2-(p-Hydroxybenzyl)indoles - Side Products Formed Upon Cleavage of Indole Derivatives from Carboxylated Wang Polymer - an NMR Study

{kind=link}
{kind=link}
Abstract
:Introduction
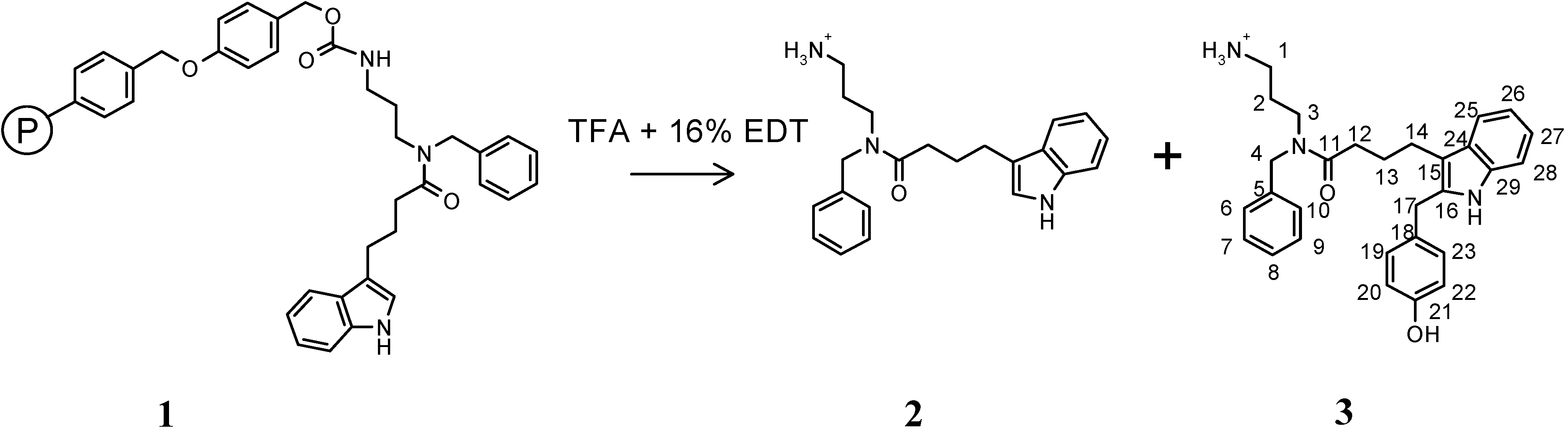
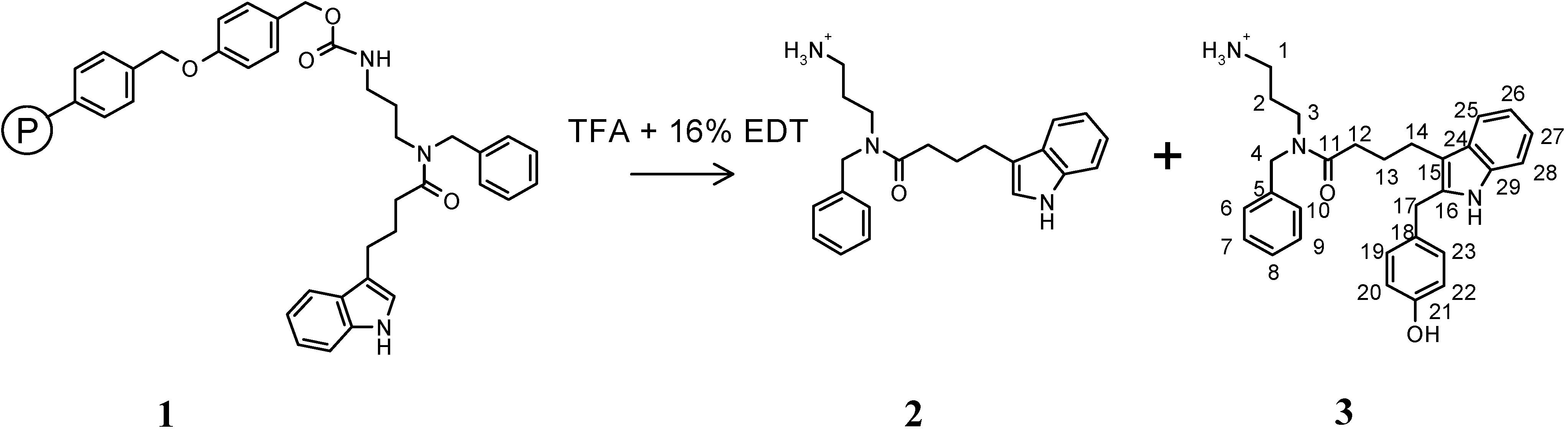
Results and Discussion
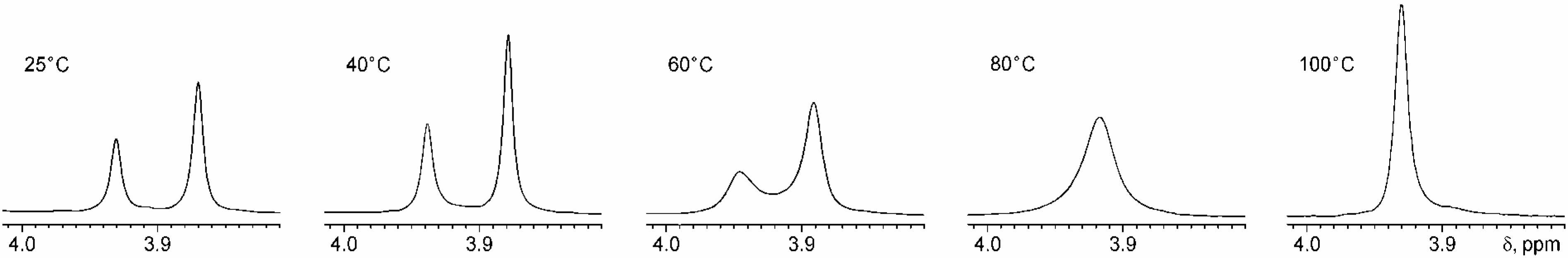
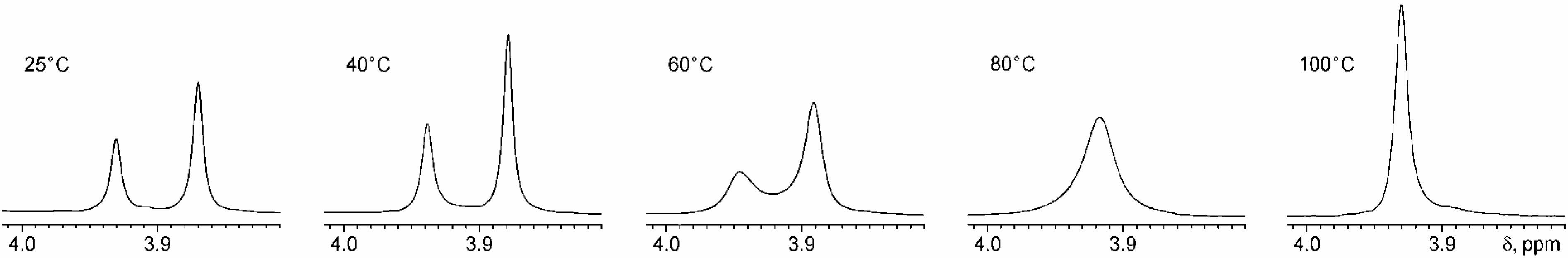
Conclusions
Experimental
General
N-(3-Amino-propyl)-N-benzyl-4-(1H-indol-3-yl)-butyramide (2) trifluoroacetate and N-(3-Amino-propyl)-N-benzyl-4-[2-(4-hydroxy-benzyl)-1H-indol-3-yl]-butyramide (3) trifluoroacetate.
Variable Temperature NMR Studies
Spectral Data
References
- Riniker, B.; Kamber, B. Peptides 1988; Jung, G., Bayer, E., Eds.; Walter de Gruyter & Co: Berlin, New York, 1989; pp. 115–117. [Google Scholar]
- Giraud, M.; Cavelier, F.; Martinez., J. A side-reaction in the SPPS of Trp-containing peptides. J. Pept. Sci. 1999, 5, 457–461. [Google Scholar]
- Prohazka, Z. Paper Chromatography. A Comprehensive Treatise; Hais, I.M., Macek, K., Eds.; Publishing House of the Czechoslovak Academy of Sciences: Prague, 1963; p. 247. [Google Scholar]
- Wokaun, A.; Ernst, R. R. Selective detection of multiple quantum transitions in NMR by two-dimensional spectroscopy. Chem Phys. Lett. 1977, 52, 407–412. [Google Scholar]
- Piantini, U.; Sörensen, O. W.; Ernst, R. R. Multiple quantum filters for elucidating NMR coupling networks. J. Am. Chem. Soc. 1982, 104, 6800–6801. [Google Scholar]
- Shaka, A.J.; Freeman, R. Simplification of NMR spectra by filtration through multiple-quantum coherence. J. Magn. Reson. 1983, 51, 169–173. [Google Scholar]
- Bax, A. Structure determination and spectral assignment by pulsed polarization transfer via long-range proton-carbon-13 couplings. J. Magn. Reson. 1984, 57, 314–318. [Google Scholar]
- Kessler, H.; Griesinger, C.; Zarbock, J.; Loosli, H. R. Assignment of carbonyl carbons and sequence analysis in peptides by heteronuclear shift correlation via small coupling constants with broadband decoupling in t1 (COLOC). J. Magn. Reson. 1984, 57, 331–336. [Google Scholar]
- Kessler, H.; Griesinger, C.; Lautz, J. Determination of connectivities via small proton-carbon couplings with a new two-dimensional NMR technique. Angew. Chem. Int. Ed. Engl. 1984, 23, 444–445. [Google Scholar]
- Kessler, H.; Griesinger, C.; Wagner, K. Peptide conformations. 42. Conformation of side chains in peptides using heteronuclear coupling constants obtained by two-dimensional NMR spectroscopy. J. Am. Chem. Soc. 1987, 109, 6927–6933. [Google Scholar]
- Günther, H. NMR spectroscopy; Basic Principles, Concepts, and Applications in Chemistry, 2nd. Edition ed; John Wiley & Sons: Chichester, England, 1994. [Google Scholar]
- Samples Availability: Samples not available.
© 2003 ( http://www.mdpi.org). Reproduction is permitted for non-commercial purposes.
Share and Cite
Mutulis, F.; Erdélyi, M.; Mutule, I.; Kreicberga, J.; Yahorava, S.; Yahorau, A.; Borisova-Jan, L.; Wikberg, J.E.S. 2-(p-Hydroxybenzyl)indoles - Side Products Formed Upon Cleavage of Indole Derivatives from Carboxylated Wang Polymer - an NMR Study. Molecules 2003, 8, 728-734. https://doi.org/10.3390/81000728
Mutulis F, Erdélyi M, Mutule I, Kreicberga J, Yahorava S, Yahorau A, Borisova-Jan L, Wikberg JES. 2-(p-Hydroxybenzyl)indoles - Side Products Formed Upon Cleavage of Indole Derivatives from Carboxylated Wang Polymer - an NMR Study. Molecules. 2003; 8(10):728-734. https://doi.org/10.3390/81000728
Chicago/Turabian StyleMutulis, Felikss, Máté Erdélyi, Ilze Mutule, Jana Kreicberga, Sviatlana Yahorava, Aleh Yahorau, Larisa Borisova-Jan, and Jarl E.S. Wikberg. 2003. "2-(p-Hydroxybenzyl)indoles - Side Products Formed Upon Cleavage of Indole Derivatives from Carboxylated Wang Polymer - an NMR Study" Molecules 8, no. 10: 728-734. https://doi.org/10.3390/81000728