On the Additions of Lithium Methyl p-Tolyl Sulfoxide to N-(PMP)Arylaldimines

Abstract
:Introduction
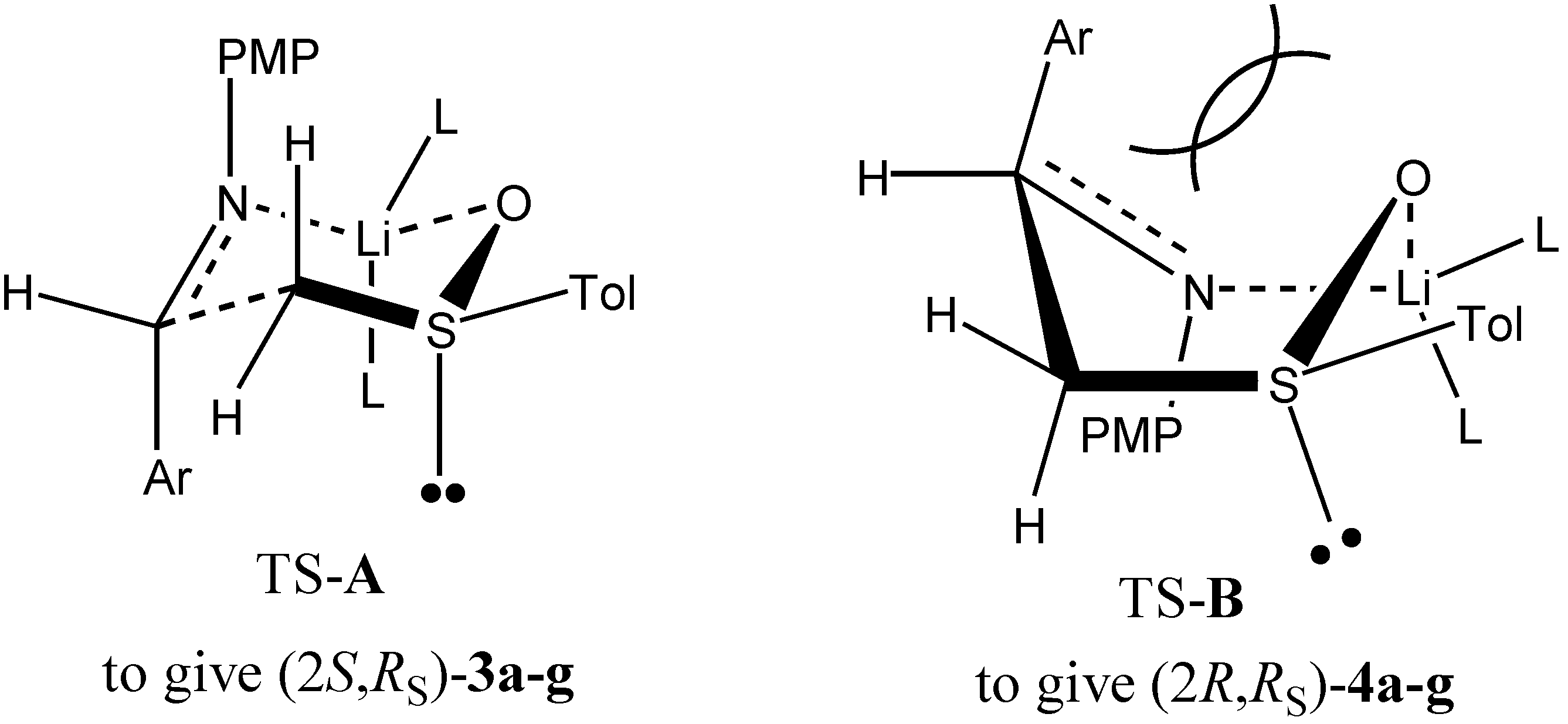
Results and Discussion
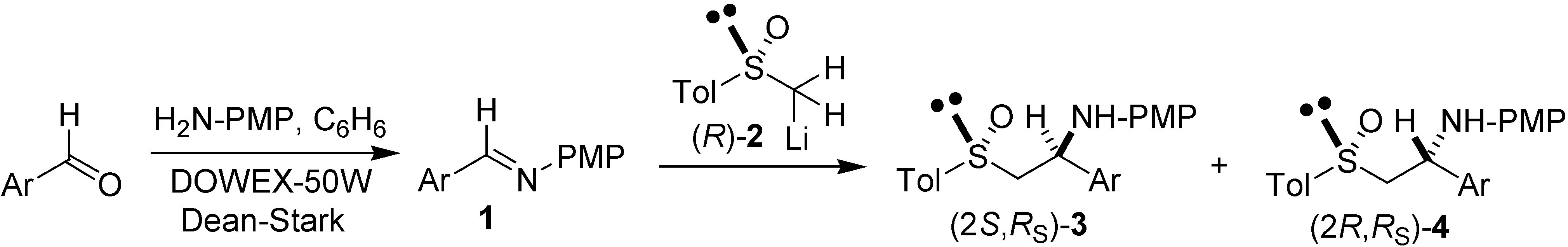

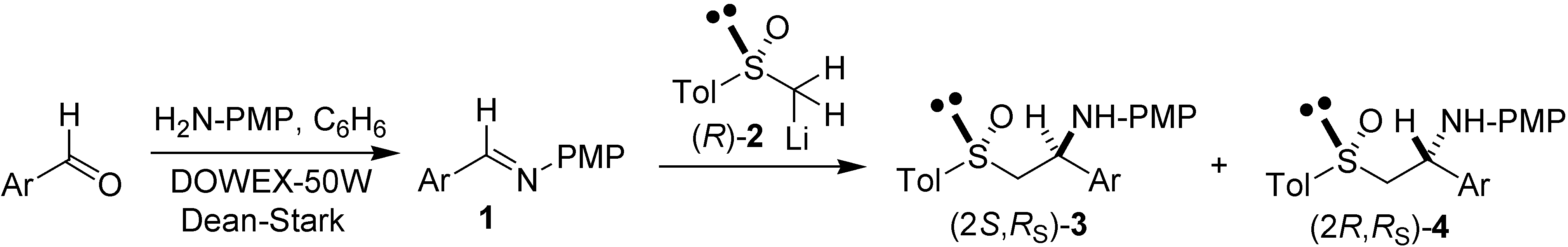
{kind=link}
{kind=link}
{kind=link}
| Entry | Ar | Time (min) | Yield (%)a | (2S,RS)-3 (%)b | (2R,RS)-4 (%)b |
| 1c (a) | 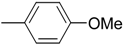 | 80 | 91 | 95.0 | 5.0 |
| 2 (a) | 80 | < 2 | Not determined | Not determined | |
| 3 (b) |  | 60 | 34 | 91.7 | 8.3 |
| 4 (c) | 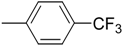 | 30 | 88 | 83.8 | 16.2 |
| 5 (d) | 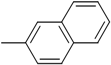 | 30 | 76 | 81.6 | 18.4 |
| 6 (e) | 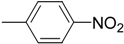 | 40 | 83 | 76.5 | 23.5 |
| 7 (f) | 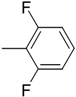 | 30 | 86 | 73.7 | 26.3 |
| 8 (g) | 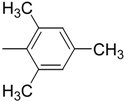 | 90 | < 2 | Not determined | Not determined |
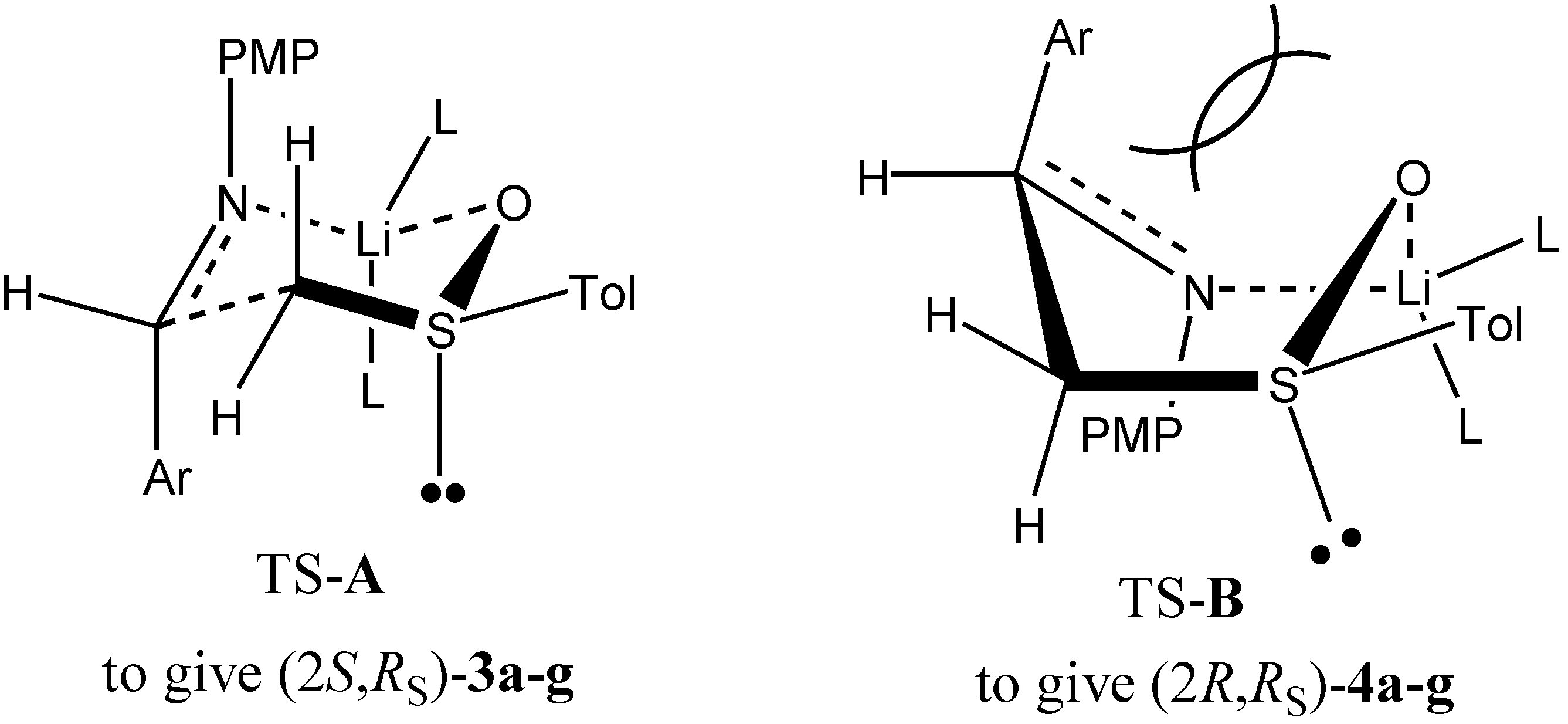
Conclusions
Experimental
General.
Spectral Data
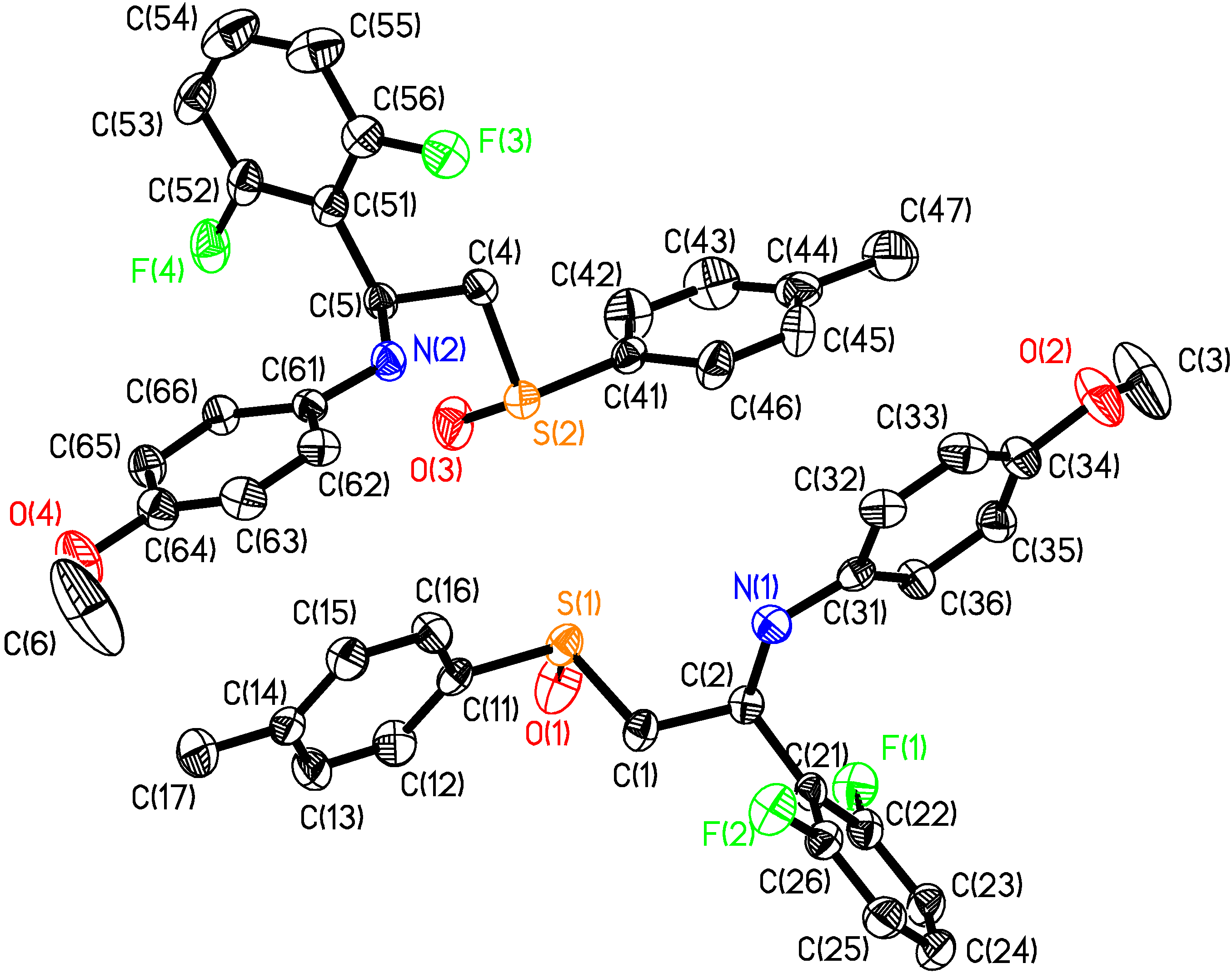
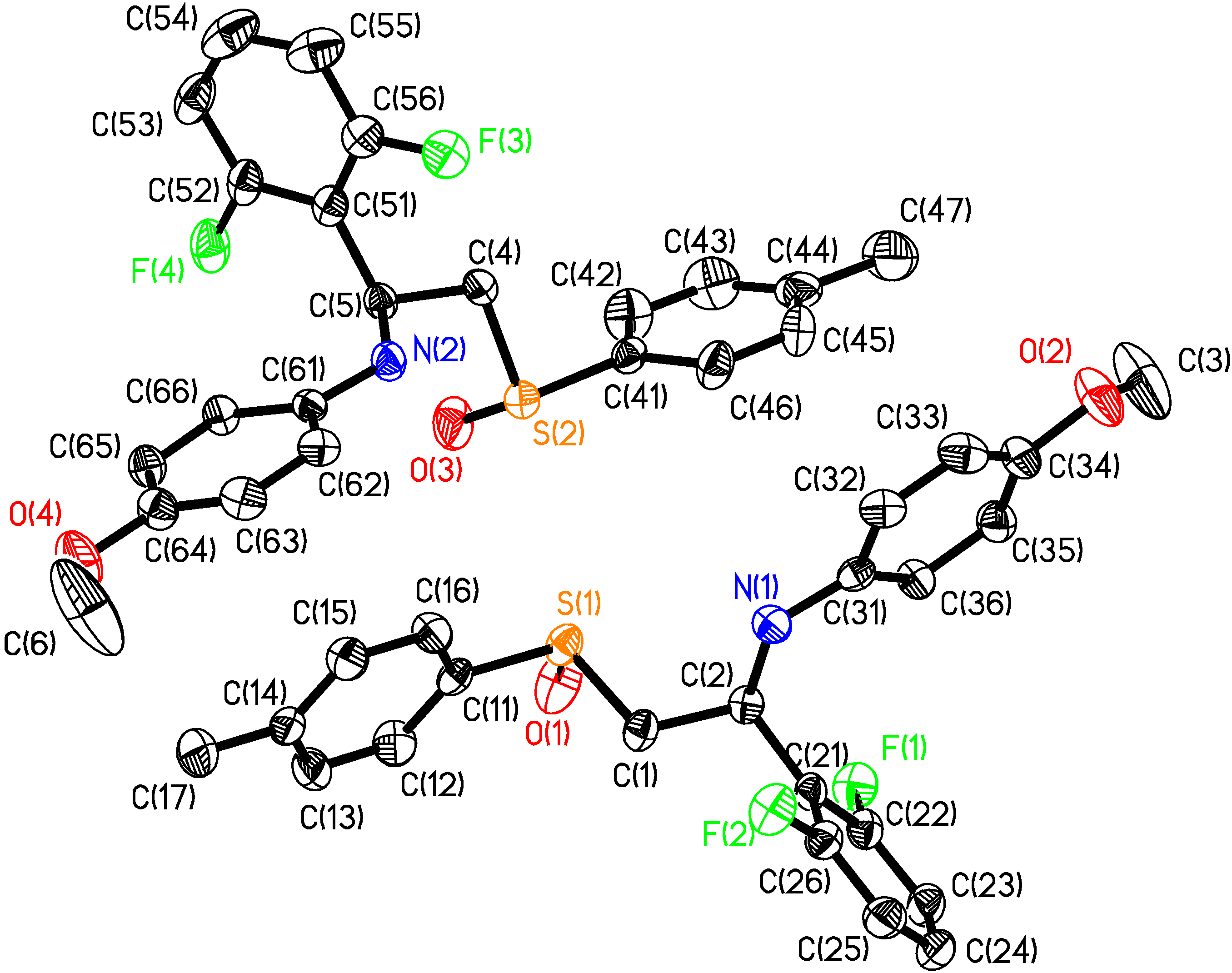
| Bond lengths (Å) | Bond angles (°) | ||
|---|---|---|---|
| S(1)-O(1) | 1.501(3) | C(31)-N(1)-C(2) | 122.9(3) |
| S(2)-O(3) | 1.479(4) | C(61)-N(2)-C(5) | 122.5(3) |
| N(1)-C(31) | 1.376(5) | O(1)-S(1)-C(11) | 107.3(2) |
| N(1)-C(2) | 1.453(5) | O(1)-S(1)-C(1) | 104.8(2) |
| N(2)-C(61) | 1.390(5) | C(11)-S(1)-C(1) | 96.69(17) |
| N(2)-C(5) | 1.442(5) | O(3)-S(2)-C(41) | 108.1(2) |
| C(52)-F(4) | 1.345(6) | O(3)-S(2)-C(4) | 105.2(2) |
| C(56)-F(3) | 1.358(5) | C(41)-S(2)-C(4) | 97.08(18) |
| C(26)-F(2) | 1.356(5) | Torsion angles (°) | |
| C(22)-F(1) | 1.351(6) | C31-N1-C2-C1 | -169.39(0.34) |
| C61-N2-C5-C4 | -164.02(0.34) | ||
| S1-C1-C2-N1 | 78.16(0.35) | ||
| S2-C4-C5-N2 | 68.33(0.38) | ||
References and Notes
- Walker, A. J. Tetrahedron: Asymmetry 1992, 3, 961–998. Carreño, M. C. Chem. Rev. 1995, 95, 1717–1760.
- Bravo, P.; Capelli, S.; Crucianelli, M.; Guidetti, M.; Markovsky, A. L.; Meille, S. V.; Soloshonok, V. A.; Sorochinsky, A. E.; Viani, F.; Zanda, M. Tetrahedron 1999, 55, 3025–3040.
- Tsuchihashi, G.; Iriuchijima, S.; Maniwa, K. Tetrahedron Lett. 1973, 14, 3389–3392. Ronan, B.; Marchalin, S.; Samuel, O.; Kagan, H. B. Tetrahedron Lett. 1988, 29, 6101–6104. Pyne, S. G.; Boche, G. J. Org. Chem. 1989, 54, 2663–2667. [CrossRef] Pyne, S. G.; Dikic, B. J. Chem. Soc., Chem. Commun. 1989, 826–827. [CrossRef] Pyne, S. G.; Dikic, B. J. Org. Chem. 1990, 55, 1932–1936. [CrossRef]
- Selected molecular dimensions of (2S,RS)-3f are reported in Table 2. Bond lengths and angles fall all in the expected range (Allen, F.M.; Kennard, O; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R.J.; J. Chem. Soc., Perkin Trans. 2, 1987, S1-S19). The two molecules in the unit cell are configurationally identical but not related by any symmetry operation. The angles found on atoms N1 and N2 suggest a planar arrangement, which is common for aromatic amines. The high thermal parameters and the corresponding elongated thermal ellipsoids of atom C(3) and C(6) suggest disorder involving these atoms. In the crystal, the molecules of (2S,RS)-3f pack arranging the aromatic rings of adjacent molecules with a favourable herringbone interaction geometry
- Ojima, I.; Kwon, H. B. J. Am. Chem. Soc. 1988, 110, 5617–5621. [CrossRef] Soloshonok, V. A.; Kacharov, A. D.; Hayashi, T. Tetrahedron 1996, 52, 245–254.
- Altomare, A.; Cascarano, G.; Guagliardi, A. J. Appl. Cryst. 1993, 26, 343–350. [CrossRef]
- Sheldrick, G. SHELXL97, Program for crystal structure refinement; University of Göttingen: Germany, 1997. [Google Scholar]
- Flack, H. D. Acta Cryst 1983, A 39, 876–880.
- Sample Availability: Available from the authors.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes
Share and Cite
Zucca, C.; Bravo, P.; Corradi, E.; Meille, S.V.; Volonterio, A.; Zanda, M. On the Additions of Lithium Methyl p-Tolyl Sulfoxide to N-(PMP)Arylaldimines. Molecules 2001, 6, 424-432. https://doi.org/10.3390/60500424
Zucca C, Bravo P, Corradi E, Meille SV, Volonterio A, Zanda M. On the Additions of Lithium Methyl p-Tolyl Sulfoxide to N-(PMP)Arylaldimines. Molecules. 2001; 6(5):424-432. https://doi.org/10.3390/60500424
Chicago/Turabian StyleZucca, Cristina, Pierfrancesco Bravo, Eleonora Corradi, Stefano V. Meille, Alessandro Volonterio, and Matteo Zanda. 2001. "On the Additions of Lithium Methyl p-Tolyl Sulfoxide to N-(PMP)Arylaldimines" Molecules 6, no. 5: 424-432. https://doi.org/10.3390/60500424