Introduction
The interactions of DNA-binding proteins and DNA-binding drugs with double-stranded DNA (dsDNA) in the genome are involved in many important biological functions, including gene transcription regulation [
1], DNA recombination [
2], restriction [
3], replication [
4] and DNA-drug intercalation [
5,
6]. Therefore, lots of techniques including nitrocellulose-binding assays [
7], gel mobility-shift analysis [
8,
9], Southwestern blotting [
10,
11], ELISA [
12], reporter constructs in yeast [
13], Chromatin immunoprecipitation (ChIP) [
14], phage display [
15], binding-site signatures [
16], in-vitro selection [
17], UV crosslinking [
18], and methylization interfering assay [
19] and X-ray crystallography [
20,
21] were developed to effectively examine sequence-specific DNA-protein interactions, and also many techniques including UV absorption [
22], melting temperature (thermodynamics) [
23], NMR [
24], X-ray crystallography [
25,
26], free solution capillary electrophoresis (FSCE) [
27], scanning force microscopy (SFM) [
28,
29,
30], atomic force microscopy (AFM) [
31], surface plasmon resonance (SPR) [
32], polymerase chain reaction (PCR) [
33,
34] and footprinting [
35,
36] were used to effectively study DNA-drug interactions. However, these techniques using non-immobilized free dsDNAs in the liquid-phase to probe dsDNA interactions with other molecules such as proteins, ligands and drugs suffered from being laborious, time-consuming and incapable of high-throughput parallel analysis. Therefore, the solid surface-coupled dsDNA has become more and more important for high-throughput examination of sequence-specific DNA-protein interactions [
37,
38,
39] and DNA-drug interactions [
40,
41], paving the way for new strategies for screening DNA-binding proteins [
42], predicting DNA-binding sites [
38,
39], assessing binding affinity [
40,
41] and screening sequence-specific DNA-binding drugs and finding drug-preferential sequences [
40,
41].
When the surface-coupled homogenous dsDNA were employed for examination of sequence-specific DNA-proteins or DNA-drug interactions, the number of classes of dsDNAs immobilized on a detectable solid entity determines how much information would be obtained in a single study. The surface-coupled homogenous dsDNAs were traditionally used in affinity chromatography to isolate the sequence-specific DNA-binding proteins [
43,
44] and the homogenous dsDNA probes immobilized on small paramagnetic particles were used to identify DNA-binding proteins by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) [
42]. These solid entities carrying homogenous dsDNAs suffered from the problem that they isolated or identified only one target molecule each time, therefore, they were not highly informative strategies for surface-coupled dsDNA applications. However, libraries of dsDNA oligonucleotides comprising a plurality of different members immobilized on a solid support much improved this limitation [
45]. These solid-immobilized libraries of dsDNA oligonucleotides provided a useful technique for the screening of numerous biological samples by sequence-specific interactions [
45]. Thereafter, the fabrication of fast, economical and high informative dsDNA-coupled solid entities became the pivotal problem for extensive surface-coupled dsDNA applications. Nevertheless, the earliest designed surface-immobilized libraries of different dsDNA oligonucleotides were fabricated by chemically synthesizing and immobilizing very long single-stranded DNA (ssDNA) oligonucleotides on solid surface and then forming unimolecular dsDNA oligonucleotides by intra-strand annealing self-complementery elements in immobilized long single-stranded oligonucleotides. This kind of surface-coupled dsDNA were high informative, but suffered from economic problems [
45].
The appearance of high density bimolecular dsDNA microarrays greatly promoted the application of solid-immobilized duplex nucleic acids [
37,
38,
39,
40,
41], and it is demonstrated that these high density bimolecular dsDNA microarrays were very effective for high throughput examination of DNA-protein interactions [
38,
39] and DNA-drug interactions. However, the developments of these bimolecular dsDNA microarrays were slowed by several innate technical and economic drawbacks. The methods to manufacture bimolecular dsDNA microarrays could be divided into two types, one was by hybridization [
40,
41,
46], and the other was enzymatic elongation [
37]. The former spotted larger numbers of chemically synthesized ssDNA oligonucleotides onto solid surfaces to firstly fabricate a ssDNA microarray and subsequently converted this ssDNA microarray into a bimolecular dsDNA microarray by hybridizing the ssDNA microarray with a mixture of complementary ssDNA oligonucleotides. This method suffered from two main problems, one was high manufacturing costs resulting from amino-modification of immobilized ssDNA oligonucleotides and the second, complementary ssDNA oligonucleotide synthesis; the other more important problem was that the method could not fabricate dsDNA microarrays carrying very sequence-similar probes such as probes with single-nucleotide variations [
40,
41]. The latter on-chip photo-addressably synthesized high density ssDNA microarray with a constant sequence at far surface-attached 3′ end of each oligonucleotides, and then annealed a general primer to constant sequence and performed enzymatic extension reactions on the array to convert ssDNA microarrays into bimolecular dsDNA microarrays. This method encountered more serious economic and technical issues. Technically, the method relied on currently expensive and proprietary technology for the surface photo addressable synthesis of oligonucleotides, but synthesis of single-stranded oligonucleotides on solid surfaces was inefficient, with per-nucleotide synthesis efficiencies thought to be only 92-96% [
47,
48], for a 40-mer oligonucleotides, only 4-20% of the sequences on a chip could be of desired length and sequence [
49]. In practice, oligonucleotide arrays constructed in this fashion are heavily contaminated with truncated molecules [
47,
50]. Moreover, the presence of so many competing truncated molecules and single-stranded oligos not accessible to the primers might strongly interfere and mislead binding experiments [
49]. Finally, considering the possible instability of bimolecular dsDNA oligos towards binding or washing reactions, the usefulness of this kind of bimolecular dsDNA microarrays might be diminished.
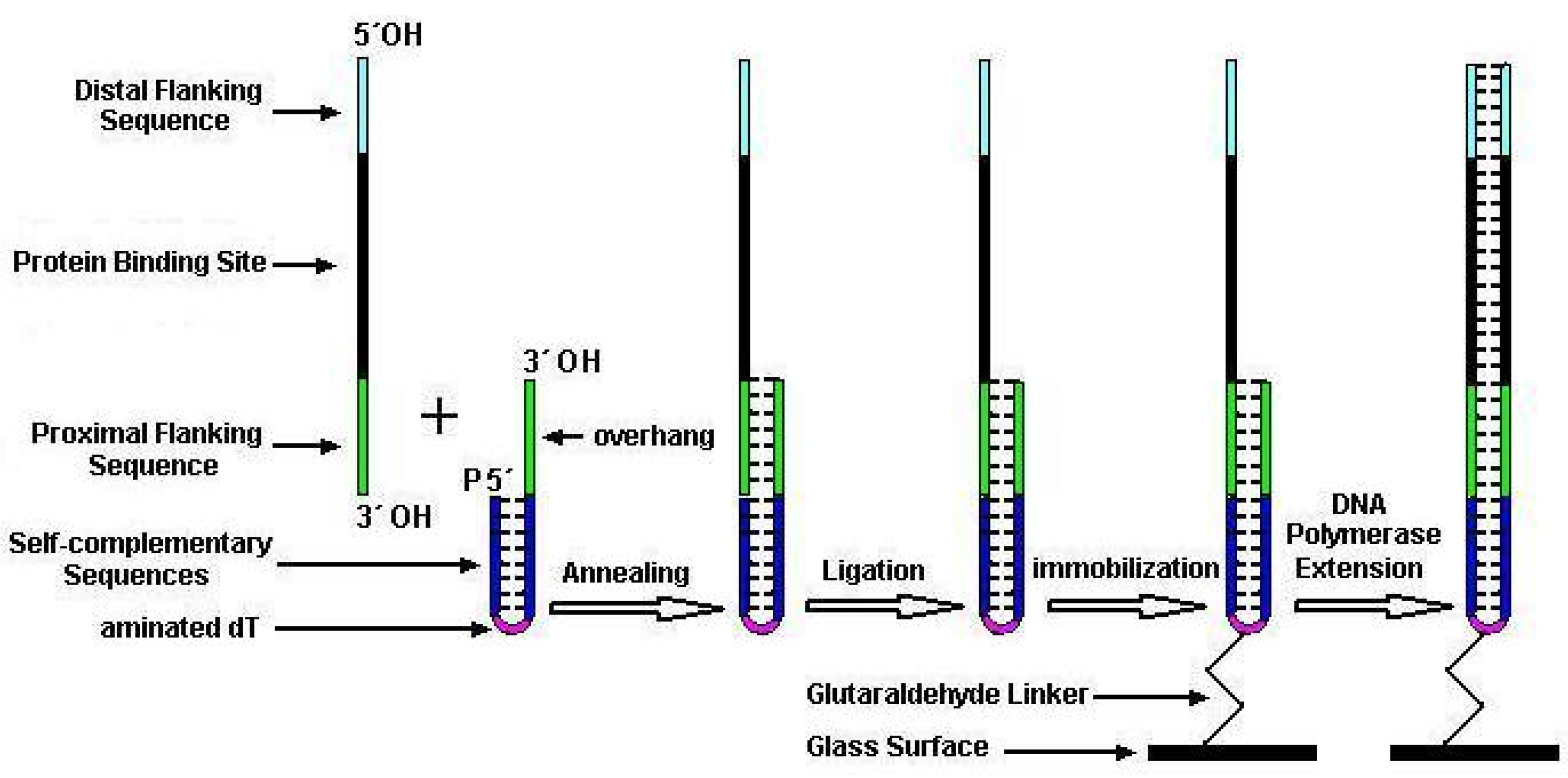
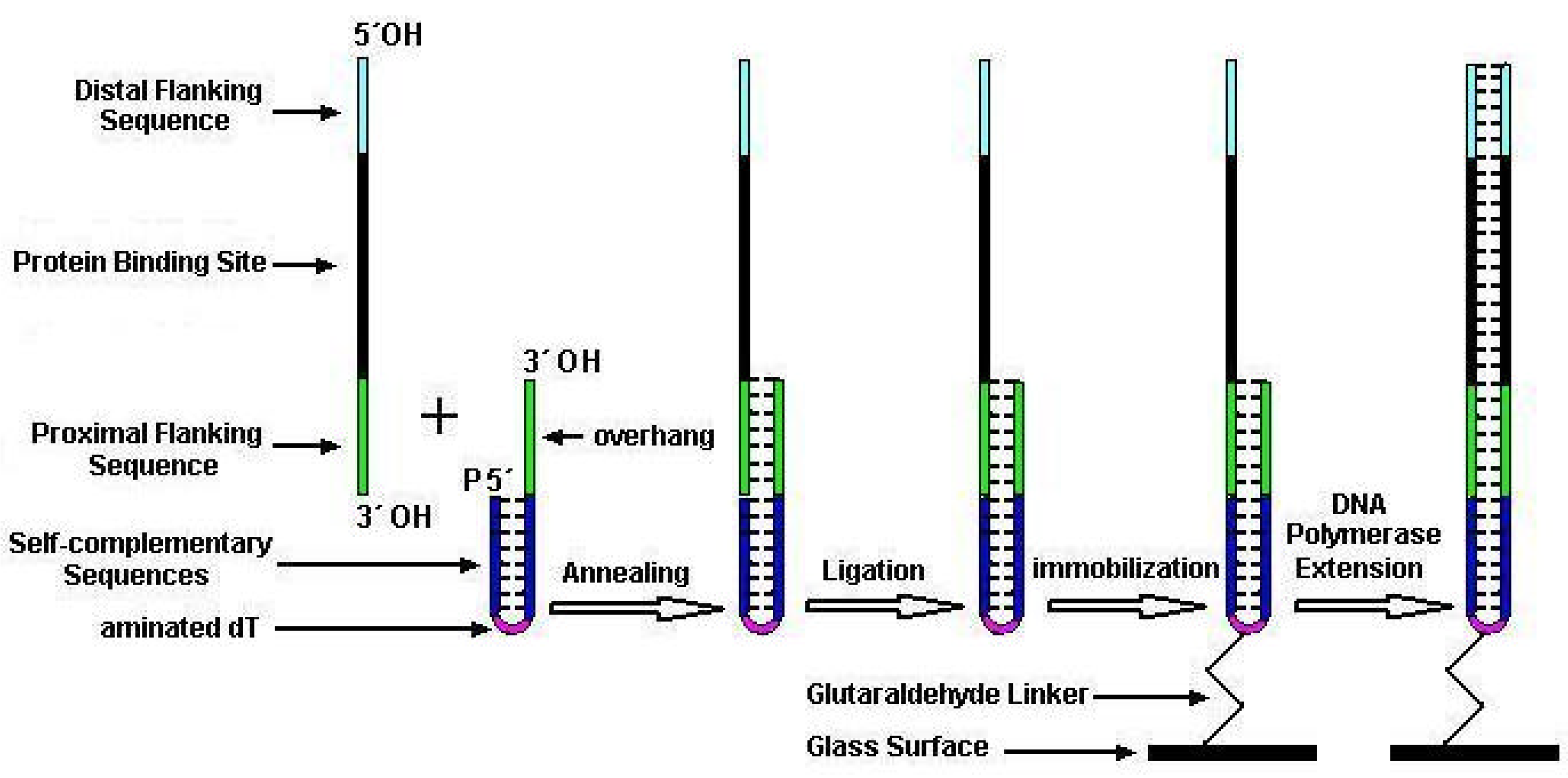
To overcome the drawbacks existing in previous dsDNA microarray techniques, we present a new method for economically fabricating unimolecular dsDNA microarrays which could be used many times. In this method, two kinds of special single-stranded oligonucleotides were firstly chemically synthesized for manufacturing unimolecular dsDNA microarrays, one was a target oligonucleotide harboring binding site of DNA-binding proteins and the other was a constant oligonucleotide for capturing, immobilizing DNA on glass slide and providing a primer for polymerization. The target oligonucleotides and the constant oligonucleotide were then annealed and ligated into the new unimolecular oligonucleotides, which were subsequently microspotted and immobilized on aldehyde-derivatized glass surfaces. Finally, we elongated the primer along the overhang template of unimolecular oligonucleotide with Klenow fragments to convert the partial-dsDNA microarrays into unimolecular complete-dsDNA microarrays. We verified the feasibility of our method and demonstrated the sensitivity and specificity of detecting DNA-binding proteins with our dsDNA microarray.
Materials and Methods
Synthesis of ssDNA Oligonucleotides
Two kinds of special single-stranded oligonucleotides were chemically synthesized by Shengyou Inc. (Shanghai, China) for manufacturing dsDNA microarrays (listed in
Table 1), one was a constant oligonucleotide (CO) consisting of a 7-base capture sequence at the 3´ end and two 10-base reverse complementary sequences linked by an internal dT with a primary NH
2 group (amino modifier C6) at the 5´ end. The other was a target oligonucleotide (TO) containing a 7-base proximal flanking sequence complementary to capture sequence of CO, a randomized-base target sequence corresponding to DNA-binding sites of the sequence-specific DNA-binding proteins, and a distal flanking sequence from the 3´ to the 5´ end. The 3´ end of CO was hydroxyl and 5´ end was phosphate. Both 3´ and 5´ ends of TO were hydroxyl.
Preparation of partial-dsDNA oligonucleotides
The variant TOs were respectively annealed and ligated with pre-self-annealed COs in 1:1 molar ratio in a ligation reaction containing 40 mM Tris-HCl (pH 7.8), 10 mM MgCl4, 10 mM DTT, 0.5 mM ATP and 0.5 U/µl T4 DNA ligase (MBI fermentas). The completed ligation reactions were exchanged into sodium carbonate buffer (0.1M, pH9.0) at a concentration of 50 µM, by using CentriSpin-10 spin columns (Princeton Separations, Adelphia, NJ).
Manufacture of partial-dsDNA Microarrays
Cleaned microscopy glass slides were silanized in 2% aminopropyltriethoxysilane (Sigma) dissolved in 95% acetone for 5 min. After washing twice with acetone and baking for 45min at 75°C, the aminosilane slides were activated in glutaraldehyde solution (5% glutaraldehyde in 0.01M PB at pH7.0) for 30 min. The glutaraldehyde-activated slides were washed 3 times with distilled water and blown to dryness with N2. A PixSys5500 pin-based spotting robot (Cartesian Technology Inc.) with a CMP3 pin was employed to print the prepared oligonucleotides dissolved in sodium carbonate buffer (0.1M, pH9.0) on glutaraldehyde-activated slides. After printing, the microarrays were kept in a humidity chamber containing sodium carbonate buffer (0.1M, pH9.0) and incubated overnight at room temperature, and then followed by a incubation at 37°C for 1 h. The rest of the aldehyde groups on slides were inactivated by a 30-min incubation in 0.28% (w/v) NaBH4/76% (v/v) PBS/24% (v/v) alcohol. After through washing in sterile double-distilled water (ddH2O), slides were spun dry in a clinical centrifuge and then stored in a close slide cassette at 4°C until use.
Fabrication of complete-dsDNA Microarrays
The slides with prepared partial-dsDNA microarrays were denatured in boiling water for 5 min and then incubated with hybridization buffer for 1 h at 50°C. Subsequently, the slides were rinsed in turn with 2°SSC/0.1% SDS, 0.2°SSC/0.1% SDS and sterile ddH2O. Thereafter, the partial-dsDNA microarrays were incubated with DNA polymerase reagent containing 50 mM Tris-HCl (pH 7.2), 10 mM MgSO4, 0.1 mM DTT, 40 µM of each dNTP, 20µg/ml acetylated BSA and 2 U/µl DNA polymerase I large (Klenow) fragment (3´ to 5´ exo–; Promega, Madison, WI). Detection of the enzymatic extension was accomplished by replacing the dTTP with Cy3-labeled dUTP (Pharmacia, Piscataway, NJ) in a DNA polymerase reaction. After extension reaction, the microarrays were respectively washed with 2°SSC/0.1% SDS, 0.2°SSC/0.1% SDS and sterile ddH2O for 10min at room temperature. At last, the microarrays were dried in a clinical centrifuge and kept in closed cassette at 4°C until use.
Digestion of dsDNA Microarray with Restriction Endonuclease
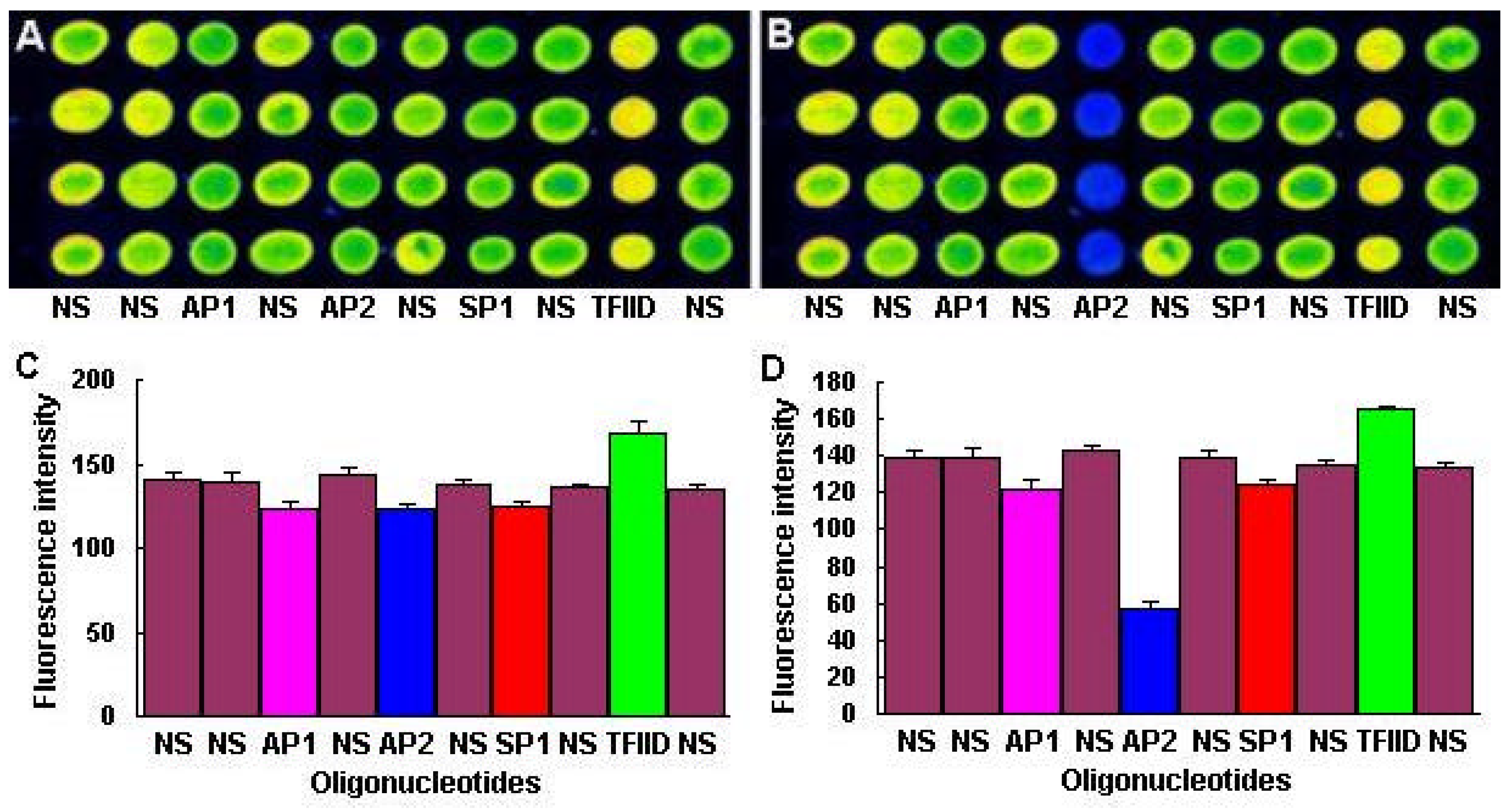
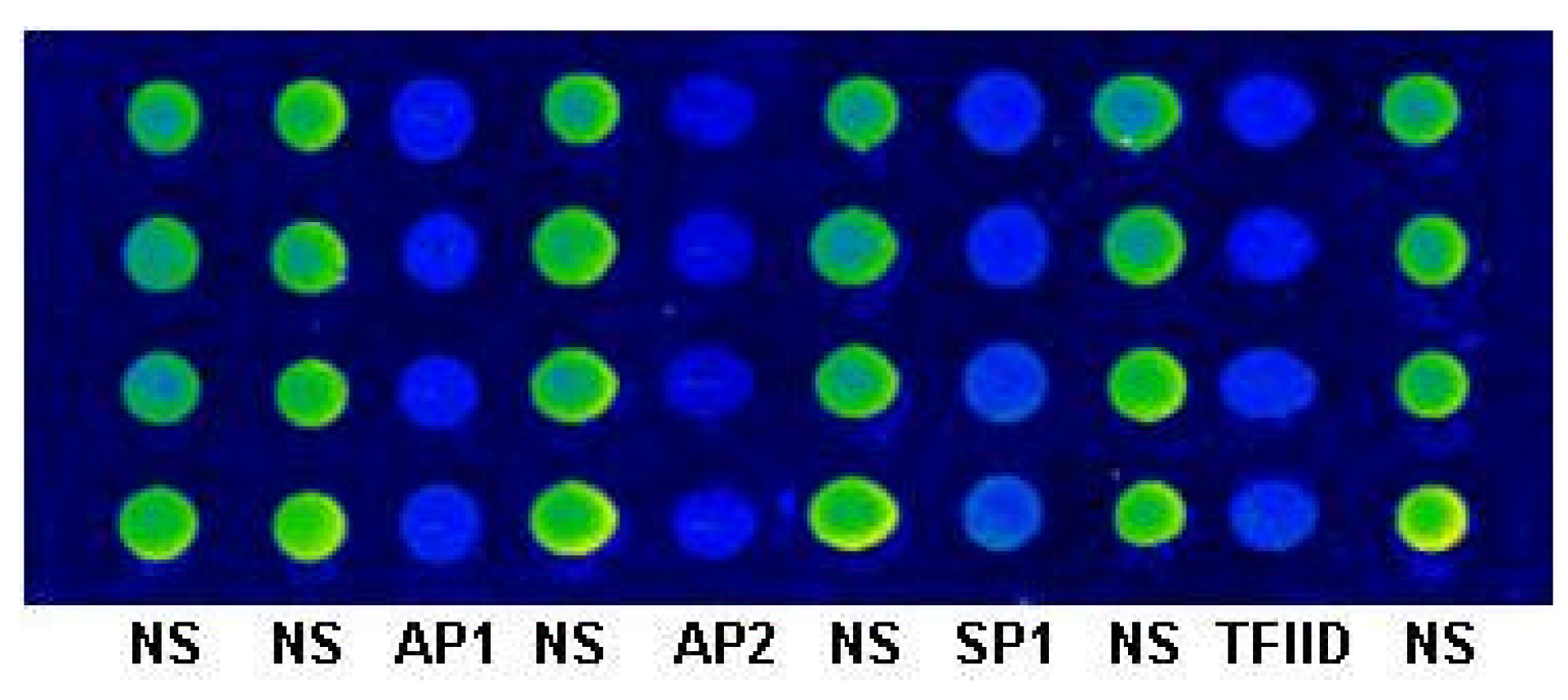
Hae IIIigestion of the dsDNA Microarray was performed at 37°C for 2h by reaction with 10 mM Tris-HCl (pH 8.5), 10 mM MgCl2, 100 mM KCl, 0.02% Triton X-100, 0.1 mg/mL BSA and 0.5 U/µL Hae III (MBI fermentas). After digestion, the slides were rinsed in turn with 2°SSC/0.1% SDS, 0.2°SSC/0.1% SDS and sterile ddH2O.
Labeling of DNA-binding Proteins
DNA-binding protein NF-κB (human recombinant p50 expressed in bacteria from a full-length cDNA encoding 453 amino acids) was purchased from Promega. The proteins provided in glycerol solutions were exchanged with BioRad Biospin P6 column into sodium carbonate-sodium bicarbonate buffer (pH9.3) at the concentration of 1mg/mL and labeled with FluoroLinkTM Cy3 monofunctional dye (Amersham Pharmacia Biotech, Piscataway, NJ) at room temperature for 30 min. After labeling, the protein solutions were exchanged with BioRad Biospin P6 column into glycerol-free, phosphate-buffer saline (PBS) solution (141 mM NaCl, 7.2 mM Na2HPO4, 2.8 mM NaH2PO4, pH7.4). The labeled proteins PBS solutions were aliquoted and kept at 4°C until use.
Binding of DNA-binding Proteins to dsDNA Microarray
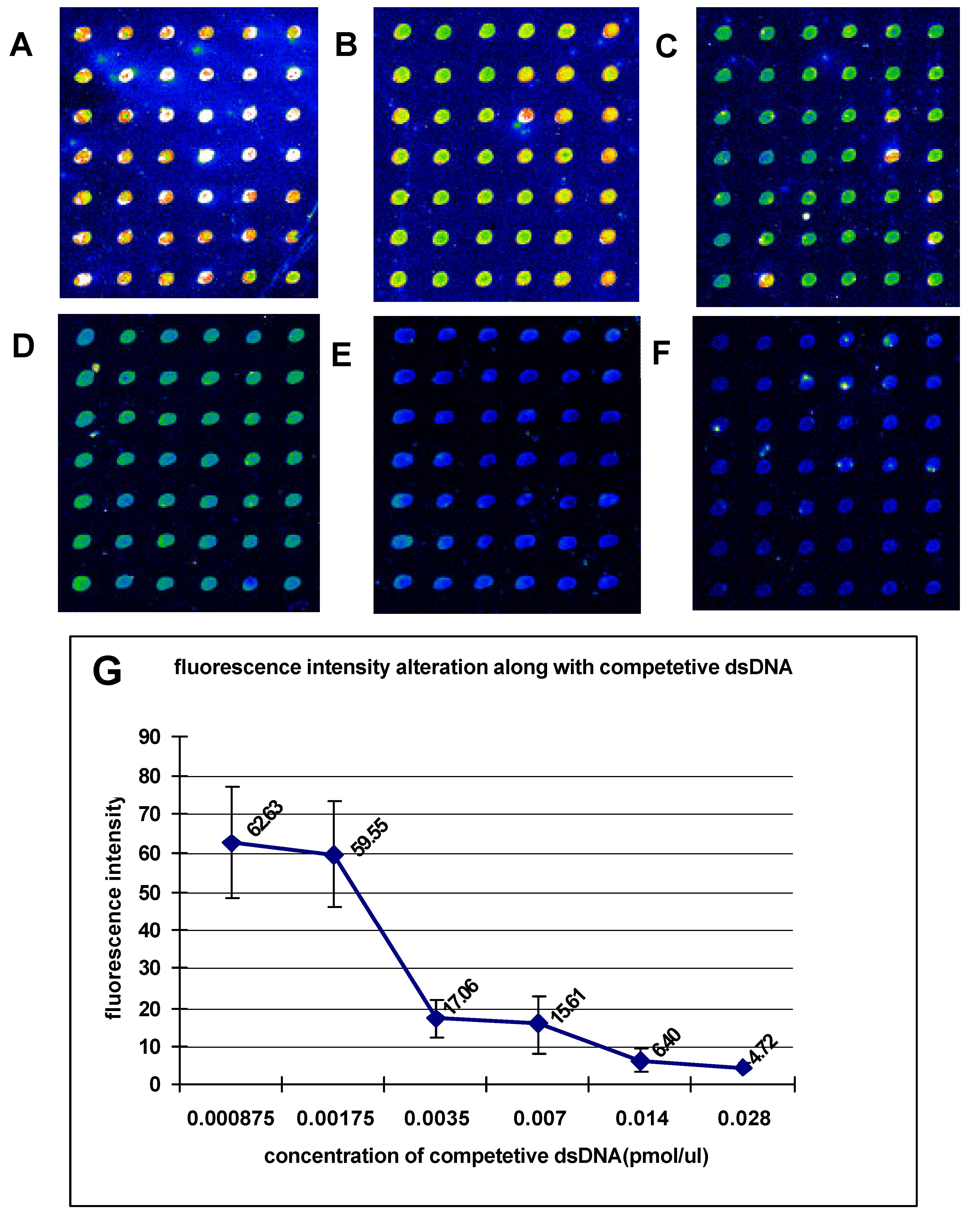
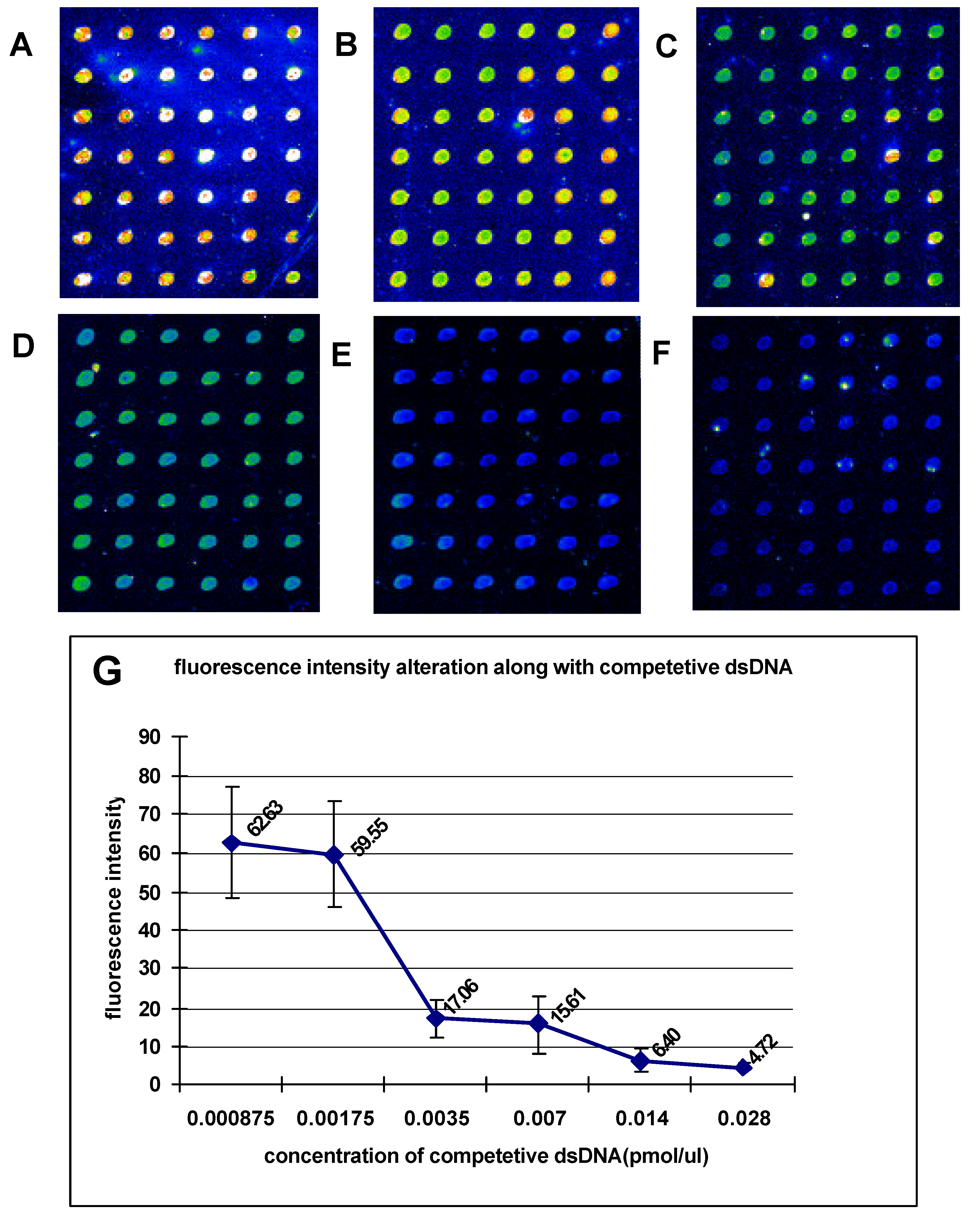
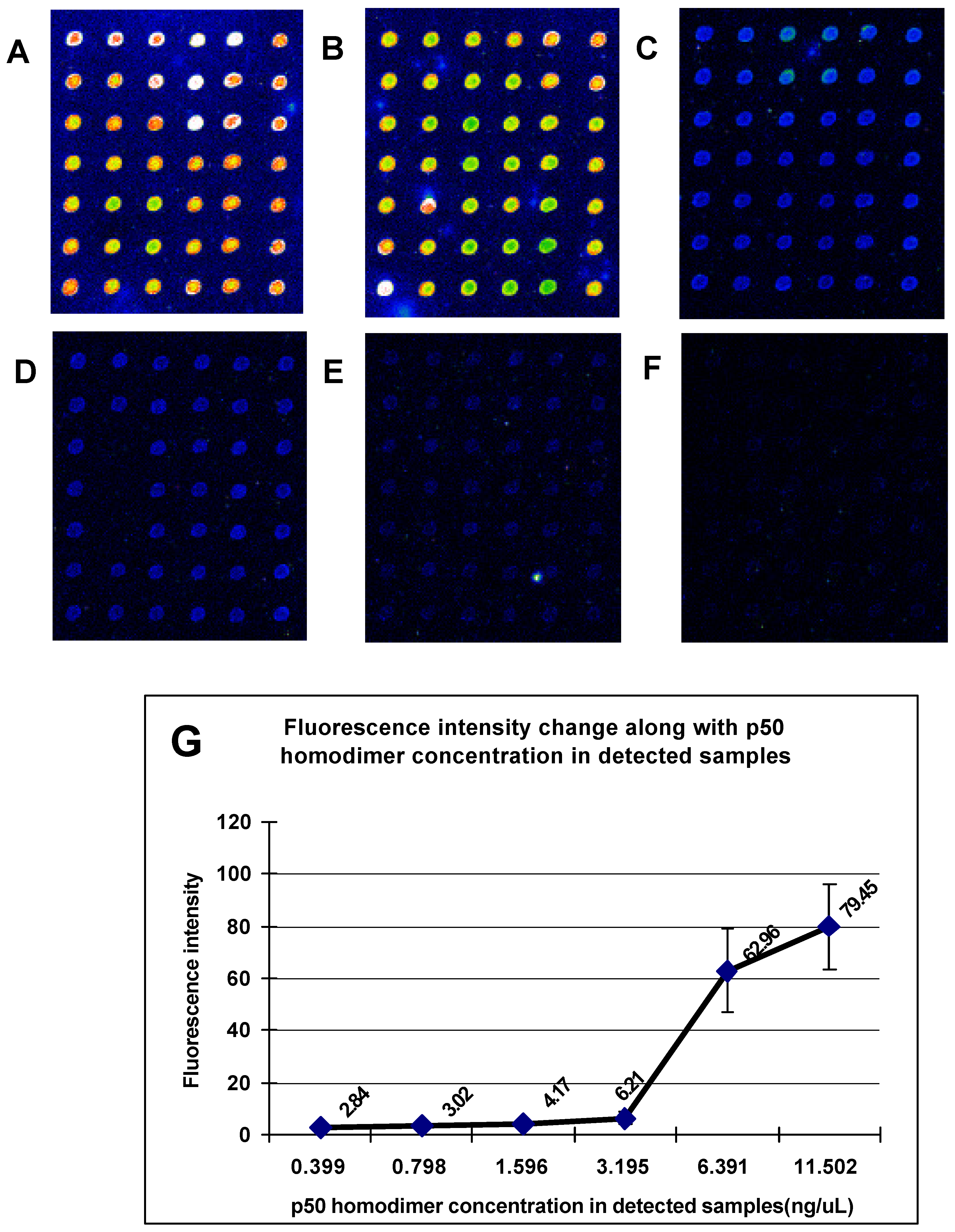
The dsDNA microarrays were blocked with 10% BSA/ 0.01 M PBS for 1 h at room temperature, then washed with 0.01M PBS and spun dry. The blocked dsDNA microarrays were incubated with DNA-binding buffer (10 mM HEPES pH7.9, 50 mM KCl, 2.5 mM DTT, 0.1 mM EDTA, 0.05% NP-40, 10% Glycerol, 5% BSA) containing Cy3 labeled NF-κB at room temperature for 1 h. After incubation, the dsDNA microarrays were washed in turn with 0.01 M PBS/0.05% Tween 20 for 15 min, and 0.01 M PBS/0.01% Triton 100 for 15 min. Subsequently, the dsDNA microarrays were briefly washed with sterile ddH2O and spun dry in a clinical centrifuge. At last, the dsDNA microarrays were scanned with ScanArray® Lite of Packard Biochip Technologies in the Cy3 channel at 90% laser power, 80% PMT gain, 5µm resolution.
dsDNA Microarray Data Analysis
The signal intensities of the spots on microarray scanned false color images were quantified with QuantArray® microarray analysis software (Packard Biochip Technologies). The signal intensity of the spots refereed to the absolute signal intensity calculated by substracting the background fluorescence intensity from the detected signal intensity of spots.
Conclusions
Church’s lab saw the great potentials of dsDNA microarrays for studying sequence-specific DNA/protein interactions [
37], and fabricated dsDNA microarray for exploring the DNA-binding specificities of zinc fingers with arrayed DNA targets [
38,
39]. Their creative work verified the feasibility and high effectiveness of dsDNA microarrays in studying sequence-specific DNA/protein interaction. Nevertheless, their dsDNA microarray fabrication replied on the Affymetrix proprietary technology of photo-addressable oligonucleotide synthesis which is unaffordable for general laboratories until now. In this paper, we have presented a novel method for fabricating unimolecular dsDNA microarray and verified its reliability. This method has several significant advantages. Firstly, considering the expensive amino-labeling of oligonucleotides which cost was almost identical to target oligonucleotide synthesis (about 25 base pairs), the free constant oligonucleotide with amino modifier C6 dT was adopted to avoid repeatedly synthesizing long constant oligonucleotide with C6dT on each target oligonucleotides. This strategy can dramatically decrease the cost of unimolecular dsDNA microarray manufacture. Secondly, with free constant C6dT oligonucleotide, we only need to chemically synthesize target oligonucleotides for fabricating unimolecular dsDNA microarray. We testified that as short as 23bp (AP2) and 24bp (NF-
kB) oligonucleotides harboring protein-binding sites were long enough for proteins binding interaction. To synthesize so short single-stranded oligonucleotides also greatly lowers the cost of unimolecular dsDNA microarray manufacture. It avoids synthesizing long self-complementary single-stranded oligonucleotides for fabricating dsDNA oligonucleotides by annealing [
45]. Thirdly, as the “second” complementary strands of oligonucleotides were enzymatically synthesized by DNA polymerase, it also greatly lowers the cost of unimolecular dsDNA microarray manufacture. Fourthly, as the exact complements of immobilized target oligonucleotides were synthesized by high-affinity Klenow DNA polymerase I, this method is competent for fabricating dsDNA microarray containing generic or homogenous dsDNA oligonucleotides with similar sequences as single nucleotide polymorphism (SNP). It overcomes the impossibility of fabricating generic or homogenous dsDNA, especially SNP dsDNA microarrays by hybridization [
40,
41]. This is very important for dsDNA microarray applications to studying sequence-specific DNA/protein interactions. Fifthly, the enzymatically synthesized ultimate dsDNA oligonucleotides immobilized on glass were unimolecular nucleic acids, which can hold their function by reanneating after heat denature. Therefore, the unimolecular dsDNA microarray can be used for many times by removing bound proteins with stringent washing or heat denaturing. This far differs from bimolecular-dsDNA microarray [
37,
38,
39,
40,
41]. Finally, this method of unimolecular dsDNA microarray fabrication accommodates with commercially available microarray spotting robots, it is reachable for extensive application.
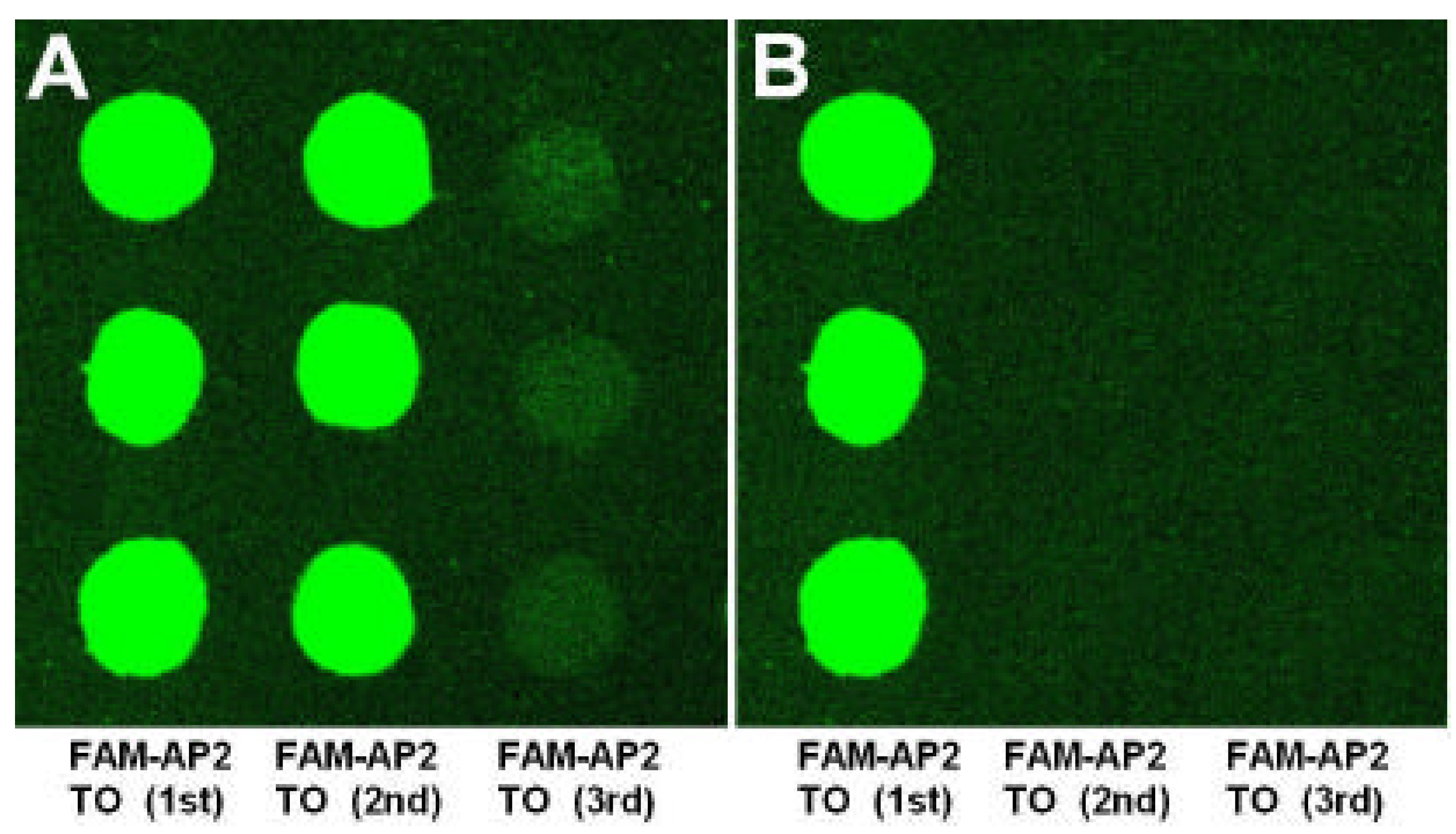
Our experiments revealed that the sequence-specific DNA-binding proteins as restriction endonuclease and transcription factor in detected samples could sensitively and specifically bind with dsDNA targets immobilized on microarray. This demonstrates that the method we presented in this paper provides a stable and affordable technique for fabricating the reusable unimolecular dsDNA microarray which could be used for high-throughput investigation of the interactions between biomolecules. The unimolecular dsDNA microarray could be potentially used to studies including (1) screening sequence-specific DNA-binding proteins, (2) predicting new DNA-binding sites of transcription factors in genome, (3) assessing importance of nucleotides in DNA-binding sites for DNA/protein interactions, (4) monitoring the expression of drug-induced DNA-binding proteins, and (5) screening sequence-specific DNA-binding drugs.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}