la Matemáticas Discretas; Revolucionaria: Havana, Cuba, 1990; pp. 237–298. [Google Scholar]
- Estrada, E.; Rodriguez, L. Matrix Algebraic Manipulation of Molecular Graphs. 2. Harary- and MTI-like Molecular Descriptors. Match 1997, 35, 157–167. [Google Scholar]
- Needham, D. E.; Wei, I-C.; Seybold, P. G. Molecular Modeling of the Physical Properties of the Alkanes. J. Am. Chem. Soc. 1998, 110, 4186–4194. [Google Scholar]
- Krenkel, G.; Castro, E. A.; Toropov, A. A. Improved Molecular Descriptors Based on the Optimization of Correlation Weights of local Graph Invariants. Int. J. Mol. Sci. 2001, 2, 57–65. http://www.mdpi.org.ijms. [Google Scholar]
- Morrison, R. T.; Boyd, R. N. Organic Chemistry; Revolucionaria: Havana, Cuba, 1970. [Google Scholar]
- Solomon, J. W. G. Química Orgánica; Limusa: Mexico, 1987. [Google Scholar]
- STATISTICA ver. 5.5. Statsoft, Inc., 1999.
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Modell. 2002, 20, 269–276. [Google Scholar]
- Rose, K.; Hall, L. H.; Kier, L. B. Modeling Blood-Brain Barrier Partitioning Using the Electrotopological State. J. Chem. Inf. Comput. Sci. 2002, 42, 651–666. [Google Scholar]
- Wold, S.; Erikson, L. Statistical Validation of QSAR Results. Validation Tools. In Chemometric Methods in Molecular Design; van de Waterbeemd, H., Ed.; VCH Publishers: New York, 1995; pp. 309–318. [Google Scholar]
- Randić, M.; Basak, S. Optimal Molecular Descriptors Based on Weighted Path Numbers. J. Chem. Inf. Comput. Sci. 1999, 39, 261–266. [Google Scholar]
- Chenzhong, C.; Zhiliang, L. Molecular Polarizability. 1. Relationship to Water Solubility of Alkanes and Alcohols. J. Chem. Inf. Comput. Sci. 1998, 38, 1–7. [Google Scholar]
- Katritzky, A. R.; Lobanov, V. S.; Karelson, M. Normal Boiling Points for Organic Compounds: Correlation and Prediction by a Quantitative Structure-Property Relationship. J. Chem. Inf. Comput. Sci. 1998, 38, 28–41. [Google Scholar]
- Estrada, E.; Ivanciuc, O.; Gutman, I.; Gutiérrez, A.; Rodríguez, L. Extended Wiener Indices. A New Set of Descriptors for Quantitative Structure-Property Studies. New J. Chem. 1998, 22, 819–822. [Google Scholar]
- Katrizky, A.; Maran, U.; Lobanov, V. S.; Karelson, M. Structurally Diverse Quantitative Structure-Property Relationship Correlations of Technologically Relevant Physical Properties. J. Chem. Inf. Comput. Sci. 2000, 40, 1–18. [Google Scholar]
- Stanton, D. T. Development of a Quantitative Structure-Property Relationship Model for Estimating Normal Boiling Points of Small Multifunctional Organic Molecules. J. Chem. Inf. Comput. Sci. 2000, 40, 81–90. [Google Scholar]
- Belsey, D. A.; Kuh, E.; Welsch, R. E. Regression Diagnostics; Wiley: New York, 1980. [Google Scholar]
- Alzina, R. B. Introduccion conceptual al análisis multivariable. Un enfoque informatico con los paquetes SPSS-X, BMDP, LISREL Y SPAD; PPU, SA: Barcelona, 1989; Chapter 8; Vol. 1, p. 202. [Google Scholar]
- Basak, S. C.; Balaban, A. T.; Grunwald, G. D.; Gute, B. D. Topological Indices: Their Nature and Mutual Relatedness. J. Chem. Inf. Comput. Sci. 2000, 40, 891–898. [Google Scholar]
- Patel, H.; Cronin, M. T. D. A Novel Index for the Description of Molecular Linearity. J. Chem. Inf. Comput. Sci. 2001, 41, 1228–1236. [Google Scholar]
- Romanelli, G. P.; Cafferata, L. F. R.; Castro, E. A. An improved QSAR study of toxicity of saturated alcohols. J. Mol. Struct. (Theochem). 2000, 504, 261–265. [Google Scholar]
- Randić, M. Orthogonal Molecular Descriptors. New J. Chem. 1991, 15, 517–525. [Google Scholar]
- Randić, M. Fitting of Nonlinear Regression by Orthogonalized Power Series. J. Comput. Chem. 1993, 14, 363–370. [Google Scholar]
- Randić, M. Resolution of Ambiguities in Structure-Property Studies by us of Orthogonal Descriptors. J. Chem. Inf. Comput. Sci. 1991, 31, 311–320. [Google Scholar]
- Randić, M. Correlation of Enthalpy of Octanes with Orthogonal Connectivities indices. J. Mol. Struct. (Theochem). 1991, 233, 45–59. [Google Scholar]
- Lučić, B.; Nikolić, S.; Trinajstić, N.; Jurić, D. The Structure-Property Models can be Improbad Using the Orthogonalized Descriptors. J. Chem. Inf. Comput. Sci. 1995, 35, 532–538. [Google Scholar]
- Cronin, M. T. D.; Schultz, T. W. Pitfalls in QSAR. J. Mol. Struct. (Theochem). 2003, 622, 39–51. [Google Scholar]
- Estrada, E.; Gonzáles, H. What Are the Limits of Applicability for Graph Theoretic Descriptors in QSPR/QSAR? Modeling Dipole Moments of Aromatic Compounds with TOPS-MODE Descriptors. J. Chem. Inf. Comput. Sci. 2003, 43, 75–84. [Google Scholar]
- Sample Availability: Not applicable.
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Ponce, Y.M. Total and Local Quadratic Indices of the Molecular Pseudograph's Atom Adjacency Matrix: Applications to the Prediction of Physical Properties of Organic Compounds. Molecules 2003, 8, 687-726. https://doi.org/10.3390/80900687
Ponce YM. Total and Local Quadratic Indices of the Molecular Pseudograph's Atom Adjacency Matrix: Applications to the Prediction of Physical Properties of Organic Compounds. Molecules. 2003; 8(9):687-726. https://doi.org/10.3390/80900687
Chicago/Turabian StylePonce, Yovani Marrero. 2003. "Total and Local Quadratic Indices of the Molecular Pseudograph's Atom Adjacency Matrix: Applications to the Prediction of Physical Properties of Organic Compounds" Molecules 8, no. 9: 687-726. https://doi.org/10.3390/80900687
APA StylePonce, Y. M. (2003). Total and Local Quadratic Indices of the Molecular Pseudograph's Atom Adjacency Matrix: Applications to the Prediction of Physical Properties of Organic Compounds. Molecules, 8(9), 687-726. https://doi.org/10.3390/80900687