Molecular Polarizability of Sc and C (Fullerene and Graphite) Clusters

Institut Universitari de Ciència Molecular, Universitat de València, Dr. Moliner 50, E-46100 Burjassot (València), Spain
Molecules 2001, 6(6), 496-509; https://doi.org/10.3390/60600496
Submission received: 8 March 2001
/
Revised: 3 April 2001
/
Accepted: 26 April 2001
/
Published: 31 May 2001
Abstract
:A method (POLAR) for the calculation of the molecular polarizability <α> is presented. It uses the interacting induced dipoles polarization model. As an example, the method is applied to Scn and Cn (fullerene and one-shell graphite) model clusters. On varying the number of atoms, the clusters show numbers indicative of particularly polarizable structures. The <α> are compared with reference calculations (PAPID). In general, the Scn calculated (POLAR) and Cn computed (POLAR and PAPID) are less polarizable than what is inferred from the bulk. However, the Scn calculated (PAPID) are more polarizable than what is inferred. Moreover, previous theoretical work yielded the same trend for Sin, Gen and GanAsm small clusters. The high polarizability of the Scn clusters (PAPID) is attributed to arise from dangling bonds at the surface of the cluster.
Introduction
Benichou et al. measured the static electric dipole polarizabilities of lithium clusters made of n (n=2-22) atoms [1]. The experiment consisted of deflecting a collimated cluster beam through a static inhomogeneous electric field. The strong decrease per atom from Li to Li3-Li4 showed that electronic delocalization was reached for very small sizes. Moreover, directly measured polarizabilities were consistent with photoabsorption data. They thus confirmed unambiguously the missing optical strength in lithium clusters. Maroulis and Xenides reported highly accurate ab initio calculations with specially designed basis sets for Li4 [2]. The molecule emerged as a particularly soft system, with very anisotroic dipole polarizability and very large second dipole hyperpolarizability. An extensive investigation of basis set and electron correlation effects led to values of = 387.01 and = 354.60 a.u. The mean hyperpolarizability was = 2394·103 a.u.. They also discussed the computational aspects of the effort in view of the extension of quantumchemical studies to larger lithium clusters. Their values for the mean dipole polarizabilitiy were systematically higher than the recently reported experimental static value (326.6 a.u.) of this important quantity [1].
Fuentealba presented a theoretical study of the static dipole polarizability of carbon clusters Cn with n ≤ 8 [3]. They calculated the dipole polarizabilities using density functionals of the hybrid type in combination with the finite field method. They investigated large basis sets in order to obtain reliable results. They showed that the dipole polarizabilities are an important quantity for the identification of clusters with different numbers of atoms and even for the separation of isomers. In particular, they predicted that the jet formed by the two isomers of C6, cyclic and linear, would split up in the presence of an electric field. Fuentealba and Reyes calculated the dipole polarizability of a series of clusters of the type LinHm using density functional methods [4]. They explained the study of the trends in the mean polarizability and the anisotropy in terms of the interplay between electronic and geometrical effects. They also discussed the changes in the polarizability for different isomers of a given cluster as well as its variations when hydrogen atoms were added to a given cluster. They also calculated a very related quantity, the hardness, in the simple approximation of hardness equal to the energy gap. They discussed their values in terms of the possible stability of the different clusters.
Jackson et al. used a first-principles, density-functional-based method to calculate the electric polarizabilities and dipole moments for several low-energy geometries of Si clusters in the size range 10 ≤ n ≤ 20 [5]. They found that the polarizability per atom is a slowly varying, nonmonotonic function of n. Over this size range the polarizability appeared to be correlated most strongly to cluster shape and not with either the dipole moment or the highest occupied—lowest unoccupied molecular-orbital gap. The calculations indicated that the polarizability per atom for Si clusters approaches the bulk limit from above as a function of size. Deng et al. calculated the polarizabilities of Si clusters with 9 to 28 atoms using a density functional cluster method [6,7]. They based the atomic geometries on those carefully optimized by energy optimization. The polarizability showed fairly irregular variation with cluster size, but all calculated values were higher than the polarizability of a dielectric sphere with bulk dielectric constant and equivalent volume.
Hohm et al. deduced an experimental value of 116.7 ± 1.1 a.u. for the static dipole polarizability of As4 from the analysis of refractivity measurements in arsenic vapour [8]. This was in close agreement with the theoretical result of 119.5 ± 3.6 a.u., obtained from ab initio finite-field many-body perturbation theory and coupled-cluster calculations.
In a previous paper, the following metal clusters and fullerenes were calculated: Scn (1 ≤ n ≤ 7 and n = 12), Cn (n = 1, 12, 60, 70 and 82) and endohedral Sc@C60 and Scn@C82 (1 ≤ n ≤ 3) [9]. In the present paper, the following metal clusters have been calculated: Sc; Sc2 linear (D∞h); Sc3 triangle (D3h); Sc4 in three conformations, square (D4h), rhomb (D2h) and tetrahedron (Td); Sc5 triangular bipyramid (D3h); Sc6 in two conformations, octahedron (Oh) and antiprism (D3d); Sc7 pentagonal bipyramid (D5h); Sc12 icosahedron (Ih); Sc17 (hexagonal close packing, HCP) and Sc74 (HCP). For some clusters, several isomers have been considered. Atom-atom contact distances have been held at 2.945Å. The following fullerene models have been studied: C, C12 (Ih), C60 (Ih), C70 (D5h) and C82 (C2). The following one-shell graphite models have been computed: Cn (n =1, 6, 10, 13, 16, 19, 22, 24, 42, 54, 84 and 96). Atom-atom contact distances have been held at the experimental distance of 1.415Å. In the next section, the description of the electrostatic properties used in this study is presented. Next, the interacting induced dipoles polarization model for the calculation of molecular polarizabilities is presented. Following that, results are presented and discussed. The last section summarizes my conclusions.
Electrostatic properties
Atomic net charges and polarizabilities have been calculated from their and contributions [10,11,12,13,14]. The net charges and polarizabilities have been calculated by the principle of electronegativity equalization [15,16,17,18] but applied bond by bond in the molecule [19,20,21,22]. The net charges and polarizabilities have been evaluated with the method of Hückel. The molecule has been brought into its principal inertial coordinate system.
It is well known that conjugation vanishes for perpendicular structures (e.g., biphenyl). Therefore, the Hückel parameter can be evaluated, in first approximation, between pz orbitals twisted from coplanarity by an angle as , where is equal to the parameter for benzene [23,24,25,26,27,28,29,30,31,32,33]. Joachim et al. [34] evaluated the electronic coupling Vab of the binuclear mixed valence MII-L-MIII complex [(NH3)5Ru-bipyridyl-Ru(NH3)5]5+. When a pyridine ring rotates around the ligand axis , Vab() was fitted by a function. From this observation, the α function is assumed universal and has the same form as Vab for this complex: .
Interacting induced dipoles polarization model for molecular polarizabilities
The calculation of molecular polarizabilities has been carried out by the interacting induced dipoles polarization model [35,36,37] that calculates tensor effective anisotropic point polarizabilities by the method of Applequist et al. [38,39,40]. One considers the molecule as being made up of N atoms (represented by i, j, k,…), each of which acts as a point particle located at the nucleus and responds to an electric field only by the induction of a dipole moment, which is a linear function of the local field. If a Cartesian component of the field due to the permanent multipole moments is , then the induced moment in atom i is:
where is the polarizability of atom i and is the symmetrical field gradient tensor,
where e is the charge of the proton and the subscripts a, b, c,… stand for the Cartesian components x, y, z. In Equation (1), the expression in parentheses is the total electric field at atom i, consisting of the external field plus the fields of all the other induced dipoles in the molecule.
The set of coupled linear Equations (1) for the induced dipole moments can be expressed conveniently in compact matrix equation form, if one introduces the 3N × 3N matrices and , with elements and being the Kronecker ), respectively. To suppress the restriction in the sum, the diagonal elements are defined as zero. Similarly and are 3N × 1 column vectors with elements and . Equation (1) is thus written in matrix form:
Where is the 3N × 3N -dimensional unit matrix. This matrix equation can be solved for the induced dipolesas
Here the symmetrical many-body polarizability matrix has been introduced:
The compact matrix equation is equivalent to the N matrix equations:
Let the molecule be in a uniform applied field, so that for all j. Then this equation becomes
The coefficient of in this equation is seen to be an effective polarizability of unit i, . The total moment induced in the molecule is:
from which it is seen that the molecular polarizability tensor is:
From energetic considerations it is known that is a Hermitian matrix [41,42] and must therefore be symmetric if all elements are real.
The significance of a polarizability of ± ∞ is that the molecule is in a state of resonance and absorbs energy from the applied field [43,44,45,46]. The following improvements have been implemented in the model:
- A damping function has been used in the calculation of the symmetrical field gradient tensor in order to prevent the polarizability from going to infinity [47].
- The interaction between bonded atoms and atoms with a distance lying in an interval defined by [rinf, rsup] has been neglected. The starting values for this interval are [0,1030] and rinf is incremented if resonance conditions are detected.
- To build up the many-body polarizability matrix the atomic polarizability tensors given by have been used instead of the scalar polarizability .
Calculation results and discussion
The molecular dipole-dipole polarizabilities <α> for the Scn clusters are reported in Table 1. Program POLAR gives <α> results that are one third of the corresponding PAPID reference values. This is due to a limitation in the current parametrization of POLAR, that will be improved in a future paper. In particular, the numerical restricted Hartree-Fock (RHF) value for Sc1 calculated by Stiehler and Hinze (22.317Å3) is significatively above the POLAR value but of the same order of magnitude as the PAPID one [50].

{kind=link}
{kind=link}
{kind=link}
| Scn | <α> (Å3)a | <α> ref.b |
|---|---|---|
| Sc | 5.631 | 16.893 |
| Sc2 | 1.418 | 13.744 |
| Sc3 | 2.103 | 11.557 |
| Sc4 D4h | 2.111 | 12.873 |
| Sc4 D2h | 2.461 | 11.163 |
| Sc4 Td | 2.657 | 10.041 |
| Sc5 | 3.116 | 9.690 |
| Sc6 Oh | 3.367 | 10.330 |
| Sc6 D3d | 3.904 | 10.330 |
| Sc7 | 3.429 | 9.321 |
| Sc12 | 3.891 | 8.724 |
| Sc17 h.c.p. | 3.590 | 25.278 |
| Sc74 h.c.p. | 3.630 | 23.471 |
a Average dipole-dipole polarizability (Å3).b Reference: calculations carried out with the PAPID program.
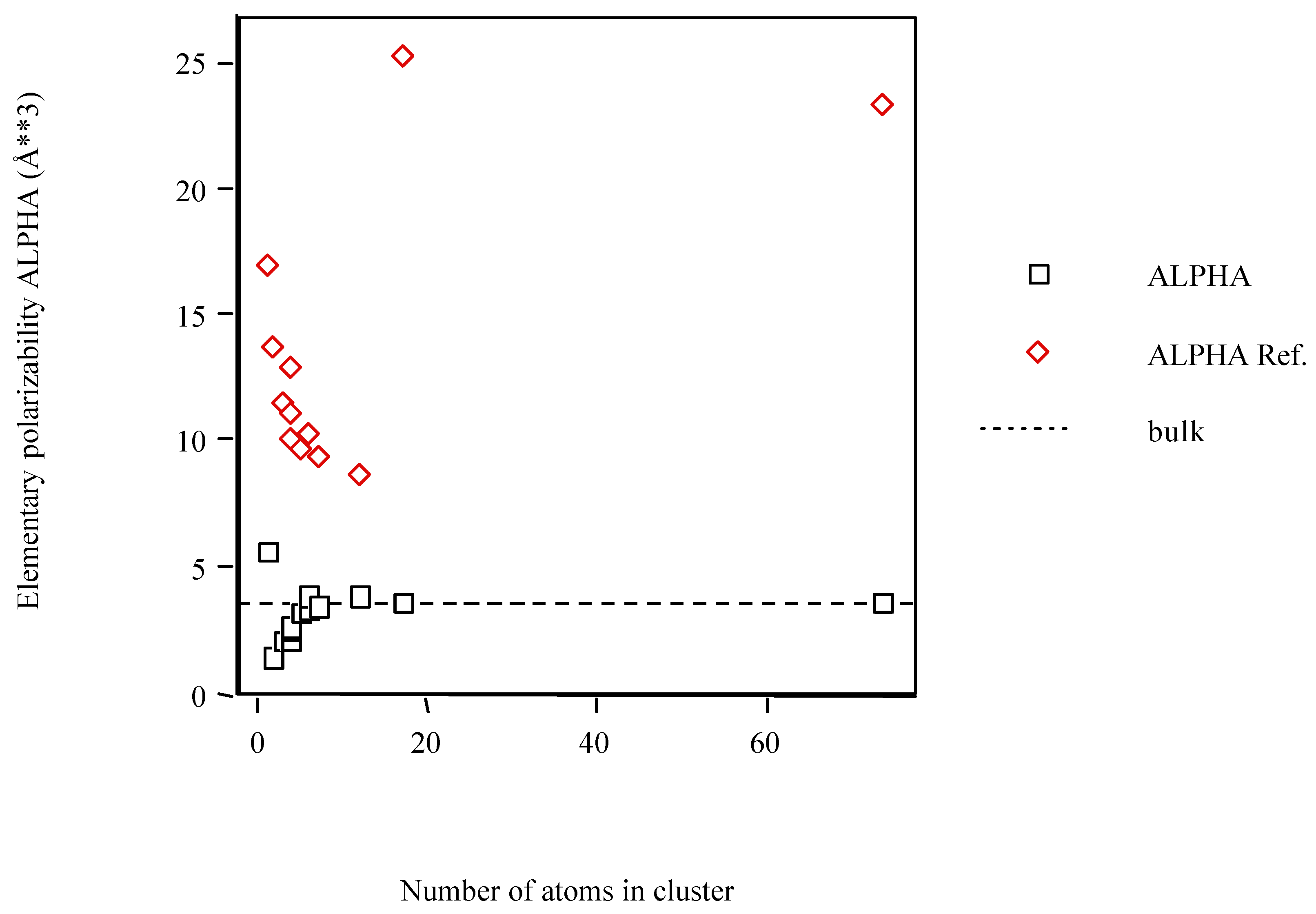
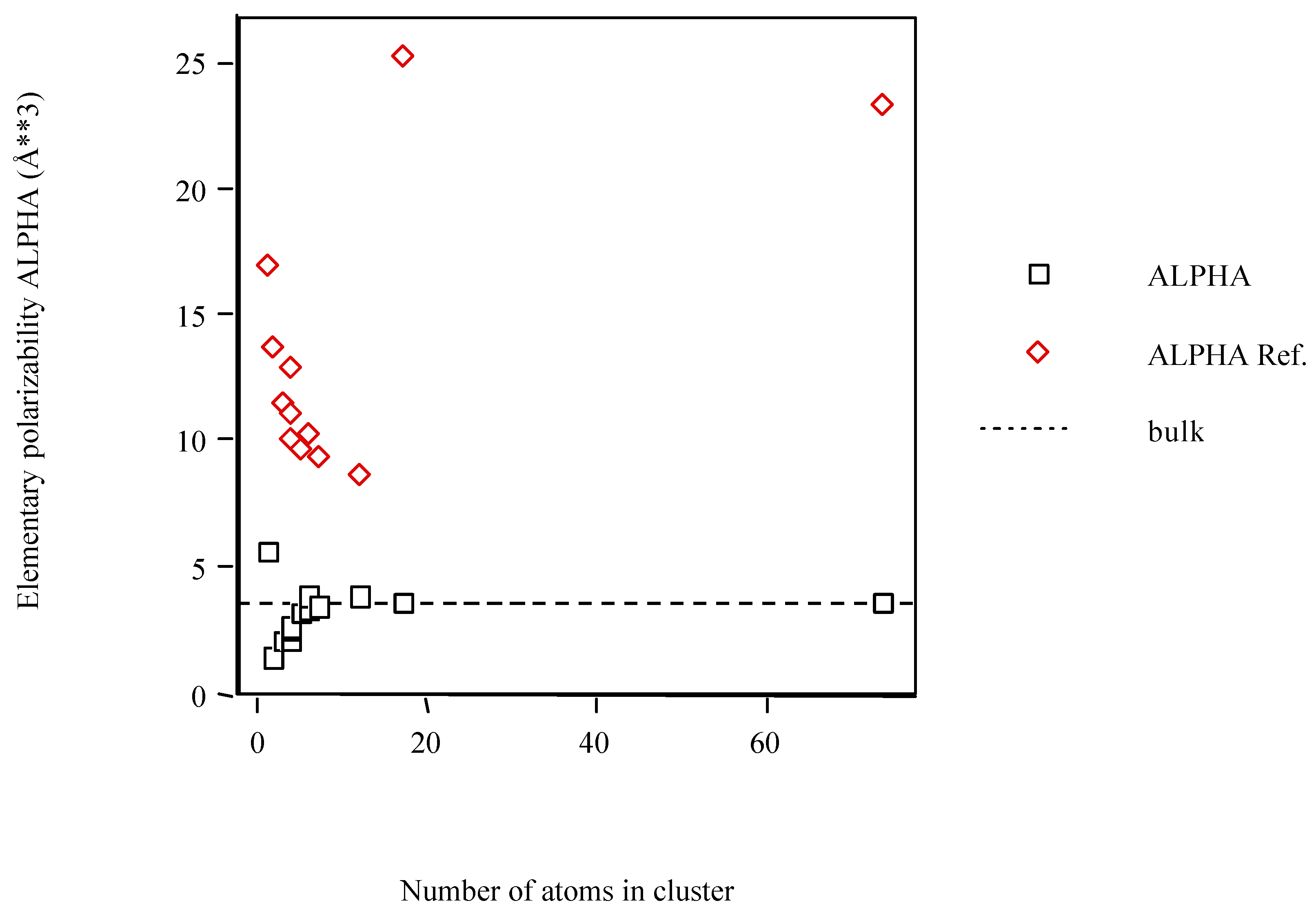
The variation of the computed values for the elementary polarizability of Scn clusters with the number of atoms is illustrated in Figure 1. The PAPID results for both Sc6 isomers are nearly equal and so they are superposed. On varying the number of atoms, the clusters show numbers indicative of particularly polarizable structures. Despite the PAPID results for the small clusters tend to the bulk limit, both clusters in the HCP structure comes away from this limit. Thus, both HCP cluster results should be taken with care.
Figure 1.
Average atom-atom polarizabilities per atom of Scn clusters vs. cluster size. Dotted lines correspond to the bulk polarizabilities.
Figure 1.
Average atom-atom polarizabilities per atom of Scn clusters vs. cluster size. Dotted lines correspond to the bulk polarizabilities.
As a reference, the bulk limit for the polarizability has been included, estimated from the Clausius-Mossotti relationship:
where v is an elementary volume per atom in the crystalline state and is the bulk dielectric constant. For metals, approaches infinite and the dependence of with disappears. In this work, the v value used for Sc is 15.0Å3 per atom, is calculated as 3.581Å3 per atom and for C, ν=5.3Å3 and is obtained as 1.265Å3.
The polarizability trend for the Scn clusters as a function of size is different from what one might have expected. In general, the Scn clusters calculated with POLAR are less polarizable than what one might have inferred from the bulk polarizability. Although an exception occurs for Sc1 the trend is clearer after this cluster. Previous experimental work [51] yielded the same trend for Sin, GanAsm and GenTem somewhat larger clusters. However, the Scn clusters computed with PAPID are more polarizable than what is inferred from the bulk, i.e., the polarizability of clusters tend to be greater than the bulk limit and approach this limit from above. Moreover, previous theoretical work with density functional theory within the one-electron approximation yielded the same trend for Sin, Gen and GanAsm small clusters [52]. At present, the origin of this difference is problematic. One might argue that smaller clusters need not behave like those of intermediate size. In addition, the error bars in the experiments are quite large.
The high polarizability of the Scn clusters (PAPID) is attributed to dangling bonds at the surface of the cluster. Indeed, most of the atoms within clusters reside on the surface. In fact, these structures are thought more closely related to the high-pressure metallic phases than to the diamond structure [53]. For example, it has been shown that the polarizabilities of alkali clusters significantly exceed the bulk limit and tend to decrease with increasing cluster size [54,55].
The geometries of the Cn fullerene models have been optimized with MMID [35,36]. The polarizabilities <α> for the fullerene models are summarized in Table 2.
| Cn fullerene | <α> (Å3)a | <α> ref.b |
|---|---|---|
| C | 0.588 | 1.322 |
| C12 | 0.978 | 0.722 |
| C60 | 0.782 | 0.904 |
| C70 | 0.781 | 0.920 |
| C82 | 0.763 | 0.911 |
a Average dipole-dipole polarizability (Å3).b Reference: calculations carried out with the PAPID program.
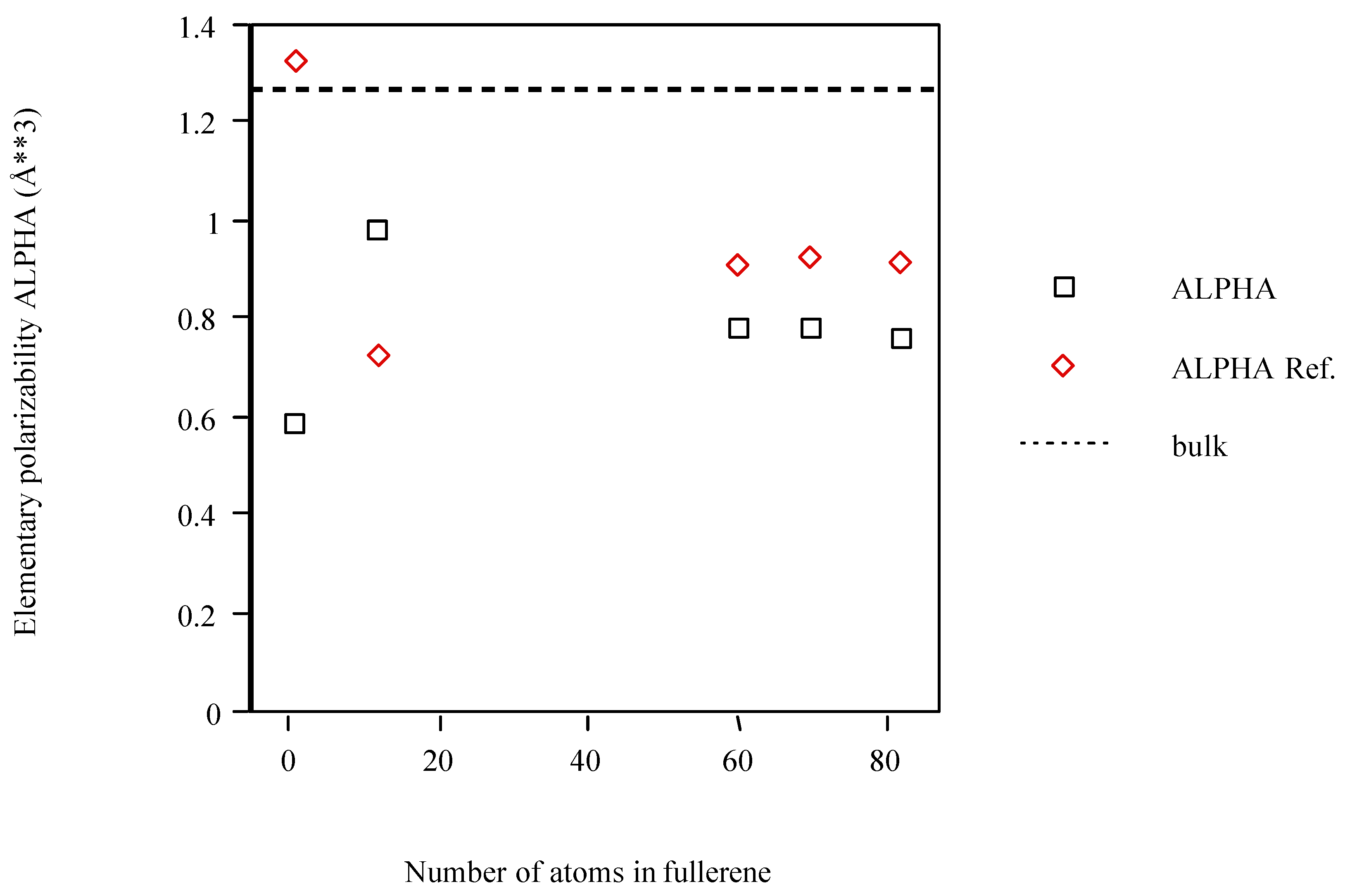
In general, POLAR underestimates <α>. In particular, the numerical RHF value for C1 (1.783Å3) [50] and the density functional theory (DFT) value calculated by Fuentealba (1.882Å3) [3] are significatively above POLAR but on the same order of magnitude as PAPID. For C60, the experimental elementary value measured by Antoine et al. (1.28±0.13Å3) [56] and the ab initio value calculated by Norman et al. (1.430Å3) [57] are somewhat greater than those obtained with POLAR and PAPID.
The fullerene models calculated with POLAR and PAPID are, in general, less polarizable than what is inferred from the bulk and approach this limit from below (see Figure 2). Although an exception occurs for C1 (PAPID) or C12 (POLAR) this trend is clearer after this structure.
Figure 2.
Average atom-atom polarizabilities per atom of fullerene models vs. cluster size.
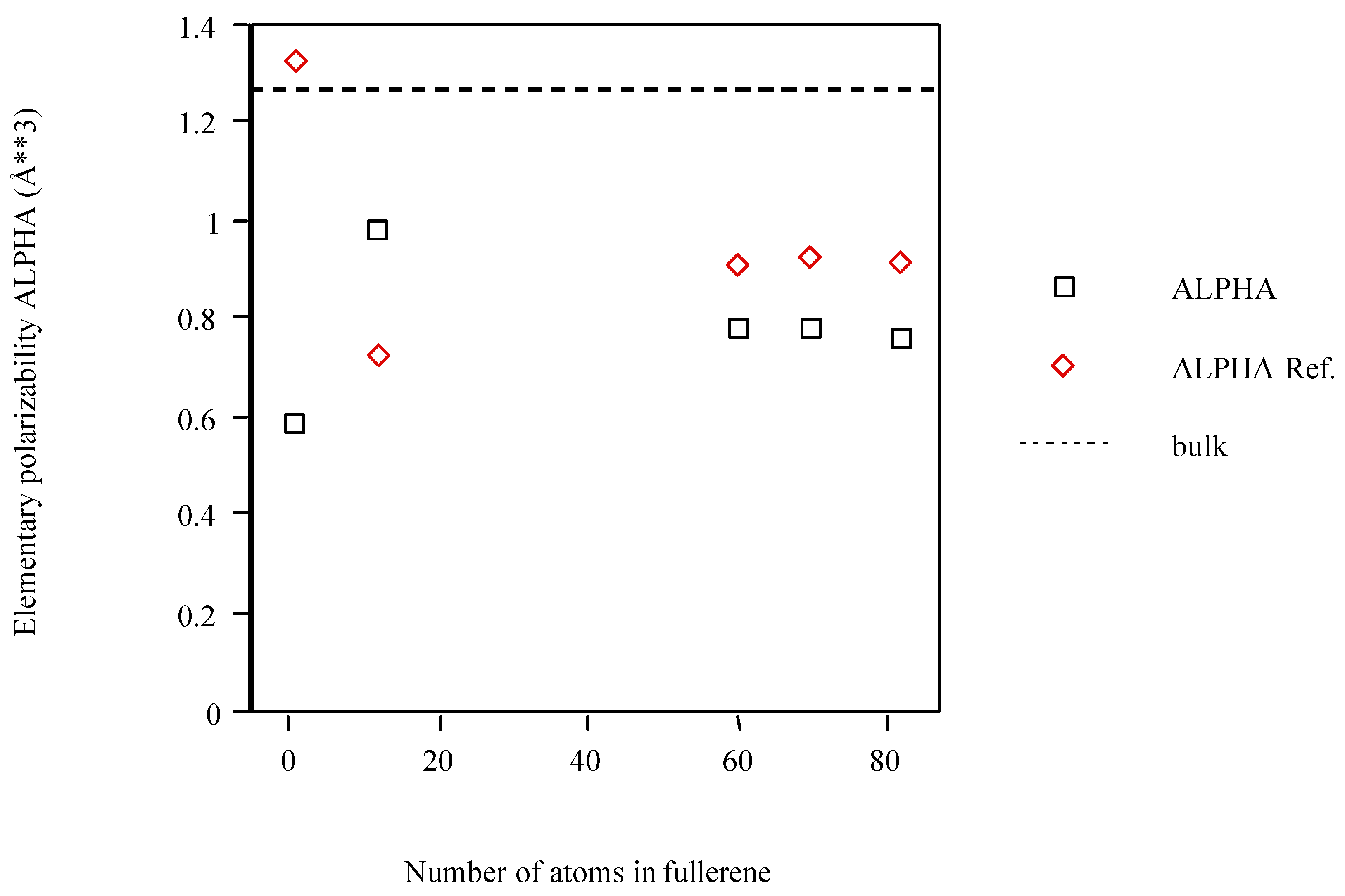
The polarizabilities <α> for the Cn one-shell graphite models are listed in Table 3.
| Cn graphite | <α> (Å3)a | <α> ref.b |
|---|---|---|
| C | 0.588 | 1.322 |
| C6 | 0.746 | 1.024 |
| C10 | 0.775 | 1.067 |
| C13 | 0.789 | 1.074 |
| C16 | 0.795 | 1.091 |
| C19 | 0.805 | 1.109 |
| C22 | 0.798 | 1.116 |
| C24 | 0.796 | 1.117 |
| C42 | 0.813 | 1.185 |
| C54 | 0.839 | 1.212 |
| C84 | 0.851 | 1.273 |
| C96 | 0.875 | 1.293 |
a Average dipole-dipole polarizability (Å3).b Reference: calculations carried out with the PAPID program.
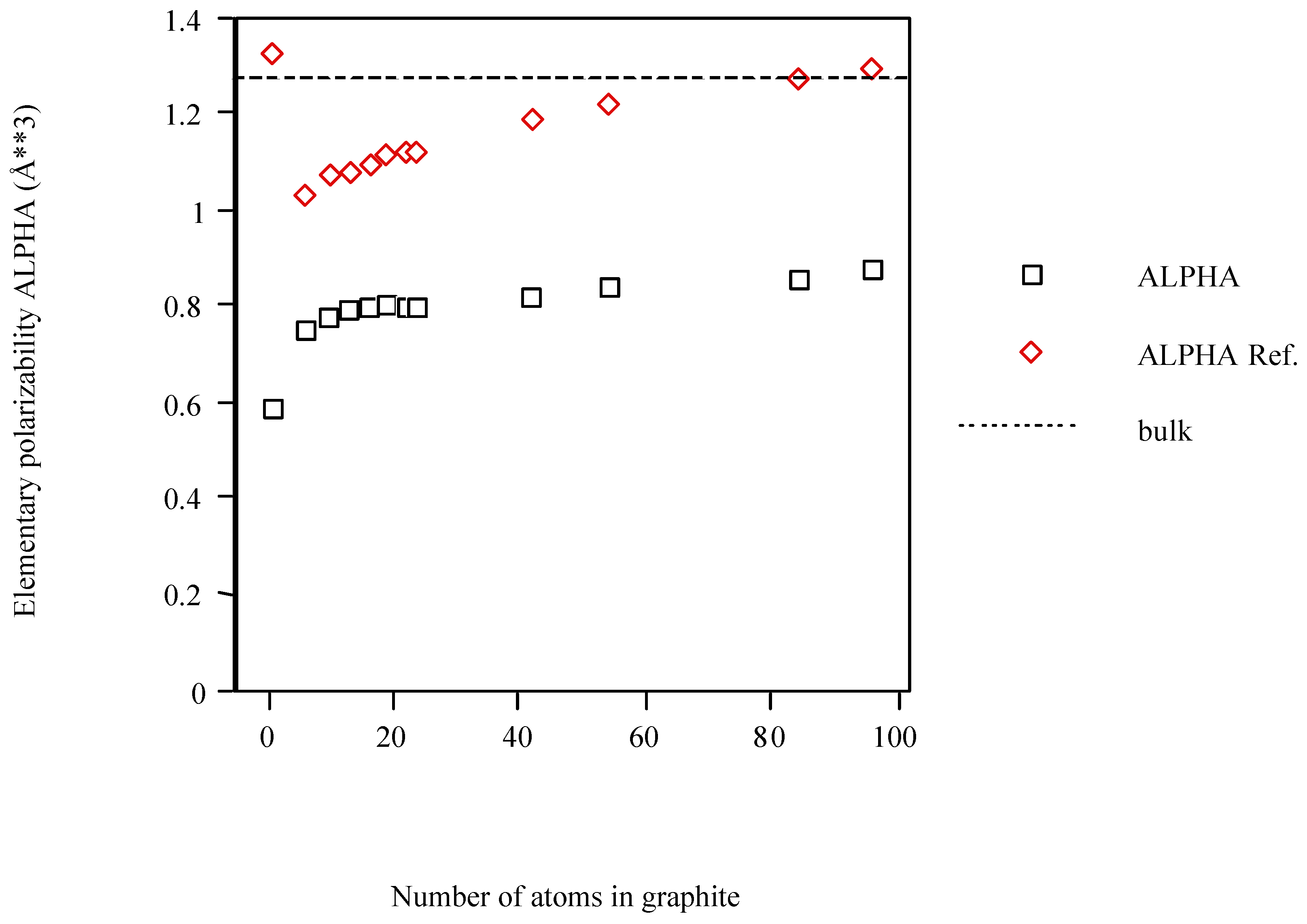
POLAR underestimates <α>. In particular, the DFT value for C6-cyclic (1.445Å3) is doubled with respect to POLAR but only somewhat greater than that obtained with PAPID [3]. The graphite models calculated with POLAR and PAPID are, in general, less polarizable than what is inferred from the bulk and approach this limit from below (see Figure 3). Although an exception occurs for C1 (PAPID) this trend is clearer after this structure.
Figure 3.
Average atom-atom polarizabilities per atom of one-shell graphite models vs. cluster size.
Figure 3.
Average atom-atom polarizabilities per atom of one-shell graphite models vs. cluster size.

When comparing Scn and Cn, <α> is greater for the three-dimensional (3D) Scn clusters than for the two-dimensional Cn clusters. This is due to the 3D character of the metallic bond in Scn. The <α> is greater for the planar Cn graphite models than for the curved fullerene models due to the weakening of the π bonds in the non-planar fullerene structure (see Section Electrostatic properties). For all the clusters in Table 1, Table 2 and Table 3, the mean relative error is -39%. It should be noted that this error improves to -34% if the exception structures are eliminated from the tables. In order to prevent these small figures caused by the algebraic sum, the mean unsigned relative error has been calculated. This error results 44% and decreases to 40% without the exception structures.
Conclusions
A method for the calculation of the molecular dipole-dipole polarizability <α> has been presented and applied to Scn and Cn (fullerene and one-shell graphite) model clusters. From the preceding results the following conclusions can be drawn:
1. On varying the number of atoms, the clusters show numbers indicative of particularly polarizable structures. The polarizability also depends on the chosen isomer.
2. The results of the present work clearly indicate that due to the differences between POLAR and PAPID results it may become necessary to recalibrate POLAR. It appears that the results of good quality ab initio calculations might be suitable as primary standards for such a calibration. Work is in progress on the recalibration of POLAR.
3. The polarizability trend for the clusters as a function of size is different from what one might have expected. The small Scn clusters (POLAR) and the large Cn fullerenes (both POLAR and PAPID) are less polarizable than what one might have inferred from the bulk polarizability. The Scn clusters (PAPID) are more polarizable than what is inferred from the bulk. The high polarizability of the Scn clusters (PAPID) is attributed to arise from dangling bonds at the surface of the cluster. Recommended elementary polarizability values are 17—22Å3 (Scn), 1.8—1.9Å3 (small Cn-fullerene), and 1.3—1.9Å3 (small Cn-graphite) and 1.3Å3 (big Cn-fullerene and Cn-graphite).
Acknowledgements
The author acknowledges the financial support of the Dirección General de Enseñanza Superior of the Spanish MEC (Project No. PB97-1383).
References
- Benichou, E.; Antoine, R.; Rayane, D.; Vezin, B.; Dalby, F. W.; Dugourd, Ph.; Broyer, M.; Ristori, C.; Chandezon, F.; Huber, B. A.; Rocco, J. C.; Blundell, S. A.; Guet, C. Measurement of static electric dipole polarizabilities of lithium clusters: Consistency with measured dynamic polarizabilities. Phys. Rev. A 1999, 59, R1–R4. [Google Scholar] [CrossRef]
- Maroulis, G.; Xenides, D. Enhanced linear and nonlinear polarizabilities for the Li4 cluster. How satisfactory is the agreement between theory and experiment for the static dipole polarizability? J. Phys. Chem. A 1999, 103, 4590–4593. [Google Scholar] [CrossRef]
- Fuentealba, P. Static dipole polarizabilities of small neutral carbon clusters Cn (n=8). Phys. Rev. A 1998, 58, 4232–4234. [Google Scholar] [CrossRef]
- Fuentealba, P.; Reyes, O. Density functional study of LinHm clusters. Electric dipole polarizabilities. J. Phys. Chem. A 1999, 103, 1376–1380. [Google Scholar] [CrossRef]
- Jackson, K.; Pederson, M.; Wang, C.-Z.; Ho, K.-M. Calculated polarizabilities of intermediate-size Si clusters. Phys. Rev. A 1999, 59, 3685–3689. [Google Scholar] [CrossRef]
- Deng, K.; Yang, J.; Chan, C. T. Calculated polarizabilities of small Si clusters. Phys. Rev. A 2000, 61, 025201-1–4. [Google Scholar] [CrossRef]
- Deng, K.; Yang, J.; Yuan, L.; Zhu, Q. Hybrid density-functional study of Si13 clusters. Phys. Rev. A 2000, 62, 045201-1–4. [Google Scholar] [CrossRef]
- Hohm, U.; Goebel, D.; Karamanis, P.; Maroulis, G. Electric dipole polarizability of As4, a challenging problem for both experiment and theory. J. Phys. Chem. A 1998, 102, 1237–1240. [Google Scholar] [CrossRef]
- Torrens, F. Molecular polarizability of Scn, Cn, and endohedral Scn@Cm clusters. Microelectronic Eng. 2000, 51-52, 613–626. [Google Scholar] [CrossRef]
- Torrens, F. Theoretical characterization of iron and manganese porphyrins for catalyzed saturated alkane hydroxylations. J. Mol. Catal. 1997, A-119, 393–403. [Google Scholar] [CrossRef]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. Interacting induced dipoles polarization model for molecular polarizabilities. Application to benzothiazole (A)-benzobisthiazole (B) oligomers: A-B13-A. J. Mol. Struct. (Theochem) 1998, 426, 105–116. [Google Scholar] [CrossRef]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. Torsional effects on the molecular polarizabilities of the benzothiazole (A)-benzobisthiazole (B) oligomer A-B13-A. J. Mol. Graphics 1996, 14, 245–259. [Google Scholar] [CrossRef]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. Interacting induced dipoles polarization model for molecular polarizabilities. Reference molecules, amino acids and model peptides. J. Mol. Struct. (Theochem) 1999, 463, 27–39. [Google Scholar] [CrossRef]
- Torrens, F.; Ortí, E.; Sánchez-Marín, J. Vectorized TOPO program for the theoretical simulation of molecular shape. J. Chim. Phys. Phys.-Chim. Biol. 1991, 88, 2435–2441. [Google Scholar]
- Mulliken, R. S. Chem. Phys. 1934, 2, 782.
- Huheey, J. E. The electronegativity of groups. J. Phys. Chem. 1965, 69, 3284–3291. [Google Scholar] [CrossRef]
- Sanderson, R. T. An interpretation of bond lengths and classification of bonds. Science 1951, 114, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Bratsch, S. G. Electronegativity equalization with Pauling units. J. Chem. Educ. 1984, 61, 588–589. [Google Scholar] [CrossRef]
- Vogel, A. I. Physical properties and chemical constitution. XXIII. Miscellaneous compounds. Investigation of the so-called coördinate or dative link in esters of oxy acids and in nitro paraffins by molecular refractivity determinations. Atomic, structural, and group parachors and refractivities. J. Chem. Soc. 1948, 1833–1855. [Google Scholar]
- Gresh, N.; Claverie, P.; Pullman, A. Intermolecular interactions: Reproduction of the results of ab initio supermolecule computations by an additive procedure. Int. J. Quantum Chem., Symp. 1979, 13, 243–253. [Google Scholar] [CrossRef]
- Applequist, J.; Carl, J. R.; Fung, K.-K. An atom dipole interaction model for molecular polarizability. Application to polyatomic molecules and determination of atom polarizabilities. J. Am. Chem. Soc. 1972, 94, 2952–2960. [Google Scholar] [CrossRef]
- Applequist, J. Atom charge transfer in molecular polarizabilities. Application of the Olson-Sundberg model to aliphatic and aromatic hydrocarbons. J. Phys. Chem. 1993, 97, 6016–6023. [Google Scholar] [CrossRef]
- Lowe, J. P. Quantum Chemistry; Academic Press: New York, 1978. [Google Scholar]
- Mulliken, R. S. The theory of molecular orbitals. J. Chim. Phys. Phys.-Chim. Biol. 1949, 46, 497–542. [Google Scholar]
- Mulliken, R. S. Magic formula, structure of bond energies, and isovalent hybridization. J. Phys. Chem. 1952, 56, 295–311. [Google Scholar] [CrossRef]
- Mulliken, R. S.; Rieke, C. A.; Orloff, D.; Orloff, H. Formulas and numerical tables for overlap integrals. J. Chem. Phys. 1949, 17, 1248–1267. [Google Scholar] [CrossRef]
- Streitwieser Jr., A. Molecular Orbital Theory for Organic Chemists; John Wiley and Sons: New York, 1961. [Google Scholar]
- Mulliken, R. S. Overlap integrals and chemical binding. J. Am. Chem. Soc. 1950, 72, 4493–4503. [Google Scholar] [CrossRef]
- Streitwieser Jr., A.; Nair, P. M. Molecular orbital treatment of hyperconjugation. Tetrahedron 1959, 5, 149–165. [Google Scholar] [CrossRef]
- Parr, R. G.; Crawford Jr., B. L. Molecular orbital calculations of vibrational force constants. I. Ethylene J. Chem. Phys. 1948, 16, 526–532. [Google Scholar] [CrossRef]
- Dewar, M. J. S. A molecular orbital theory of organic chemistry. II. The structure of mesomeric systems. J. Am. Chem. Soc. 1952, 74, 3345–3350. [Google Scholar] [CrossRef]
- Simonetta, M.; Winstein, S. Neighboring carbon and hydrogen. XVI. 1,3-interactions and homoallylic resonance. J. Am. Chem. Soc. 1954, 76, 18–21. [Google Scholar]
- Kreevoy, M. M. A theoretical study of 1,4-dithiadiene by the L.C.A.O.-M.O. method. J. Am. Chem. Soc. 1958, 80, 5543–5547. [Google Scholar] [CrossRef]
- Joachim, C.; Treboux, G.; Tang, H. A model conformation flip-flop molecular switch. In Molecular Electronics: Science and Technology; AIP Conference Proceedings No. 262. AIP: New York, 1992; pp. 107–117. [Google Scholar]
- Torrens, F.; Ruiz-López, M.; Cativiela, C.; García, J. I.; Mayoral, J. A. Conformational aspects of some asymmetric Diels-Alder reactions. A molecular mechanics + polarization study. Tetrahedron 1992, 48, 5209–5218. [Google Scholar] [CrossRef]
- Torrens, F.; Sánchez-Marín, J.; Rivail, J.-L. Interacting induced dipoles polarization in a force field for dipeptide models (glycine derivative). An. Fís. (Madrid) 1994, 90, 197–204. [Google Scholar]
- Torrens, F. Polarization force fields for peptides implemented in ECEPP2 and MM2. Mol. Simul. 2000, 24, 391–410. [Google Scholar] [CrossRef]
- Silberstein, L. Philos. Mag. 1917, 33, 92.
- Silberstein, L. Philos. Mag. 1917, 33, 215.
- Silberstein, L. Philos. Mag. 1917, 33, 521.
- Born, M. Optik, (Springer-Verlag, Berlin, 1933). 308.
- Stuart, H. A. Die Struktur des freien Moleküls; Springer-Verlag: Berlin, 1952; p. 363. [Google Scholar]
- Kauzmann, W. Quantum Chemistry; Academic Press: New York, 1957; p. 568. [Google Scholar]
- Mahan, G. D. Davydov splittings in anthracene. J. Chem. Phys. 1964, 41, 2930–2933. [Google Scholar] [CrossRef]
- Rhodes, W.; Chase, M. Generalized susceptibility theory. I. Theories of hypochromism. Rev. Mod. Phys. 1967, 39, 348–356. [Google Scholar] [CrossRef]
- Philpott, M. R. Dipole Davydov splittings in crystalline anthracene, tetracene, naphthalene, and phenanthrene. J. Chem. Phys. 1969, 50, 5117–5128. [Google Scholar] [CrossRef]
- Voisin, C.; Cartier, A.; Rivail, J.-L. Computation of accurate electronic molecular polarizabilities. J. Phys. Chem. 1992, 96, 7966–7971. [Google Scholar] [CrossRef]
- Allinger, N. L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977; 99, 8127–8134. [Google Scholar]
- Némethy, G.; Pottle, M. S.; Scheraga, H. A. Energy parameters in polypeptides. 9. Updating of geometrical parameters, nonbonded interactions, and hydrogen bond interactions for the naturally occurring amino acids. J. Phys. Chem. 1983, 87, 1883–1887. [Google Scholar] [CrossRef]
- Stiehler, J.; Hinze, J. Calculation of static polarizabilities and hyperpolarizabilities for the atoms He through Kr with a numerical RHF method. J. Phys. B 1995, 28, 4055–4071. [Google Scholar] [CrossRef]
- Schäfer, R.; Schlecht, S.; Woenckhaus, J.; Becker, J. A. Polarizabilities of isolated semiconductor clusters. Phys. Rev. Lett. 1996, 76, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Chelikowsky, J. R. The electronic and structural properties of semiconductor clusters and nanostructures. Res. Rep. UMSI 97/132; University of Minnesota Supercomputing Institute, 1997. [Google Scholar]
- Jarrold, M. F. Nanosurface chemistry on size-selected silicon clusters. Science 1991, 252, 1085–1092. [Google Scholar] [CrossRef]
- De Heer, W. A. The physics of simple metal clusters: Experimental aspects and simple models. Rev. Mod. Phys. 1993, 65, 611–675. [Google Scholar] [CrossRef]
- Brack, M. The physics of simple metal clusters: Self-consistent jellium model and semiclassical approaches. Rev. Mod. Phys. 1993, 65, 677–732. [Google Scholar] [CrossRef]
- Antoine, R.; Dugourd, Ph.; Rayane, D.; Benichou, E.; Broyer, M.; Chandezon, F.; Guet, C. Direct measurement of the electric polarizability of isolated C60 molecules. J. Chem. Phys. 1999, 110, 9771–9772. [Google Scholar] [CrossRef]
- Norman, P.; Luo, Y.; Jonsson, D.; Ågren, H. Ab initio calculations of the polarizability and the hyperpolarizability of C60. J. Chem. Phys. 1997, 106, 8788–8791. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes
Share and Cite
MDPI and ACS Style
Torrens, F. Molecular Polarizability of Sc and C (Fullerene and Graphite) Clusters. Molecules 2001, 6, 496-509. https://doi.org/10.3390/60600496
AMA Style
Torrens F. Molecular Polarizability of Sc and C (Fullerene and Graphite) Clusters. Molecules. 2001; 6(6):496-509. https://doi.org/10.3390/60600496
Chicago/Turabian StyleTorrens, Francisco. 2001. "Molecular Polarizability of Sc and C (Fullerene and Graphite) Clusters" Molecules 6, no. 6: 496-509. https://doi.org/10.3390/60600496