Results and Discussion
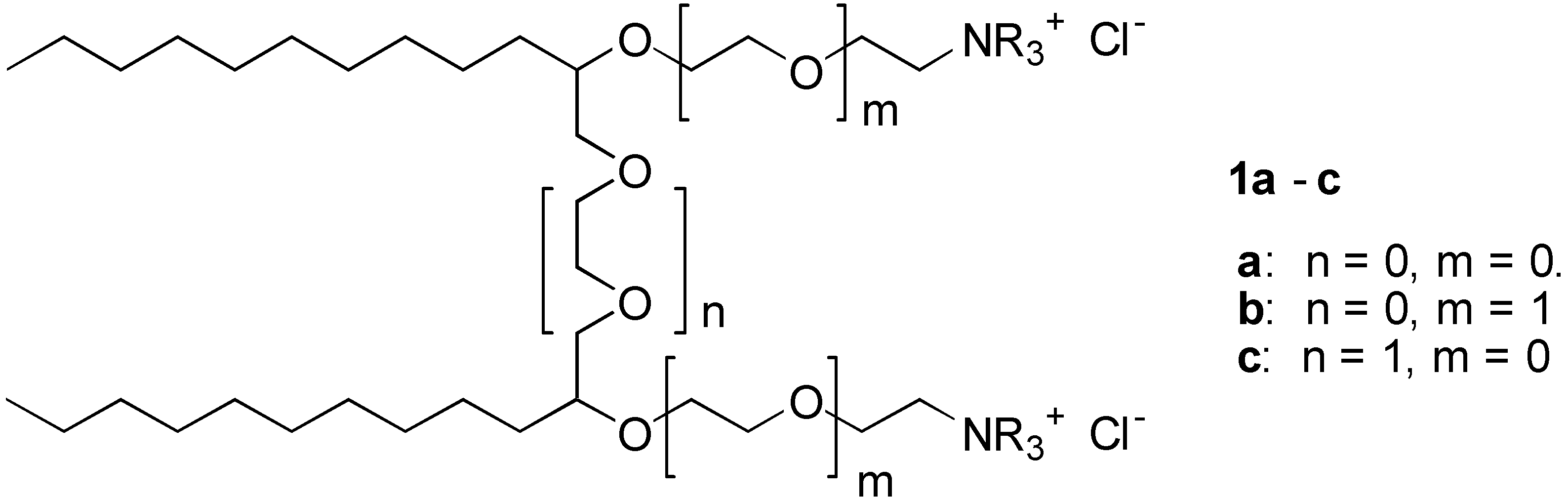
We report here the synthesis of a series of cationic surfactant molecules with the general structure
1 shown in
Figure 1:
These general structures 1a-c belongs to the class of gemini surfactants. Common for products 1 is that they consist of two hydrophobic alkyl chains connected with ether spacer groups. These spacer group may contain ethyleneoxy (EO) ether groups (i.e., n = 0,1.. etc.). The hydrophilic heads consist of ethyleneamine- or the corresponding quaternary ammonium group, which is attached to the hydrophobic part of the molecule by ether bonds. The hydrophilic head may be further extended with EO-groups (i.e., m = 0,1, ..).
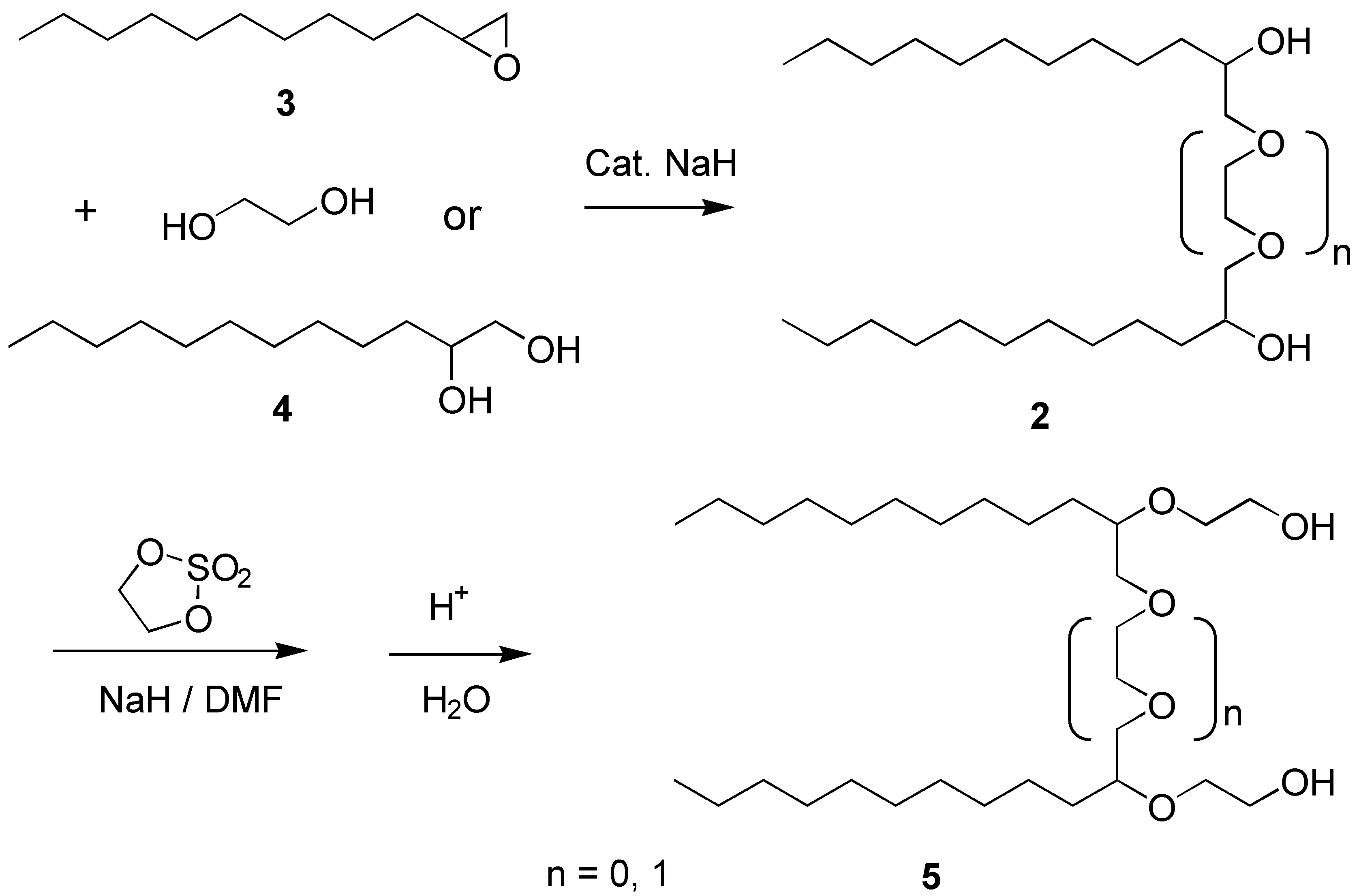
The basic framework of molecules,
2, was constructed from the readily available 1,2-epoxy-dodecane (
3) and 1,2-dodecanediol (
4) which in the presence of a catalytic amounts of base (NaH) readily formed the lipophilic part of one target molecules, (n = 0). If the same transformation was carried out using 1,2-epoxydodecane and ethylene glycol the spacer sections were readily extended with one ethyleneoxy unit (n = 1). These reactions, in general, gave the pure products in approximately 60% yields after recrystallization. The selective extension for the hydrophilic head with the EO-groups in a controlled manner was best accomplished by the reaction of the alcohol
2 with the cyclic ethylene sulfate and subsequent hydrolysis [
5] yielding diol
5. These reactions were carried out as has been described earlier [
6].
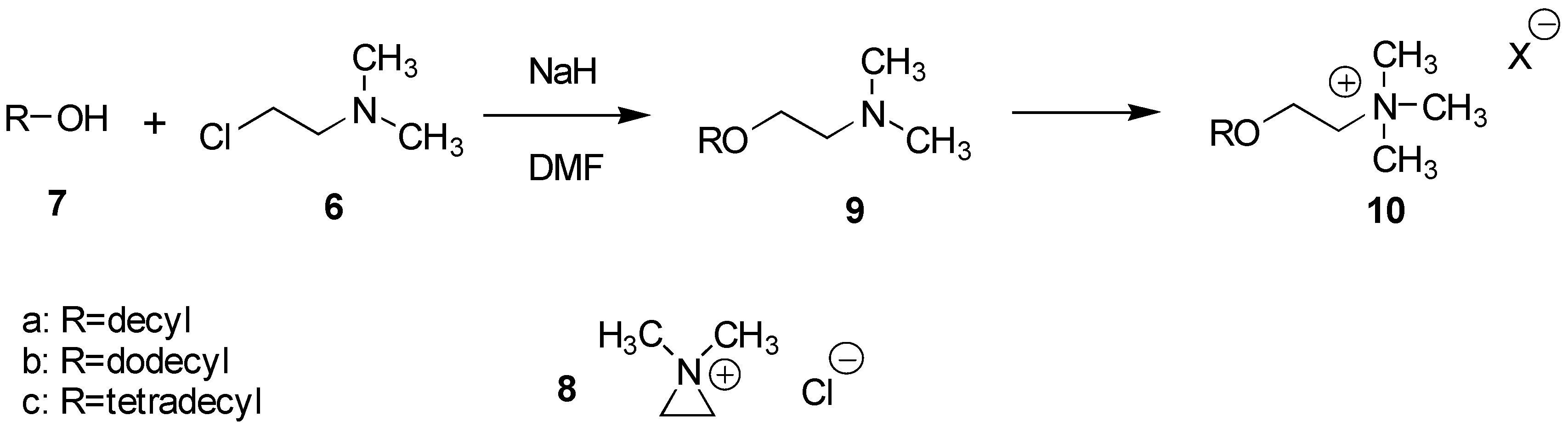
The introduction of the ethylene amine function was furnished in a short and efficient, one step procedure in which the alcohol was reacted with 2-chloro-
N,N-dimethylethylamine (
6) in the presence of a base,
Scheme 2. This reaction was first studied for the corresponding C
10-, C
12- and C
14-alcohols,
7a-
c, and proceeds presumably through the formation of the reactive
N,N-dimethylaziridinium ion intermediate,
8. Thus, reacting these alcohols,
7a -c, with 3 equivalents of
6 as its hydrochloride in DMF at 50
oC for 3 h in the presence of 7 equivalents of base (NaH), readily afforded the desired products
9a-c in 80-90% yields. The hydrochloride of
6 was very hygroscopic and was therefore always dried by azeotropic distillation with toluene prior to use. After extraction, products
9a -
c of better than 97% purity (GC and NMR) were routinely obtained.
The corresponding quaternary ammonium salts, 10a-c, were easily obtained by reaction of the amines 9a -c with methyl iodide at room temperature without a solvent. The purity of the salts were generally > 95% purity, as judged from their NMR spectra. The corresponding chlorides were obtained by reaction of the amines, 9a-c, with methyl chloride in dichloromethane at room temperature in a closed reaction flask for 2 days. These reactions were not optimized, but to ensure complete conversion 5 equivalents of the alkylating agent was used. These salts were obtained in nearly quantitative yields and were more than 95% pure by NMR.
The same reaction sequence was applied on the gemini diols,
2 and
5 to synthesize the desired gemini surfactants
1a-
c (
Scheme 3). Due to the difficulties usually experienced when attempting to dialkylate a molecule in one step, a large excess of reagent
6 as well as base (NaH) was applied. Attempts to reduce the excess of the reagents resulted in lower yields of the desired products. Therefore, 20 mol equivalents of NaH was used relative to diols
2 and
5 in DMF. The reaction mixture was heated in DMF at 50
oC for 1 h, followed by addition of 10 mol equivalents of
6 as its hydrochloride. Complete conversion was obtained after 2-3 h as indicated by TLC analysis. The products were obtained after extraction as described above in yields ranging between 70 and 99%. Purities of the products were all between 90 and 95%, with DMF as the main impurity. The formation of the gemini amine surfactants
1a-
c proceeded slower for the large diols (
e.g., n = 1, m = 0) than for the smaller ones. NMR analysis indicated that the degree of dialkylation in general was better than 95‑98%. The quarternization was carried out as described in the above. The products were obtained in 78-90% yields. The purity of these cationic surfactants was better than 95%. Flash chromatography on silica gel and ethanol as the eluent, improved the purity only slightly but also resulted in some loss of product. These reactions were not optimized.
The NMR spectra of the single chain surfactants 10a-c were recorded in CDCl3 and in D2O, and were in agreement with the proposed structures. However, the NMR-spectra looked different in the two solvents. Thus, when compound 10a, for example, was recorded in D2O, the signals corresponding to the hydrophobic part of the molecule appeared as broad, poorly separated peaks, while the signals corresponding to the hydrophilic part displayed well resolved peaks. When the spectra were recorded in CDCl3, the signals corresponding to the hydrophobic part of the molecule were well resolved, while the signals for the hydrophilic part appeared as broad signals with poor resolution. This can be rationalized by the formation of micelles. In D2O, formation of normal micelles restricts the movement of the alkyl groups in the core of the micelles, causing line broadening and poor resolution due to relaxation phenomena. The opposite was the case for the experiments in CHCl3, where formation of inverse micelles caused fewer restrictions on the movements of the alkyl groups. The 1H-NMR spectra were in all cases in accordance with the proposed structures 1a-c and 10a-c as was also the case for the 13C-NMR spectra of compounds 10a-c. However, 13C-NMR spectra are not reported for compounds 1a-c as no signals exceeding the noise level were recorded using standard pulse sequences. This can also explained by micelle formation and the associated relaxation phenomena. Molecules 1a-c contain two stereogenic carbon atoms, resulting in the formation of diastereomers. In the 13C-NMR spectrum of the precursor 11a there was indication of the two diastereomeric forms, as the signals corresponding to the stereogenic carbons were observed as double peaks. The physiochemical properties of these new surfactants are currently being investigated.
Experimental
General
All chemicals and solvents applied were of synthesis quality unless otherwise stated. The 1,2-dodecanediol and 1,2-epoxydodecane were of bulk quality, (97-98% purity). N,N-dimethylformamide was dried over 4Å molecular sieves. NMR-spectra were recorded (as CDCl3 solutions, unless stated otherwise) on a JEOL JNM-EX 400 FT NMR System, and Bruker Avance DPX300 or 400 instruments. IR spectra were obtained with a Nicolet 20SXC FT-IR Spectrometer. Mass spectra were obtained on a AEI MS 902 double focusing high resolution instrument equipped with electron impact ionization (EI, 70 eV). Melting points are uncorrected. GLC analyses were recorded on a Perkin Elmer Auto System with a Chrompack CP-5 CB column.
General Procedure for Synthesis of Monoamines (9a-c)
Solutions of alcohols 9a-c (0.175 mol) in dry DMF (200 mL) were added under nitrogen at room temperature to sodium hydride (1.25 mol, 30.0 g) and dry DMF (300 mL) over a 30 minute period. 2‑Chloro-N,N-dimethylet hylamine hydrochloride (6) (0.53 mol; 76 g), dried by azeotropic distillation with toluene and stored over phosphorus pentoxide, was then added portionwise over 60 minutes. The reaction mixture was heated at 50°C for 3 hours, and then quenched with ethanol (50 mL). The solvent was removed under reduced pressure, and the crude product then dissolved in water (500 mL). The aqueous phase (its pH was approximately 12) was extracted with diethyl ether (5 x 200 mL). The ether phase was then extracted with 6M hydrochloric acid (4 x 30 mL). The pH of the water phase was adjusted to 12-13 by adding 5M aqueous NaOH (approx.100 mL). This aqueous phase was finally extracted with ether (4 x 200 mL) and the ether phase then was washed with brine (100 mL) and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure to give the products.
N,N-Dimethyl-3-oxa-1-tridecylamine (9a). Obtained from 1-decanol as an oil (37 g, 92% yield) of > 98% purity (GLC). 1H-NMR: δ 0.88 (t, J = 6.7 Hz, 3H), 1.26 (m, 14H), 1.58 (m, 2H), 2.27 (s, 6H), 2.49 (t, J = 5.9, 2H), 3.42 (t, J = 6.8 Hz, 2H), 3.50 (t, J = 6.0 Hz, 2H) ppm. 13C‑NMR: δ 14.0, 22.6, 26.2, 29.2 29.4, 29.5, 29.5, 29.6, 31.8, 45.8, 58.8, 68.8, 71.2 ppm. IR (neat): 2925, 2855, 2817, 1465, 1118, 1042 (m) cm-1. MS [m/z (% rel. int.)]: 229 (2, M+), 58 (100).
N,N-Dimethyl-3-oxa-1-pentadecylamine (9b). Obtained from 1-dodecane in a yield of 35.6 g (80%) and 97% purity (GLC). 1H-NMR: δ 0.88 (t, J = 6.7 Hz, 3H), 1.26 (m, 18H), 1.58 (m, 2H), 2.27 (s, 6H), 2.49 (t, J = 5.9 Hz, 2H), 3.42 (t, J = 6.8 Hz, 2H), 3.51 (t, J = 5.9 Hz, 2H) ppm. 13C-NMR: δ 14.1, 22.7, 26.2, 29.4, 29.5, 29.6, 29.7, 29.7, 29.7, 29.9, 31.9, 45.9, 59.0, 68.9, 71.4 ppm. IR (neat): 2925, 2854, 2817, 2768, 1465, 1119, 1042 cm-1. MS [m/z (% rel. int.)]: 257 (1.4, M+), 58 (100).
N,N-Dimethyl-3-oxa-1-heptadecylamine (9c). Obtained from 1-tetradecane in 41.4 g (83%) yield of >98% purity (GLC). 1H-NMR: δ 0.88 (t, J = 6.7 Hz, 3H), 1.27 (m, 22H), 1.57 (m, 2H,), 2.26 (s, 6H), 2.49 (t, J = 5.9 Hz, 2H), 3.42 (t, J = 6.8 Hz, 2H), 3.50 (t, J = 5.9 Hz, 2H) ppm. 13C-NMR: δ 14.0, 22.6, 26.0, 29.3, 29.4, 29.5, 29.5, 29.5, 29.5, 29.6, 29.6, 29.6, 31.8, 45.7, 58.8, 68.8, 71.2 ppm. IR (neat): 2924, 2854, 2817, 2768, 1465, 1119, 1042 cm-1. MS [m/z (% rel. int.)]: 285 (1.3, M+), 58 (100).
General Procdure for the Synthesis of N,N,N-Trimethyl-3-oxa-1-alkylammonium halides (10a-c iodides or chlorides).
N,N-Dimethyl-3-oxa-1-alkylamine (approx. 50 mmol) was added methyl iodide (220 mmol; 15 mL) or methyl chloride (11 g) in a sealed tube. The mixture was kept at room temperature for 24 h. Pale yellow solids were formed. The products were washed with several portions of ether and then dried in vacuum.
N,N,N-Trimethyl-3-oxa-1-tridecylammonium iodide (10a-iodide). Obtained in 96% yield and > 98% purity (by 1H-NMR). 1H-NMR: δ 0.88 (t, J = 6.7 Hz, 3H), 1.26 (m, 14H), 1.58 (m, 2H), 3.48 (m, 2H), 3.50 (s, 9H), 3.90 (m, 2H), 3.97 (m, 2H) ppm. 13C-NMR: δ 14.2, 22.8, 26.3, 29.4, 29.5, 29.5, 29.7, 29.7, 32.0, 55.1, 64.9, 66.1, 72.1 ppm. IR (KBr): 2916, 2850, 1478, 1370, 1139, 1120, 1056, 955, 877 cm-1.
N,N,N-Trimethyl-3-oxa-1-pentadecylammonium iodide (10b-iodide). Obtained in 96% yield and >98% purity (1H-NMR). 1H-NMR: δ 0.88 (t, J = 6.7 Hz, 3H), 1.26 (m, 18H), 1.57 (m, 2H), 3.48 (m, 2H), 3.49 (s, 9H) 3.91 (m, 2H), 3.98 (m, 2H) ppm. 13C-NMR: δ 14.3, 22.9, 26.3, 29.5, 29.6, 29.6, 29.7, 29.8, 29.8, 29.9, 32.1, 55.1, 64.9, 66.1, 72.1 ppm. IR (KBr): 2917, 2849, 1466, 1140, 1120, 954, 866, 720 cm-1.
N,N,N-Trimethyl-3-oxa-1-heptadecylammonium iodide (10c-iodide). Obtained in 90% yield and >98 % purity (by 1H-NMR). 1H-NMR: δ 0.88 (t, J = 7 Hz, 3H), 1.26 (m, 22H), 1.56 (m, 2H), 3.49 (t, J = 6.6 Hz, 2H), 3.52 (s, 9H) 3.91 (s, 2H), 3.97 (m, 2H) ppm. 13C-NMR: δ 14.3, 22.9, 26.3, 29.5, 29.6, 29.6, 29.7, 29.8, 29.8, 29.8, 29.9, 29.9, 32.1, 55.2, 64.9, 66.1, 72.2 ppm. IR (KBr): 2917, 2849, 1468, 1400, 1139, 1120, 1054, 954, 879, 721 cm-1.
N,N,N-Trimethyl-3-oxa-1-tridecylammonium chloride (10a-chloride). Obtained in 96% yield and >98 % purity (by 1H-NMR). 1H-NMR: δ 0.88 (t, J = 6.8 Hz, 3H), 1.26 (m, 14H), 1.58 (m, 2H), 3.47 (t, J = 6.6 Hz, 2H), 3.50 (s, 9H), 3.88 (m, 2H), 4.00 (m, 2H) ppm. 13C-NMR: δ 14.5, 23.0, 26.5, 29.6, 29.7, 29.8, 29.9, 29.9, 32.2, 54.8, 65.2, 66.0, 72.2 ppm. IR (KBr): 2958, 2916, 2851, 1483, 1418, 1370, 1156, 1124, 1073, 987, 965, 880, 719 cm-1.
N,N,N-Trimethyl-3-oxa-1-pentadecylammonium chloride (10b-chloride). Obtained in 90% yield and >98% purity (by 1H-NMR). 1H-NMR: δ 0.84 (t, J = 6.7 Hz, 3H), 1.22 (m, 18H), 1.51 (m, 2H), 3.43 (t, J = 6.6 Hz, 2H), 3.46 (s, 9H) 3.84 (s, 2H), 3.95 (m, 2H) ppm. 13C-NMR: δ 14.3, 22.8, 26.3, 29.5, 29.5, 29.6, 29.7, 29.8, 29.8, 29.8, 32.1, 54.7, 65.0, 65.8, 72.0 ppm. IR (KBr): 2957, 2916, 2851, 1470, 1418, 1370, 1123, 1072, 961, 879, 719 cm-1.
N,N,N-Trimethyl-3-oxa-1-heptadecylammonium chloride (10c-chloride). Obtained in 95% yield and >98% purity (by 1H-NMR). 1H-NMR: δ 0.88 (t, J = 6.8 Hz, 3H), 1.26 (m, 22H), 1.56 (m, 2H), 3.47 (m, 2H), 3.48 (s, 9H), 3.89 (m, 2H), 4.00 (m, 2H) ppm. 13C-NMR: δ 14.3, 22.9, 26.4, 29.6, 29.6, 29.6, 29.7, 29.8, 29.8, 29.9, 29.9, 29.9 32.1, 54.7, 65.0, 65.8, 72.1 ppm. IR (KBr): 2958, 2916, 2850, 1484, 1470, 1419, 1394, 1157, 1124, 1087, 1072, 1007, 966, 880, 718 cm-1.
General Procedure for the Synthesis of diamines (11a-c).
The diol 2 and 5 (25 mmol) were dried by azeotropic distillation with toluene before use. The diol was then dissolved in dry DMF (300 mL). The reaction was heated at 50°C, sodium hydride (12.4 g, 517 mmol) was added slowly and the reaction was stirred for 1 hour. Dry 2-chloro-N,N-dimethyl-ethylamine hydrochloride (37.0 g, 257 mmol) was then added and the reaction mixture stirred for 3 hours. After cooling to room ethanol (100 mL) was added. The solvent was removed by evaporation under reduced pressure, and water (300 mL) was added. The pH of the aqueous phase was adjusted to 12-13, and it was then extracted with dichloromethane (6 x 50 mL). The organic phase was extracted with 1 M hydrochloric acid (3 x 50 mL). The pH of the aqueous phase was adjusted to 12-13 with 5 M sodium hydroxide (50 mL) and was then extracted with dichloromethane (5 x 50 mL). The organic phase was dried over MgSO4, filtered and concentrated under reduced pressure.
N,N,N’,N’-Tetramethyl-4,8-didecyl-3,6,9-trioxa-1,11-undecanediamine (11a). Obtained from 13-oxa-11,15-pentacosanediol (2a), and isolated as an oil in quantitative yield (104%) and 95% purity (GLC). 1H‑NMR : δ 0.88 (t, J = 6.7 Hz, 6H), 1.26-1.48 (m, 36H), 2.27 (s, 12H), 2.50 (m, 4H), 3.37-3.84 (m, 10H) ppm. 13C-NMR: δ 14.5, 23.0, 26.0, 29.7, 30.0, 30.0, 30.1, 30.1, 32.2, 32.3, 45.9, 59.4, 68.0, 74.4, 74.5 (two diastereomers), 79.5, 79.6 (two diastereomers) ppm. IR (neat): 2925, 2854, 2817, 2768, 1460, 1117 cm-1. MS [m/z (% rel. int.)]: 529 (M+1, 0.2), 528 (0.5, M+), 527 (1, M-1), 458 (8), 73 (22), 72 (40), 71 (24), 58 (100).
N,N,N’,N’-Tetramethyl-7,11-didecyl-3,6,9,12,15-pentaoxa-1,17-heptadecanediamine (11b). Obtained from 4,8-didecyl-3,6,9-trioxa-1,11-undecanediol (5) in 98% yield and 88% purity (GLC). 1H-NMR: δ 0.88 (t, J = 6.8 Hz, 6H), 1.26 (m, 32H), 1.46 (m, 4H), 2.36+2.42 (s, 12H), 2.65 (m, 4H), 3.39-3.55 (m, 8H), 3.59-3.70 (m, 8H), 3.76-3.81 (m, 2H) ppm. 13C-NMR: δ 14.5, 23.0, 25.9, 29.7 29.9, 29.9, 30.0, 30.1, 32.2, 32.3, 46.0, 59.4, 68.1, 69.2, 71.1, 74.4, 79.6 ppm. IR (neat): 2924, 2854, 2769, 1676, 1461, 1384, 1263, 1121, 963, 732 cm-1. MS [m/z (% rel. int.)]: 616 (1, M+), 615 (4, M-1), 116 (13), 72 (28), 71 (22), 58 (100).
N,N,N’,N’,-Tetramethyl-4,11-didecyl-3,6,9,12-tetraoxa-1,14-tetradecanediamine (11c). Obtained from 13,16-dioxa-11,18-octacosanediol (2c) in 80% yield and 95% purity. 1H-NMR: δ 0.88 (t, J = 6.8 Hz, 6H), 1.26 (m, 32H), 1.46 (m, 4H), 2.36+2.39 (s, 12H), 2.65 (m, 4H), 3.39-3.52 (m, 6H), 3.59-3.67 (m, 6H), 3.77-3.81 (m, 2H) ppm. 13C-NMR: δ 14.5, 23.0, 25.9, 29.7, 29.9, 30.0, 30.0, 30.0, 30.1, 32.3, 46.0, 59.4, 68.1, 71.1, 74.4, 79.6 ppm. IR (neat): 2925, 2854, 2817, 2768, 1684, 1462, 1116, 1049 cm-1. MS [m/z (% rel. int.)]: 572 (0.4, M+), 571 (1 M-1), 73 (19), 72 (33), 17 (16), 58 (100).
4,8-Didecyl-3,6,9-trioxa-undecane-1,11-di(trimethylammonium) dichloride (1a). Prepared from 11a as described above and obtained in 82% yield and 96% purity (by 1H-NMR). A sample was purified by flash chromatography (SiO2/ethanol) gave a yield of 44% of product of >99 % purity (by 1H-NMR). 1H-NMR (D2O): δ 0.85 (t, J = 6.7 Hz, 6H), 1.27 (m, 32H), 1.53 (m, 4H), 3.19 (s, 18H), 3.28-3.30 (m, 2H), 3.37-3.72 (m, 16H), 3.95-4.04 (m, 4H) ppm. IR (neat): 2954, 2922, 2853, 1480, 1467, 1415, 1378, 1345, 1271, 1244, 1188, 1119, 955, 877, 831 cm-1.
7,11-Didecyl-3,6,9,12,15-pentaoxa-heptadecane-1,17-di(trimethylammonium) dichloride (1b). Obtained from 11b in 93% yield and >96% purity. 1H-NMR (D2O): δ 0.87 (t, J = 6.8 Hz, 6H), 1.28 (m, 32H), 1.58 (m, 4H), 3.21 (s, 18H), 3.30-3.33 (m, 2H), 3.47-3.69 (m, 8H), 3.96-4.08 (m, 4H) ppm. IR (neat): 2953, 2924, 2853, 1471, 1462, 1116, 950, 876 cm-1.
4,11-Didecyl-3,6,9,12-tetraoxa-tetradecane-1,14-di(trimethylammonium) dichloride (1c). Obtained from 11c in 78% yield and 90% purity. Flash-chromatography (SiO2/ethanol) gave the product in 40% yield and >96% (+ 4% ethanol) (by 1H-NMR). 1H-NMR (D2O): δ 0.85 (t, J = 6.8 Hz, 6H), 1.26 (m, 32H), 1.53 (m, 4H), 3.19 (s, 18H), 3.29-3.30 (m, 2H), 3.36-3.64 (m, 12H), 3.94-4.02 (m, 4H) ppm. IR (neat): 2955, 2923, 2853, 1471, 1116, 957, 878, 721 cm-1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}