Introduction
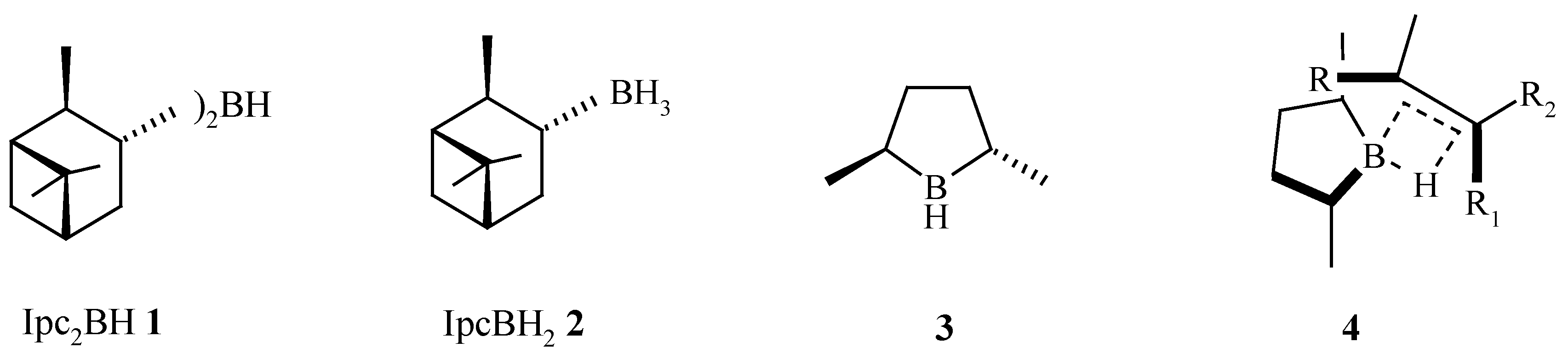
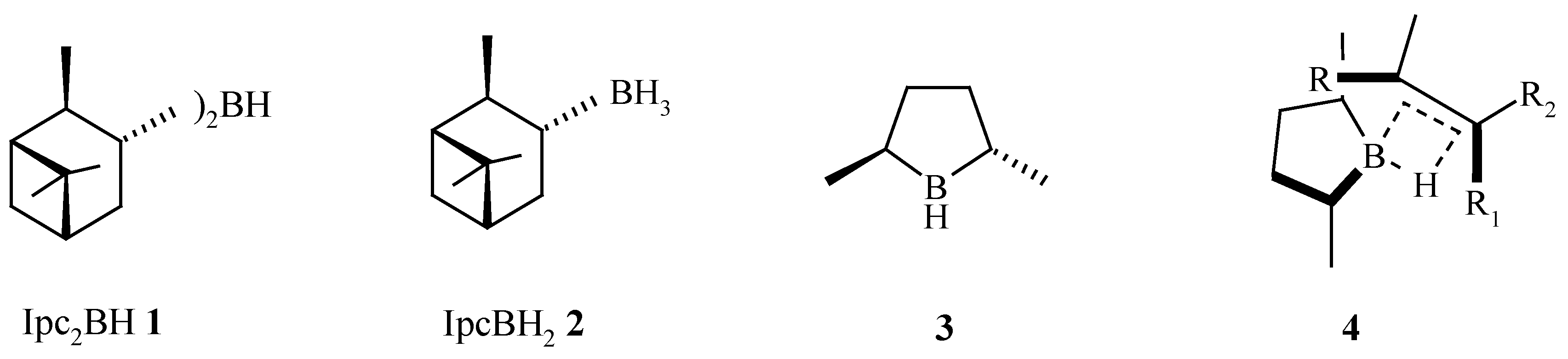
In 1961, Brown described the first synthesis of a chiral hydroborating reagent, diisopinocampheylborane Ipc2BH (
1), a reagent that has been shown to hydroborate sterically less demanding prochiral
cis-alkenes in high e.e. [
1]. In later years, monoalkylboranes such as monoisopinocampheylborane IpcBH
2 (
2) were developed [
2]. The reduced steric requirements of IpcBH
2 2 facilitates the hydroboration of tri-substituted and
trans-alkenes in good to excellent e.e. [
3,
4]. In 1985, Masamune [
5] introduced the C
2 symmetric
trans-2,5-dimethylborolane (
3) [
6] as a rationally designed hydroboration reagent that gave very high e.e.’s for
cis-,
trans- and tri-substituted alkenes [
7]. The extent and directionality of the asymmetric induction is consistent with the proposed 4-membered transition state model
4 (
Scheme 1).
Scheme 1.
Hydroboration reagents and typical % e.e. for the hydroboration of prochiral alkenes.
Scheme 1.
Hydroboration reagents and typical % e.e. for the hydroboration of prochiral alkenes.
These results would suggest Masamune’s C
2 symmetric
trans-2,5-dimethylborolane (
3) to be the reagent of choice for asymmetric hydroboration, however,
3 has found almost no use as a reagent for asymmetric hydroboration. This is presumably because of the rather lengthy and tedious sequence of reactions and separations required for its preparation [
5]. We wished to prepare new reagents for asymmetric hydroboration that retained the structural features of Masamune’s reagent
3 but were easier and more practical to prepare [
8].
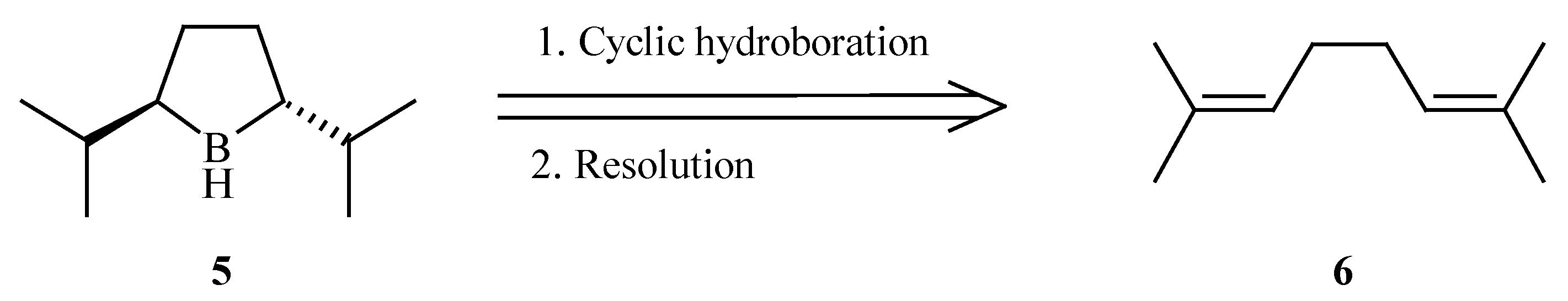
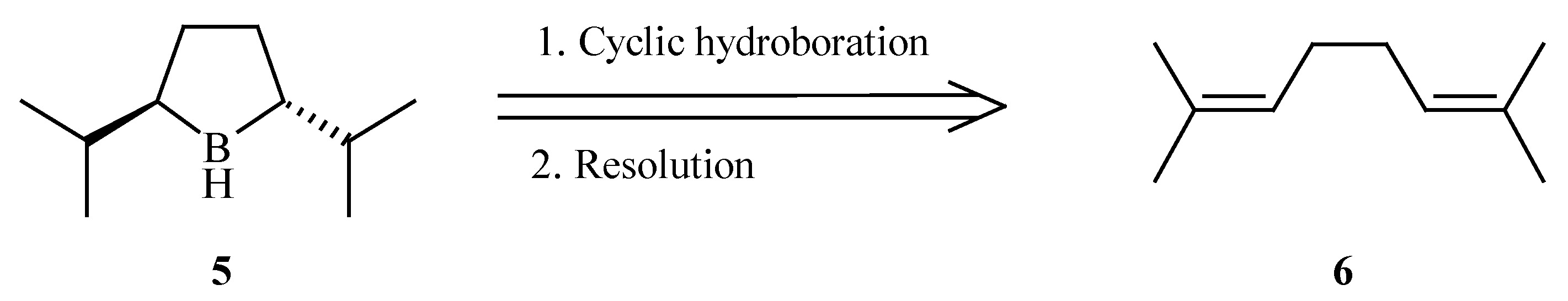
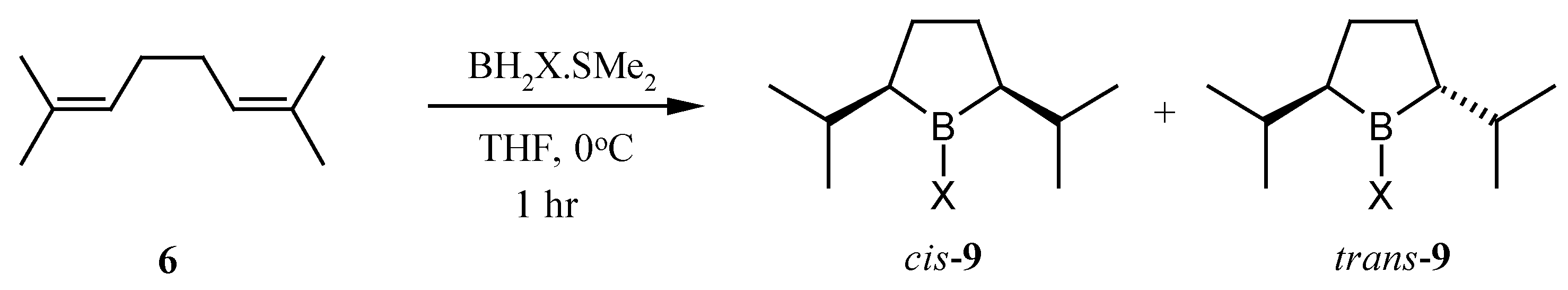
Trans-2,5-diisopropylborolane (
5), having a greater steric demand than its methyl predecessor was identified as our target. We envisioned that the
trans-borolane might be selectively formed
via the cyclic hydroboration [
9] of 2,7-dimethyl-2,6-octadiene (
6) (
Scheme 2).
Results and Discussion
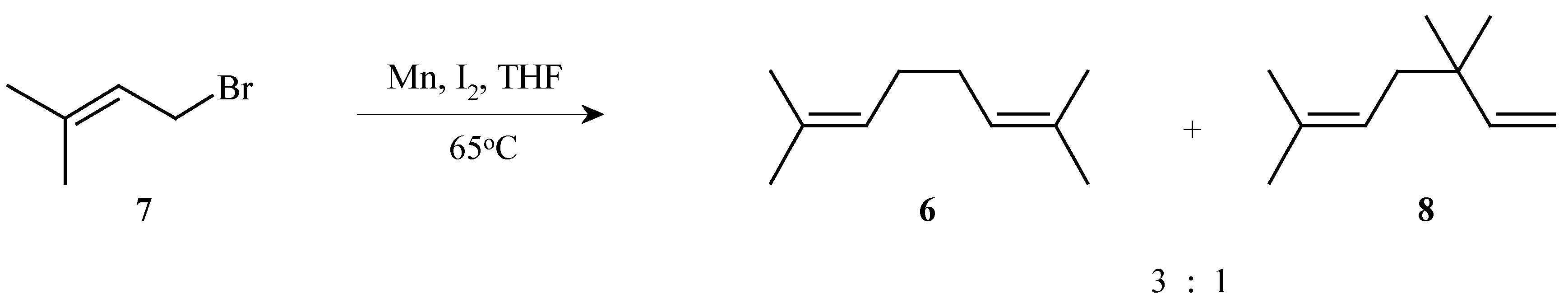
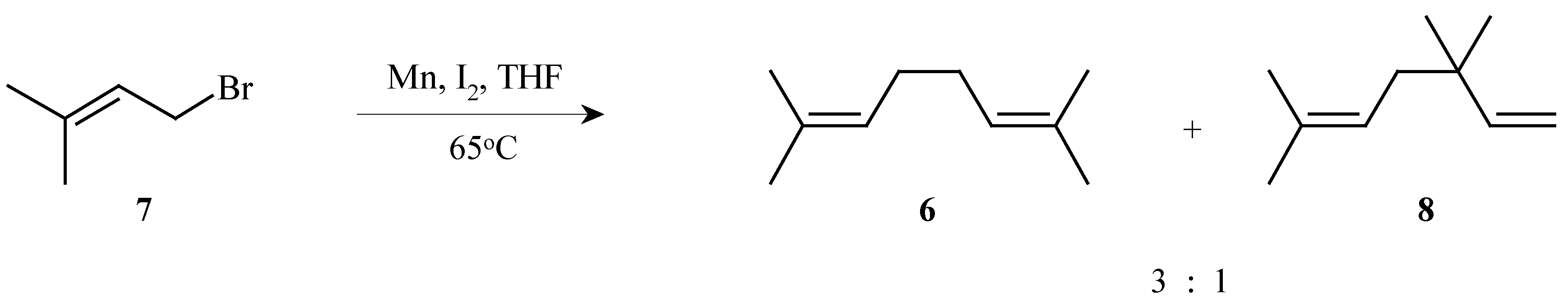
2,7-Dimethyl-2,6-octadiene (
6) was conveniently prepared by the dimerization of 4-bromo-2-methyl-2-butene (
7) with manganese powder and iodine. A 75% yield of a 3:1 mixture of the desired diene
6 and the isomeric 3,3,6-trimethyl-1,5-heptadiene (
8) was obtained. The pure diene
6 was isolated following distillation through a Vigreaux column in 39% yield (
Scheme 3).
With a large quantity of 2,7-dimethyl-2,6-octadiene (
6) in hand we were in position to examine its cyclic hydroboration. Still has previously reported that hydroboration of
6 with thexylborane and oxidative work up gave predominantly
meso-2,7-dimethyl-3,6-octanediol (
10) [
10]. We anticipated that replacement of the bulky thexyl group of the hydroboration reagent with a smaller group would lead to greater selectivity for the desired
trans-2,5-diisopropyllborolane (
9). Indeed, monobromoborane gave a 2:1 ratio of the
cis : trans-borolanes in THF at 0 °C. Under the same conditions monochloroborane gave a 1:1 ratio and borane itself gave a ratio slightly in favor of the desired
trans-borolane
9 (
Scheme 4).
Scheme 4.
Hydroboration of the diene 6
Scheme 4.
Hydroboration of the diene 6
| Entry | X | Cis : Trans* | Yield 10 + 11 |
|---|
| 1 | thexyl | 20 : 1.0 | 39% |
| 2 | Br | 2 : 1.0 | 62% |
| 3 | Cl | 1 : 1.0 | 65% |
| 4 | H | 1 : 1.5 | 65% |
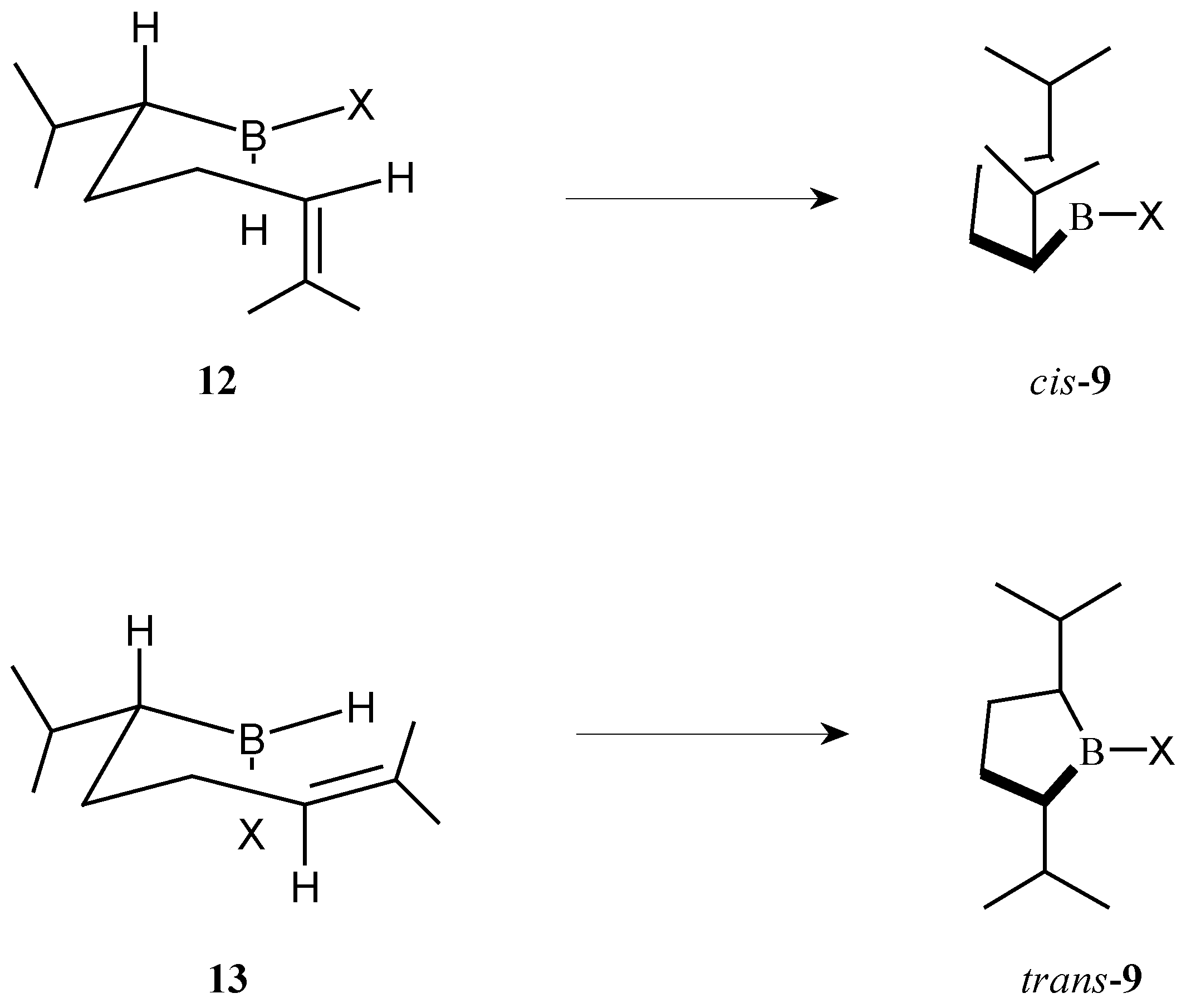
The preference of cyclic hydroboration for the
cis- or
trans-borolane can be explained by considering the intermediates
12 and
13 in which the isopropyl group is in an equatorial position (
Scheme 5). To produce the
cis-borolane
9, hydroboration must proceed across the axial double bond with the X group occupying an equatorial position as depicted in
12. To produce the
trans-borolane
9 hydroboration proceeds across the equatorial double bond with the X group occupying an axial position as depicted in
13. These intermediates are consistent with the results observed. For thexylborane the large thexyl group adopts the equatorial position and therefore gives the
cis-borolane
9 via intermediate
12. As the X group decreases in size (thexyl>Br>Cl>H) the intermediate
13 becomes more important and more of the
trans-borolane
9 is produced under the conditions studied.
Although there is a trend towards the desired
trans-borolane the ratios are not much better than those obtained by Masamune’s ‘double’ Grignard reaction in his synthesis of
trans-2,5-dimethyl-borolane (
3). Since hydroboration is a reversible reaction we postulated that under equilibration conditions the
trans-borolane
9 might be the more favored product. We therefore studied the cyclic hydroboration of
6 at the refluxing temperature of several solvents with monobromo- and monochloroborane (
Scheme 6) [
11]. The hydroboration of
6 with monobromoborane in THF at 0 °C for 1 h gave as reported earlier a
trans : cis ratio of 1:2. Increasing the reaction time to 8 h at 0 °C gave the same product ratio. However, increasing the reaction temperature to 65 °C gave a 1:1 product ratio after 1 h and after 8 h the ratio was 1.5:1 in favor of the desired
trans-borolane. Changing the solvent to ether, dichloromethane or toluene gave, after 8 h at the refluxing temperature of the solvent,
trans : cis ratios of 2.5-3.0:1. The best
trans : cis ratio of 4.0:1 was found when the reaction was carried out in refluxing carbon tetrachloride for 8 h. Similar product ratios were obtained when monochloroborane was used as the hydroboration reagent. Increasing the reaction times further or carrying out the reaction in sealed tubes at higher temperatures failed to improve the
trans : cis ratios and generally resulted in extensive decomposition of the products. Nevertheless the
trans : cis ratio of 4:1 from the carbon tetrachloride reaction was a significant improvement and we next investigated the resolution of the borolane isomers.
Scheme 6.
| Entry | Borane | Solvent | Temp. (°C) | Time (h) | Yield (%) | Trans: Cis |
|---|
| 1 | H2BBr.SMe2 | THF | 0 | 1 | 63 | 1.0:2 |
| 2 | H2BBr.SMe2 | THF | 0 | 8 | 59 | 1.0:2 |
| 3 | H2BBr.SMe2 | THF | 65 | 1 | 57 | 1.0:1 |
| 4 | H2BBr.SMe2 | THF | 65 | 8 | 55 | 1.5:1 |
| 5 | H2BBr.SMe2 | Et2O | 34 | 8 | 69 | 2.5:1 |
| 6 | H2BBr.SMe2 | CH2Cl2 | 40 | 8 | 67 | 3.0:1 |
| 7 | H2BBr.SMe2 | PhMe | 110 | 8 | 49 | 2.7:1 |
| 8 | H2BBr.SMe2 | CCl4 | 76 | 8 | 70 | 4.0:1 |
| 9 | H2BCl.SMe2 | THF | 65 | 8 | 57 | 1.0:1 |
| 10 | H2BCl.SMe2 | CCl4 | 76 | 8 | 62 | 2.8:1 |
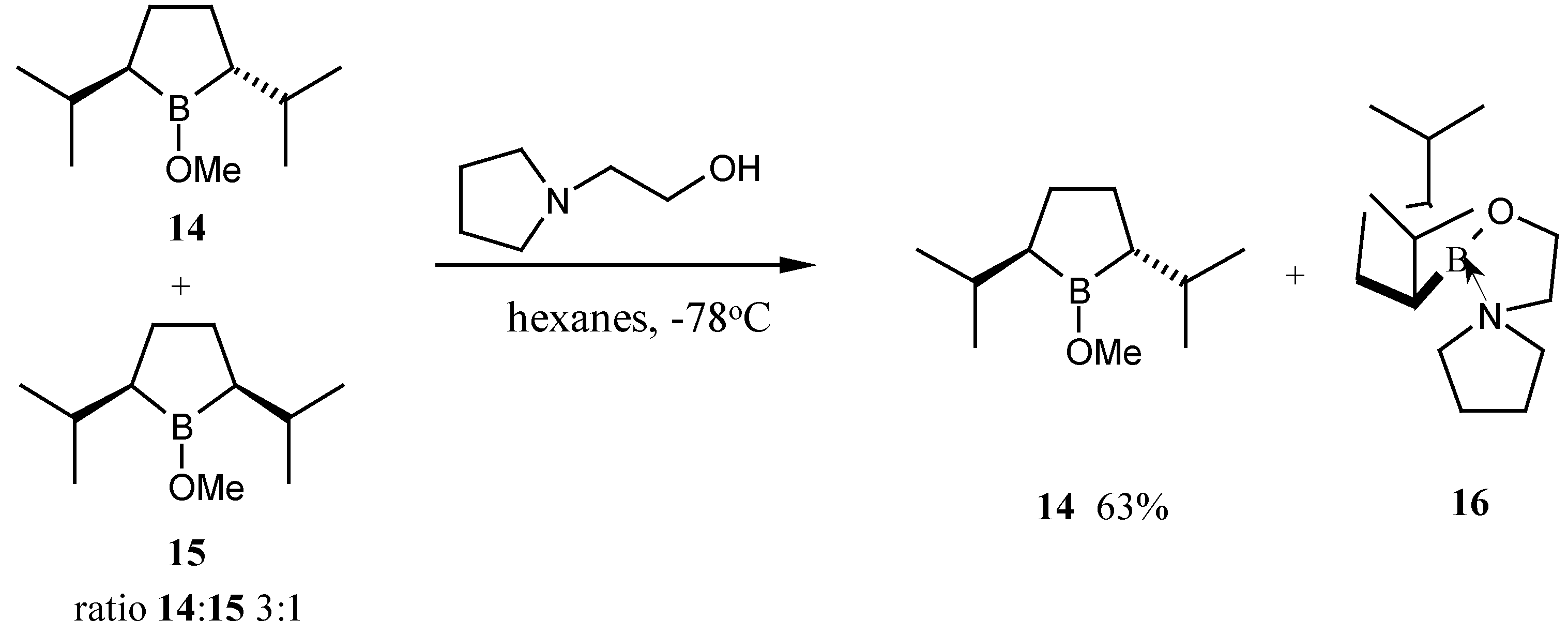
In Masamune’s work the
cis-dimethylborolane was removed by complexation with
N,N-dimethylaminoethanol and the
trans-dimethylborolanes then resolved by complexation with (
S)-prolinol and (
S)-valinol respectively. Initially we tried to directly resolve our 4:1 mixture by complexation with the appropriate amount of (
S)-prolinol, however, the small amount of complex formed was identified as the
cis-borolane complex. Attempted resolution with various other amino alcohols also failed to precipitate the
trans-borolane. It therefore appears to be necessary to remove the offending 20% of the
cis-borolane
15 first. Replication of Masamune’s work with
N,N-dimethyl-aminoethanol failed to produce a separable complex. Various primary, secondary and tertiary amino-alcohols were screened in the complexation process. Gratifyingly the use of pyrrolidinoethanol in hexane at low temperatures gave a precipitate of the
cis-complex
16 (
Scheme 7). Storage of the mixture at –78 °C for 4 h and removal of the solution
via cannula left behind essentially pure
cis-complex
16. The decanted solution was concentrated and distilled at reduced pressure to give the racemic
trans-borolane
14 in >95% purity and 63% yield.
Experimental
General
1H-, 13C- and 11B-NMR spectra were obtained using a Varian Gemini 300 NMR and were recorded at 300, 75 and 96 MHz respectively. Melting points were determined using a Thomas-Hoover capillary melting apparatus and are uncorrected. Electron ionization mass spectra (MS) were recorded on a Hewlett-Packard 5890 mass spectrometer. All reagents, chemicals and starting materials were obtained from commercial sources and were used as received unless otherwise noted. Column chromatography was performed using silica gel and the flash technique.
2,7-Dimethyl-2,6-octadiene (6)
Manganese powder (10 μm, 10.6 g, 193 mmol) suspended in THF (350 mL) containing iodine (6 g, 24 mmol) was heated under nitrogen at reflux for 2 h. The mixture was cooled to room temperature and 4-bromo-2-methyl-2-butene (7) (24 g, 161 mmol) in THF (250 mL) was added. The reaction mixture was heated to reflux for 12 h, cooled and filtered through Celite. Ether (250 mL) was added and the organic layer washed with water (250 mL), brine (250 mL), dried (MgSO4) and evaporated to a dark yellow oil. Distillation of the oil through a Vigreaux column under reduced pressure (60 mmHg) gave first 3,3,6-trimethyl-1,5-heptadiene (8) (1.6 g, 15%); 1H-NMR (300 MHz, CDCl3) 0.96 (6 H, s), 1.59 (3 H, s), 1.66 (3 H, s), 1.89 (2H, d, J = 7.5 Hz), 4.71 (2 H, m), 5.07 (1 H, t, J = 7.5 Hz), 5.69 (1 H, dd, J = 10 and 18 Hz). Later fractions contained 2,7-dimethyl-2,6-octadiene (6) (4.33 g, 39%), b.p. 85o C at 60 mmHg; 1H NMR (300 MHz, CDCL3) 1.58 (6 H, s), 1.66 (6 H, s), 1.97 (4 H, brs), 5.10 (2 H, brs); m/z 137 (m-1).
Representative procedure for the hydroboration and oxidation of 2,7-dimethyl-2,6-octadiene (6)
To a solution of 2,7-dimethyl-2,6-octadiene (6) (138 mg, 1.0 mmol) in dry THF (5 mL) at 0 °C under nitrogen was added monochloroborane-dimethylsulphide complex (1.0 M, 1.0 mL, 1.0 mmol). The reaction mixture was stirred at 0 °C for 1 h and oxidized by adding 1M NaOH (1 mL) and 30% hydrogen peroxide (1 mL). After stirring for 0.5 h the solution was extracted with dichloromethane (3 x 10 mL), washed with brine (15 mL), dried (MgSO4) and evaporated. The crude mixture was dissolved in dichloromethane (5 mL) and treated with acetic anhydride (302 mg, 3 mmol), pyridine (237 mg, 3 mmol) and a crystal of DMAP. After stirring at room temperature for 3 h, the mixture was poured into saturated aqueous NaHCO3 (5 mL). The mixture was extracted with dichloromethane (3 x 10 mL), dried (MgSO4) and evaporated. The crude mixture was analyzed by capillary column G.C. and shown to be a 1:1 mixture of 3,6-diacetoxy-2,7-dimethyl octane isomers. For the mixture 1H-NMR (300 MHz, CDCl3) 0.84 (12 H, d, J = 7.0 Hz), 1.45-1.60 (6 H, m), 2.02 (6 H, s), 4.67 (2 H, m); υmax (thin film) 1740 and 1020 cm-1; m/z 258 (m+1).
cis- and trans-2,5-Diisopropyl-B-methoxyborolanes (14) and (15)
To a solution of 2,7-dimethyl-2,6-octadiene (6) (10 g, 73 mmol) in dichloromethane (50 mL), under a nitrogen atmosphere, was added monobromoborane-dimethyl sulphide complex (11.3 g, 73 mmol). The reaction mixture was heated to reflux for 8 h, cooled and the volatiles removed under reduced pressure. The yellow residue was distilled to give 1-bromo-2,5-diisopropylborolane, a colorless oil (12.8 g, 76%) b.p. 42°C at 0.2 mm; 11B-NMR (96 MHz, CD2Cl2) 77; 1H-NMR (300 MHz) 0.7-2.1 (brm).
To a solution of 1-bromo-2,5-diisopropylborolane (12.8 g, 56 mmol) in dichloromethane (100 mL), under a nitrogen atmosphere, at 0 °C, was slowly added dry methanol (4.6 mL, 112 mmol) followed by 2,4,6-collidine (7.4 mL, 56 mmol). The mixture was stirred at room temperature for 4 h and the volatiles removed under reduced pressure. The residue was distilled to give a mixture of cis- and trans-2,5-diisopropyl-B-methoxyborolanes (14) and (15), as a colorless oil (6.4 g, 48%); b.p. 47 °C at 1 mmHg; 11B-NMR (96 MHz, CD2Cl2) 58. Oxidation of a sample and acetylation indicated a trans : cis ratio of 3:1 by G.C.
(±)-trans-2,5-Diisopropyl-B-methoxyborolane (14)
To a stirred mixture of cis- and trans-diisopropylborolanes (14) and (15) (3:1) (5.43 g, 30 mmol), under an argon atmosphere, in hexane, was added 1-(2-hydroxyethyl)-pyrrolidine (863 mg, 7.5 mol). Approximately 50% of the hexane was then removed by distillation at atmospheric pressure and methanol (0.025 mL, 1.0 mmol) was added. The reaction was stored in a freezer overnight and a white solid was formed. The solution was removed from the solid via cannula, the hexane distilled off at atmospheric pressure and the residue distilled to give trans-2,5-diisopropyl-B-borolane (14) (3.42 g, 63%); b.p. 47 °C at 1 mmHg; 1H-NMR (300 MHz, C6D6) 0.7-1.02 (m, 2 H), 0.82 (d, 6 H, J = 7 Hz), 0.94 (d, 6H, J = 7 Hz), 1.15-1.30 (m, 2 H), 1.70-1.91 (m, 2 H), 1.93-2.10 (2H, m), 3.83 (s, 3H); 13C-NMR (75 MHz, C6D6) 22.2, 23.3, 26.9, 27.6, 38.1, 56.2; 11B (96 MHz, C6D6) 57 (br).
The white solid 16 was collected: mp 32-34o C; 1H-NMR (300 MHz, CDCl3) 0.3-0.45 (m, 2H), 0.85 (d, 6H, J = 6.5 Hz), 0.92 (d, 6 H, J = 7 Hz), 1.12-1.59 (m, 4H), 1.61-2.04 (m, 6H), 2.81-3.00 (m, 4H), 2.87 (t, 2 H, J = 6.5 Hz), 4.02 (t, 2H, J = 6.6 Hz); 13C NMR (75 MHz, CDCL3) 21.7, 22.9, 24.5, 28.1, 29.0, 38.2, 54.2, 56.9, 63.1; 11B-NMR (96 MHz, CDCl3) 28.8.
(S*, R*) 2,7-Dimethyl-3,6-octanediol (10)
A sample of 16 was oxidized with alkaline hydrogen peroxide as described earlier and purified by flash chromatography (elution with 1:1 hexane:ether) to give pure (S*, R*)-2,7-dimethyl-3,6-octanediol (10); 1H-NMR (300 MHz; CDCl3) 0.95 (12H, d, J = 7 Hz), 1.60 (6H, m), 2.05 (2H, br), 3.40 (2H, m); 13C-NMR (75 MHz, CDCl3) 17.5, 18.9, 20.5, 33.8, 75; m/z 175 (M+1).
(S*, S*)-2,7-Dimethyl-3,6-octanediol (11)
A sample of trans-2,5-diisopropyl borolane (14) was oxidized with alkaline hydrogen peroxide as described earlier. The product was purified by flash chromatography (elution with 1:1 hexane:ether) to give pure (S*, S*)-2,7-dimethyl-3,6-octanediol (11); 1H-NMR (300 MHz; CDCl3) 0.89 (d, J = 7 Hz, 12H), 1.43 (m, 2H), 1.62 (m, 4H), 2.02 (br, 2H,), 3.36 (m, 2H); 13C-NMR (75 MHz, CDCl3) 17.4, 18.7, 31.2, 34.0, 77.1; m/z 175 (M+1).





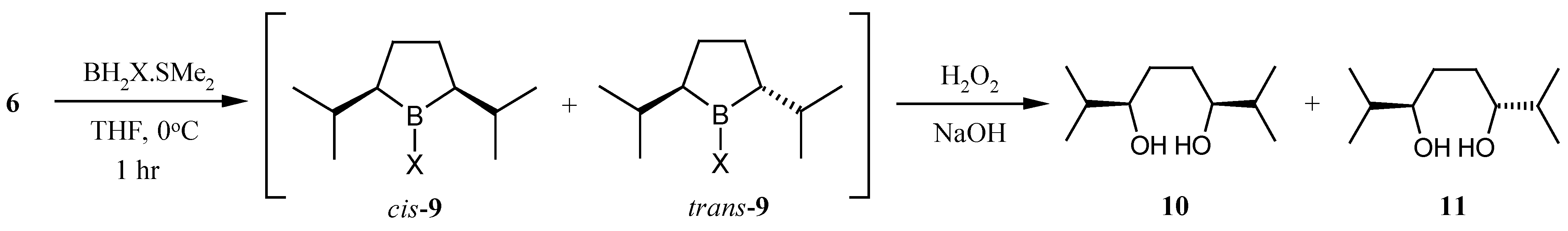




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}