Results and Discussion
In this communication we describe the syntheses of a series of hitherto unreported diaryl sulphides and diaryl sulphones containing carbazone, thiazoline and thiazolidinone moieties. The starting material for these syntheses and subsequent studies is 4,4'-diacetyldiphenyl sulphide (
2), which was prepared according to the literature method [
4], and was also obtained during our attempts to acetylate diphenyl sulphoxide (
1) using the Friedel Crafts reaction [
4]. Compound
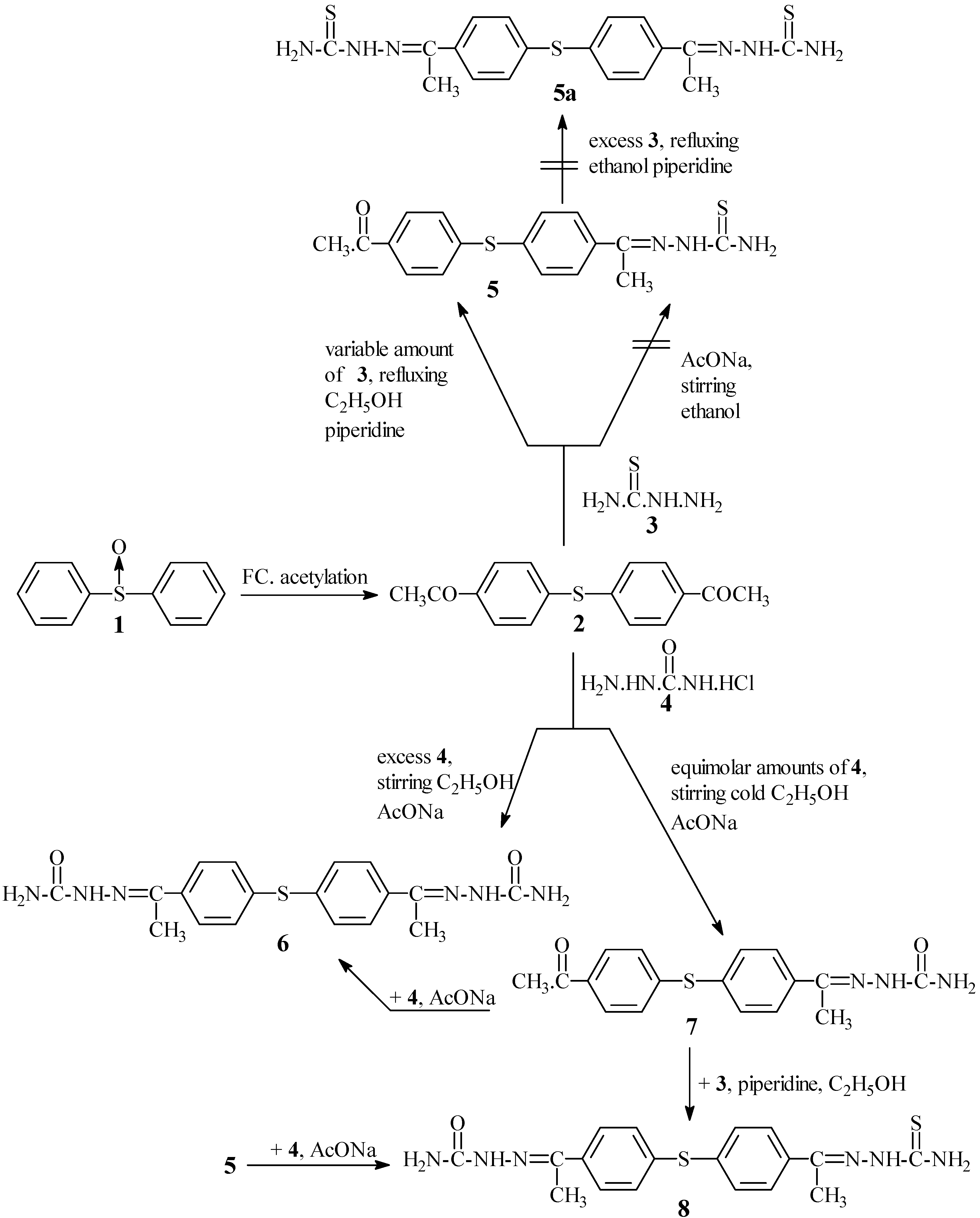
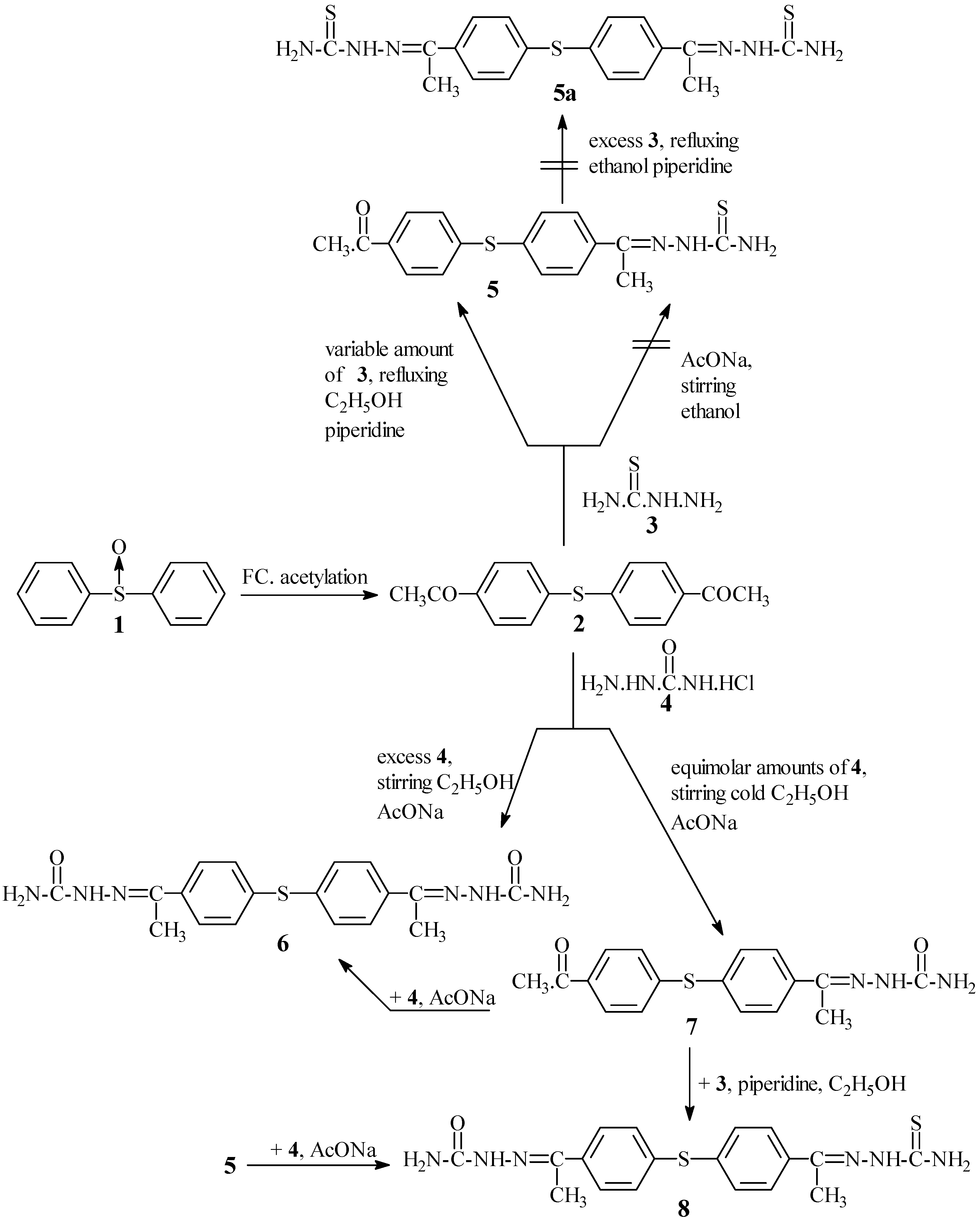
2 condensed with excess of thiosemicarbazide
3 in refluxing ethanol in the presence of a catalytic amount of piperidine as a basic catalyst to afford only 4-acetylthiosemicarbazone-4'-acetyldiphenyl sulphide (
5). Attempts to prepare 4,4'-diacetylthiosemicarbazone diphenyl sulphide (
5a) starting from
2 and/or
5 using the same condensation with
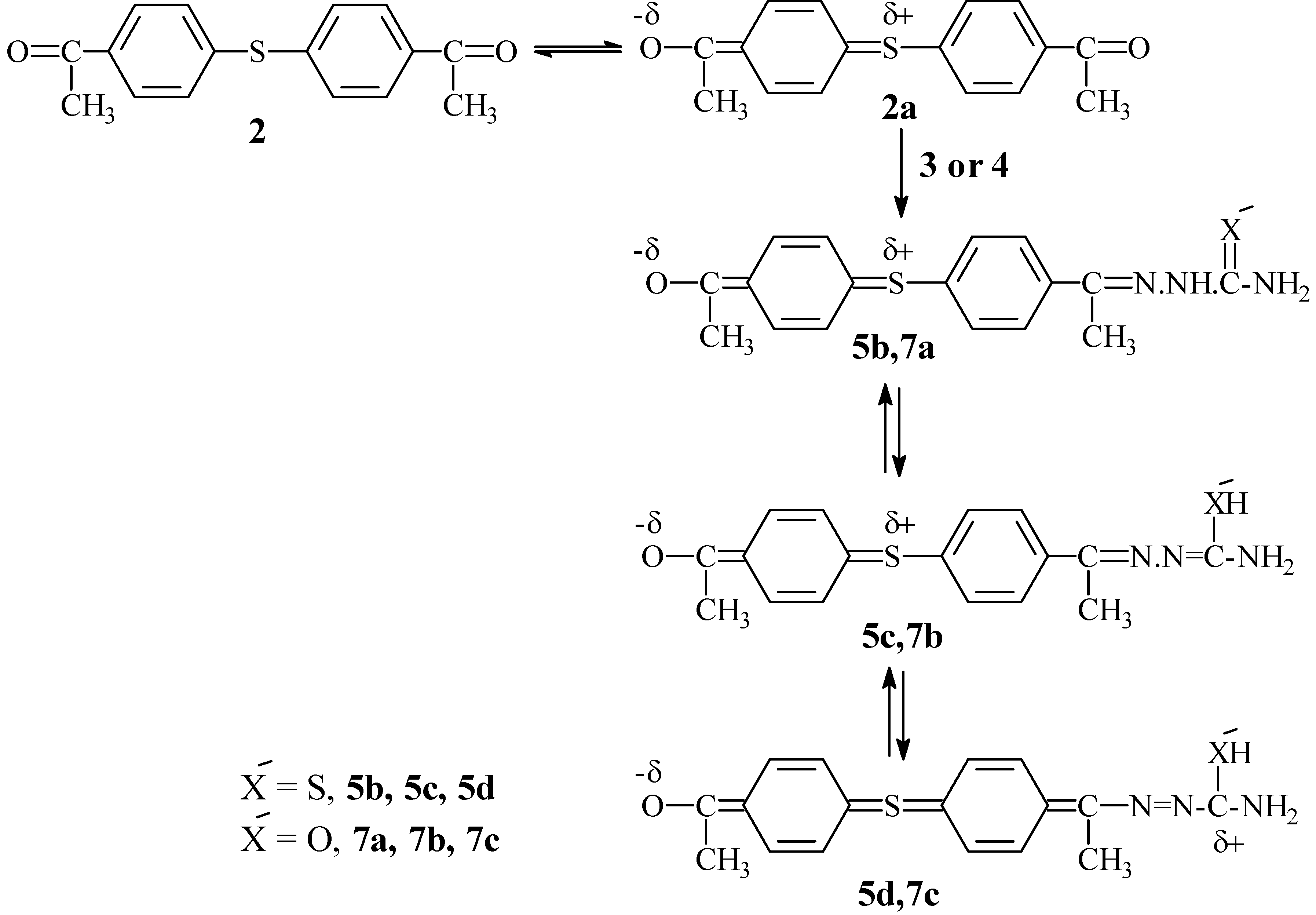
3 under a variety of conditions were unsuccessful. As an explanation for this unexpected behaviour the mechanism illustrated in
Scheme I is suggested. Thus, it is believed that structure
5d, which has an incompletely polarized carbonyl group in 4'-acetyl group, probably represents one of the contributing charged resonance structures of
5. The positive charge of that C=O is shifted through π conjugation from the migration origin (which is its carbon atom) to the migration terminus (which is the carbon atom attached to extreme -NH
2 ) (with the sulphur atom acting as a relay for conjugation) [
25].
The development of positive charge at that new position is rigidly localized by the +I of -SH group (according to Ingold terminology). It is believed that structure
5d affects the behaviour of the whole molecule of
5 and is responsible for the previously mentioned local incomplete polarization of its 4'-C=O group, which now has low nucleophilicity; hence this decreases to a large extent the subsequent nucleophilic attack of
3 on
5 [
20,
21], to a degree that prevents the second step condensation and/or simultaneous condensation (actually an
in situ stepwise reaction) to form the expected symmetrical sulphide
5a (
Scheme II).
The proposed mechanism depicted in
Scheme I is supported by the following evidence: (i) literature precedents [
24,
25]; (ii) a molecular modeling study on the more stable structure of
2, which is not coplanar (c.f. Fig. A) [
26]; (iii) the
1H-NMR of
2 (c.f. Experimental), which shows a singlet at δ 2.55-2.59 (6H, 4,4'-dimethyl) with some splitting at the top of that signal into a doublet with a difference of ≈ δ 0.04 ppm between its two singlet peaks. This difference may be attributable to different environments of the 4,4'-diacetyl groups of
2, which may in turn be related to
cis-
trans geometrical isomerism or to the fact that the resonance structure of
2 (i.e.
2a) is not coplanar or both. Similar results were obtained from the
1H-NMR spectrum of
13 (c.f. Experimental and Fig. B) [
7,
23]; (iv) the lower IR stretching frequency of the remote carbonyl group in
5 due its enolization (c.f.
Table III); (v) the
13C-NMR chemical shift of compound
5 shows a signal at δ 55 ppm that reveals that structure
5d is the more predominant one for
5.
![Molecules 08 00622 i001]()
In contrast to the behaviour of
2 in its condensation with
3, the former compound smoothly condensed with excess semicarbazide hydrochloride (
4) in the presence of a catalytic amount of fused AcONa and/or piperidine to give the expected 4,4'-diacetylsemicarbazone diphenyl sulphide (
6). This formation could be interpreted
via inspection of the charged contributing resonance structure
7c (
Scheme I) which has comparatively less rigidly localized developed positive charge than the corresponding one
5d, clearly due to the lower basicity of the -OH group compared to that of the -SH group. As a result, the 4-acetylsemicarbazone moiety in structures
7a,
b,
c has a comparatively little effect on the localized polarisation of the 4'- C=O of
7, to such an extent that it permits the second step condensation of
7 with
4 and/or simultaneous condensation of
2 with excess
4 to occur, thus with forming
6 under a variety of conditions (
Scheme II).
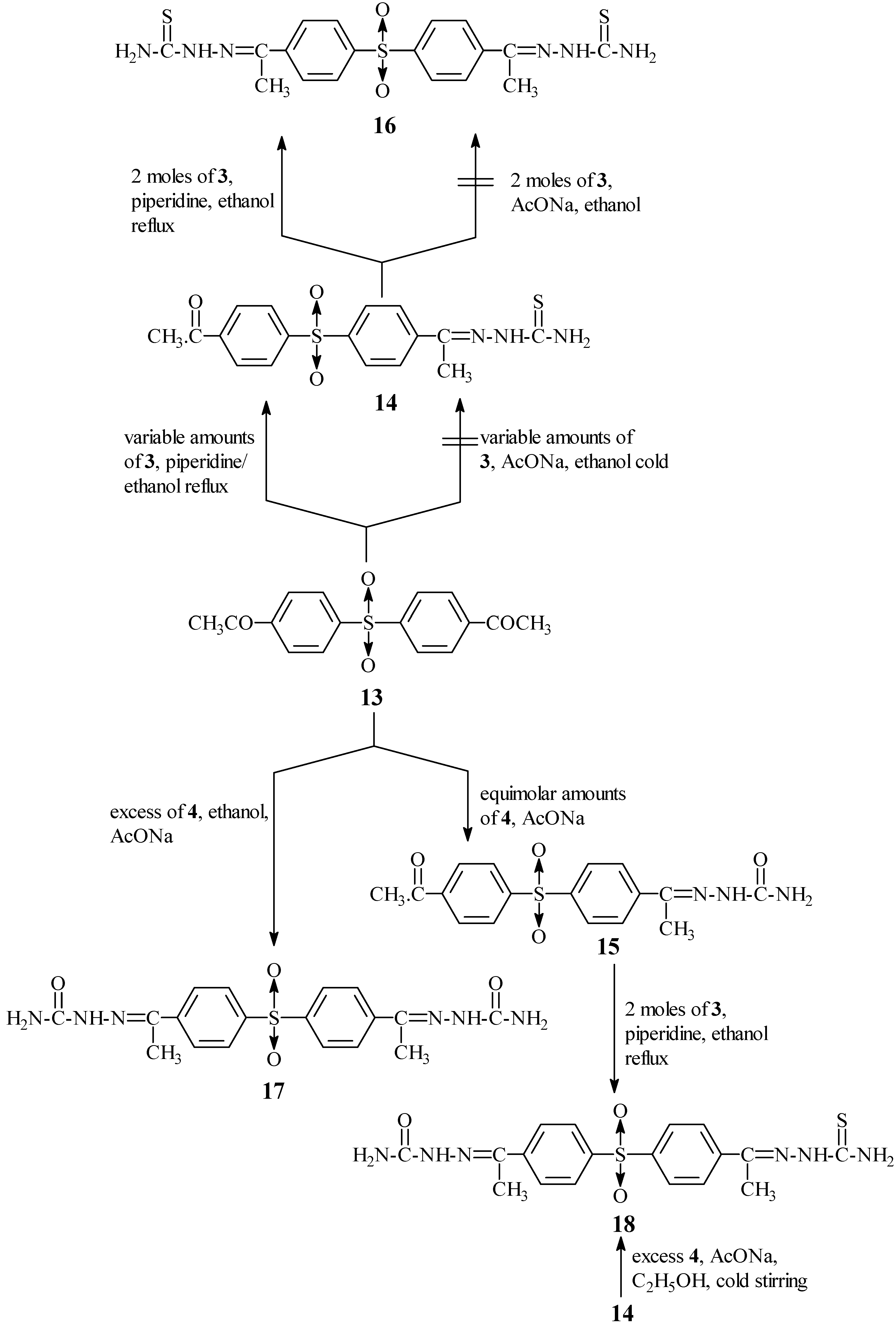
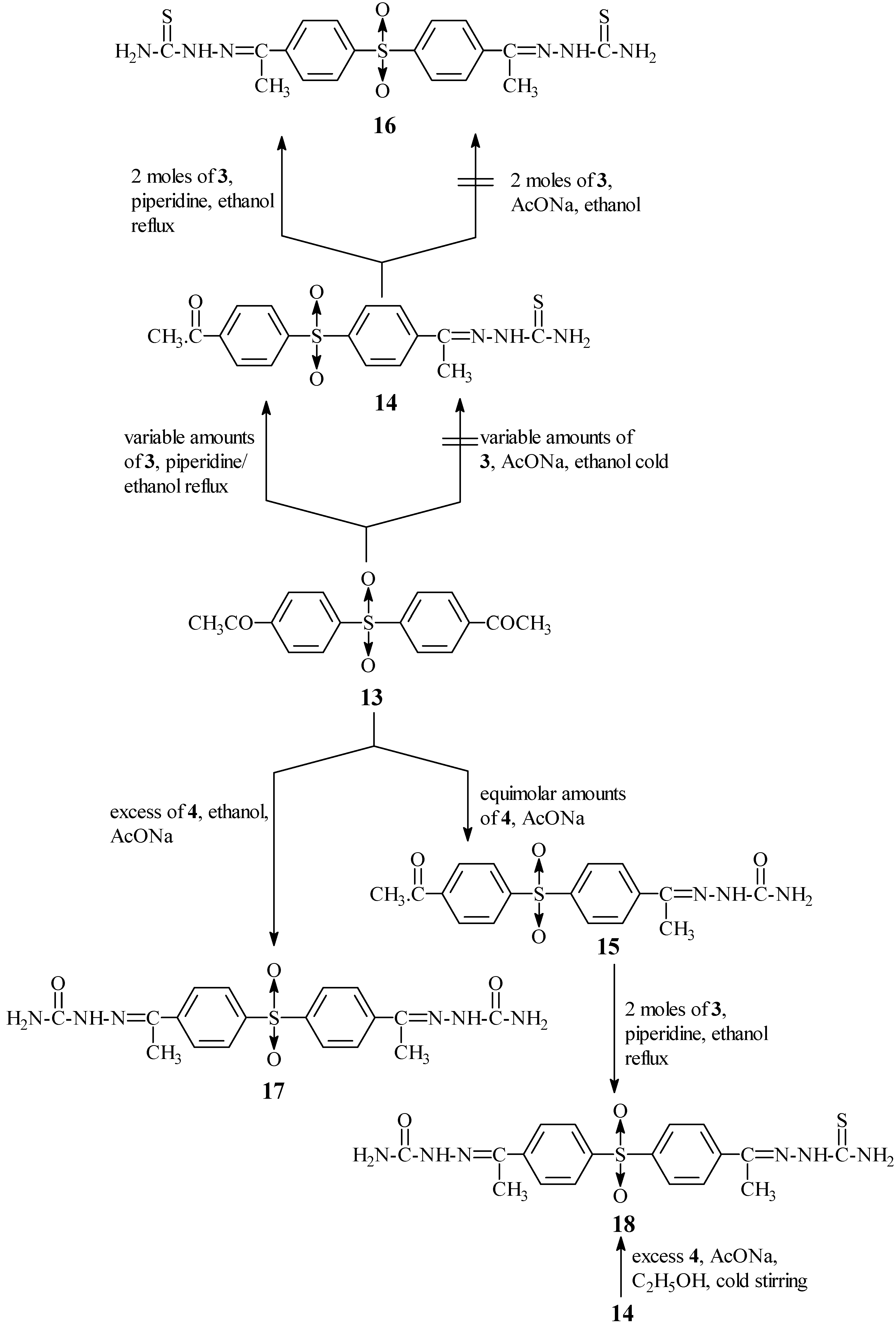
It is worth noting that two additional pieces of evidence could also clarify the previous statements. The first chemical evidence was achieved by replacing the sulphide linkage in
5 by a sulphone one to produce 4-acetylthiosemicarbazone-4'-acetyldiphenyl sulphone (
14) (
Scheme III) which was easily condensed (second step) with 3 in refluxing ethanol in the presence of a catalytic amount of piperidine to form 4,4'-diacetylthiosemicarbazone diphenyl sulphone (
16).
Here the sulphone group prevents the complete π conjugation through 14, which as a result displays a rather different carbonyl group behaviour than that of 5 and even 2, and accordingly it does not experience any effects from the carbazone moiety.
The second piece of chemical evidence comes from examination of the results when the part containing the thione group in the thiosemicarbazone moiety of
5 was used in assembling the heterocyclic moieties of sulphides
9-12. In these reactions the effect of the thiosemicarbazone group was totally cancelled to a degree that permits the 4'-C=O to condense easily with
3 to produce the sulphides
23-26 under the same conditions previously used (
Scheme IV). It is interesting to note that sulphide
5 smoothly condensed with
4 in cold ethanol containing fused AcONa to give 4-acetylthio-semicarbazone-4'-acetylsemicarbazone diphenyl sulphide (
8) which alternatively was formed by condensation of sulphide
7 with
3 using the previously mentioned method (c.f. Experimental). The two sulphides obtainable by two different routes (
Scheme II) are identical (m.p., mixed m.p. and spectral data). The chemical behaviour of
5 in its condensation with
4 is different than that observed with
3. The theoretical reasoning for that difference is that it apparently depends upon the different reactivity of
3 and
4 towards addition on 4'- C=O [
22].
Condensation of 4,4'-diacetyldiphenyl sulphone
13 (
vide infra) with an excess of
3 in refluxing ethanol containing a catalytic amount of piperidine afforded only 4-acetylthiosemicarbazone-4'-acetyl diphenyl sulphone (
14) (this condensation was not accomplished using fused AcONa). A separate second step condensation of
14 with
3 using the same conditions gave 4,4'-diacetylthiosemicarbazone diphenyl sulphone (
16). The latter stepwise formation could be explained as previously discussed and also based on the fact that the energy needed for formation of
16 via simultaneous condensation (E= 155.5505 kcal/mol, G= 1.4838) is more than that needed for formation of
14 (which is the monothiosemicarbazone derivative of
13)
via a one step condensation (E= 17.7515; G= 14.4536) [
26]. Condensation of
13 with an equimolar amount of
4 in the presence of fused AcONa smoothly yielded 4-acetylsemicarbazone-4'-acetyldiphenyl sulphone (
15), while using an excess of
4, it afforded 4,4'-diacetylsemicarbazone diphenyl sulphone (
17). 4- Acetylthiosemicarbazone-4'-acetylsemi- carbazone diphenyl sulphone (
18) was prepared as previously mentioned either by condensation of
15 with
3 and/or
14 with
4 (
Scheme III). The two sulphones obtained by the two different routes are identical (m.p, mixed m.p and spectral data). The structures of the prepared compounds
5-8 and
13-18 were established by elemental analysis, I.R,
1H-NMR,
13C-NMR and mass spectral data (c.f. Experimental). It is interesting to note that the electron attracting properties of the 4,4'-diacetyl groups in
13 are nearly equal to that of the SO
2 group and that this affects the NMR data of the aromatic protons (nearly a singlet) which is completely different from the data of the corresponding protons in 2. The same environmental comparison between sulphides
6,
8 and sulphones
17,
18, respectively, is also valid. In the latter sulphones the electron attracting properties of the 4,4'-bis-azomethine groups are nearly equal to that of SO
2 groups in the same molecules (c.f. Experimental).
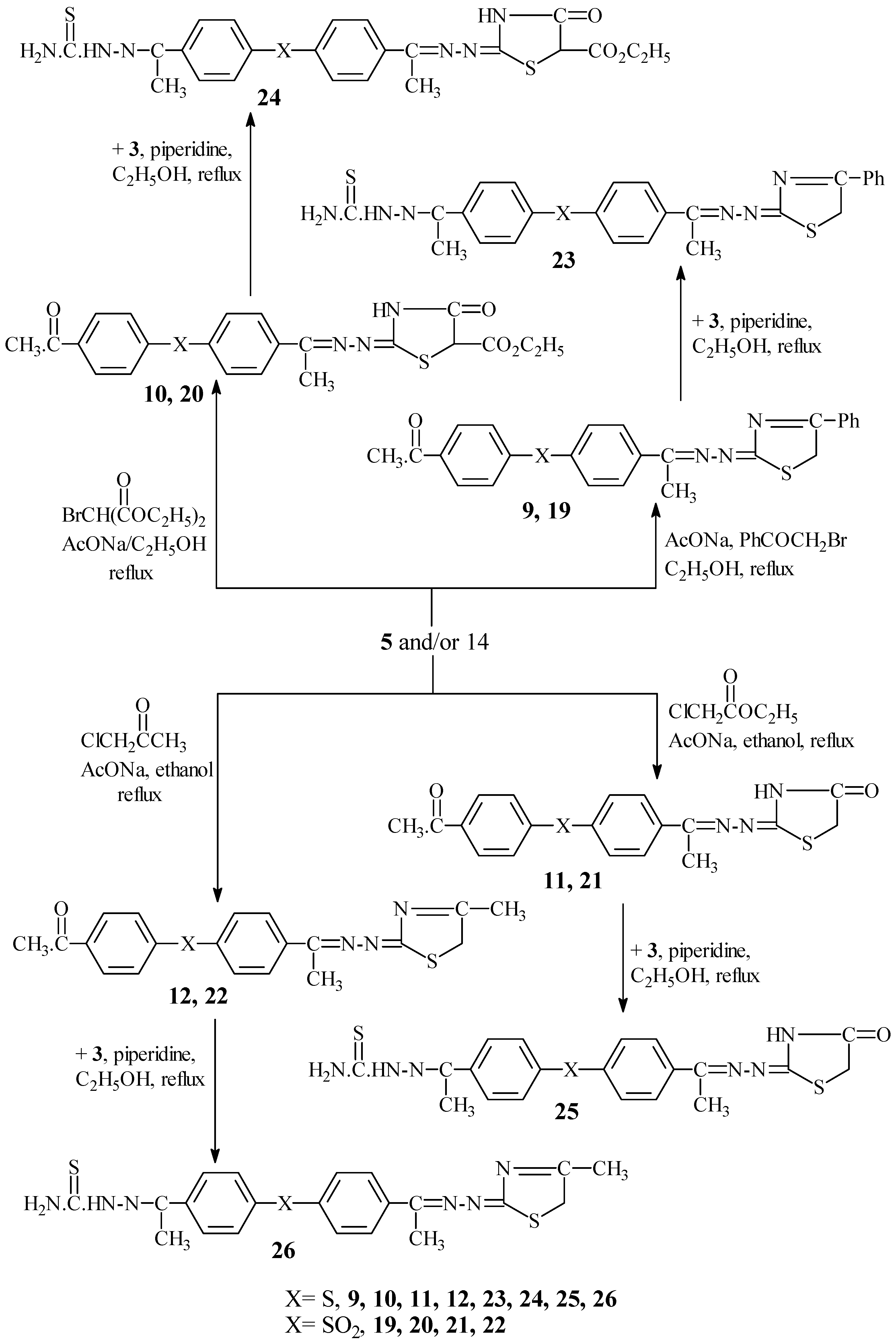
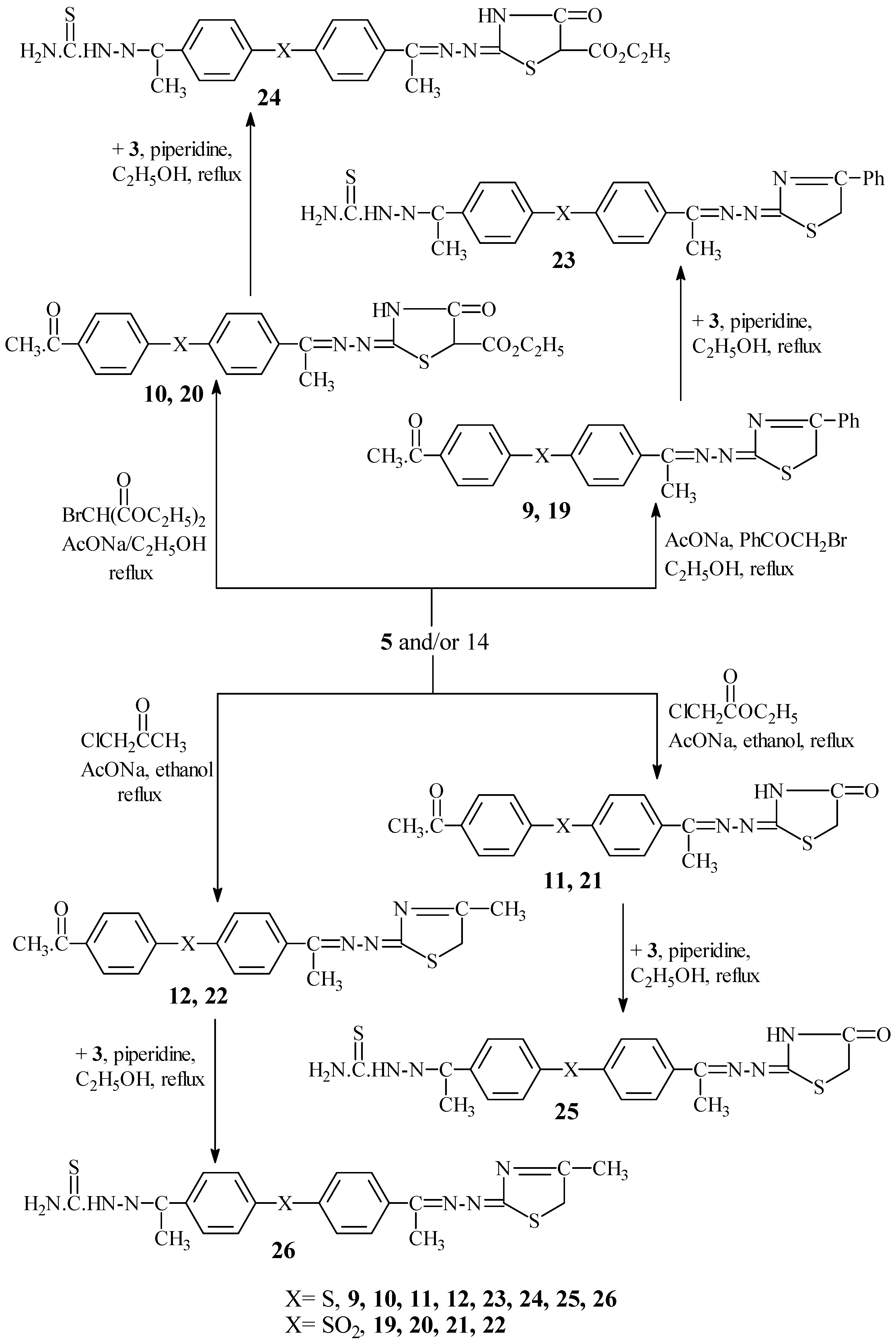
Interaction of
5 and/or
14 with phenacyl bromide in the presence of fused AcONa afforded 4-(4"-phenyl-Δ
3-thiazoline-2"-acetylazino)-4'-acetyldiphenyl sulphide (
9) and 4-(4"-phenyl-Δ
3-thiazoline-2"-acetylazino)-4'-acetyl-diphenyl sulphone (
19), respectively. Similarly, reaction of
5 and/or
14 with bromo diethylmalonate in the presence of fused AcONa gave 4-(5"-carboxyethyl-4"-thiazolidinone-2"-acetylazino)-4'-acetyldiphenyl sulphide (
10) and 4-(5"-carboxyethyl-4"-thiazolidinone-2"-acetylazino)-4'-acetyldiphenyl sulphone (
20), respectively, while reaction with chloroethylacetate in the presence of fused AcONa gave 4-(4"-thiazolidinone-2"-acetylazino)-4'-acetyldiphenyl sulphide (
11) and 4-(4"-thiazolidinone-2"-acetylazino)-4'-acetyldiphenyl sulphone (
21), respectively. Finally, reactions with chloroacetone in the presence of fused AcONa gave 4-(4"-methyl-Δ
3-thiazoline-2"-acetylazino)-4'-acetyldiphenyl sulphide (
12) and 4-(4"-methyl-Δ
3-thiazoline-2"-acetylazino)-4'-acetyldiphenyl sulphone (
22) respectively (
Scheme IV). The 4'-acetylthiosemicarbazone derivatives of sulphides
9-12 were used to obtain sulphides 4-(4"-phenyl-Δ
3-thiazoline-2"-acetylazino)-4'-acetyl-thiosemicarbazone diphenyl sulphide (
23), 4-(5"-carboxyethyl-4"-thiazolidinone-2"-acetylazino)-4'-acetylthiosemi-carbazonediphenyl sulphide (
24); 4-(4"-carboxyethyl-4"-thiazolidinone-2"-acetylazino)-4'-acetylthio-semicarbazone diphenyl sulphide (
25) and 4-(4"-thiazolidinone-2"-acetylazino)-4'-acetylthio-semicarbazone diphenyl sulphide (
26), respectively (
Scheme IV). A shortcut to achieving that goal was by interaction of sulphides
9-12 with
3 in the presence of piperidine to afford compounds
23-26 in moderate yields,
Oxidation of the prepared sulphides
5-12 and
23-26 using glacial AcOH/H
2O
2 mixtures at room temperature for at least one week yielded only
13 as the main product from every sulphide examined, although other compounds were isolated in some cases (see
Table I). The structures
19-26 were elucidated on the basis of elemental analyses, IR and NMR data (
Table II,
Table III).
Table I.
Oxidation products of the prepared sulphides using glacial AcOH/H2O2 mixtures.
Table I.
Oxidation products of the prepared sulphides using glacial AcOH/H2O2 mixtures.
| Prepared sulphides | Oxidation product(s) | Prepared sulphides | Oxidation product(s) |
|---|
| 5 | 13 + 3 | 11 | 13 + unidentified substance |
| 6 | 13 + 4 | 12 | 13 + 3 + unidentified substance |
| 7 | 13 + 4 | 23 | 13 + 3 + unidentified substance |
| 8 | 13 + 3 + 4 | 24 | 13 + 3 + unidentified substance |
| 9 | 13 + unidentified substance | 25 | 13 + 3 + unidentified substance |
| 10 | 13 + unidentified substance | 26 | 13 + 3 + unidentified substance |
Along with the experimental investigation and as further confirmation of the structures of the compounds synthesized, we also carried out some theoretical studies on selected prepared compounds with the help of molecular modeling software [
26]. From the obtained data (
Table IV) the parent compound
2 (Fig. A) has energy (E)= 15.7673 kcal, Gradient, (G)= 0.099593; compound
5 (Fig. C), the monosubstituted thiosemicarbazone derivative of
2, has E= 33.71358, G= 0.089813. By comparison of
5 with
5a (the disubstituted dithiosemicarbazone derivative of
2, Fig. D), which has E= 24.24438, G= 0.09677 it was noted that despite the fact that
5a was not experimently obtained in the present work under a variety of conditions for reasons were previously discussed, the theoretical values of the latter are less than that of the former, i.e.
5a is more stable than
5. Comparing
7 (the monosubstituted semicarbazone derivative of
2), which has E= 19.878, G= 0.0946 with
6, which has E= 97.59393, G= 0.09596 it was found that
7 is more stable than
6 (Figs. E, F). Furthermore, comparisons were carried out between the structures
10 (keto-form), E= 101.8926, G= 0.0957, and its tautomeric forms
10a (OH-form), E= 35.82579, G= 0.08946 and
10b (HN-(HO)C= form), E= 29.69458, G= 0.09995. These results proved that
10 is more stable than
10a and/or
10b and are in accordance with the data obtained experimentally (IR, NMR, c.f.
Table III).
![Molecules 08 00622 i002]()
On the other hand, when the same comparison was repeated between the structures
11 (keto-form), E= 27.941029, G= 0.08989 and
11a (HN-(HO)C= form), E= 35.82579, G= 0.08946, and
11b (HN-(HO)C= form), E= 29.69458, G= 0.0995, these results proved that
11 is more stable than 11a and/or
11b and are also in accordance with experimental data (IR, NMR, cf.
Table III).
Experimental
General
The times required for the completion of the reactions and the purity of the prepared compounds were monitored by thin layer chromatography (TLC). Melting points were determined on a Fisher-Johns melting point apparatus and are uncorrected. Elemental analyses were performed on a Perkin-Elmer 240C elemental analyser and a GmbH VARIOEL V
23 elemental analysis system in CHNS mode. IR spectra [
27] were recorded on a Pye-Unicam SP3-100 spectrophotometer using KBr wafer technique.
1H-NMR and
13C-NMR spectra were recorded on JNM-LA400-MHZ NMR spectrophotometer using the appropriate deuterated solvent and TMS as internal standard (chemical shifts expressed in δ ppm). Mass spectra were recorded on Jeol JMS-600 mass spectrometer.
4,4'-Diacetyldiphenyl sulphide (2).
This compound was prepared according to a literature method [
4], or by the following procedure: Anhydrous AlCl
3 (6.66 g, 0.049 mole) was added in small portions to a conical flask containing diphenylsulphoxide (
1, 2.02 g, 0.01 mole) and acetyl chloride (2.8 mL, 0.04 mole) dissolved in carbon disulphide (30 mL), The reaction mixture was stirred in ice bath for 6 hr. The CS
2 was evaporated under vacuum, the residue poured onto a mixture of ice and conc. HCl and the resulting pale yellow precipitate was collected and recrystallized from pet. ether (60-80°C) as colourless flakes. Yield: 85%; m.p 90-92°C; IR (ν, cm
-1): 1690 (C=O);
1H-NMR (CDCl
3): δ 2.57 (s, 6H, 2-CO
CH3, split into two bands at the top with δ 2.55 and 2.59), 7.34-7.93 (m, 8H, Ar-
H); Anal. Calc. for C
16H
14O
2S: C, 70.93; H, 5.39; S, 11.85; Found: C, 70.81; H, 5.25; S, 11.92; MS: M
+, m/z (%): 270 (78%).
4-Acetylthiosemicarbazone-4'-acetyldiphenyl sulphide (5).
A mixture of 2 (1.0 g, 0.0037 mole) and 3 (0.675 g, 0.0074 mole) in ethanol (30 mL) was refluxed for 7 hr in presence of two drops of piperidine. On cooling, yellow crystals separated from the reaction mixture and were recrystallized from C2H5OH. Yield 74.8%; m.p 190-192°C; IR (ν, cm-1): 3400, 3300, 3200 (NH and NH2), 1640 (C=O, the lower frequency due to enolization of the remote carbonyl group), 1600 (C=N); 1H-NMR (DMSO-d6): δ 3.22 (s, 6H, 2CH3), 4.48 (s, 2H, NH2), 7.1-7.5 (m, 8H, Ar-H), 8.62 (s, 1H, NH); 13C-NMR: δ 18.429 (CH3-C=N); δ 27.010 (CH3-C=O); 55.928 (-C δ+); 127.109-145.571 (C-aromatic); 181.038 (-C=N-) and 197.220 (-C=O); Anal. Calc. for C17H17N3OS2: C, 59.47; H, 4.95; N, 12.24; S, 18.65; Found: C, 59.40; H, 4.55; N, 12.11; S, 18.61; MS: M+, m/z (%): 343 (2.2).
4,4'-Diacetylsemicarbazone diphenyl sulphide (6).
To a mixture of semicarbazide hydrochloride (0.92 g, 0.00824 mole) and AcONa (1.04 g, 0.01264 mole) dissolved in water (8 mL) a solution of 2 (0.58 g, 0.00214 mole) in ethanol was added with continuous shaking. Ethanol was added if necessary to obtain a clear solution. Shaking was continued for further one hour, and the mixture was cooled in the refrigerator. The separated crystals were filtered off, washed with cold water, dried and crystallized from acetic acid. Yield 85.70%; m.p >300°C (decomp.); IR (ν, cm-1): 3450, 3350, 3200 (NH, NH2), 1680 (C=O), 1580 (C=N); 1H-NMR (DMSO-d6): δ 2.15 (s, 6H, 2CH3), 6.49 (s, 4H, 2NH2), 7.27-7.93 (m, 8H, Ar-H), 9.39 (s, 2H, 2NH). Anal. Calc. for C18H20N6O2S: C, 56.25; H, 5.20; N, 21.87; S, 8.33; Found: C, 56.20; H, 5.10; N, 21.66; S, 8.10.
4-Acetyl-4'-acetylsemicarbazone diphenyl sulphide (7).
A solution of compound 2 (0.58 g, 0.00214 mole) in ethanol was added with continuous shaking to a mixture of 4 (0.23 g, 0.00206 mole) and AcONa (0.26 g, 0.00316 mole) dissolved in water (8 mL). The reaction mixture was worked up as in the case of 6. The separated crystals were filtered off, washed with cold water, dried and crystallized from ethanol, m.p 190-191°C, yield 85%; IR (ν, cm-1): 3470, 3150, 3160 (NH, NH2), 1695, 1665 (C=O), 1580 (C=N); 1H-NMR (DMSO-d6): δ 2.25 (s, 3H, CH3-CN); 2.6 (s, 3H, O=C-CH3); 6.70 (s, 2H, NH2); 7.30-8.20 (m, 8H, Ar-H); 9.70 (s, 1H, NH); Anal. Calc. for C17H17N3O2S: C, 63.38; H, 5.19; N, 12.84; S, 9.78; Found: C, 63.48; H, 5.13; N, 12.67; S, 9.90.
4-Acetylthiosemicarbazone-4'-acetylsemicarbazone diphenyl sulphide (8).
This compound was obtained by the following two methods:
Method A: To a mixture of 4 (0.08 g, 0.00582 mole) and AcONa (0.35 g, 0.0042 mole) dissolved in water (≈ 8 mL), a solution of 5 (1.0 g, 0.00291 mole) in ethanol was added with continuous shaking. The reaction mixture was worked up as in the case of 6. The crystals formed were filtered off, washed with cold water, dried and recrystallized from AcOH, m.p >300°C (decomp.), yield 75%; IR (ν, cm-1): 3400, 3200, 3150 (NH, NH2), 1680 (C=O), 1580 (C=N), 1480 (C=S); 1H-NMR (DMSO-d6): δ 2.15 (s, 3H, CH3, acetyl thiosemicarbazone); 2.26 (s, 3H, CH3, acetylsemicarbazone); 6.48 (s, 4H, 2NH2); 7.25-8.28 (m, 8H, Ar-H); 9.36 (s, 1H, NH) and 10.26 (s, 1H, NH); Anal. Calc. for C18H20N6OS2: C, 54.00; H, 5.00; N, 21.00; S, 16.00; Found: C, 53.88; H, 4.96; N, 20.79; S, 15.93.
Method B: A mixture of 7 (1.0 g, 0.003 mole) and 3 (0.55 g, 0.0061 mole) in ethanol (30 mL) containing two drops of piperidine was refluxed for 7 hr. On cooling the crystals that separated were filtered and recrystallized from AcOH, m.p >300°C (decomp.), yield 71%. The physical and spectral data of the two products from the two methods were identical.
4-Acetylthiosemicarbazone-4'-acetyldiphenyl sulphone (14).
A mixture of 13 (1.0 g, 0.0033 mole) and 3 (0.675 g, 0.0074 mole) in ethanol (30 mL) containing a few drops of piperidine was refluxed for 7 hr. After cooling the pale yellow crystals formed were separated and recrystallized from ethanol, m.p 185°C, yield 80.5%; IR (ν, cm-1): 3380, 3280, 3200 (NH, NH2); 1680 (C=O); 1640 (C=N); 1335, 1160 (SO2); 1H-NMR (DMSO-d6): δ 2.58 (s, 6H, 2CH3); 4.48 (s, 2H, NH2); 7.17-8.12 (m, 8H, Ar-H) and 8.61 (s, 1H, NH); Anal. Calc. for C17H17N3O3S2: C, 54.11; H, 4.50; N, 11.11; S, 16.97; Found: C, 54.38; H, 4.36; N, 11.35; S, 16.85.
4-Acetylsemicarbazone-4'-acetyldiphenyl sulphone (15).
To a mixture of compound 5 (0.36 g, 0.0012 mole) and AcONa (0.26 g, 0.003) dissolved in water (8 mL) a solution of 13 (1.0 g, 0.0031 mole) in ethanol was added with continuous shaking. The reaction mixture was worked up as previously mentioned in the case of 6. The separated crystals were filtered off, washed with cold water, dried and crystallized from ethanol, m.p 200°C, yield 82%; IR (ν, cm-1): 3470, 3250, 3170 (NH, NH2); 1690, 1665 (C=O); 1580 (C=N), 1340, 1160 (SO2); 1H-NMR (DMSO-d6): δ 2.60 (s, 6H, 2CH3); 4.56 (s, 2H, NH2); 7.30-8.20 (m, 8H, Ar-H) and 8.60 (s, 1H, NH); Anal. Calc. for C17H17N3O4S: C, 56.82; H, 4.73; N, 11.69; S, 8.91; Found: C, 56.66; H, 4.59; N, 11.39; S, 8.63.
4,4'-Diacetylthiosemicarbazone diphenyl sulphone (16).
A mixture of 14 (1.0 g, 0.0026 mole) and 3 (0.972 g, 0.01 mole) in ethanol (30 mL) containing a few drops of piperidine was refluxed for 7 hr. After cooling the pale yellow crystals formed were separated, crystallized from AcOH, m.p >350 (decomp.), yield 65%; IR (ν, cm-1): 3400, 3280, 3200 (NH, NH2); 1680 (C=O); 1620 (C=N); 1510 (C=S); 1H-NMR (DMSO-d6): δ 2.32 (s, 6H, 2CH3); 4.42 (s, 4H, 2NH2); 8.22 (s, 8H, Ar-H) and 10.80 (s, 2H, 2NH); Anal. Calc. for C18H20N6O2S3: C, 48.21; H, 4.46; N, 18.75; S, 21.42; Found: C, 48.11; H, 4.43; N, 18.53; S, 21.30.
4,4'-Diacetylsemicarbazone diphenyl sulphone (17).
To a mixture of 4 (1.47 g, 0.013 mole) and AcONa (0.81 g, 0.0099 mole) dissolved in water (8 mL) a solution of 13 (1.0 g, 0.00316 mole) in ethanol was added with continuous shaking. The reaction mixture was worked up as in the case of 6. The separated crystals were filtered off, washed with cold water, dried and crystallized from AcOH, m.p >350°C (decomp.), yield 65%; IR (ν, cm-1): 3400, 3250, 3100 (NH, NH2); 1680 (C=O); 1620 (C=N); 1340, 1170 (SO2); 1H-NMR (DMSO-d6): δ 2.23 (s, 6H, 2CH3); 6.68 (s, 4H, 2NH2); 8.12 (s, 8H, Ar-H) and 11.00 (s, 2H, 2NH); Anal. Calc. for C18H20N6O4S:C, 51.92; H, 4.80; N, 20.19; S, 7.69; Found: C, 51.69; H, 4.21; N, 20.40; S, 7.88.
4-Acetylthiosemicarbazone-4'-acetylsemicarbazone diphenyl sulphone (18).
This compound was prepared by the following two methods:
Method A: To a mixture of 4 (0.59 g, 0.00533 mole) and AcONa (0.32 g, 0.004 mole) dissolved in water (≈ 8 mL), a solution of 14 (1.0 g, 0.00266 mole) in ethanol was added with continuous shaking. The reaction mixture was worked up as in the case of 6. The separated crystals were filtered off, washed with cold water, dried and crystallized from AcOH, m.p >350°C (decomp.), yield 77%; IR (ν, cm-1): 3450, 3200, 3150 (NH, NH2); 1675 (C=O), 1580 (C=N); 1485 (C=S); 1340, 1160 (SO2); 1H-NMR (DMSO-d6): δ 2.23 (s, 3H, CH3); 2.52 (s, 3H, CH3); 6.50 (s, 4H, 2NH2); 7.28-7.59 (m, 8H, Ar-H); 9.56 (s, 1H, NH) and 10.30 (s, 1H, NH); Anal. Calc. for C18H20N6O3S2: C, 50.00; H, 4.62; N, 19.44; S, 14.81; Found: C, 49.98; H, 4.55; N, 19.35; S, 14.63.
Method B: A mixture of 15 (1.0 g, 0.0027 mole) and 3 (0.50 g, 0.005 mole) in ethanol (30 mL) containing a catalytic amount of dry piperidine (two drops) was refluxed for 6 hr. On cooling the separated crystals were collected, dried and recrystallized from AcOH, m.p >350°C (decomp.), yield 75%. The physical and spectral data of the two products obtainable by both methods were identical.
General procedure for the oxidation of sulphides 5-12,23-26 and preparation of 13.
A solution of diarylsulphide (0.02 mole) in glacial AcOH (≈ 20 mL) was warmed if necessary, cooled, filtered and 30% H2O2 (≈ 30 mL) was added. The mixture was kept at room temperature for 7 days and the deposited crystalline product from each oxidation was filtered, purified as usual and identified as 4,4'-diacetyldiphenylsulphone (13); IR (ν, cm-1): 1690 (C=O); 1310, 1150 (SO2); 1H-NMR (DMSO-d6): δ 2.57 (s, 6H, 2CH3); 8.10 (s, 8H, Ar-H). MS: M+, m/z (%): 302 (23.5); Anal. Calc. for C16H14O4S: C, 63.57; H, 4.63; S, 10.59; Found: C, 63.44; H, 4.35; S, 10.51.
Isolation of 3 and/or 4 from the oxidation mixtures of 5 and/or 6 respectively (as representative examples):
After filtration of 13 from the oxidation mixture of 5 and/or 6, the filtrate was neutralized with NaHCO3 solution, the precipitate formed was filtered, purified and identified as 3. To a filtrate from the reaction mixture of 6, conc. HCl was added (3 mL) and the reaction mixture was evaporated to a small volume, cooled, the formed precipiate was filtered, purified and identified as 4.
General procedure for the preparation of compounds 9-12.
Compound
5 (0.5 g, 0.00145 mole) was mixed separately with (0.0014 mole) of phenacyl bromide, bromodiethylmalonate, chloroethylacetate and chloroacetone in the presence of anhydrous AcONa (3.0 g) in ethanol (30 mL). Each mixture was refluxed for 5-7 hr, then allowed to cool and poured onto ice/water mixture. The precipitate solid product from every reaction was collected, crystallized from the appropriate solvent. The results are given in
Table II and
Table III.
General procedure for the preparation of compounds 19-22.
Compound
14 (1.0 g, 0.00266 mole) was mixed separately with (0.0026 mole) of phenacylbromide, bromodiethylmalonate, chloroethylacetate and chloroacetone in the presence of anhydrous sodium acetate (3.0 g) in ethanol (30 mL). Each mixture was refluxed for 5-7 hr, then allowed to cool and poured onto ice/water mixture. The precipitated solid product of every reaction was collected and purified from the proper solvent. The yields and characterization details for the products of these reactions are given in
Table II and
Table III.
General procedure for the preparation of compounds 23-26.
A mixture of each sulphide
9-12 (0.0011 mole) and thiosemicarbazide (0.2 g, 0.002 mole) in ethanol (30 mL) containing two drops of piperidine was refluxed for 7 hr. On cooling yellow crystals were separated from the reaction mixtures and recrystallized from the appropriate solvent. The results are summarized in
Table II and
Table III.
Table II.
Yields and Physical data of compounds 9-12, 19-26.
Table II.
Yields and Physical data of compounds 9-12, 19-26.
Compd.
No. | M.p.
recrystallization solvent | Yield
% | Molecular
formulae | Elemental analysis
Calcd/Found % |
|---|
| (Mol. wt.) | C | H | N | S |
|---|
| 9 | 160°C, decomp. EtOH/H2O
(1:1) | 54 | C25H21N3OS2
(443) | 67.72
67.90 | 4.74
4.52 | 9.48
9.70 | 14.44
14.32 |
| 10 | 260°C,
decomp.EtOH/H2O
(1:1) | 53 | C22H21N3O4S2
(455) | 58.02
58.12 | 4.61
4.52 | 9.23
9.32 | 14.06
13.90 |
| 11 | 160°C
EtOH | 53 | C19H17N3O2S2
(383) | 59.53
59.40 | 4.43
4.32 | 10.96
10.80 | 16.71
16.88 |
| 12 | 120°C, EtOH | 52.5 | C20H19N3OS2
(381) | 62.99
62.81 | 4.98
4.80 | 11.02
11.11 | 16.79
16.60 |
| 19 | >300°C
AcOH | 62 | C25H21N2O3S2
(475) | 63.15
63.11 | 4.42
4.32 | 8.84
8.71 | 13.47
13.40 |
| 20 | >300°C
AcOH | 65 | C22H21N3O6S2
(487) | 54.20
54.10 | 4.31
4.22 | 8.62
8.51 | 13.11
12.99 |
| 21 | >300°C
AcOH | 61.5 | C19H17N3O4S
(415) | 54.93
54.12 | 4.09
4.01 | 10.12
10.11 | 15.42
14.99 |
| 22 | >300°C
AcOH | 60 | C20H19N3O3S2
(413) | 58.11
58.10 | 14.60
14.30 | 10.16
10.15 | 15.49
15.32 |
| 23 | 300°C,
EtOH/H2O
(1:1) | 70 | C26H24N6S3
(516) | 60.46
60.31 | 4.65
4.55 | 16.27
16.11 | 18.60
18.40 |
| 24 | 300°C
EtOH | 75 | C23H24N6O3S3
(528) | 52.27
52.17 | 4.54
4.06 | 15.90
15.80 | 18.18
18.11 |
| 25 | 300°C
EtOH | 73 | C20H20N6OS3
(456) | 52.63
52.61 | 4.38
4.10 | 18.42
18.22 | 21.05
20.75 |
| 26 | 300°C
EtOH-water
(1:1) | 71 | C21H23N6S3
(455) | 55.38
55.14 | 5.05
4.84 | 18.46
18.40 | 21.09
20.93 |
Table III.
Spectral data of compounds
9-12.
![Molecules 08 00622 i003]()
Table III.
Spectral data of compounds 9-12. ![Molecules 08 00622 i003]()
Compd.
No. | X2 | X1 | Ar | Spectral data |
|---|
| 9 | -S- | Acetyl | ![Molecules 08 00622 i004]() | IR (ν, cm-1): 1660 (C=O), 1600 (C=N); 1H-NMR (DMSO-d6): δ 2.27 (s, 3H, CH3-C=N), 2.30 (s, 3H, CH3CO), 4.35 (s, 2H, CH2 of thiazoline), 7.27-7.94 (m, 13H, Ar-H). |
| 10 | -S- | Acetyl | ![Molecules 08 00622 i005]() | IR (ν, cm-1): 3400 (NH), 1700 (ester C=O), 1680 (C=O), 1580 (C=N); 1H-NMR (DMSO-d6): δ 1.13 (t, 3H, CH2 ester), 2.25 (s, 3H, CH3-C=N), 2.52 (s, 3H, COCH3), 4.02 (q, 2H, CH2 ester), 5.36 (s, 1H, -CH of thiazoline ring), 7.30-7.99 (m, 8H, Ar-H), 8.28 (s, 1H, NH). |
| 11 | -S- | Acetyl | ![Molecules 08 00622 i006]() | IR (ν, cm-1): 3450 (NH), 1700, 1705 (C=O), 1605 (C=N); 1H-NMR (DMSO-d6): δ 2.24 (s, 3H, CH3-C=N), 2.40 (s, 3H, CH3CO), 3.92 (s, 2H, CH2 of thiazolidenone ring), 7.00-8.00 (m, 8H, Ar-H), 8.25 (s, 1H, NH). |
| 12 | -S- | Acetyl | ![Molecules 08 00622 i007]() | IR (ν, cm-1): 1670 (C=O), 1590 (C=N); 1H-NMR (CDCl3): δ 2.25 (s, 6H, CH3 of thiazoline, CH3=C=N), 2.45 (s, 3H, CH3CO), 6.20 (s, 2H, CH2 of thiazoline ring), 7.30-7.85 (m, 8H, Ar-H). |
| 19 | ![Molecules 08 00622 i008]() | Acetyl | ![Molecules 08 00622 i004]() | IR (ν, cm-1): 1670 (C=O), 1600 (C=N), 1340, 1160 (SO2); 1H-NMR (DMSO-d6): δ 2.28 (s, 3H, CH3-CN), 2.35 (s, 3H, CH3CO), 6.30 (s, 2H, CH2 of thiazoline ring), 7.85-7.99 (m, 13H, Ar-H). |
| 20 | ![Molecules 08 00622 i008]() | Acetyl | ![Molecules 08 00622 i005]() | IR (ν, cm-1): 3450 (NH), 1710 (ester C=O), 1680 (C=O), 1340, 1160 (SO2); 1H-NMR (DMSO-d6): δ 1.30 (t, 3H, CH3 ester), 2.12 (s, 3H, CH3-C=N), 2.65 (s, 3H, CH3CO), 4.40 (q, 2H, CH2 ester), 7.99-8.35 (m, 8H, Ar-H), 8.80 (s, 1H, CH of thiazolidinone), 9.95 (s, 1H, NH). |
| 21 | ![Molecules 08 00622 i008]() | Acetyl | ![Molecules 08 00622 i006]() | IR (ν, cm-1): 3450 (NH), 1710, 1700 (C=O), 1600 (C=N); 1H-NMR (DMSO-d6): δ 2.21 (s, 3H, CH3CN), 2.41 (s, 3H, CH3CO), 4.00 (s, 2H, thiazolinone CH2), 7.70-7.90 (m, 8H, Ar-H), 8.95 (s, 1H, NH). |
| 22 | ![Molecules 08 00622 i008]() | Acetyl | ![Molecules 08 00622 i007]() | IR (ν, cm-1): 1680 (C=O), 1580 (C=N); 1H-NMR (CDCl3): δ 2.20, 2.30 (s, 6H, CH3 of thiazoline ring and CH3-C=N), 2.60 (s, 3H, CH3CO), 6.20 (s, 2H, CH2 of thiazoline ring), 7.85-7.90 (m, 8H, Ar-H). |
| 23 | -S- | Acetyl-thiosemi-carbazone | ![Molecules 08 00622 i004]() | IR (ν, cm-1): 3450, 3260, 3150 (NH2, NH), 1600 (C=N); 1H-NMR (DMSO-d6): δ 2.3 (s, 6H, 2CH3), 4.7 s, 2H, NH2), 6.30 (s, 2H, CH2 of thiazoline ring), 7.00-7.44 (m, 19H, Ar-H), 8.85 (s, 1H, NH). |
| 24 | -S- | Acetyl-thiosemi-carbazone | ![Molecules 08 00622 i005]() | IR (ν, cm-1): 3420, 3300, 3170 (NH, NH2), 1700 (C=O), 1600 (C=N); 1H-NMR (DMSO-d6): δ 1.3 (t, 3H, CH3 ester), 2.56 (s, 6H, 2CH3), 4.40 (q, 2H, CH2 ester), 5.14 (s, 2H, NH2), 5.40 (s, 1H, CH of thiazolidinone ring), 7.24-8.90 (m, 8H, Ar-H), 9.14 (s, 1H, NH). |
| 25 | -S- | Acetyl-thiosemi-carbazone | ![Molecules 08 00622 i006]() | IR (ν, cm-1): 3450, 3250, 3200 (NH, NH2), 1720 (C=O), 1600 (C=N); 1H-NMR (DMSO-d6): δ 2.24 (s, 6H, 2CH3), 3.92 (s, 2H, CH2 of thiazolidinone), 4.86 (s, 2H, NH2), 7.00-8.00 (m, 8H, Ar-H), 8.3 (s, 1H, NH), 9.7 (s, 1H, NH). |
| 26 | -S- | Acetyl-thiosemi-carbazone | ![Molecules 08 00622 i007]() | IR (ν, cm-1): 3450, 3250, 3170 (NH, NH2), 1600 (C=N); 1H-NMR (DMSO-d6): 2.3 (s, 6H, 2CH3-C=N), 2.50 (s, 3H, CH3 of thiazoline ring), 4.7 (s, 2H, NH2), 6.20 (s, 2H, CH2), 7.20-7.90 (m, 8H, Ar-H), 8.7 (s, H, NH). |
Table IV.
Theoretical data of compounds 2, 5, 5a, 6-11.





{kind=link}
{kind=link}
{kind=link}
{kind=link}













