Introduction
Immobilization of reactive species on a polymer support could provide many important advantages over analogous homogeneous systems; for example separation of the support from the reaction mixture can be achieved by simple filtration aiding isolation and purification procedures, reactive species can become more active and/or more selective due to changes in the microenvironment of the active sites, excess of a polymeric reagent can be readily employed without incurring a penalty in work-up, transition metal complexes and optically active catalysts might be efficiently retained for re-use, and noxious or toxic species might be encapsulated when bound to a macromolecule, with obvious advantages in environmental terms [
1]. Though the limited commercial availability and the initial extra costs do not favour the use of polymer supports, the numerous advantages are well recognised [
1]. Unfortunately in the case of asymmetric catalysts the supported systems often suffer from a significant drop in enantioselectivity compared to the corresponding low molecular weight species [
2]. Recently attention has focused on polymer-supported asymmetric alkene oxidation catalysts [
3]. Interestingly, there are no reports of the successful immobilisation of the Sharpless Ti−tartrate ester-based asymmetric alkene epoxidation catalyst, despite this being a relatively long-standing and well used methodology. Some approaches to the production of a heterogeneous Sharpless−type epoxidation catalyst have been reported, but the level of asymmetric induction achieved was generally only modest [
4].
We have previously reported on the synthesis of a group of poly(tartrate ester) ligands and their use with titanium tetraisopropoxide and
tert-butyl hydroperoxide (tBHP) as the oxidant in epoxidising
trans-hex-2-en-1-ol in high chemical yield and with good enantioselectivity [
5]. Although the enantiomeric excesses are in general a little lower than those achieved using monomeric tartrate esters, they are still significantly better than those from previously reported polymer-supported systems [
4]. We believe therefore that our results are important in methodological and potentially technological terms, and offer the prospect of new information regarding the structure of the active species and its mechanistic role. We have also synthesized in common organic solvents insoluble branched and/or crosslinked poly(tartrate ester)s and have studied the use of both homogeneous and heterogeneous optically active poly(tartrate ester) ligands in asymmetric epoxidations of a number of
trans-allylic alcohols with titanium tetraisopropoxide and tBHP [
6]. We have investigated the effect of the ratio of polymer ligand to titanium on the enantioselectivity and the yields, and the influence of the polymer backbone molecular architecture on asymmetric induction.
Experimental Section
General
The 1H NMR and 13C NMR spectra were obtained on a Bruker AM200 MHz or Bruker DPX400 MHz spectrometers. Chemical shifts (δ) are given in ppm relative to TMS. Gas chromatographic (GC) analysis were carried out on Carlo Erba GCHR 5300 Mega Series employing a SGE BPX5 column or Perkin Elmer 8500 equipped with FID detector employing a 25-m packed with fused silica column (phase layer 0.25 μm). FTIR spectra were measured on a Bruker IFS 66- or Nicolet Impact 400d-instruments, wawenumbers are in cm−1. Enantiomeric ratios measured by HPLC were determined using a Daicel Chiralcel OB column (flow rate = 0.5 ml/min, hexane/isopropanol = 97.5-2.5). High resolution GC/MS analysis were performed by the Mass Spectrometry Laboratory of the University of Oulu, Finland. Optical rotations were determined on Perkin-Elmer WM250 or Perkin-Elmer 243 B polarimeters using a 1-cm3 capacity, 1 dm path length, quartz cell. Elemental analyses were performed by the Microanalytical Laboratory of the University of Strathclyde, Scotland or by the Microanalytical Laboratory of the University of Oulu, Finland. Molecular weights were measured by gel permeation chromatography (polystyrene standards in THF) by RAPRA, UK. Flash chromatography was performed using Merck Silica gel 60 (230-400 mesh) with diethyl ether/petrol ether (40 °C−60°C) in various proportions as eluent. Absolute configurations were determined by comparison of the observed rotation by polarimetry with the literature value. General preparation and shift study analysis of acetates followed the procedure described in reference 15.
Materials
Pre-activated and powdered 4Å molecular sieves were available from Aldrich Chemical Co. and they were used as received. Cooling was accomplished through the use of the following bath: acetone/liquid nitrogen. The dichloromethane used did not contain methanol and therefore was not distilled but was stored over CaCl2. Aqueous 70 % tert-butyl hydroperoxide (TBHP) was obtained from the Aldrich Chemical Co. Preparation and molarity determination of anhydrous TBHP followed the procedure described in reference 21. Reagents handled by syringe were measured by weight or by volume.
General work-up procedure
The work-up followed the literature procedure [
15] with slight modifications. The work-up procedure is described for reactions utilizing 0.1 mol of substrate, 3.0 g of sieves, 5 mmol of Ti(O-i-Pr)
4, 6 mmol of tartrate and 0.2 mol of TBHP. The amounts were scaled proportionally.
The cold ( −20 °C) reaction mixture was quenched with a 30 % aqueous solution of sodium hydroxide saturated with sodium chloride. After 10 w-% of diethyl ether was added, the cold bath was removed and the stirred mixture was allowed to warm to 10 °C. Stirring was maintained for an additional 10 min at 10 °C, whereupon MgSO4 and Celite were added. After a final 15 min of stirring, the mixture was allowed to settle and the solution was filtered through a pad of Celite and washed with diethyl ether. Excess of TBHP was removed by azeotropic distillation with toluene.
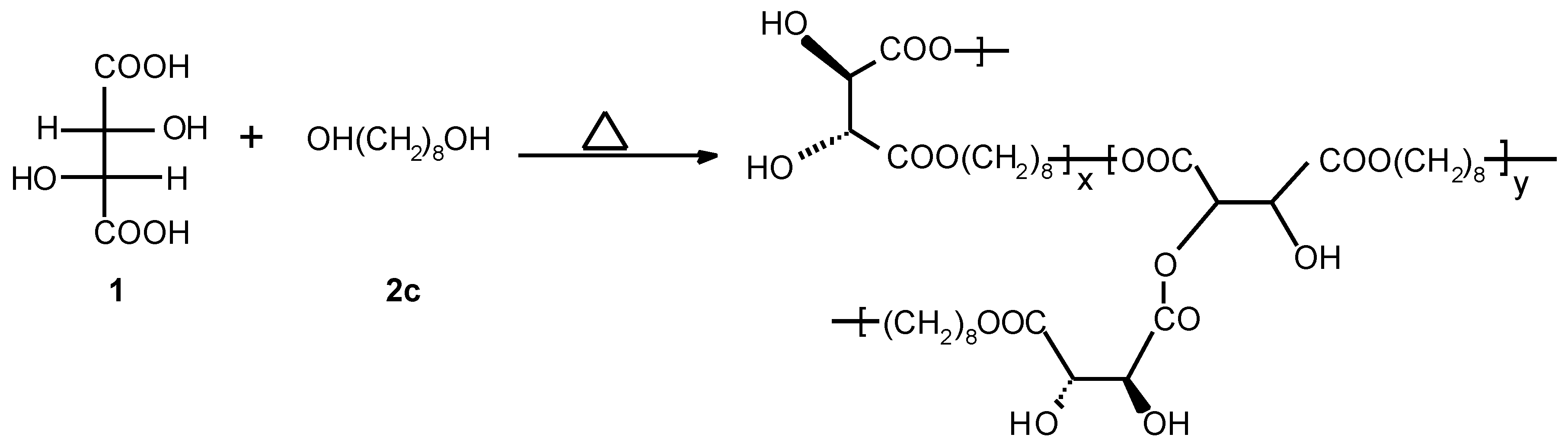
Linear Optically Active Polyesters 3d
An oven-dried 100 ml three-necked round-bottomed flask equipped with a magnetic stir-bar was charged with L-(+)-tartaric acid (5 g, 0.033 mol), 1,12-dodecanediol (6.74 g, 0.033 mol) and ~3 w-% p-toluene sulphonic acid (0.3 g). The reaction mixture was stirred at 120 °C for 3 days. Water and unreacted diol were removed by distillation under high vacuum at the end of reaction. The resulting solid was crushed, washed with acetone/hexane in a Soxhlet apparatus for ca. 16-20 hours and dried in vacuum oven (50 °C) for ca. 15 hours to yield 5.70 g, 55 % C12-poly(tartrate ester) 3d.
FTIR (KBr) 3470, 3308, 2916, 2849, 1755, 1722, 1468, 1287, 1195, 1132, 1070, 971. 1H NMR (400MHz, DMSO-d6, 70 °C) δ4.36 (s, 2H), 4.09 (t, J = 6.6 Hz, 4H), 1.59 (m, 4H), 1.30 (m, 16H). 13C NMR (400MHz, DMSO-d 6, 70°C) δ171.1, 72.5, 64.5, 60.8, 28.8, 28.5, 28.1, 25.2.
Polymers
3 a-c were obtained analogously (
Table 1).
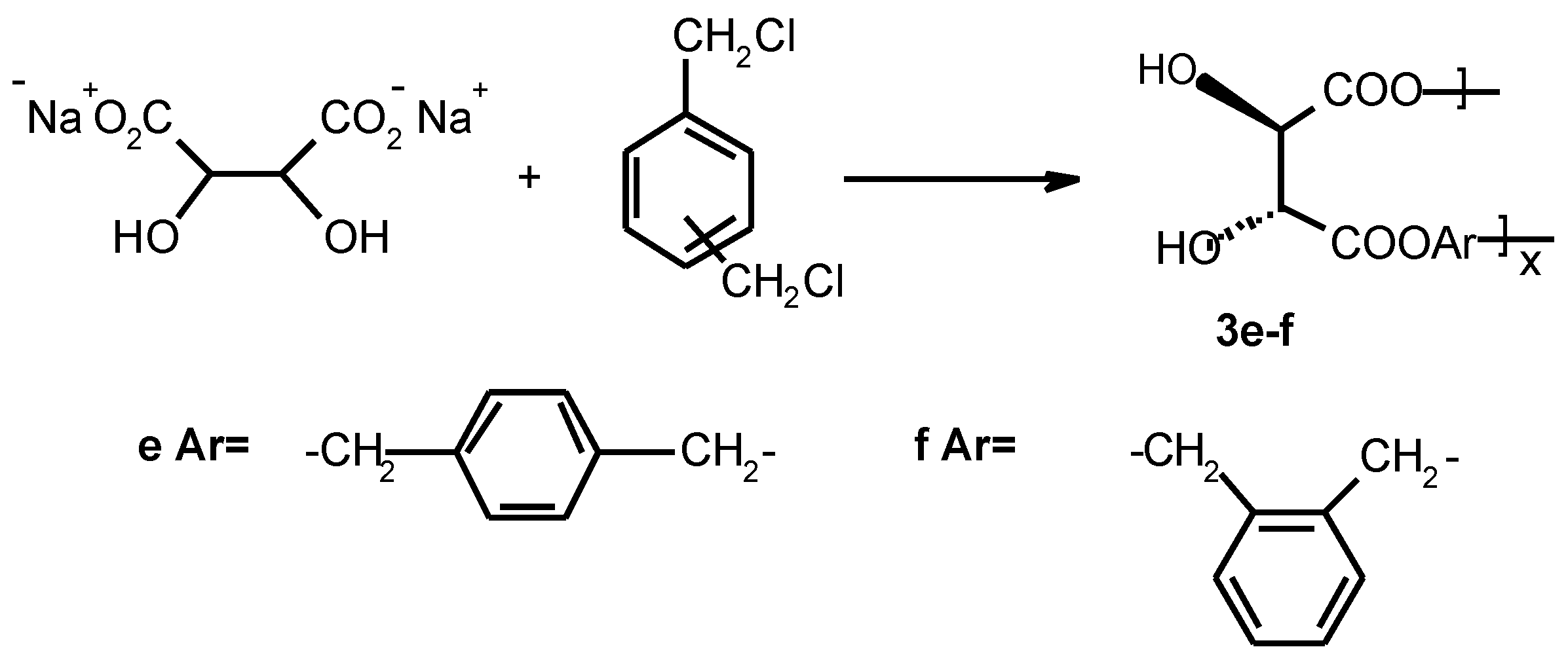
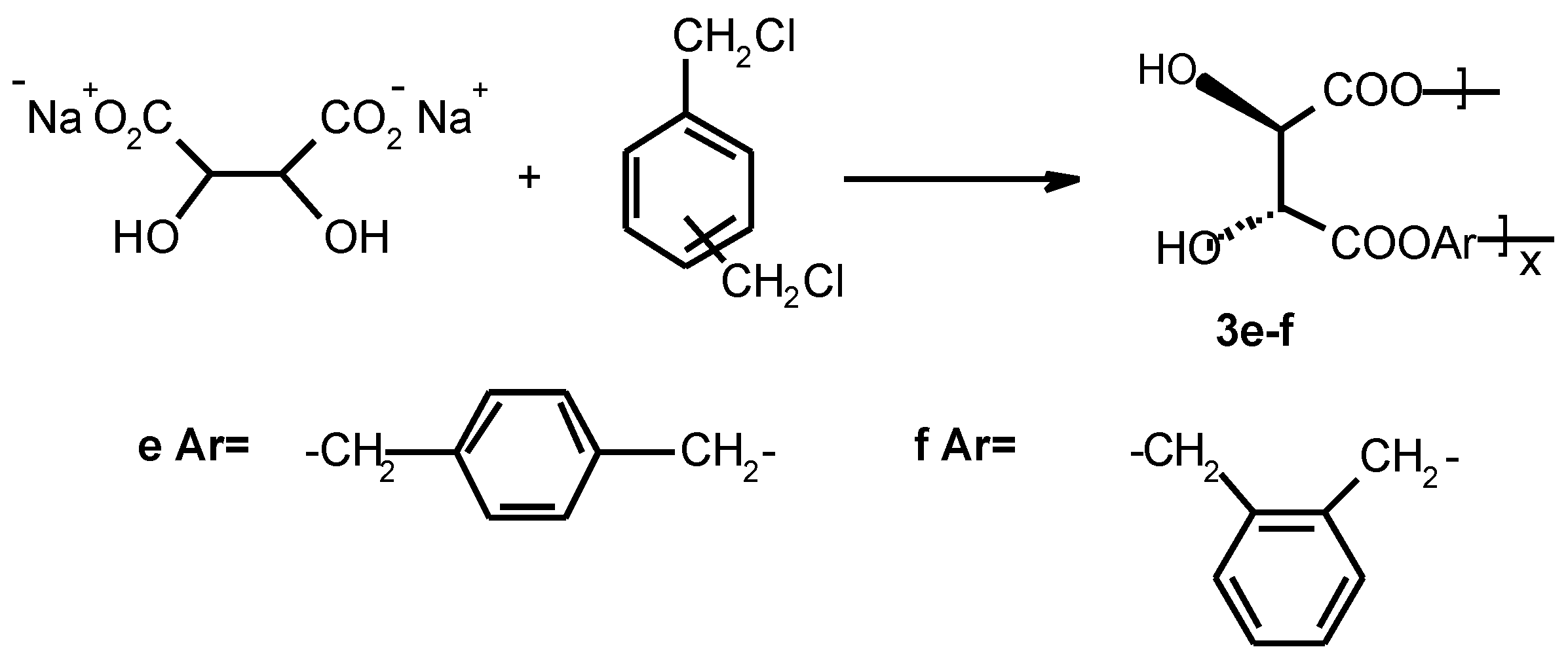
Preparation of linear optically active polyesters containing aromatic spacer 3f
An oven-dried 100 ml three-necked round-bottomed flask equipped with a magnetic stir-bar and a reflux condenser was charged with 5g of L-(+)-tartaric acid disodium salt (0.022 mol) dissolved in 10 ml of water, 3.8 g of α, α’-dichloro-o-xylene (0.022 mol) dissolved in 20 − 30 ml of chloroform and ~20 mol-% tetrabutyl ammonium bromide hydrate (1.42 g, 0.0044 mol) as a phase transfer catalyst. The reaction mixture was reflux for 7 days. Solvents were evaporated to dryness and white solid was washed with hot water and diethyl ether and dried in a vacuum oven (+50 °C) for 2 days to yield 2.65 g, 48 % poly(tartrate ester) 3f. FTIR (KBr) 3545, 3394, 2967, 2873, 1747, 1445, 1421, 1374, 1290, 1266, 1225, 1128, 1089, 970, 818, 677. 1H NMR (400MHz, DMSO-d6, 70 °C) δ 7.39 (m, 4H), 5.21 (s, 4H), 4.56 (s, 2H). 13C NMR (400MHz, DMSO-d6, 70 °C) δ170.10, 128.82, 128.62, 127.86, 127.70, 72.21, 65.78.
Polymer
3e was obtained analogously (
Table 1).
Preparation of branched/crosslinked optically active polyester 4
An oven-dried 100 ml three-necked round-bottomed flask equipped with a magnetic stirbar, nitrogen inlet and bubbler was charged with L-(+)-tartaric acid (10 g, 0.067 mol), 20 mol-% excess of 1,8-octane diol (11.67 g, 0.07996 mol) and ~3 w-% p-toluene sulphonic acid (0.6 g). Temperature was raised to 140-150 °C to get a homogeneous mixture and then stirred at 120-130 °C for 3-4 days under nitrogen. Water and unreacted diol were removed by distillation under high vacuum at the end of reaction. The resulting solid was swelled by refluxing in ethyl acetate and then poured in n-hexane. The solvents were decanted and the polymer dried in high vacuum at + 40°C for 6 hours and at room temperature for 2 days to yield 16.6 g, 95 % of C8-poly(tartrate ester) 4. [α]D25 + 9° (c 1.6, THF). FTIR (KBr) 3450, 2932, 2857, 1743, 1466, 1395, 1128, 1091, 1011, 956. Anal. Found: C, 55.26; H, 8.47. 1H NMR (400MHz, DMSO-d6, 70 °C) δ 5.75 (br s), 5.41 (d, J = 3.15 Hz), 4.62 (d, J = 2.83 Hz), 4.37 (s, 2H), 4.09 (t, J = 6.49 Hz, 4H), 1.58 (m, 4-5H), 1.30 (m, 9H). 13C NMR (400MHz, DMSO-d6, 70 °C) d 172.42 (s), 171.28 (s), 171.03 (s), 80.41 (d, JCH = 142.04 Hz), 73.45, 72.45, 72.19 (d, JCH = 146.24 Hz), 64.47 (t, JCH = 147.2 Hz), 28.32 (t, JCH = 125.64 Hz), 28.04 (t, JCH = 125.92 Hz), 25.07 (t, J = 125.66 Hz).
General procedure for the catalytic asymmetric epoxidation using homogeneous polymer-ligand
The literature procedure [
15] for the epoxidation of allylic alcohols was followed with modifications. An oven-dried three-necked round-bottomed flask equipped with a magnetic stir-bar, nitrogen inlet, septum and bubbler was charged with 4Å powdered, activated molecular sieves, polymer-ligand
3 and dry CH
2Cl
2. The flask was cooled to -20 °C and Ti(O-i-Pr)
4 (via syringe) was added sequentially with stirring. The reaction mixture was stirred at - 20 °C and after about one hour 2 equiv. of TBHP in
iso-Octane was added with a syringe at a moderate rate. The resulting mixture was stirred at −20 °C at least for hour. Substrate (dissolved in dry CH
2Cl
2) was added dropwise with a syringe, being careful to maintain the reaction temperature between −15 °C and −20 °C. The mixture was stirred for an additional 3-7 hours at −15 °C to −20 °C and the yield was determined using gas chromatographic analysis. The reaction mixture was stored in a freezer overnight, the GC yield was then determined and work-up was performed. The crude product was purified by Kugelrorh-distillation and the enantiomeric purity of the epoxide was measured by chiral HPLC analysis.
(2S-trans)-3-Propyloxiranemethanol (6)
The epoxidation was performed as described above. C8- poly(tartrate ester) 3c (0.52 g, 0.002 mol) was swelled in 30 ml of CH2Cl2 containing 0.6 g of powdered, activated 4Å molecular sieves. Ti(OPri)4 (0.28 g, 0.001 mol), 10.1 ml of a 3.97 M solution of TBHP in iso-Octane and 2 g (0.020 mol) of trans-hex-2-en-1-ol (dissolved in 10 ml CH2Cl2) were added. The reaction mixture was stirred at - 20 °C for 3 h. The reaction mixture was stored at –20 °C over the weekend. Workup was then performed and crude product was purified by Kugelrorh-distillation to give a colourless oil 5 (1.36 g, 59 % yield, 91 % purity, 79 %ee removed by distillation under high vacuum at the end of by HPLC, Chiralcel OB column): [α]25D - 32° (c 1.95, CHCl3), GC analysis after 3 h: 22 % epoxide, GC analysis before work-up: 61 % epoxide. FTIR (CHCl3) 3030, 3013, 2977, 2952, 2883, 2856, 1237, 1196, 1083, 1029, 938, 879. 1H NMR (200 MHz, CDCl3) δ3.82 (dd, J = 2, 13 Hz, 1H), 3.53 (dd, J = 2, 13 Hz, 1H), 2.91-2-83 (m, 2H), 2.68 (s, 1H), 1.55-1.43 (m, 4H), 0.90 (t, J = 7 Hz, 3H). MS (EI, m/z, relative intensity) 99 (2, M-17)+, 81 (13), 73 (48), 55 (100), 43 (36). HRMS (EI) Calcd for C6H12O2 116.0837, found 116.0832.
General procedure for the catalytic asymmetric epoxidation using heterogeneous polymer-ligand
The literature procedure [
15] for the epoxidation of allylic alcohols was followed with modifications. An oven-dried three-necked round-bottomed flask equipped with a magnetic stirbar, nitrogen inlet, septum and bubbler was charged with 4Å powdered, activated molecular sieves, polymer-ligand
4 and dry CH
2Cl
2. The flask was cooled to −20°C and Ti(O-i-Pr)
4 (via syringe) was added sequentially with stirring. The reaction mixture was stirred at - 20 °C and after about one hour 2 equiv. of TBHP in
iso-Octane was added with syringe at a moderate rate. The resulting mixture was stirred at −20 °C at least for one hour. Substrate (dissolved in dry CH
2Cl
2) was added dropwise with a syringe, being careful to maintain the reaction temperature between −15°C and −20°C. The mixture was stirred for an additional 3−12 hours at −15°C to −20°C. The reaction mixture was stored in a freezer up to weeks. The reaction was monitored by gas chromatography (GC) using dodecane as a GC internal standard. The polymer was filtered off the reaction mixture and washed troughly with dichloromethane. Workup was then performed. The crude product was purified by flash-chromatography (eluent: petrol ether (40°C−60°C):diethyl ether, 50 %) and enantioselectivity of epoxide was measured by 400 MHz NMR using chiral Eu(hfc)
3-reagent (as acetates).
(2S-trans)-3-Propyloxiranemethanol (6)
The epoxidation was performed as described above. C8- poly(tartrate ester) 4 (2.6 g, 0.01 mol) was swelled in 40 ml of CH2Cl2 containing 0.3g of powdered, activated 4Å molecular sieves. Ti(OPri)4 (1.42 g, 0.005 mol), 12.5 ml of a 3.2 M solution of TBHP in iso-Octane and 2 g (0.02 mol) of trans-hex-2-en-1-ol (dissolved in 10 ml CH2Cl2) were added. The reaction mixture was stirred at -20 °C for 6 h and stored at – 20 °C overnight. Polymer was filtered off and washed throughly with CH2Cl2. Workup was then performed and crude product was purified by flash-chromatography (eluent: petrol ether (40°C − 60°C):diethyl ether, 50 %) to give colourless oil (1.22 g, 53 % yield, 100 % purity, 87 %ee by 1H NMR analysis of the derived acetate with Eu(hfc)3): [α]25D −38° (c 2.01, CHCl3), GC analysis before work-up: 78 % epoxide. FTIR (CHCl3) 2977, 2952, 1717, 1457, 1237, 1196, 933, 881, 806, 796. 1H NMR (200 MHz, C6D6) δ3.72 (ddd, J = 3, 6, 12 Hz, 1H), 3.46 (ddd, J = 5, 7, 12 Hz, 1H), 2.79-2.85 (m, 1H), 2.70-2.74 (m, 1H), ), 2.33 (t, J = 6 Hz, 1H), 1.34-1.45 (m, 4H), 0.93 (t, J = 7 Hz, 3H). MS (EI, m/z, relative intensity) 99 (0.8, M-17)+, 81 (7), 73 (20), 61 (8), 55 (100), 43 (86). HRMS (EI) Calcd for C6H12O2 116.0837, found 116.0882.

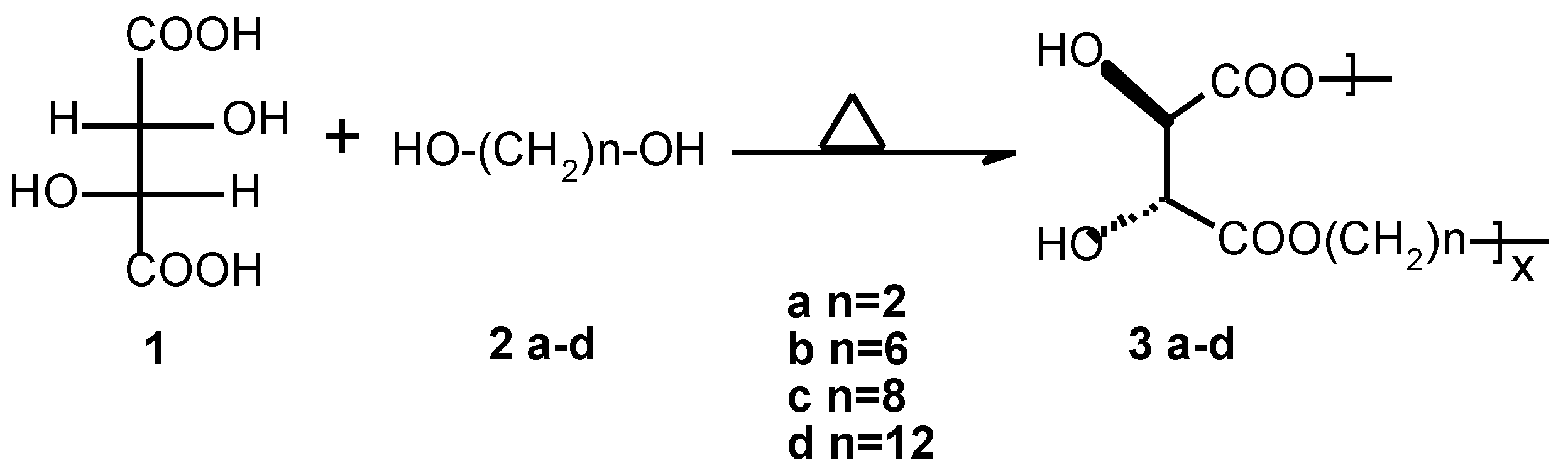

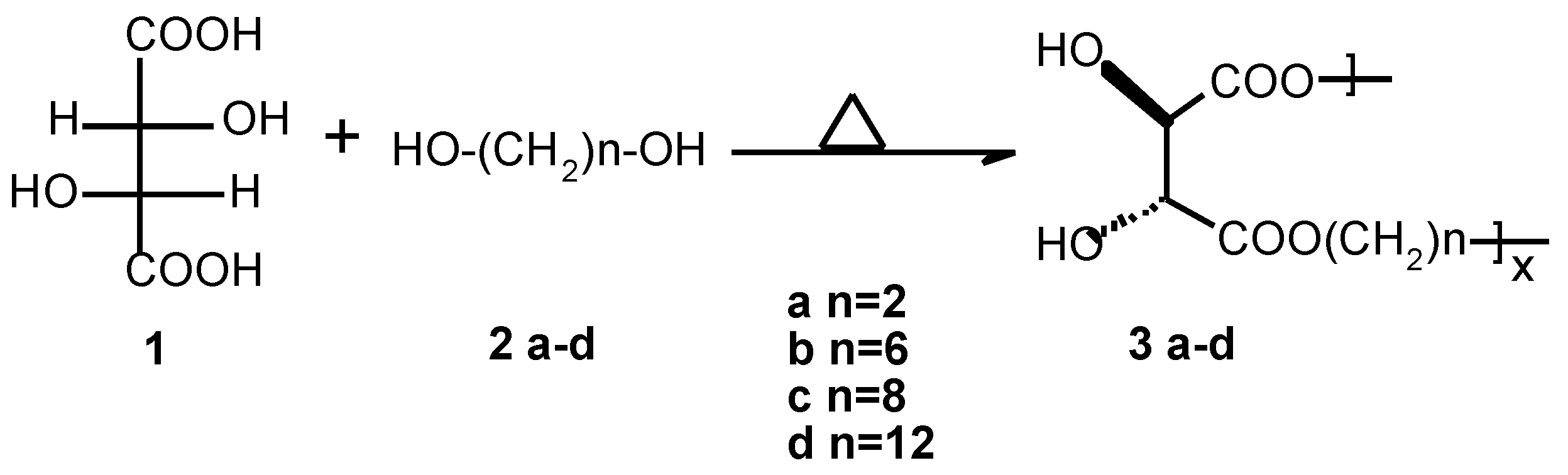{kind=link}
{kind=link}
{kind=link}
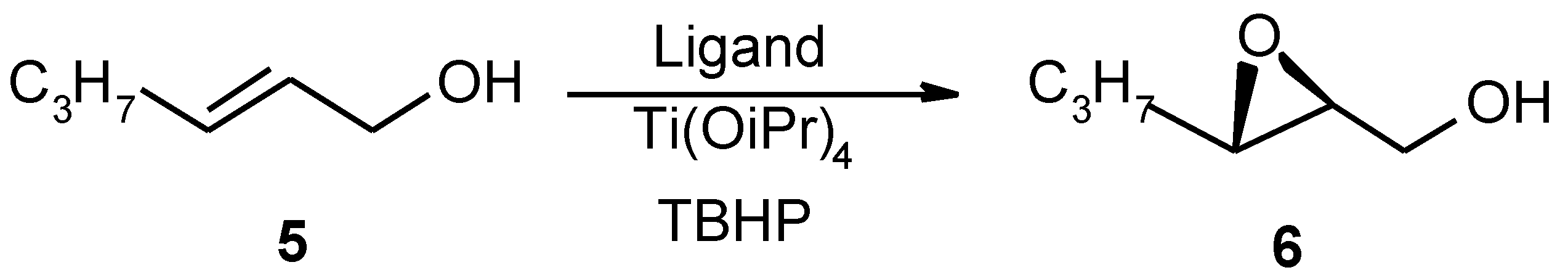{kind=link}

