1H and 13C NMR spectroscopy
The chemical shifts of compounds
9-
11 are reported in
Table 1 (
1H NMR) and
Table 2 (
13C NMR). The assignments have been made by
1H-
1H and
1H-
13C bidimensional experiments. Although the spectrum of the parent biacridine
7 has been recorded in CDCl
3 [
14], it is possible to calculate the Substituent Chemical Shifts (SCS) of compounds
9-
11 and compare them with those reported by Ewing for monosubstituted benzenes [
18].
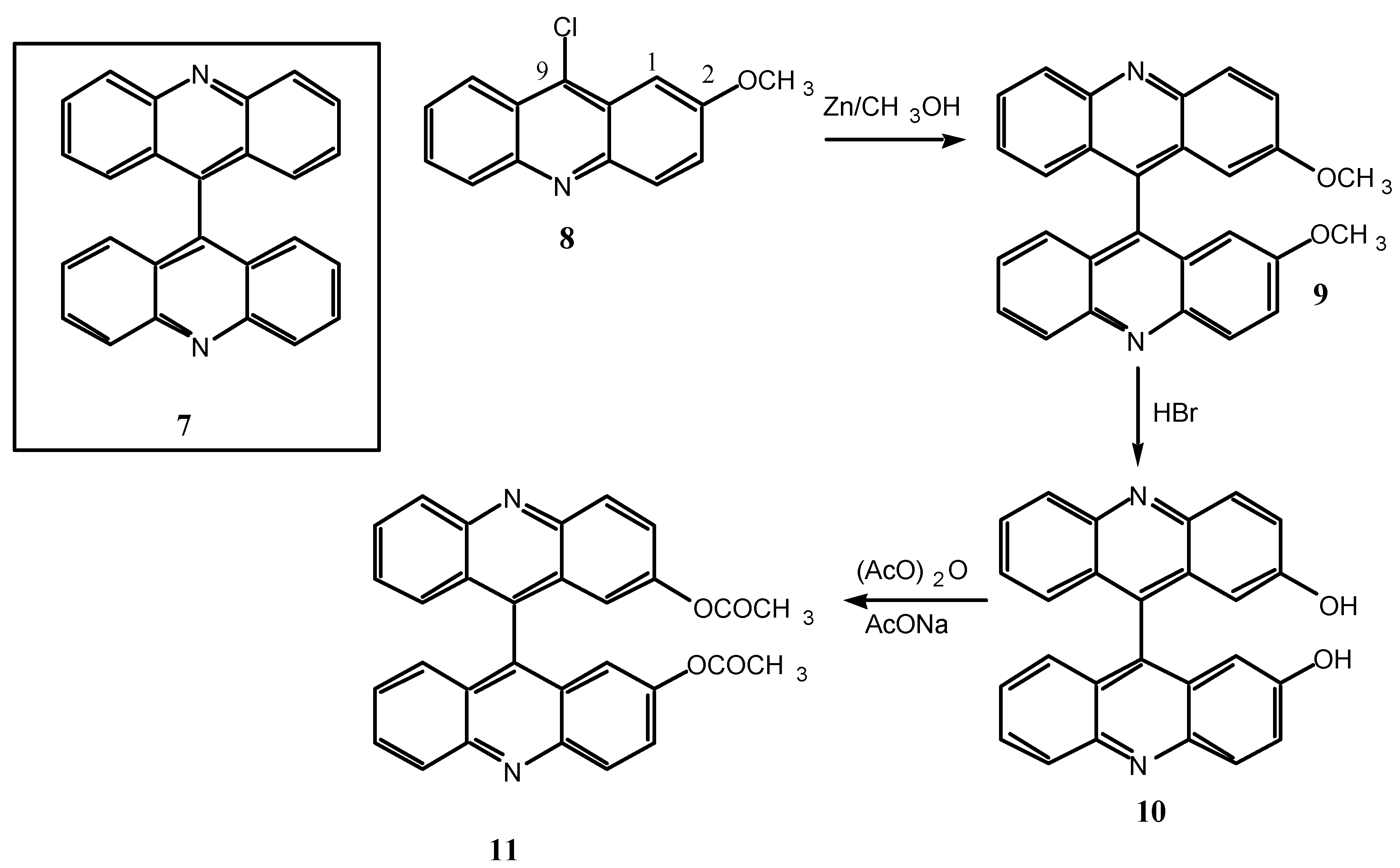
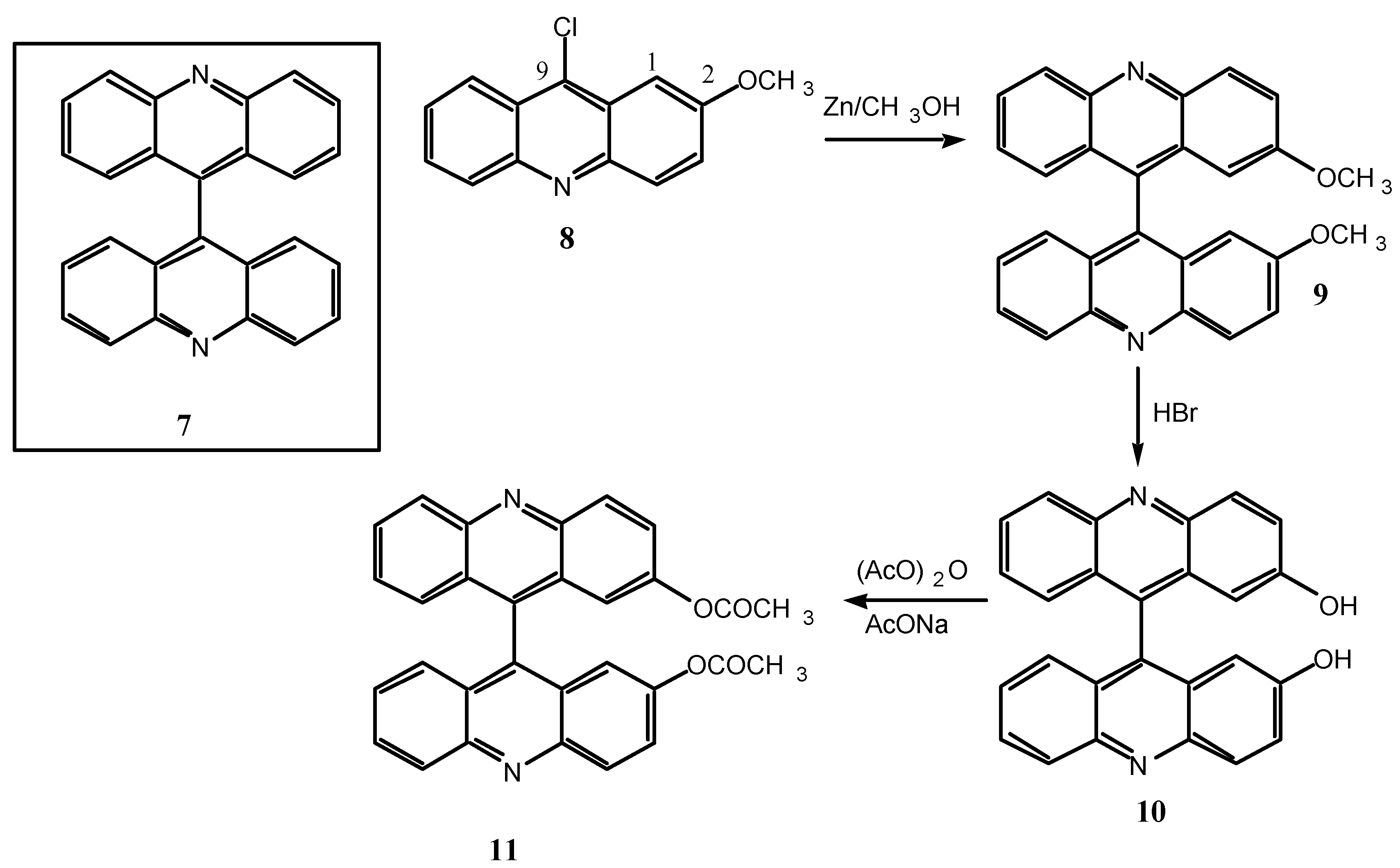
Scheme 4.
9, R= OCH3; 10, R= OH, 11, R= OCOCH3.
Scheme 4.
9, R= OCH3; 10, R= OH, 11, R= OCOCH3.
Table 1.
1H chemical shifts (in ppm) and 1H-1H coupling constants (in Hz) in DMSO-d6.
Table 1.
1H chemical shifts (in ppm) and 1H-1H coupling constants (in Hz) in DMSO-d6.
| No | 9 | 10 | 11 |
| H1 | 6.18, 4J=2.5 | 6.17, 4J=2.0 | 6.76 |
| H3 | 7.59, 3J=9.4, 4J=2.5 | 7.49, 3J=9.4, 4J=2.0 | 7.73, 3J=9.4 |
| H4 | 8.29, 3J=9.4 | 8.25, 3J=9.4 | 8.40, 3J=9.4 |
| H5 | 8.31, 3J=8.5 | 8.29, 3J=8.8 | 8.36, 3J=8.8 |
| H6 | 7.79, 3J=7.8, 3J=8.5 | 7.76, 3J=7.4, 3J=8.8 | 7.83, 3J=7.5, 3J=8.8 |
| H7 | 7.35, 3J=7.8, 3J=8.5 | 7.34, 3J=7.4, 3J=8.6 | 7.41, 3J=7.5, 3J=8.6 |
| H8 | 6.92, 3J=8.5 | 6.95, 3J=8.6 | 7.00, 3J=8.6 |
| R | 3.33 (OCH3) | 9.96 (OH) | 2.07 (OCOCH3) |
Table 2.
13C chemical shifts (in ppm) in DMSO-d6.
Table 2.
13C chemical shifts (in ppm) in DMSO-d6.
| No | 9 | 10 | 11 |
| C1 | 101.13 | 103.93 | 115.56 |
| C2 | 157.35 | 155.75 | 148.81 |
| C3 | 125.14 | 125.88 | 127.72* |
| C4 | 131.76 | 131.76 | 131.56 |
| C4a | 145.19 | 145.27 | 147.15 |
| C5 | 129.80 | 129.89 | 129.98 |
| C6 | 129.40 | 129.10 | 130.81 |
| C7 | 127.18 | 127.07 | 127.81* |
| C8 | 125.28 | 125.33 | 125.70 |
| C8a | 125.28 | 125.26 | 125.28 |
| C9 | 137.52 | 137.04 | 139.50 |
| C9a | 125.87 | 126.56 | 125.28 |
| C10a | 146.61 | 146.38 | 146.55 |
| R | 56.02 (CH3) | ----- | 169.21 (CO) |
| | | | 20.84 (CH3) |
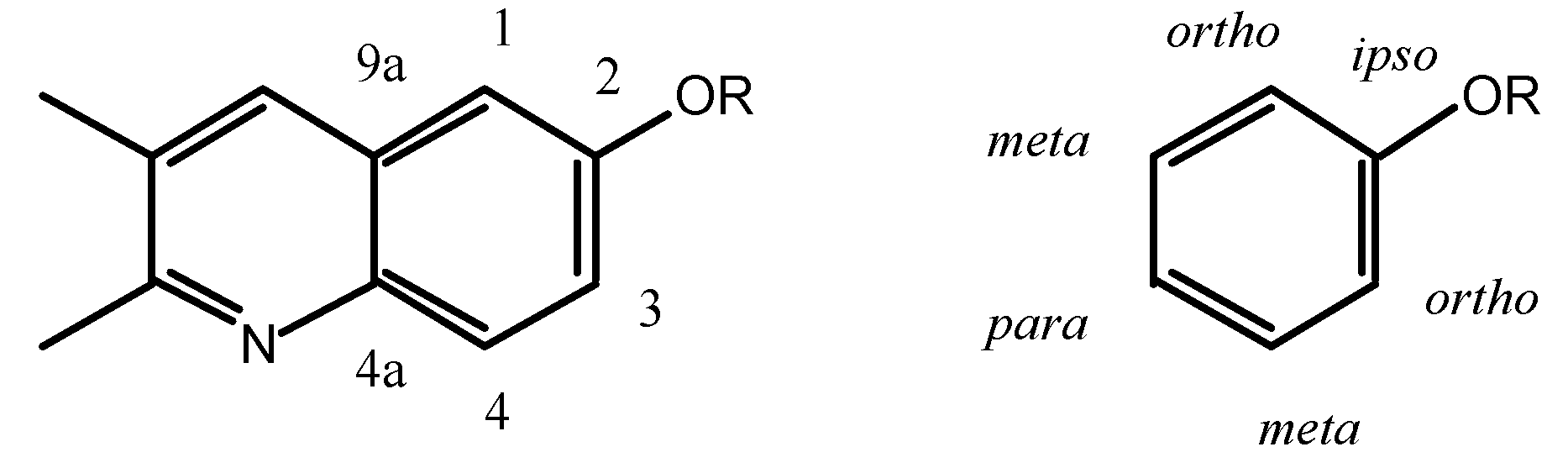
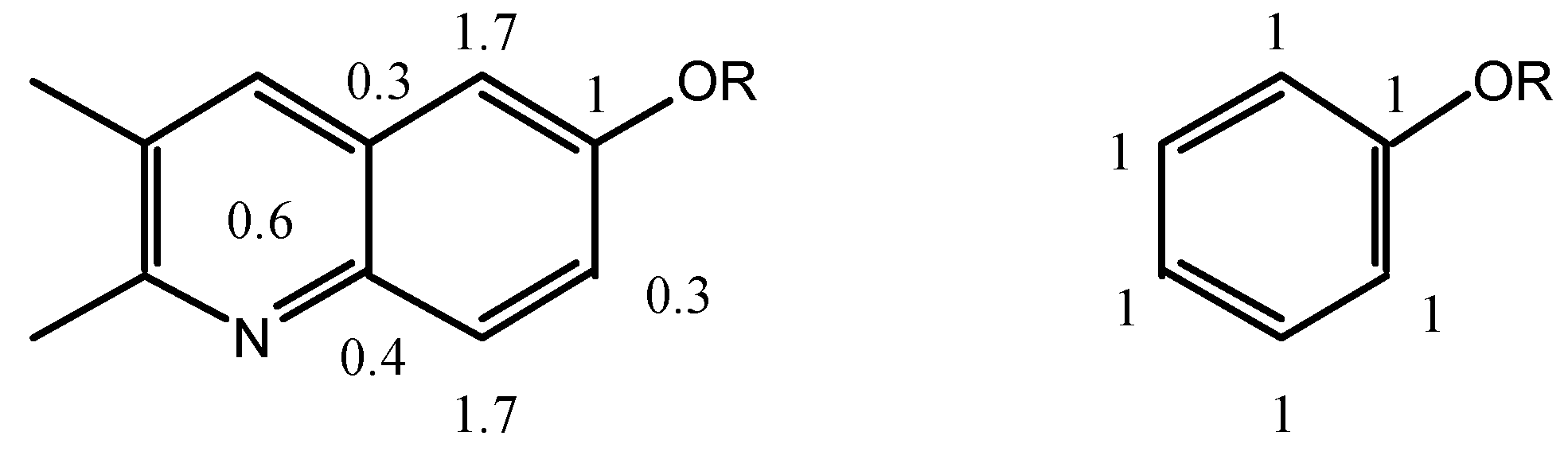
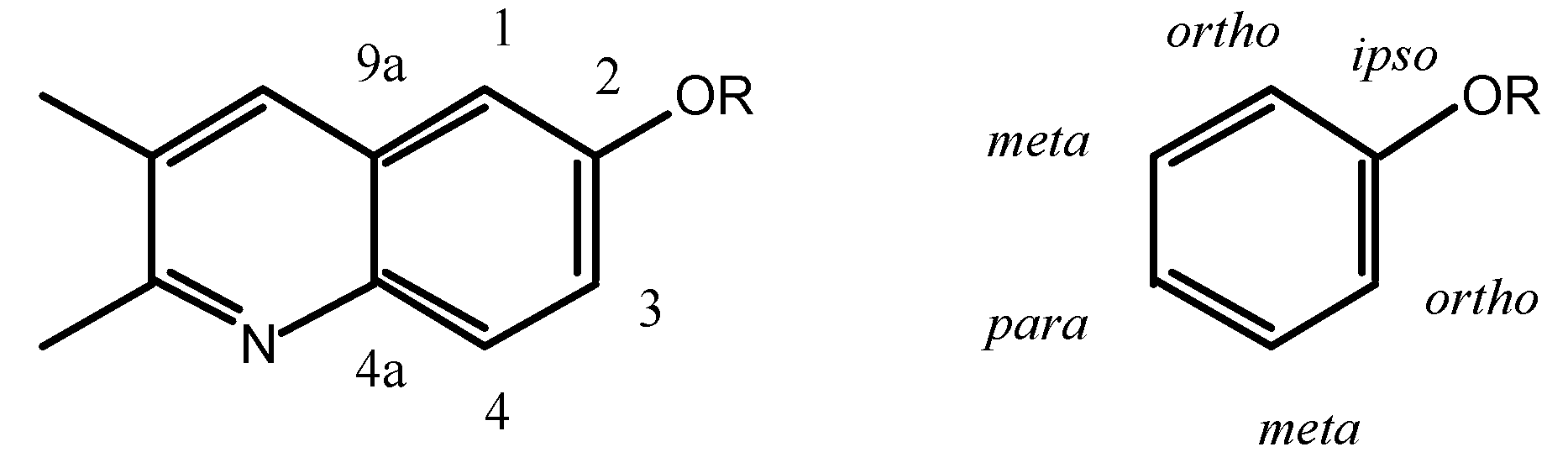
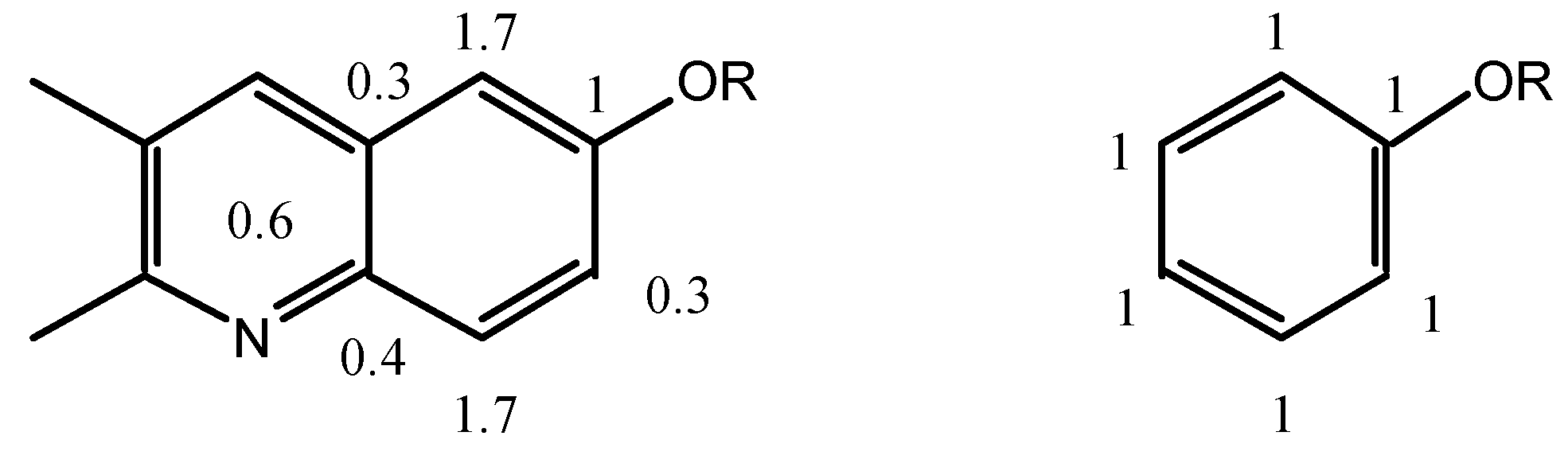
The
ortho and
meta positions of biacridines have to be treated separately. Even if the number of points (n = 3) is clearly insufficient, the following equations are obtained assuming that there is no intercept (SCS = 0 for the unsubstituted compounds):
The meta carbons (C-4 and C-9a) are not sensitive enough to SCS effects and the correlations are not good, nevertheless the effects on C-4 (slope 1.7) are larger than on C-9a (slope 0.3) and more similar to those observed in benzenes. If we make the assumption that in benzenes all the SCS effects correspond to a slope of 1 (total 6) and that in biacridines the overall effect is the same (total 6), then we can represent the two cases as follows:
The fusion to the heterocyclic moiety produces a localization of the π system in the sense indicated in the above resonance structure for 9-11 (the perturbation extends to this moiety which could account for the remaining 0.6).
1H NMR experiments with lanthanide shifts reagents (LSR)
The synthesis used to prepare
9 must result in a racemic mixture and, since racemization of this kind of compounds is highly improbable, the same should happen to
10 and
11. We carried out some experiments to find out if
1H NMR in the presence of either LSR* or Pirkle's alcohol [
19] could be used to determine the enantiomeric composition in the special case of axial chirality.
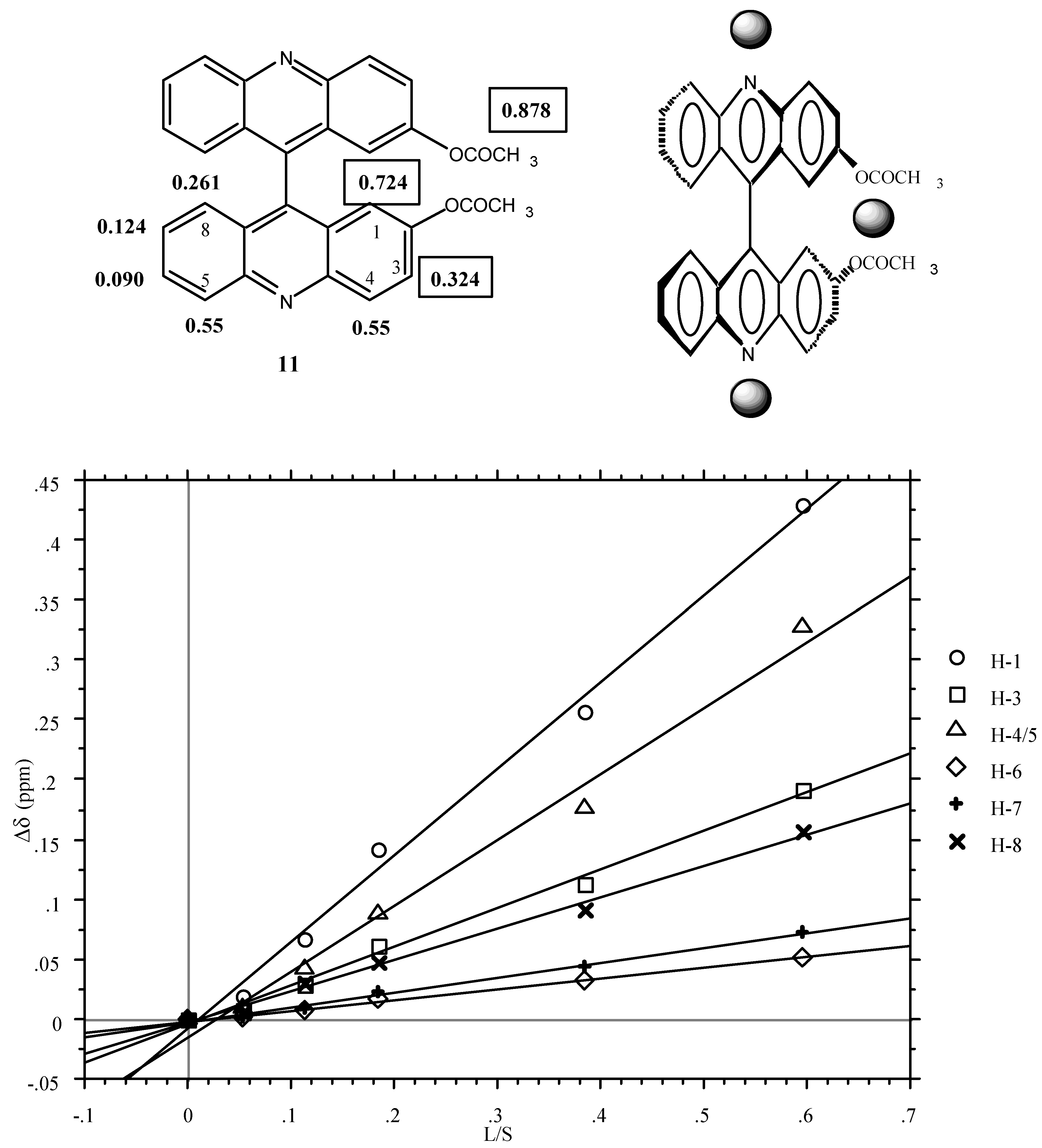
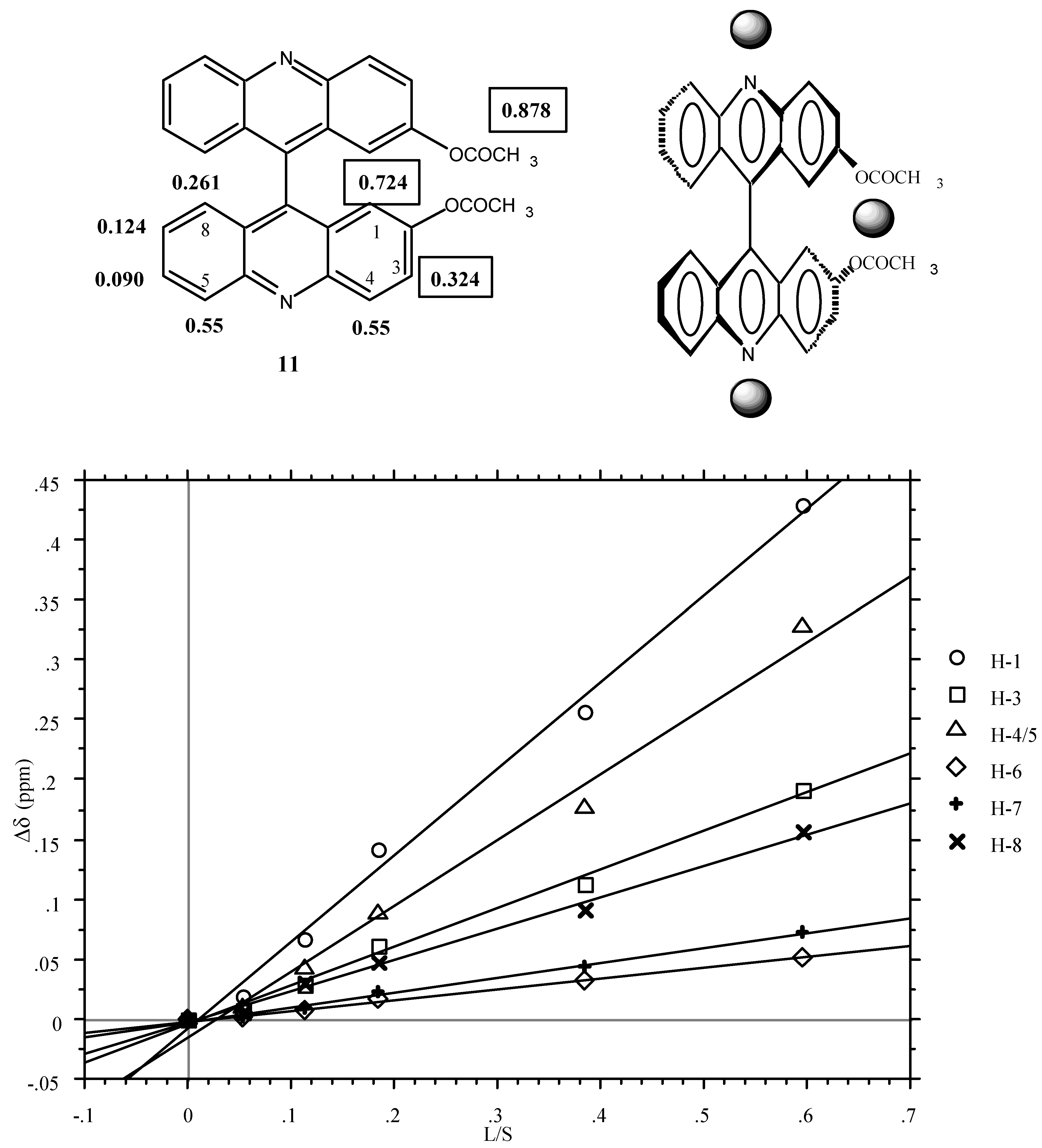
The LSR experiments were carried out on the diacetyl derivative
11. First the achiral Eu(fod)
3 reagent was used in order to find which were the most sensitive signals. The result is represented graphically in
Figure 1 (LIS, Δδ in ppm versus lanthanoid-substrate molar ratios) and the corresponding equations are in the Experimental part. The slopes are given below:
Figure 1.
Plot of Δδ [δ(11)+Eu(fod)3] - δ(11)] versus the molar ratio [Lanthanide Eu(fod)3]/ [Substrate (11)] in 0.2 mL of CDCl3 (the CH3 signal of the acetoxy group is not shown).
Figure 1.
Plot of Δδ [δ(11)+Eu(fod)3] - δ(11)] versus the molar ratio [Lanthanide Eu(fod)3]/ [Substrate (11)] in 0.2 mL of CDCl3 (the CH3 signal of the acetoxy group is not shown).
In a first approximation (for quantitative analysis see ref. [
20]), the LSR (grey spheres) occupies two positions, one near the acetoxy groups and the other near the pyridine nitrogen atoms.
The use of the chiral LSR reagent [LSR*] Eu(tfc)3 (see experimental part) produces a splitting of two signals those corresponding to H1 and (using larger concentrations) to H3. As expected, the intensities of both signals are the same. The fact that the phenomenon affects H1 and H3 but not H5 points out that it is the LSR* located near the acetoxy groups which is responsible for the diasterotopicity.
1H NMR experiments with R-Pirkle's alcohol in CDCl3
With
R-Pirkle's alcohol [
19], preliminary results (see Experimental Section) show that the methyl (Δδ = 0.105 ppm), H4 (Δδ = 0.024 ppm) and H6 (Δδ = 0.011 ppm) signals of compound
11 split (Δδ is the value of the splitting). For small amounts of the reagent, the two methyl group singlets are the most useful for determining possible enantiomeric excesses.
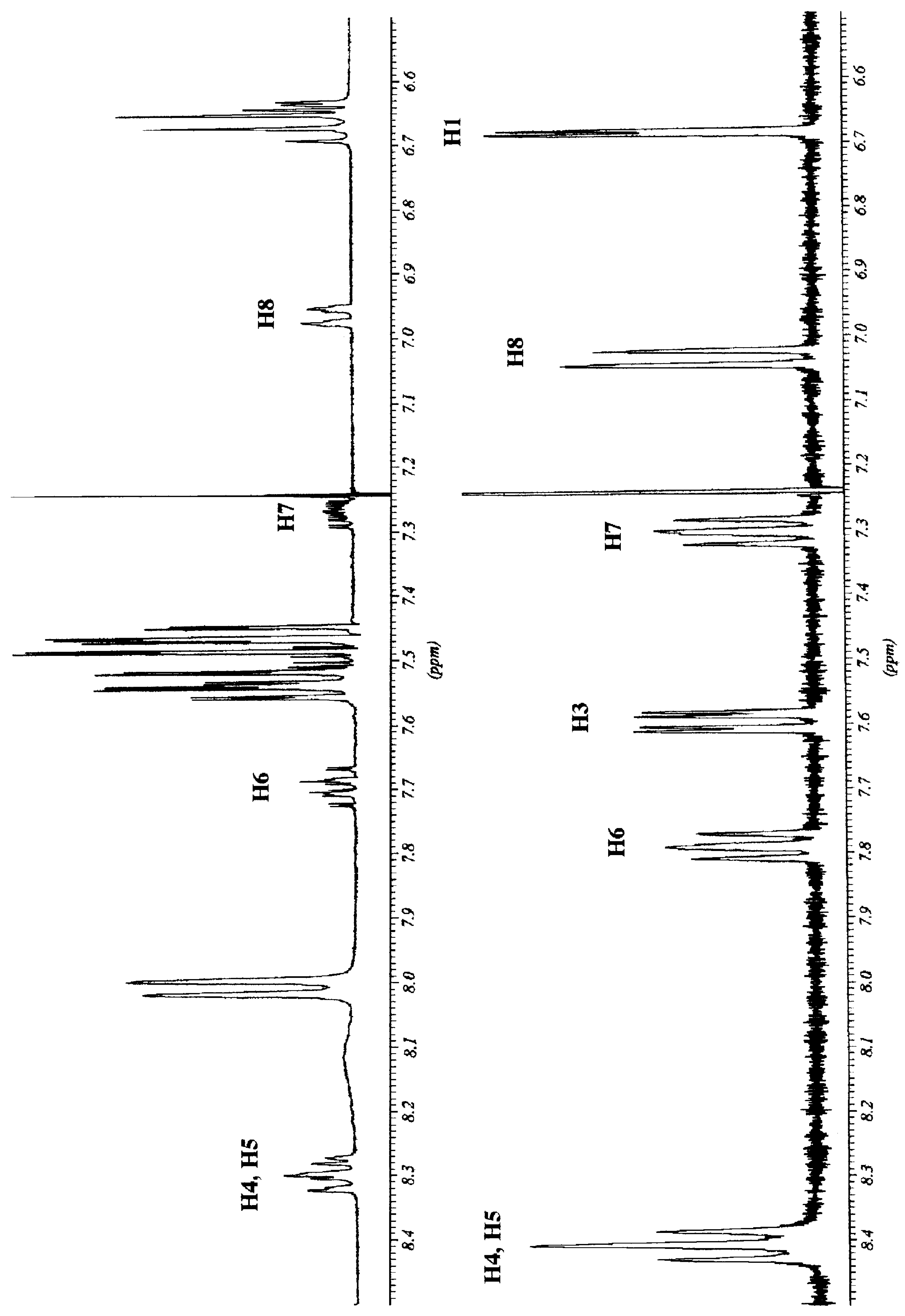
We decided to examine in detail the behaviour of
11, chiral by virtue of a "chiral axis", in the presence of
R-Pirkle's alcohol. In
Figure 2 the aromatic part of the
1H NMR spectra of compound
11 with and without
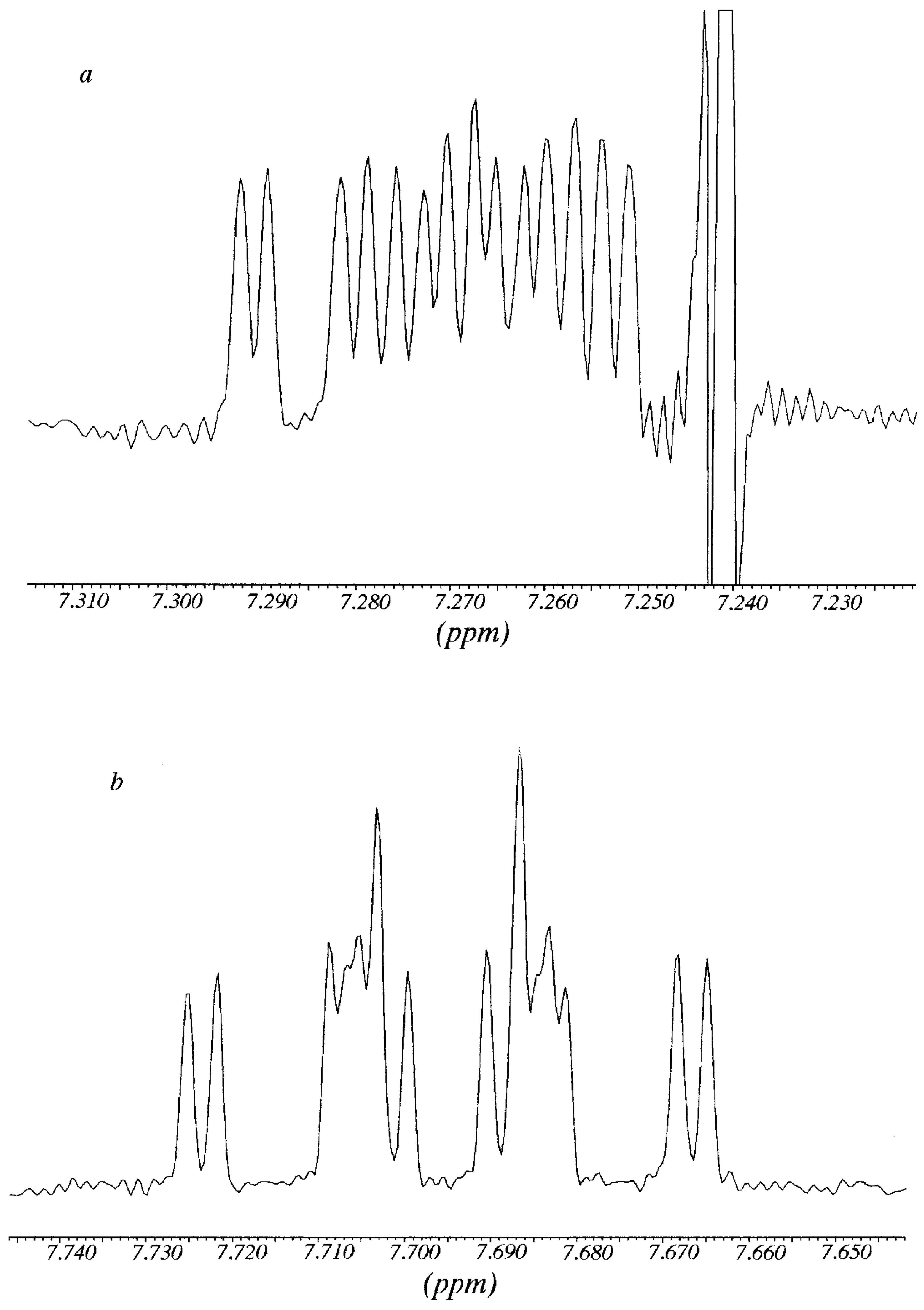
R-Pirkle's alcohol are represented. The splitting affects the methyl group, H4, H6 and H7 aromatic protons. Since H4 and H5 almost overlapped, the analysis was carried out on the signals of H6 and H7 protons.
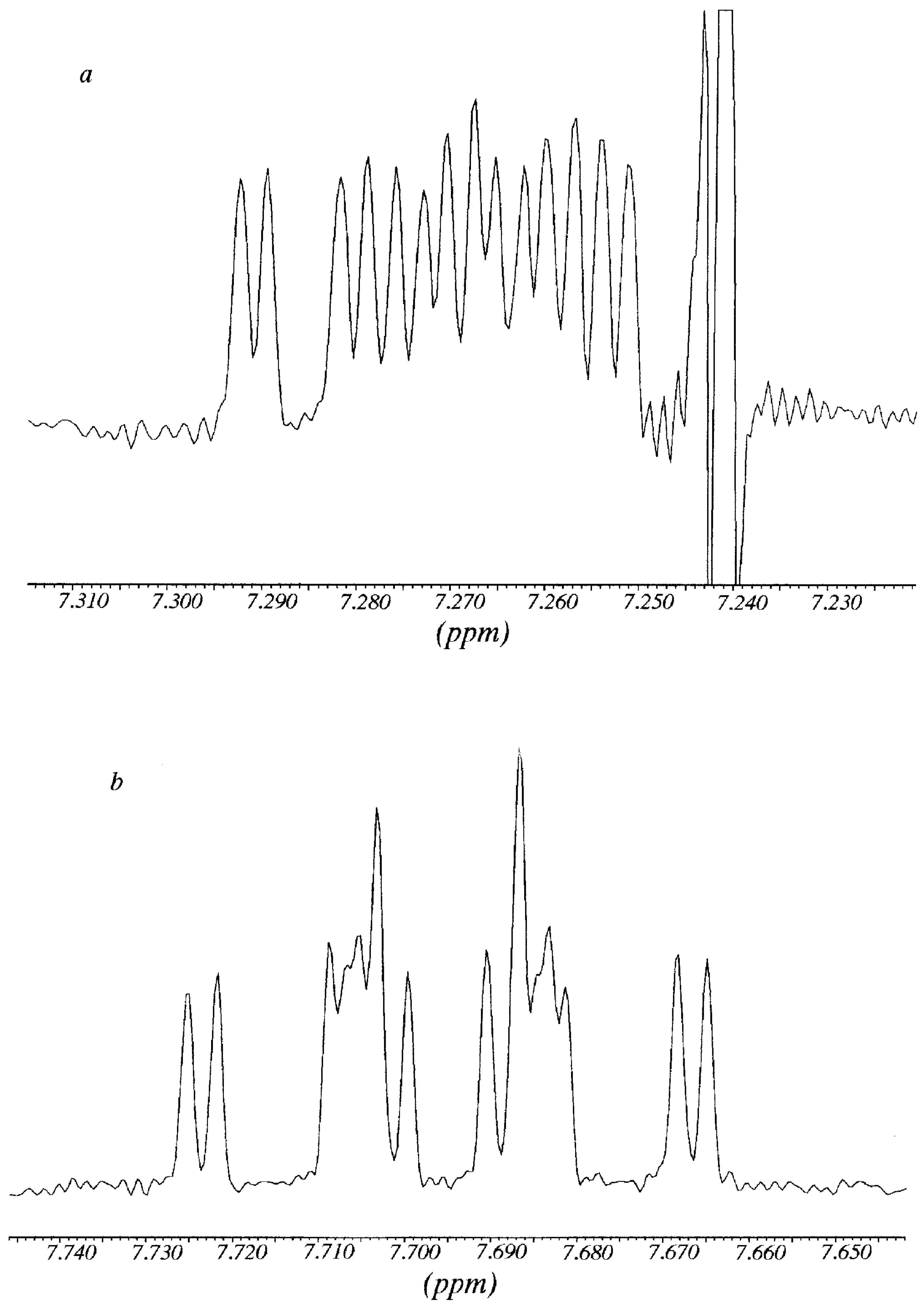
The signal of H6 at 7.792 ppm appeared as an overlapped double doublet that by deconvolution analysis showed a
3J = 8.9 Hz (with H5) and
3J = 6.65 Hz (with H7); in the same way, from the signal of H7 at 7.304 ppm it was possible to determine a
3J = 8.4 Hz (with H8) and a
3J ≈ 6.65 Hz (with H7). After addition of
R-Pirkle's reagent, protons H6 and H7 gave rise to sixteen signals (
Figure 3). Each signal was decomposed and simulated as the addition of
two double double doublets (
3J
ortho+
3J
ortho+
4J
meta), with the same coupling constants that in absence of Pirkle's alcohol plus a
4J
meta = 1.1 Hz (not observed in the free
11), obtaining two chemical shifts for each one: H7 7.262 and 7.272 (
Figure 3a); H6 7.686 and 7.704 (
Figure 3b). A possible explanation of why the effect of Pirkle's alcohol is observed mainly on H6 and H7, is that these two "external" protons are the most accessible to the chiral reagent.
Figure 2.
1H NMR spectra (400 MHz) of compound 11 before (bottom) and after addition of R-Pirkle`s alcohol (top).
Figure 2.
1H NMR spectra (400 MHz) of compound 11 before (bottom) and after addition of R-Pirkle`s alcohol (top).
Figure 3.
Multiplicity of the signals of H7 (
Figure 3a) and H6 (
Figure 3b) of compound
11 in the presence of R-Pirkle`s alcohol (corresspond to
Figure 2 top).
Figure 3.
Multiplicity of the signals of H7 (
Figure 3a) and H6 (
Figure 3b) of compound
11 in the presence of R-Pirkle`s alcohol (corresspond to
Figure 2 top).
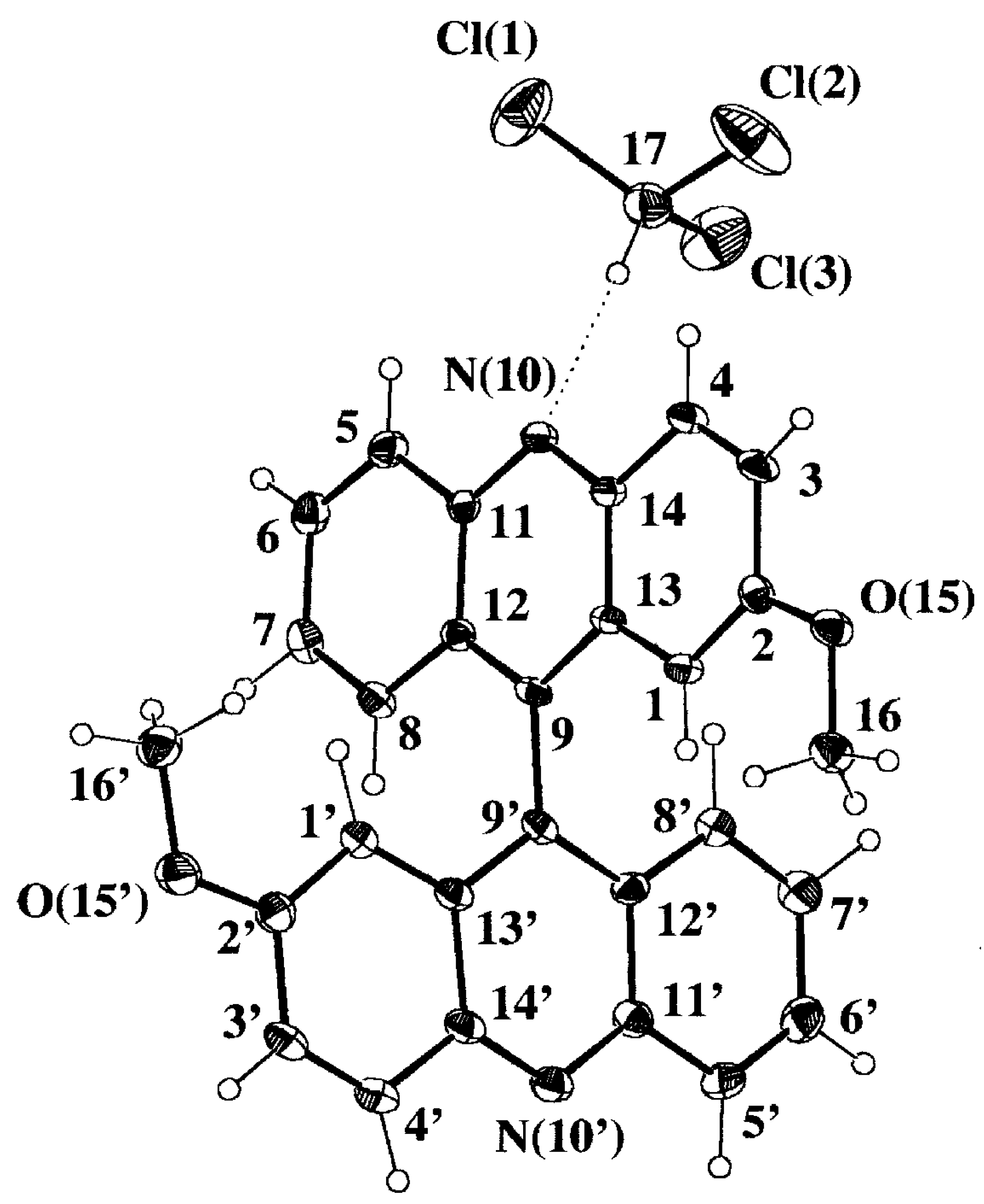
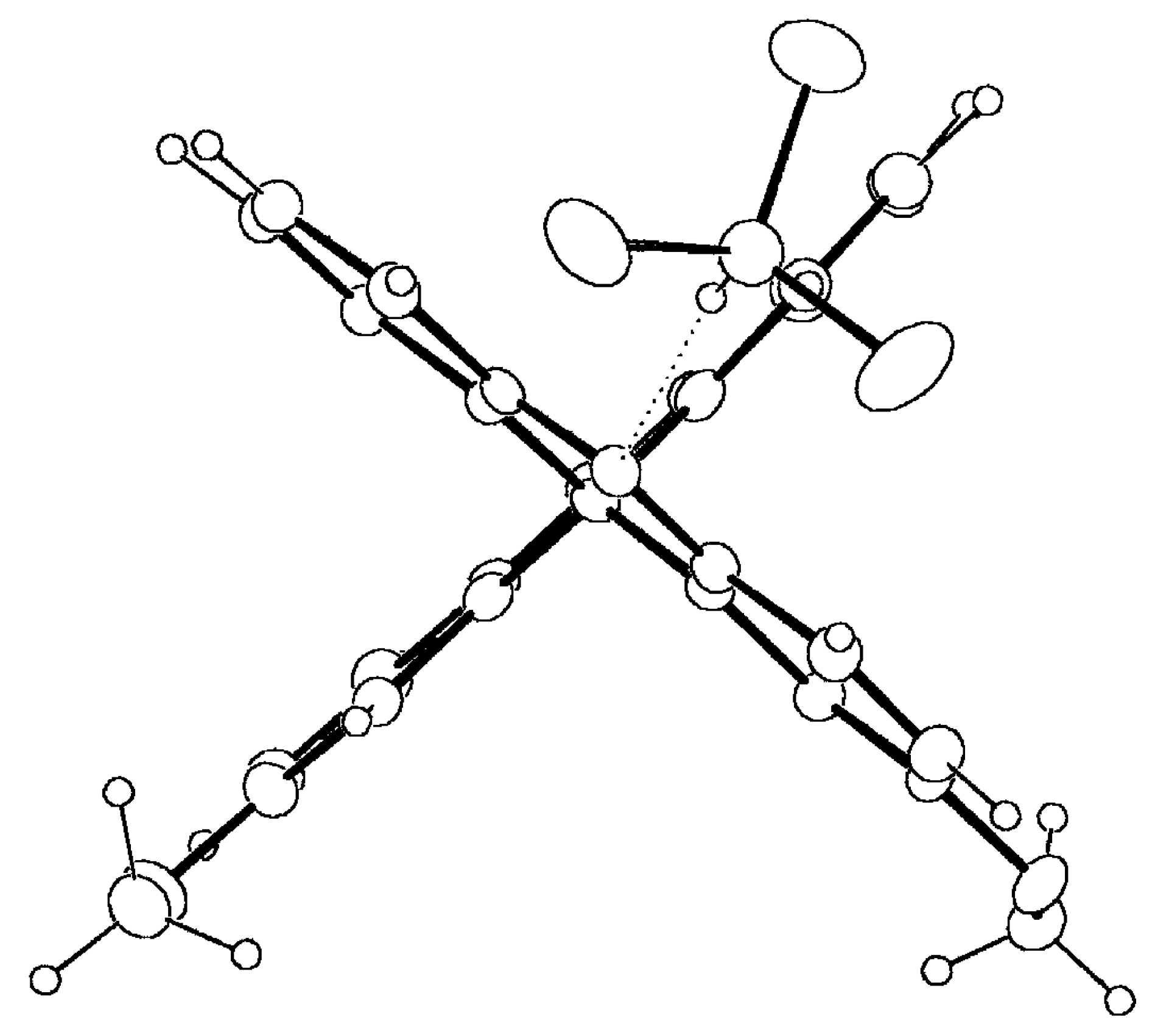
Single Crystal X-Ray Analysis
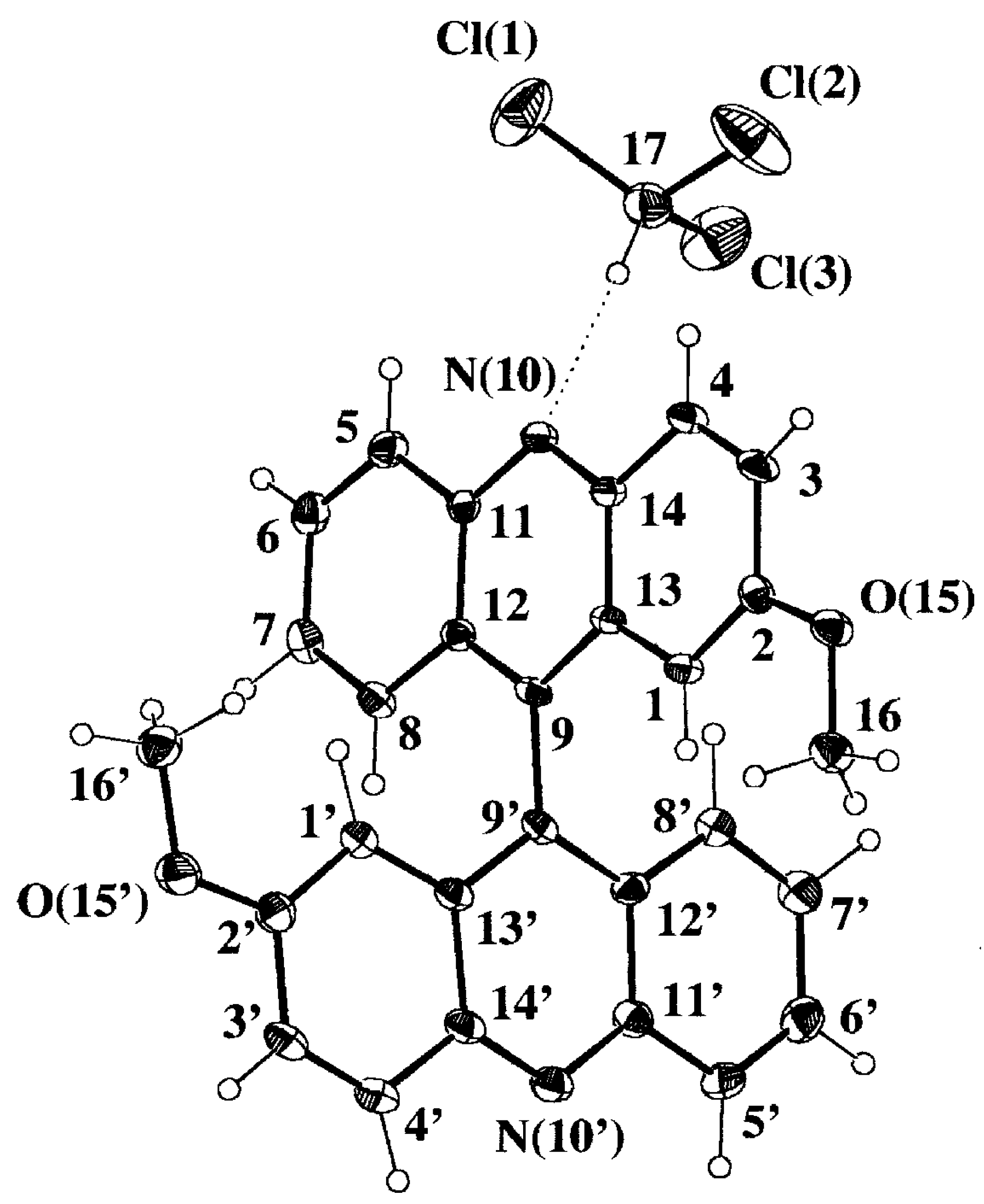
Selected geometrical parameters are gathered in
Table 3 according to the numbering scheme displayed in
Figure 4a. The pattern of bond lengths and angles of the host molecule presents the same trends as the mean values obtained for seven acridine molecules (two polymorphic forms and several host-guest complexes) retrieved from the Cambridge Structural Database (CSD hereinafter, October 1997 Version) [
22]. Both acridine moieties display a significant lack of planarity that is described by the angles between the mean least-squares planes of the central pyridine ring and each lateral phenyl ring,
Table 3. The twist between both acridine planes is close to 90°, that is slightly bigger than the mean value of 83(6)° [in parentheses the estimated standard deviation of the sample] observed for six 9,9'-bianthryl derivatives not substituted in positions 1, 8, 1' and 8' and retrieved from CSD. There are no significant differences between both halves of the molecule, including the methoxy substituents, when they are overlapped according to a least-square procedure based on their atomic coordinates [
23] [χ
2 values for x, y, z coordinates of 10.95, 15.31 and 17.91 versus a tabulated value of 26.30 at 95% probability level]. However, the host molecule is somehow distorted as can be seen in the Newman projection along the C(9)-C(9') bond shown in
Figure 4b. One acridine moiety lacks its parallelism with the central C(9)-C(9') bond and is bent towards the CHCl
3 guest molecule, 6.0(2) versus 1.9(2)°,
Table 3. This could be an indication of the strength of the intermolecular CH...N hydrogen bond.
Both methoxy groups are almost coplanar with regard to the phenyl rings they are attached to,
Table 3, in agreement with the mean value of 5(5)° obtained after averaging 1797 hits retrieved from CSD. The search fragment was a methoxyphenyl moiety with no substitution at the
ortho positions. Moreover, the C(Ph)-O and O-CH
3 distances and the C(Ph)-O-CH
3 angle do not differ significantly from the mean values 1.371(16), 1.419(25) Å and 117.8(12)° respectively,
Table 3.
Table 3.
Selected geometrical parameters and hydrogen bond interactions (Å, °).
Table 3.
Selected geometrical parameters and hydrogen bond interactions (Å, °).
| C(2)-O(15) | 1.356(6) | | C(2')-O(15') | 1.356(5) |
| O(15)-C(16) | 1.426(7) | | O(15')-C(16') | 1.420(7) |
| C(9)-C(9') | 1.501(6) | | | |
| C(1)-C(2)-C(3) | 120.3(4) | | C(1')-C(2')-C(3') | 120.4(4) |
| C(6)-C(7)-C(8) | 120.7(4) | | C(6')-C(7')-C(8') | 119.7(4) |
| C(12)-C(9)-C(13) | 118.7(3) | | C(12')-C(9')-C(13') | 119.5(4) |
| C(11)-N(10)-C(14) | 118.3(3) | | C(11')-N(10')-C(14') | 118.6(4) |
| C(2)-O(15)-C(16) | 117.3(4) | | C(2')-O(15')-C(16') | 116.9(4) |
| C(1)-C(2)-O(15)-C(16) | -3.7(6) | | C(1')-C(2')-O(15')-C(16') | 1.8(7) |
| C(12)-C(9)-C(9')-C(12') | 91.6(5) | | C(12)-C(9)-C(9')-C(13') | -85.2(5) |
| C(13)-C(9)-C(9')-C(12') | -83.7(5) | | C(13)-C(9)-C(9')-C(13') | 99.5(5) |
| Angles between lines[a] and mean least-square planes[b] |
| L1^L2 | 1.9(2) | | L2^L3 | 6.0(2) |
| P1^P2 | 1.3(1) | | P4^P5 | 1.8(1) |
| P2^P3 | 1.0(1) | | P5^P6 | 3.3(1) |
| Hydrogen interactions | D-H | H⋯A | D⋯A | D-H⋯A |
| C(3)-H(3)⋯N(10')(x,y-1,z) | 0.91(6) | 2.89(7) | 3.440(6) | 120(5) |
| C(4)-H(4)⋯N(10')(x,y-1,z) | 0.93(6) | 2.68(6) | 3.339(5) | 129(4) |
| C(16')-H(16'1)⋯O(15)(-x,-y,-z+1) | 1.06(7) | 2.56(7) | 3.583(6) | 164(5) |
| C(17)-H(17)⋯N(10)(-x+1,-y,-z+1) | 0.90(7) | 2.45(7) | 3.329(7) | 168(5) |
| Cl3C-H⋯N(acridine)[c] | 1.00(11) | 2.25(18) | 3.228(97) | 167(6) |
Figure 4a.
Perspective view of the complex 9·HCCl3 displaying the numbering scheme and the host-guest intermolecular hydrogen bond (dotted line).
Figure 4a.
Perspective view of the complex 9·HCCl3 displaying the numbering scheme and the host-guest intermolecular hydrogen bond (dotted line).
Figure 4b.
Newman projection of 9·HCCl3 along the C(9)-C(9') bond, showing the molecular conformation of the biacridine moiety and the coplanarity of the metoxy substituents. Anisotropic displacement parameters are drawn at 30% probability level.
Figure 4b.
Newman projection of 9·HCCl3 along the C(9)-C(9') bond, showing the molecular conformation of the biacridine moiety and the coplanarity of the metoxy substituents. Anisotropic displacement parameters are drawn at 30% probability level.
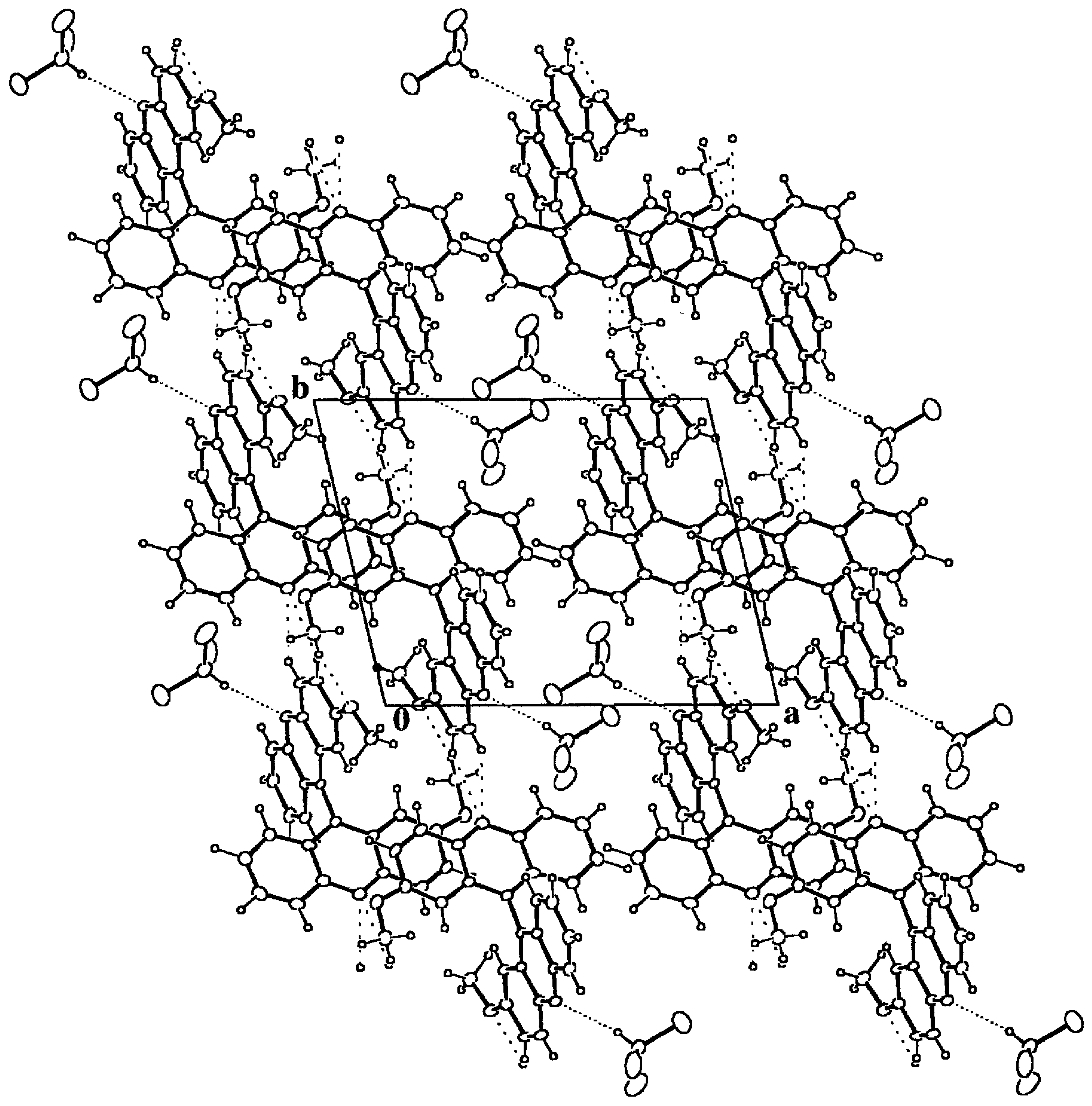
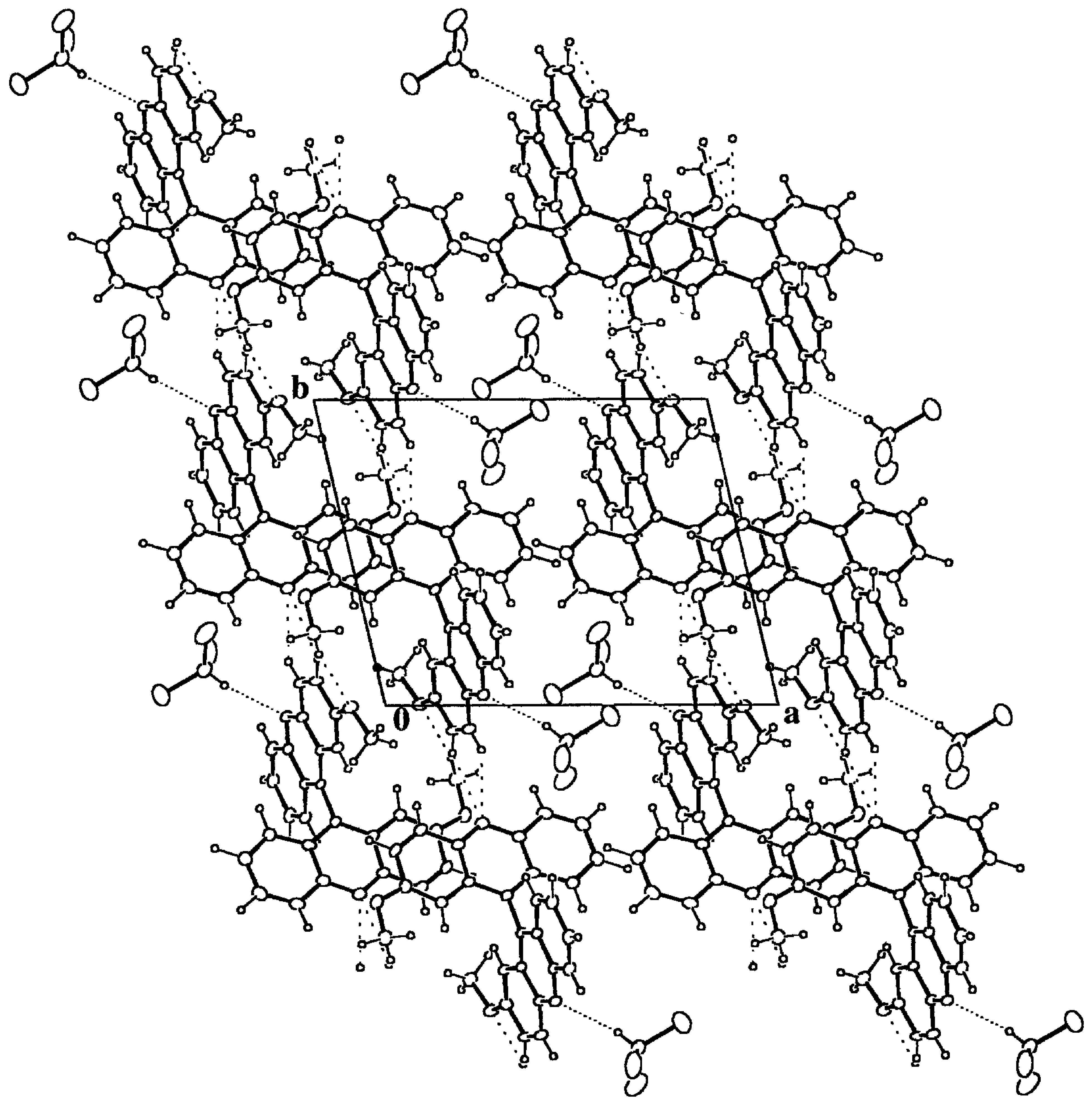
Figure 6.
Packing diagram of 9·HCCl3 down the c axis. The most compact dotted lines mark the hydrogen bonds between the host and guest moieties. The remaining dotted lines stand for intermolecular hydrogen bonds between host molecules. The resulting structure is due to the packing of chains along b formed by centrosymmetric dimers around (0, 0, 1/2) composed by host-guest pairs.
Figure 6.
Packing diagram of 9·HCCl3 down the c axis. The most compact dotted lines mark the hydrogen bonds between the host and guest moieties. The remaining dotted lines stand for intermolecular hydrogen bonds between host molecules. The resulting structure is due to the packing of chains along b formed by centrosymmetric dimers around (0, 0, 1/2) composed by host-guest pairs.
From fifty-one structures containing an acridine fragment (CSD), there are three that include CHCl
3 as guest, one of them with two crystallographically independent molecules, and in all cases, there is a strong and linear Cl
3CH...N(acridine) hydrogen bond interaction,
Table 3. The intermolecular hydrogen bond is almost as strong as that reported for the Cl
3CH...O system [
24], 2.22(3) and 3.16(2) Å for the H...O and C...O distances respectively. According to this observation, the title compound also presents this kind of host-guest interaction, which is the strongest one found in the crystal structure and that gives rise to pairs of host-guest bonded molecules,
Table 3 and Figure 5. These pairs form dimers around inversion centers at (0, 0, 1/2) and symmetry related ones, by means of C-H...O hydrogen bonds. Finally, the dimers give rise to chains along the
b axis through two contiguous C(Ph)-H...N hydrogen bonds, which constitute the final secondary structure motives that make up the whole crystal structure.
There are no voids left in the structure and the total packing coefficient [
25] is 0.71 (C
kall = V
molecules/Unit Cell Volume). However, the CHCl
3 molecules are packed together and enclosed in channels along the
c axis left by the host matrix, Figure 5. In spite of the quite strong hydrogen bond interaction between the host and guest molecules, the local packing coefficient for this moiety is significantly lower than the overall one, C
klocal = 0.56.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}