Experimental
General
THF was dried by distillation from sodium / benzophenone. All other reagents and solvents were purchased from Acros Organics and used without further purification. Each new compound was fully characterized by mass spectrometry (Perkin Elmer, EI 70 eV and ES) in addition to 1H-NMR and 13C-NMR spectroscopy (Bruker AMX 400 MHz or Brucker WM 250 MHz). Chemical shifts are relative to TMS as an internal reference.
Synthetic procedures and spectral data
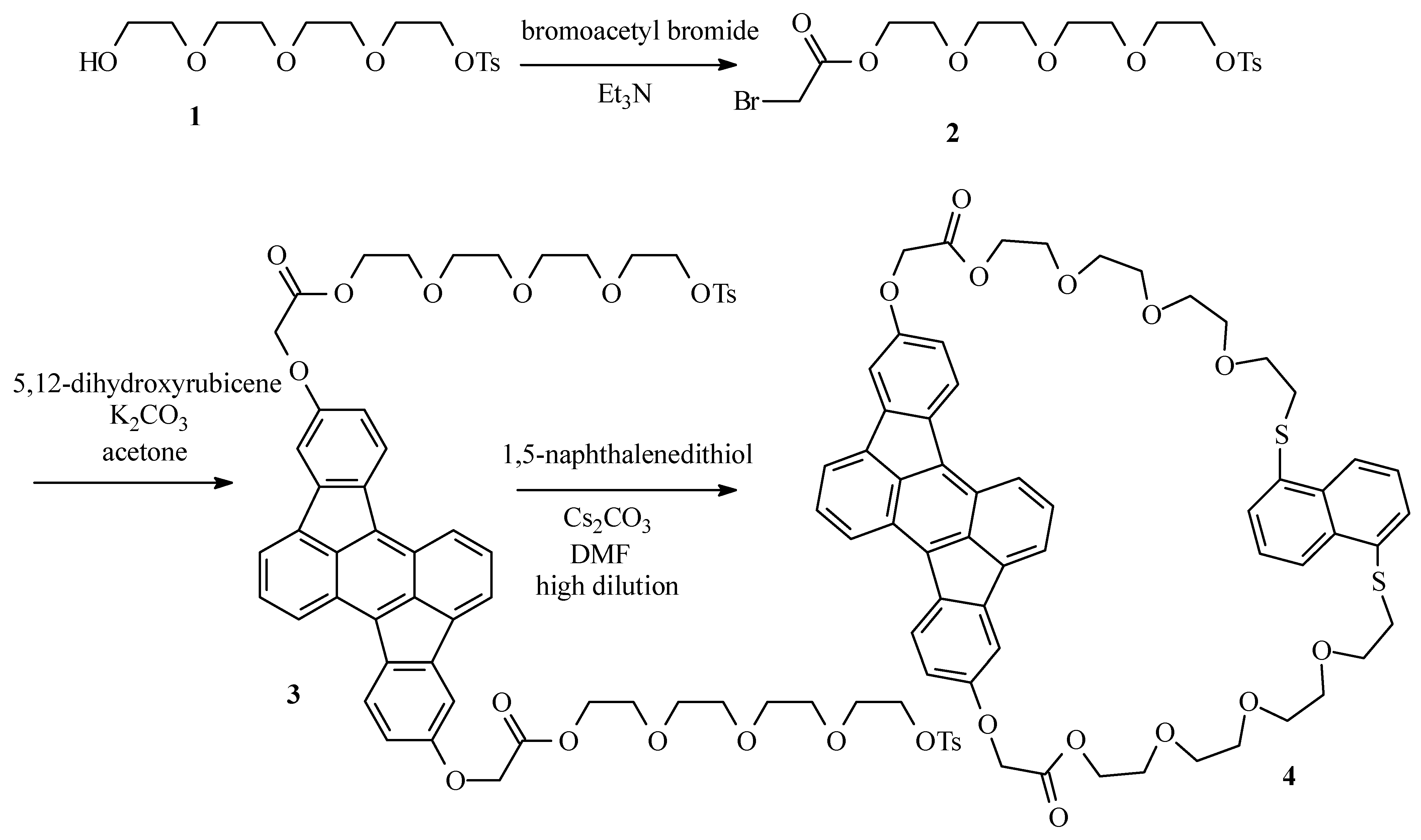
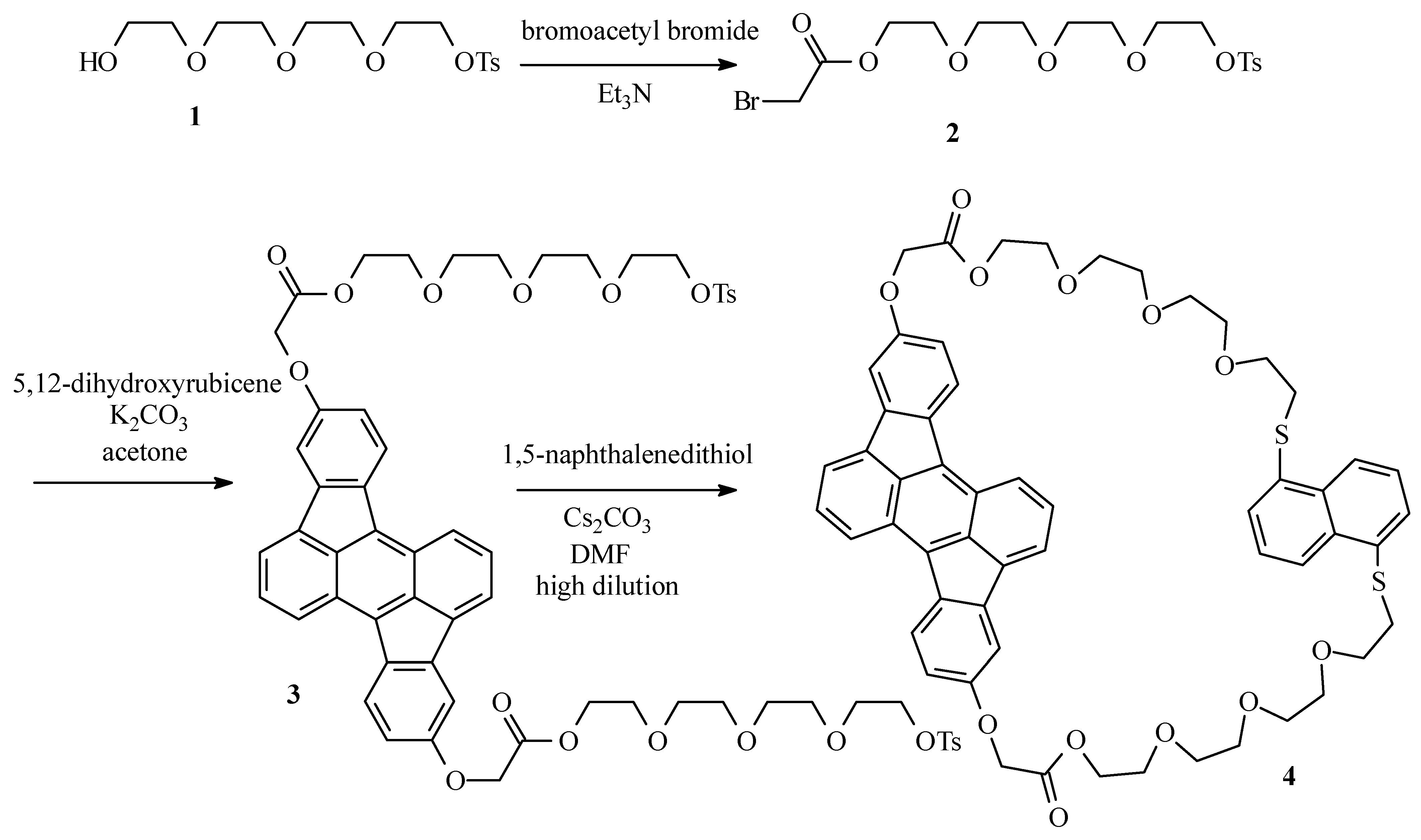
Bromoacetate 2
Tetraethyleneglycol monotosylate (1) (22g; 63 mol) and Et3N (9.6 g; 95 mmol) were dissolved in CH2Cl2 ( 130 mL) and placed under argon atmosphere. The mixture was cooled to -20°C. Through a septum, bromoacetyl bromide (19 g; 95 mmol) was added and the resulting dark suspension was left at room temperature for 1 h. The suspension was poured on crushed ice (ca. 150 g) and the mixture was washed with a solution of HCl (3 x 50 mL; 2 M) and water (2 x 50 mL). The organic layer was dried over MgSO4 and the solvent was evaporated in vacuum. The desired bromoacetate 2 was obtained as a light brown oil (24 g; 81%) after column chromatography (SiO2; CH2Cl2-ethyl acetate 1:1): 1H NMR (250 MHz, CDCl3) δ 7.78 (d, J = 8 Hz, 2H, HCCSO2), 7.33 (d, J = 8 Hz, 2H, HCCCSO2), 4.31 (m, 2H, CH2OTs), 4.14 (m, 2H, CH2OCOCH2Br), 3.89 (s, 2H, CH2Br), 3.72-3.56 (m, 12H), 2.43 (s, 3H, CH3); MS (EI) m/z: 468 (M+).
Ditosylate 3
5,12-Dihydroxyrubicene (0.25 g; 0.70 mmol), bromide 2 (0.82 g; 1.7 mmol) and K2CO3 (0.25 g; 1.7 mmol) were suspended in acetone (20 mL) and the resulting mixture was refluxed overnight under argon atmosphere. After cooling to room temperature, the solvent was evaporated in vacuum and the desired compound 3 was obtained as a dark red oil (0.65 g; 82%) after column chromatography (SiO2; CH2Cl2-ethyl acetate 1:1): 1H NMR (250 MHz, CDCl3) δ 8.01 (d, J = 8.5 Hz, 2H), 7.76 (d, J = 8.5 Hz, 2H), 7.73 (d, J = 8.5 Hz, 4H), 7.59 (d, J = 6 Hz, 2H), 7.39 (dd, J = 8.5 Hz, J = 6 Hz, 2H), 7.28 (d, J = 1.5 Hz, 2H), 6.81 (dd, J = 8.5 Hz, J = 1.5 Hz, 2H), 4.74 (s, 4H), 4.42 (t, J = 7 Hz, 4H), 4.10 (t, J = 7 Hz, 4H), 3.79-3.58 (m, 16H), 3.52 (s, 8H), 2.33 (s, 6H); 13C NMR (400 MHz, CDCl3) δ 168.87, 157.08, 144.64, 140.82, 137.08, 133.57, 132.87, 132.60, 131.72, 129.67, 127.94, 127.78, 124.31, 124.15, 123.66, 119.76, 113.33, 108.47, 71.02, 70.54, 70.44, 70.35, 69.12, 68.82, 68.51, 65.48, 64.23, 30.28, 21.42; MS (ES) (m/z): 1136 (M+).
Cyclophane 4
Ditosylate 3 (0.55 g; 0.48 mmol) and 1,5-naphthalenedithiol (92 mg; 0.48 mmol) were dissolved in DMF (50 mL). The resulting solution was added over a period of 24 h to a vigourously stirred suspension of Cs2CO3 (0.47 g; 1.4 mmol) in DMF (300 mL) at 70°C under argon atmosphere by means of an infusion pump. After complete addition, the mixture was stirred for another 12 h at 70°C. After cooling to room temperature, the solvent was evaporated in vacuum and the cyclophane 4 was obtained as a dark red oil (0.13 g; 27%) after column chromatography (SiO2; CH2Cl2-ethyl acetate 1:1): 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 8.6 Hz, 2H), 8.00 (d, J = 8.6 Hz, 2H), 7.98 (d, J = 7.6 Hz, 2H), 7.80 (d, J = 6.6 Hz, 2H), 7.58 (dd, J = 6.6 Hz, J = 8.5 Hz, 2H), 7.42 (d, J = 2.4 Hz, 2H), 7.25 (d, J = 7.6 Hz, 2H), 7.13 (t, J = 7.6 Hz, 2H), 6.88 (dd, J = 2.4 Hz, J = 8.5 Hz, 2H), 4.79 (s, 4H), 4.42-4.40 (m, 4H), 3.72-3.70 (m, 4H), 3.54-3.52 (m, 4H), 3.45-3.43 (m, 4H), 3.37-3.34 (m, 8H), 3.30-3.28 (m, 4H), 2.87-2.83 (m, 4H); 13C NMR (400 MHz, CDCl3) δ 169.11, 157.45, 141.07, 137.53, 133.85, 133.79, 133.05, 132.88, 132.20, 128.43, 127.35, 125.60, 124.70, 124.65, 123.98, 123.56, 120.24, 113.69, 108.79, 70.60, 70.42, 70.36, 70.06, 69.34, 68.80, 65.80, 64.45, 33.23; MS (ES) (m/z): 983 (M+).
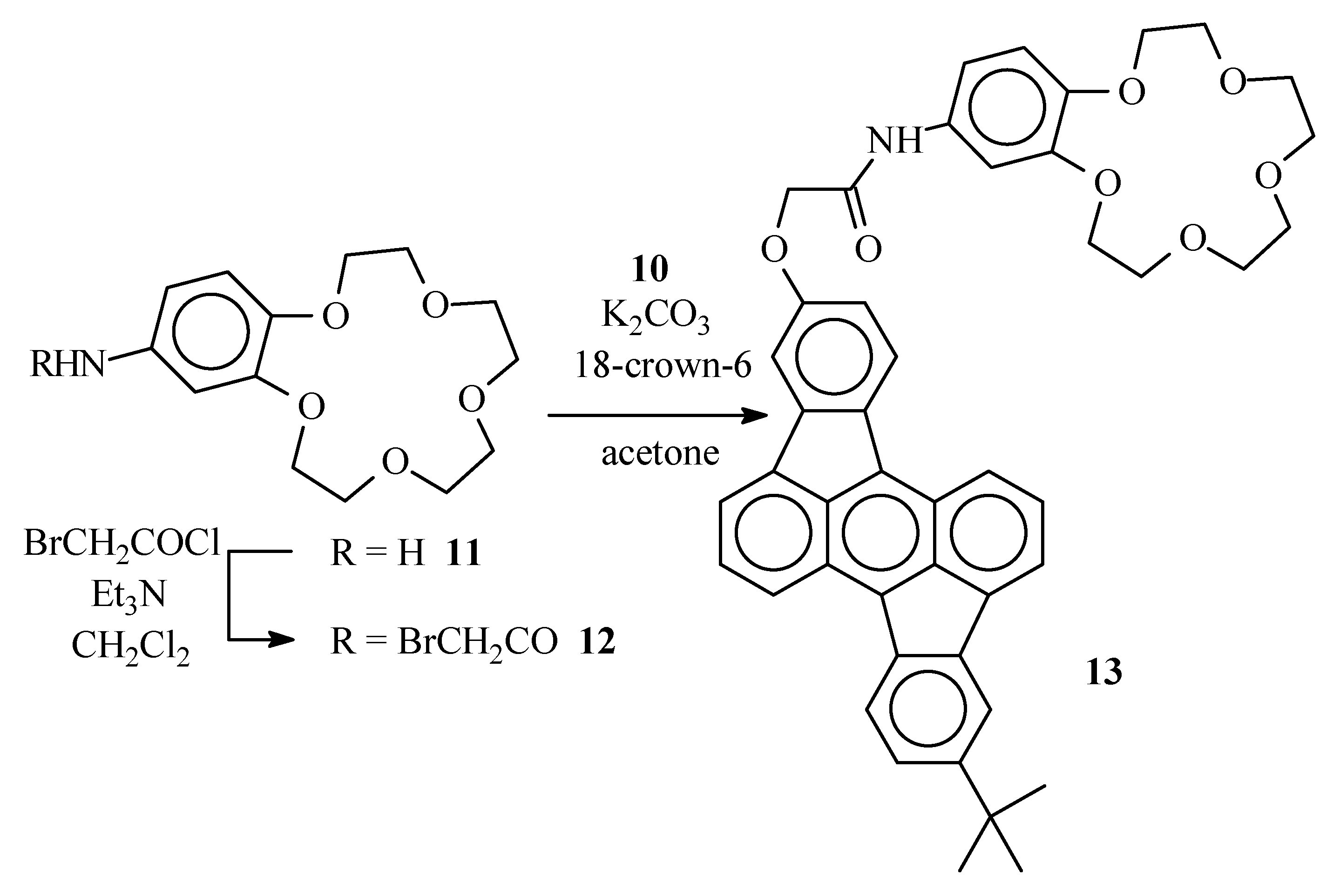
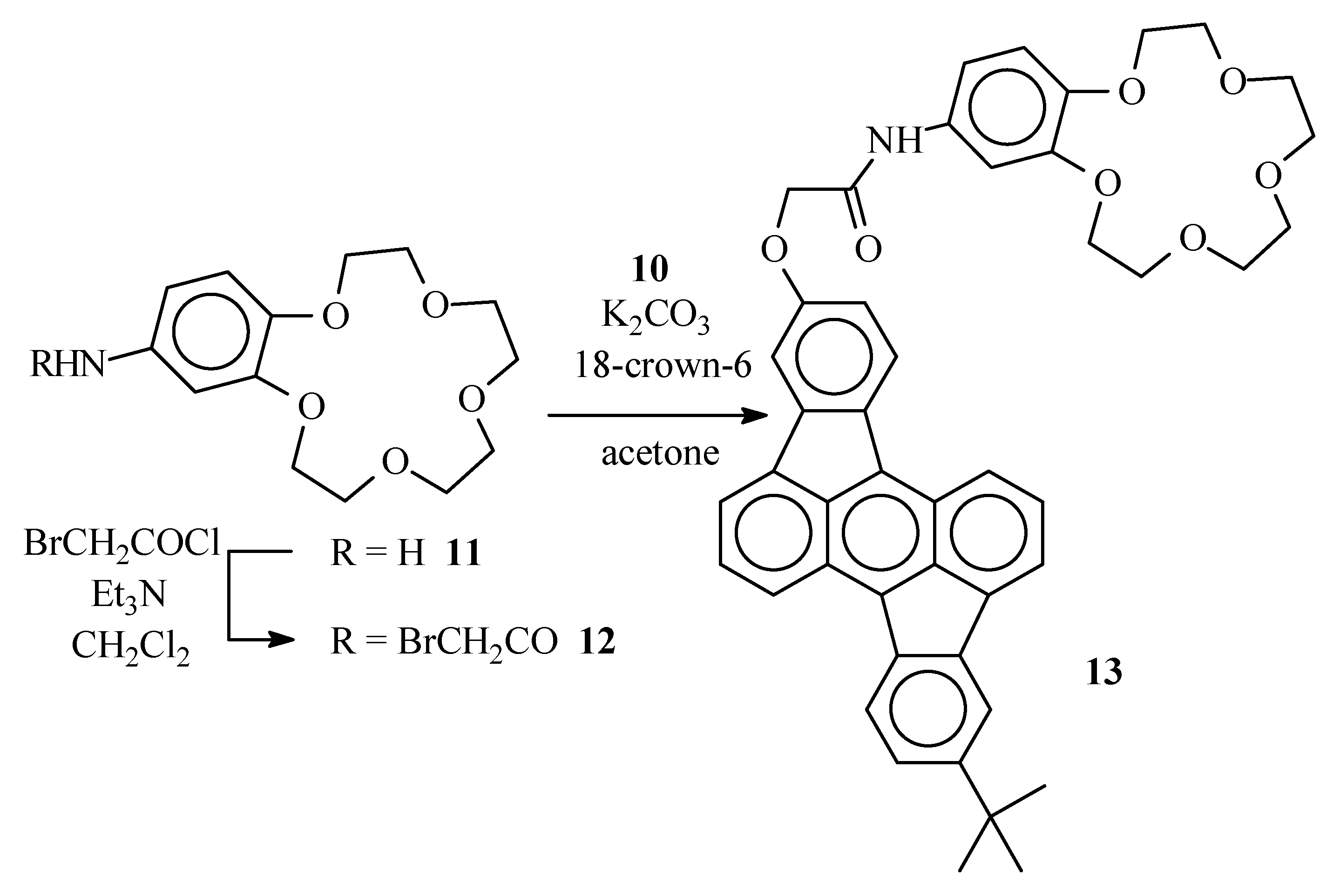
Bromoacetamide 12
A solution of 4-aminobenzo-15-crown-5 (11) (0.50 g, 1.8 mmol) and Et3N (1.1 g, 11 mmol) in CH2Cl2 (15 mL) was placed under argon and cooled to 0°C in an ice bath. Through a septum, bromoacetyl bromide (1.1 g, 5.3 mmol) was added and the resulting dark solution was left for 30 min at 0°C. The reaction mixture was poured on crushed ice (ca. 20 g). The organic layer was separated and washed with diluted (1N) HCl (2 x 20 mL) and water (20 mL). The organic layer was dried over MgSO4 and evaporated in vacuum. Bromoacetamide 12 was obtained after column chromatography (SiO2, EtOAc-CH3OH 1:1) as an amorphous solid (0.49g, 67%), νmax (KBr)/cm−1 3443 (NH), 1670 (C=O), 1631 (C=O) ca. 1100 (broad) (C-O); δH (400 MHz ; CDCl3) 3.73-3.79 (8 H, br m, Ph(O(CH2)2O(CH2)2)2O), 3.86-3.92 (4 H, br m, Ph(OCH2CH2O(CH2)2)2O), 4.07 (2 H, s, -CH2Br), 4.09 (4 H, br m, Ph(OCH2CH2O(CH2)2)2O), 6.76 (1 H, d, J 8.5, benzo 6-H), 7.04 (1 H, dd, J 1.5 and 8.5, benzo 5-H), 7.30 (1 H, d, J 1.5, benzo 3-H), 8.56-8.63 (1 H, br s, NH) ; δC (100 MHz ; CDCl3) 163.8, 148.5, 145.6, 131.6, 114.1, 113.0, 106.9, 70.39, 70.38, 69.94, 69.93, 69.19, 68.99, 68.98, 68.42, 29.78; m/z 405 (M+, 20%), 403 (M+, 20), 273 (49), 271 (49), 151 (100).
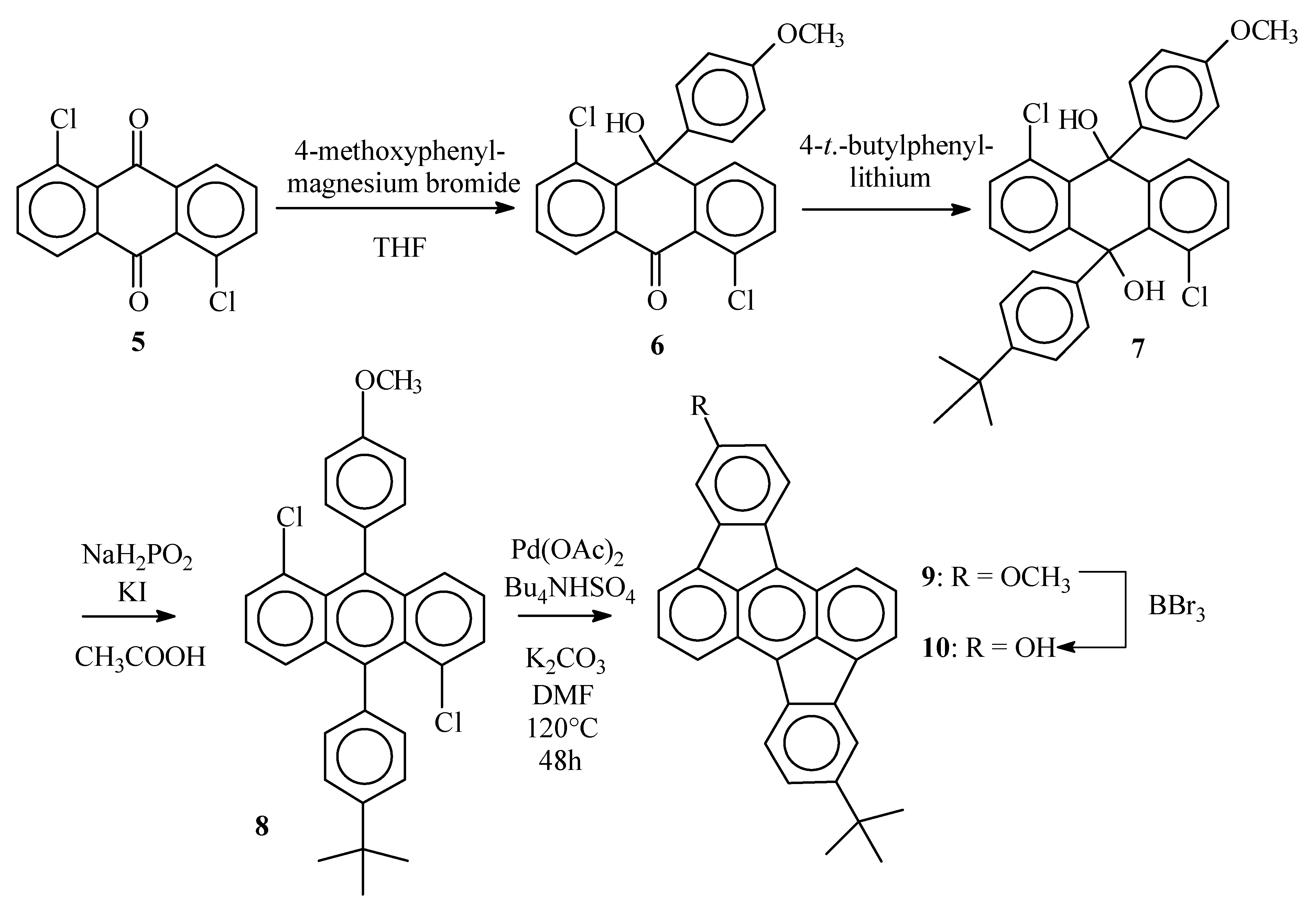
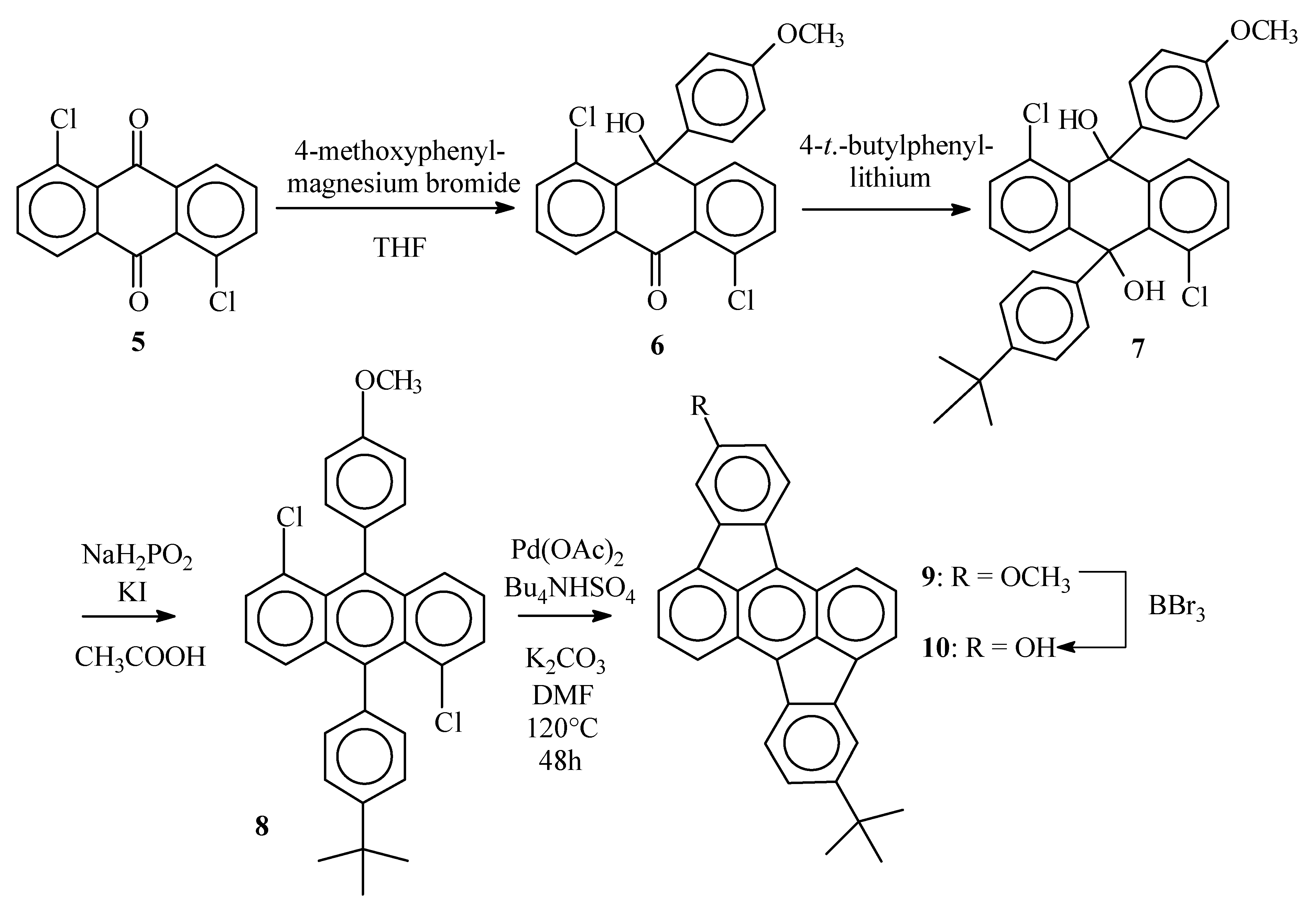
1,5-Dichloro-10-hydroxy-10’-(4-methoxyphenyl)(10H)anthracene–9-one (6)
1,5-Dichloroanthraquinone (5) (7.0 g, 25 mmol) was suspended in dry THF (200 mL) and placed under argon. A solution of 4-methoxyphenylmagnesium bromide was prepared from 4-bromoanisol (5.7 g, 28 mmol) and magnesium turnings (0.73 g, 30 mmol) in dry THF (25 mL) and added dropwise (during 30 min) to the anthraquinone suspension. After the addition was complete, the resulting clear solution was stirred for 12 h at room temperature. Water was added (100 mL) and after vigorous shaking, the organic layer was separated. The water layer was extracted with CH2Cl2 (2 x 80 mL). The combined organic layers were dried over MgSO4 and evaporated in vacuum. To the pasty residue, toluene (150 mL) was added and the resulting suspension was refluxed for 15 min. The hot solution was filtered and the precipitate purified by column chromatography (SiO2) with CH2Cl2/petroleum ether (4 :1) as the eluent. The title compound was obtained as a white solid (4.62 g, 48%), mp 199°C ; νmax (KBr)/cm−1 3445 (OH), 3078 (sp2 C-H), 2927 (sp3 C-H), 1649 (C=O), 1580 (C=C); δH (400 MHz ; DMSO d6) 3.67 (3 H, s, OCH3), 6.79 (2 H, d, J 9, phenyl 3,5-H), 6.96 ( 1H, s, OH), 7.21 (2 H, d, J 9, phenyl 2,6-H), 7.47 (1 H, dd, J 7.7 and 1.3, 2-H anthrone), 7.56 (1 H, t, J 7.7, 7-H anthrone), 7.56 (1 H, t, J 7.7, 3-H anthrone), 7.63 (1 H, dd, J 7.7 and 1.3, 4-H anthrone), 7.68 (1 H, dd, J 7.7 and 1.3, 6-H anthrone), 8.16 (1 H, dd, J 7.7 and 1.3, 8-H anthrone) ; δC (100 MHz ; DMSO d6) 181.5, 157.6, 153.4, 142.2, 136.7, 136.2, 134.2, 133.4, 133.3, 131.9, 130.6, 129.4, 128.6, 126.7, 125.6, 124.0, 113.4, 71.6, 54.9; m/z 386 (M+, 55), 387 (M+, 21), 384 (M+, 82), 351 (23), 349 (63), 333 (31), 332 (21), 331 (82), 279 (66), 277 (100).
1,5-Dichloro-9,10-dihydroanthracene-9-(4-(1,1-dimethylethyl)phenyl)-10-(4-methoxy-phenyl)-9,10- diol (7)
1-Bromo-4-(1,1-dimethylethyl)benzene (1.1 g, 5.2 mmol) was dissolved in dry THF (10 mL). The solution was placed under argon and cooled to 0°C. BuLi (2.1 mL 2.5 M solution in hexanes, 5.2 mmol) was added through a septum and the mixture was allowed to warm to room temperature. The anthrone 6 (0.50 g, 1.3 mmol) was added and the resulting suspension was stirred overnight. The reaction mixture was quenched with water (20 mL) and extracted with CH2Cl2 (3 x 15 mL). The combined organic layers were dried over MgSO4 and the solvent was evaporated in vacuum. The title compound was obtained after column chromatography (SiO2, CH2Cl2) as a white solid (0.57 g, 85%), δH (250 MHz ; CDCl3) 1.23 (9 H, s, C(CH3)3), 3.72 (3 H, s, OCH3), 4.48 (1 H, s, OH), 4.53 (1 H, s, OH), 6.73 (2 H, d, J 8, CHCOCH3), 7.15-7.35 (10 H, m), 7.88-7.93 (2 H, 2 x dd, J 7.5 and 1.3, 4-H and 8-H anthracenediol) ; δC (100 MHz ; CDCl3) 158.4, 149.8, 143.8, 142.9, 142.6, 139.2, 134.2, 134.1, 132.5, 130.7, 130.6, 129.1, 127.8, 127.7, 127.2, 125.6, 125.3, 125.1, 124.9, 113.3, 74.3, 74.1, 55.1, 34.3, 31.3.
5-(1,1-Dimethylethyl)-12-methoxyrubicene (9)
The diol 7 (0.57 g, 1.1 mmol), KI (0.91 g, 5.5 mmol) and NaH2PO2 (0.48 g, 5.5 mmol) were suspended in acetic acid (6 mL) and the stirred mixture was heated to reflux for 1 h. The resulting yellowish suspension was allowed to cool to room temperatue and the solids were collected by filtration, washed with water (2 x 5 mL) and methanol ( 2 x 5 mL) and dried in vacuum. The desired anthracene 8 (0.39 g, 73%) thus obtained was used without further purification, m/z 487 (24%), 486 (72), 485 (37), 484 (100), 393 (33), 358 (27), 357 (26), 57 (44). This anthracene 8 ( 0.39 g, 0.80 mmol), K2CO3 (0.98 g, 7.0 mmol), Bu4NHSO4 (0.62 g, 1.6 mmol) and Pd(OAc)2 (38 mg, 0.16 mmol) were suspended in DMF (15 mL) and the stirred mixture was heated at 120°C under argon for 24 h. After cooling to room temperature, water (20 mL) was added and the precipitate was collected by filtration. Rubicene 9 was obtained after Soxhlet extraction (toluene) as dark red crystals (0.30 g, 91%), mp > 300°C ; νmax (KBr)/cm−1 3067 (sp3 C-H), 2957 (sp2 C-H), 1614 (C=C), 1458, 1097 (C-O), 1025 (C-O); δH (400 Mhz ; CDCl3) 1.48 (9 H, s, C(CH3)3), 3.98 (3 H, s, OCH3), 6.98 (1H, dd, J 8.3 and 2.2, 13-H), 7.49 (1 H, dd, J 8.0 and 2.0, 6-H), 7.53 (1 H, d, J 2.2, 11-H), 7.73-7.77 (2 H, 2 x t, J 8.5 and 6.7, 2-H and 9-H), 7.98 (1 H, d, J 6.7, 3-H or 10-H), 8.02 (1 H, d, J 2.0, 4-H), 8.03 (1 H, d, J 6.7, 3-H or 10-H) 8.20 (1 H, d, J 8.3, 14-H), 8.22 (1 H, d, J 8.0, 7-H), 8.52 (1 H, d, J 8.5, 1-H or 8-H), 8.58 (1 H, d, J 8.5, 1-H or 8-H); m/z 413 (35%), 412 (100), 398 (26), 397 (76).
5-(1,1-Dimethylethyl)-12-hydroxyrubicene (10)
Rubicene 9 (0.15 g, 0.36 mmol) was dissolved in CH2Cl2 (15 mL) and the resulting solution was cooled to 0°C. Through a septum, BBr3 was added (0.55 mL 1M solution in CH2Cl2, 0.55 mmol). The mixture was stirred overnight at room temperature and then poured on crushed ice (ca. 10 g). The organic layer was separated and evaporated. Methanol (5 mL) was added to the residue and the precipitate was collected by filtration, yielding the desired compound 10 as a dark red amorphous solid (0.12 g, 84%), mp > 300°C, δH (400 MHz ; DMSO d6) 1.44 (9 H, s, C(CH3)3), 6.91 (1 H, dd, J 8.2 and 2.3, 13-H), 7.51 (1 H, dd, J 8.0 and 2.0, 6-H), 7.54 (1 H, d, J 2.3, 11-H), 7.85 (1 H, t, J 8.8 and 6.6, 2-H or 9-H), 7.86 (1 H, t, J 8.8 and 6.6, 2-H or 9-H), 8.18 (1 H, d, J 6.6, 3-H or 10-H), 8.20 (1 H, d, J 2.0, 4-H), 8.30 (1 H, d, J 8.2, 14-H), 8.31 (1 H, d, J 6.6, 3-H or 10-H), 8.37 (1 H, d, J 8.0, 7-H), 8.68 (1 H, d, J 8.8, 1-H or 8-H), 8.71 (1 H, d, J 8.8, 1-H or 8-H), 9.75-9.88 (1 H, br s, OH) ; δC (100 MHz ; DMSO d6) 157.8, 149.8, 140.9, 138.5, 137.6, 137.5, 136.4, 133.4, 132.3, 131.9, 130.8, 130.3, 129.4, 129.1, 125.2, 124.85, 124.78, 124.61, 124.60, 123.8, 123.0, 121.1, 102.9, 119.1, 115.0, 109.8, 34.8, 31.2 ; m/z 398 (28%), 383 (29), 73 (54), 44(100), 43 (34).
Crown ether derivative 13
Hydroxyrubicene 10 (0.11 g, 0.28 mmol), bromoacetamide 12 (0.13 g, 0.33 mmol) and K2CO3 (92 mg, 0.66 mmol) were suspended in acetone (4 mL) and the mixture was placed under argon. The stirred suspension was maintained at reflux temperature for 72 h. After cooling to room temperature, the reaction mixture was poured in water (5 mL) and extracted with CH2Cl2 (2 x 5 mL). The organic layers were combined and dried over MgSO4. After column chromatography (SiO2) with EtOAc/CH3OH 1:1 as the eluent, the desired crown ether 13 was obtained as an amorphous solid (79 mg, 39%) (Found: C, 76.65, H, 5.95. C46H43NO7 requires C: 76.54; H: 6.00%); δH (400 MHz ; CDCl3) 1.48 (9 H, s, C(CH3)3), 3.72-3.74 (8 H, br m, Ph(O(CH2)2O(CH2)2)2O), 3.89 (4 H, br m, Ph(OCH2CH2O(CH2)2)2O), 4.12-4.16 (4 H, br m, Ph(OCH2CH2O(CH2)2)2O), 4.71 (2 H, s, OCH2CON), 6.81 (1 H, d, J 8.4, 6-H benzo), 6.97 (1 H, d, J 8.2, 13-H rubicene), 7.08 (1 H, d, J 8.4, 5-H benzo), 7.42 (1 H, s, 3-H benzo), 7.47 (1 H, d, J 8.1, 6-H rubicene), 7.50 (1 H, s, 11-H rubicene), 7.66 (2 H, br m, 2-H and 9-H rubicene), 7.88 (1 H, d, J 6.6, 3-H or 10-H rubicene), 7.92 (1 H, d, J 6.6, 3-H or 10-H rubicene), 7.96 (1 H, s, 4-H rubicene), 8.07 (1 H, d, J 8.2, 14-H rubicene), 8.13 (1 H, d, J 8.1, 7-H rubicene), 8.35 (1 H, d, J 8.3, 1-H or 8-H rubicene), 8.47 ( 1H, d, J 8.3, 1-H or 8-H rubicene); m/z 721 (M+ ; 5%), 664 (48), 663 (100).

{kind=link}
{kind=link}
{kind=link}