
Synthesis, Pharmacological Evaluation, and Molecular Modeling of Lappaconitine–1,5-Benzodiazepine Hybrids
 , ,
, ,
Abstract
:1. Introduction
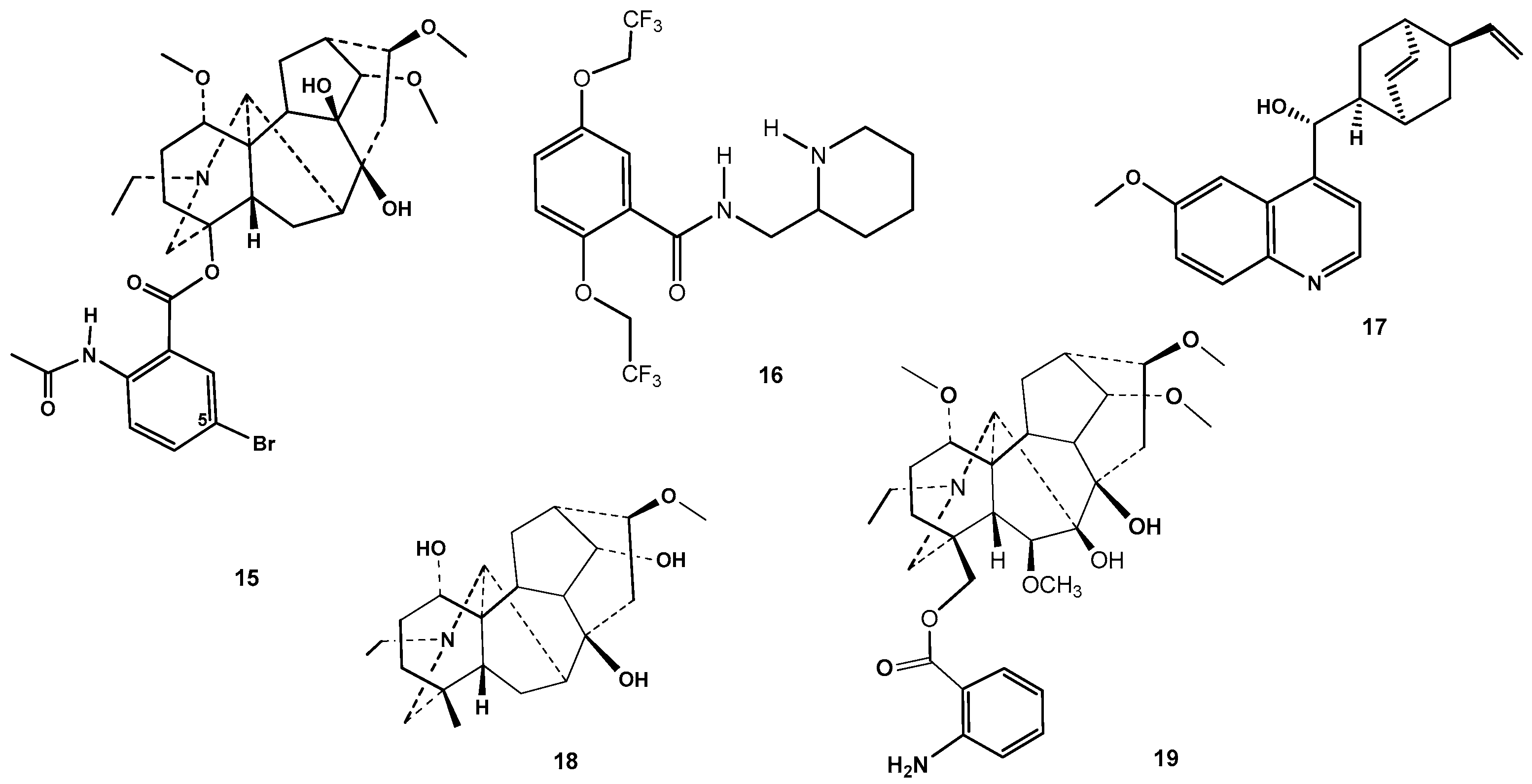
2. Results and Discussion
2.1. Chemical Synthesis
2.2. Biological Study
2.2.1. Analgesic Activity and Toxicity
2.2.2. Studying the Antiarrhythmic Activity of Compounds 8 and 10
The Antiarrhythmic Effect on In Vivo Models of Calcium Chloride and Epinephrine-Induced Arrhythmia
Ex Vivo Research
2.3. Molecular Modeling of a Possible Mechanism of Antinociceptive and Antiarrhythmic Potency of Lappaconitine-1,5-Benzodiazepine Hybrids 8 and 10
3. Materials and Methods
3.1. Chemistry
General Information
3.2. Synthesis and Spectral Data
3.2.1. Methyl 2-acetylamino-5-(3-(4-bromophenyl)propioloyl)benzoate (3)
3.2.2. Methyl 2-Acetylamino-5-(4-(4-bromophenyl)-3H-benzo[b][1,4]diazepin-2-yl)benzoate [2-(Methyl 2-acetylaminobenzoate-5-yl)-4-(4-bromophenyl)-3H-1,5-benzodiazepine] (7)
3.2.3. General Procedure for Preparation of Lappaconitine-1,5-benzodiazepene Hybrids 8–12
4β-{2′-Acetylamino-5′-(4″-(4-bromophenyl)-3H-1,5-benzodiazepine-2″-yl)benzoate}-1α, 14α,16β-Trimethoxy-20-ethylaconitane-8,9-diol (8)
4β-{2′-Acetylamino-5′-(4″-(4-chlorophenyl)-3H-1,5-benzodiazepine-2″-yl)benzoate}-1α, 14α,16β-Trimethoxy-20-ethylaconitane-8,9-diol (9)
4β-{2′-Acetylamino-5′-(4″-(4-fluorophenyl)-3H-1,5-benzodiazepine-2″-yl)benzoate}-1α, 14α,16β-Trimethoxy-20-ethylaconitane-8,9-diol (10)
4β-{2′-Acetylamino-5′-(4″-(4-trifluoromethylphenyl)-3H-1,5-benzodiazepine-2″-yl)-benzoate}-1α, 14α,16β-Trimethoxy-20-ethylaconitane-8,9-diol (11)
4β-{2′-Acetylamino-5′-(4″-(4-cyanophenyl)-3H-1,5-benzodiazepine-2″-yl)benzoate}-1α, 14α,16β-Trimethoxy-20-ethylaconitane-8,9-diol (12)
3.3. Biological Evaluation
3.3.1. Animals
3.3.2. Analgesic Tests
3.3.3. Antiarrhythmic Activity
3.4. Molecular Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wang, Y.Z.; Xiao, Y.Q.; Zhang, C. Study of analgesic and anti-inflammatory effects of lappaconitine gelata. J. Tradit. Chin. Med. 2009, 29, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Thawabteh, A.M.; Thawabteh, A.; Lelario, F.; Bufo, S.A.; Scrano, L. Classification, Toxicity and Bioactivity of Natural Diterpenoid Alkaloids. Molecules 2021, 26, 4103. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.-L.; Ao, J.-P.; Wang, Y.-R.; Li, T.-N.; Li, H.-Y.; Wang, Y.-H. Lappaconitine, a C18-diterpenoid alkaloid, exhibits antihypersensitivity in chronic pain through stimulation of spinal dynorphin A expression. Psychopharmacology 2018, 235, 2559–2571. [Google Scholar] [CrossRef]
- Ou, S.; Zhao, Y.D.; Xiao, Z.; Wen, H.Z.; Cui, J.; Ruan, H.Z. Effect of lappaconitine on neuropathic pain mediated by P2X3 receptor in rat dorsal root ganglion. Neurochem. Int. 2011, 58, 564–573. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, Y.-X.; Tang, X.-C. Anti-inflammatory and analgesic activities of N-deacetyllappaconitine and lappaconitine. Acta Pharmacol. Sin. 1987, 8, 301–305. [Google Scholar]
- Ono, M.; Satoh, T. Pharmacological studies on lappaconitine: Possible interaction with endogenous noradrenergic and serotonergic pathways to induce antinociception. Jpn. J. Pharmacol. 1992, 58, 251–257. [Google Scholar] [CrossRef]
- Dzhakhangirov, F.N.; Sultankhodzhaev, M.N.; Tashkhodzhaev, B. Diterpenoid alkaloids as a new class of antiarrhythmic agents. Structure-activity relationship. Chem. Nat. Compd. 1997, 33, 190–202. [Google Scholar] [CrossRef]
- Ameri, A. The effects of Aconitum alkaloids on the central nervous system. Prog. Neurobiol. 1998, 56, 211–235. [Google Scholar] [CrossRef]
- Ono, M.; Satoh, T. Pharmacological studies of lappaconitine. Analgesic activities. Arzneimittelforschung 1988, 38, 892–895. [Google Scholar]
- Li, X.; Wang, X.; Li, Z.; Mao, Y.; Liu, Z.; Liu, X.; Zhu, X.; Zhang, J. A Metabolomic study of the analgesic effect of lappaconitine hydrobromide (LAH) on inflammatory pain. Metabolites 2022, 12, 923. [Google Scholar] [CrossRef]
- Heubach, J.F.; Schüle, A. Cardiac effects of lappaconitine and N-deacetyllappaconitine, two diterpenoid alkaloids from plants of the Aconitum and Delphinium species. Planta Med. 1998, 64, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Guster, U.T.; Friese, J.; Heubath, J.F.; Mathiesen, T.; Selve, N.; Gleity, J. Mode of antinociceptive and toxic action of alkaloids of Aconitum species. Naunyn Schmiedeberg’s Arch. Pharmacol. 1998, 357, 39–48. [Google Scholar] [CrossRef]
- Valeev, A.E.; Verkhratskiĭ, A.N.; Dzhakhangirov, F.N. The effect of allapinine on the sodium currents of isolated trigeminal ganglion neurons and cardiomyocytes of rats. Neirofiziologiia 1990, 22, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Friese, J.; Gleitz, J.; Gutser, U.T.; Heubach, J.F.; Mathiesen, T.; Wilffert, B.; Selve, N. Aconitum sp. alkaloids: The modulation of voltage-dependent Na+ channels, toxicity and antinociceptive properties. Eur. J. Pharmacol. 1997, 337, 165–174. [Google Scholar] [CrossRef]
- Wright, S.N. Irreversible block of human heart (hH1) sodium channels by the plant alkaloid lappaconitine. Mol. Pharmacol. 2001, 59, 183–192. [Google Scholar] [CrossRef]
- Zhang, J.; Li, D.; Zhong, D.; Shou, Q.; Yin, Y.; Gao, J.; Peng, C. Processed lateral root of Aconitum carmichaelii Debx.: A review of cardiotonic effects and cardiotoxicity on molecular mechanisms. Front. Pharmacol. 2022, 13, 1026219. [Google Scholar] [CrossRef]
- Erlikh, A.D. The study of evidence base for the use of lappaconitine hydrobromide in patients with atrial fibrillation. Kardiologiia 2016, 56, 48–53. [Google Scholar] [CrossRef]
- Li, Y.-F.; Zheng, Y.-M.; Yu, Y.; Gan, Y.; Gao, Z.-B. Inhibitory effects of lappaconitine on the neuronal isoforms of voltage-gated sodium channels. Acta Pharmacol. Sin. 2019, 40, 451–459. [Google Scholar] [CrossRef]
- Quintans, J.S.S.; Antoniolli, A.R.; Almeida, J.R.G.S.; Santana, V.J.; Quintans, L.J. Natural products evaluated in neuropathic pain models—A systematic review. Basic Clin. Pharmacol. Toxicol. 2014, 114, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Nyirimigabo, E.; Xu, Y.; Li, Y.; Wang, Y.; Agyemang, K.; Zhang, Y. A review on phytochemistry, pharmacology and toxicology studies of Aconitum. J. Pharm. Pharmacol. 2015, 67, 1–19. [Google Scholar] [CrossRef]
- Pereira, F. Polypharmacology of Aconitum and Delphinium sp. Diterpene alkaloids: Antiarrhythmic, analgesic and anti-inflammatory effects. Mini Rev. Org. Chem. 2017, 14, 304–310. [Google Scholar] [CrossRef]
- Zhao, C.; Li, S.; Zhang, J.; Huang, Y.; Zhang, L.; Zhao, F.; Du, X.; Hou, J.; Zhang, T.; Shi, C.; et al. Current state and future perspective of cardiovascular medicines derived from natural products. Pharmacol. Ther. 2020, 216, 107698. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Li, Y.-C.; Sun, M.-R.; Zhang, P.-L.; Li, Y.; Yang, H. A systematic review of pharmacological activities, toxicological mechanisms and pharmacokinetic studies on Aconitum alkaloids. Chin. J. Nat. Med. 2021, 19, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Miao, X.; Li, Y.; Hu, F.; Ma, D.; Zhang, Z.; Sun, Q.; Zhu, Y.; Zhu, Q. Traditional processing, uses, phytochemistry, pharmacology and toxicology of Aconitum sinomontanum Nakai: A comprehensive review. J. Ethnopharmacol. 2022, 293, 115317. [Google Scholar] [CrossRef] [PubMed]
- McCabe, D.J. Clinical effects from ingestion of lappaconitine, an Aconitum alkaloid with sodium channel blocking effects. J. Med. Toxicol. 2022, 18, 243–247. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, Y.; Zhao, J.; Zhu, C.; Feng, N. Nanostructured lipid carriers for percutaneous administration of alkaloids isolated from Aconitum sinomontanum. J. Nanobiotech. 2015, 13, 47. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Ke, B.-W.; Qin, Y.; Wang, F.-P. The diterpenoid alkaloids. Alkaloids Chem. Biol. 2022, 87, 1–360. [Google Scholar]
- Bello-Ramírez, A.M.; Nava-Ocampo, A.A. The local anesthetic activity of aconitum alkaloids can be explained by their structural properties: A QSAR analysis. Fund. Clin. Pharmacol. 2004, 18, 157–161. [Google Scholar] [CrossRef]
- Sun, W.; Shi, Z.; Wang, H. Synthesis, characterization and antinociceptive properties of the lappaconitine salts. Med. Chem. Res. 2015, 24, 3474–3482. [Google Scholar] [CrossRef]
- Teng, G.; Zhang, F.; Li, Z.; Zhang, C.; Zhang, L.; Chen, L.; Zhou, T.; Yue, L.; Zhang, J. Quantitative electrophysiological evaluation of the analgesic efficacy of two lappaconitine derivatives: A window into antinociceptive drug mechanisms. Neurosci. Bull. 2021, 37, 1555–1569. [Google Scholar] [CrossRef]
- Qu, D.N.; Zhang, X.M.; Sang, C.Y.; Zhou, Y.Q.; Ma, J.Y.; Hui, L. Lappaconitine sulfate induces apoptosis in human colon cancer HT-29 cells and downregulates PI3K/AKT/GSK3β signaling pathway. Med. Chem. Res. 2019, 28, 907–916. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, J.; Song, N.; Guo, Y.; Hui, L.; Sang, C. Lappaconitine sulfate inhibits proliferation and induces apoptosis in human hepatocellular carcinoma HepG2 cells through the reactive oxygen species-dependent mitochondrial pathway. Pharmacology 2020, 105, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Luan, S.X.; Gao, Y.Y.; Liang, X.X.; Zhang, L.; Yin, L.Z.; He, C.L.; Liu, S.X.; Yin, Z.Q.; Yue, G.Z.; Zou, Y.F.; et al. Synthesis and structure–activity relationship of lipo–diterpenoid alkaloids with potential target of topoisomerase IIα for breast cancer treatment. Bioorg. Chem. 2021, 109, 104699. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-L.; Shen, X.-L.; Chen, Q.-H.; Qi, G.; Wang, W.; Wang, F.-P. Structure–analgesic activity relationship studies on the C18- and C19-diterpenoid alkaloids. Chem. Pharm. Bull. 2009, 57, 801–807. [Google Scholar] [CrossRef]
- Xu, J.-B.; Li, Y.-Z.; Huang, S.; Chen, L.; Luo, Y.-Y.; Gao, F.; Zhou, X.-L. Diterpenoid alkaloids from the whole herb of Delphinium grandiflorum L. Phytochemistry 2021, 190, 112866. [Google Scholar] [CrossRef]
- Pang, L.; Liu, C.Y.; Gong, G.H.; Quan, Z.S. Synthesis, in vitro and in vivo biological evaluation of novel lappaconitine derivatives as potential anti-inflammatory agents. Acta Pharm. Sin. B 2020, 10, 628–645. [Google Scholar] [CrossRef]
- Li, Y.; Shang, Y.; Li, X.; Zhang, Y.; Xie, J.; Chen, L.; Gao, F.; Zhou, X.-L. Design, synthesis, and biological evaluation of low-toxic lappaconitine derivatives as potential analgesics. Eur. J. Med. Chem. 2022, 243, 114776. [Google Scholar] [CrossRef]
- Bryzgalov, A.O.; Romanov, V.E.; Tolstikova, T.G.; Shults, E.E. Lappaconitine: Influence of halogen substituent on the antiarrhythmic activity. Cardiovasc. Hematol. Agents Med. Chem. 2013, 11, 211–217. [Google Scholar] [CrossRef]
- Tolstikova, T.G.; Shults, E.E.; Bryzgalov, A.O.; Khvostov, M.V.; Romanov, V.E.; Osadchiy, S.A.; Tolstikov, G.A. Effect of the structure of lappaconitine derivatives on antiarrhythmic activity. Chem. Sustain. Dev. 2007, 15, 599–607. [Google Scholar]
- Cheremnykh, K.P.; Savelyev, V.A.; Borisov, S.A.; Ivanov, I.D.; Baev, D.S.; Tolstikova, T.G.; Vavilin, V.A. Hybrides of alkaloid lappaconitine with pyrimidine motif on the anthranilic acid moiety: Design, synthesis, and investigation of antinociceptive potency. Molecules 2020, 25, 5578. [Google Scholar] [CrossRef]
- Teli, S.; Teli, P.; Soni, S.; Sahiba, N.; Agarwal, S. Synthetic aspects of 1,4- and 1,5-benzodiazepines using o-phenylenediamine: A study of past quinquennial. RSC Adv. 2023, 13, 3694–3714. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.; Dhiman, P.; Kumar, S.; Singh, G.; Monga, V. Recent advances in synthesis and medicinal chemistry of benzodiazepines. Bioorg. Chem. 2020, 97, 103668. [Google Scholar] [CrossRef] [PubMed]
- Willy, B.; Dallos, T.; Rominger, F.; Schönhaber, J.; Müller, T.J.J. Three-component synthesis of cryofluorescent 2,4-disubstituted 3H-1,5-benzodiazepines. Conformational control of emission properties. Eur. J. Org. Chem. 2008, 2008, 4796–4805. [Google Scholar] [CrossRef]
- Albano, G.; Aronica, L.A. Acyl Sonogashira cross-coupling: State of the art and application to the synthesis of heterocyclic compounds. Catalyst 2020, 10, 25. [Google Scholar] [CrossRef]
- Niedballa, J.; Müller, T.J.J. Heterocycles by Consecutive Multicomponent Syntheses via Catalytically Generated Alkynoyl Intermediates. Catalyst 2022, 12, 90. [Google Scholar] [CrossRef]
- Osadchii, S.A.; Shults, E.E.; Polukhina, E.V.; Shakirov, M.M.; Vasilevskii, S.F.; Stepanov, A.A.; Tolstikov, G.A. Study of alkaloids of the Siberian and Altai flora 14. Synthesis of alkaloid-based tertiary N-(3-arylprop-2-ynyl)amines. Russ. Chem. Bull. 2007, 56, 1261–1267. [Google Scholar] [CrossRef]
- Cheremnykh, K.P.; Savelyev, V.A.; Pokrovskii, M.A.; Baev, D.S.; Tolstikova, T.G.; Pokrovskii, A.G.; Shults, E.E. Design, synthesis, cytotoxicity, and molecular modeling study of 2,4,6-trisubstituted pyrimidines with anthranilate ester moiety. Med. Chem. Res. 2019, 28, 545–558. [Google Scholar] [CrossRef]
- Cheremnykh, K.P.; Savelyev, V.A.; Shults, E.E. An efficient access to 3,5-disubstituted isoxazoles with anthranilate ester moiety: Alkaloid lappaconitine–aryl conjugates with an isoxazole linker. Asian J. Org. Chem. 2021, 10, 2638–2643. [Google Scholar] [CrossRef]
- Osadchii, S.A.; Shul’ts, E.E.; Polukhina, E.V.; Vasil’ev, V.G.; Tolstikov, G.A. Study of alkaloids of the flora of Siberia and Altai: Synthesis of bivalent ligands of the aconitane type. Dokl. Chem. 2007, 416, 251–256. [Google Scholar] [CrossRef]
- Ahmad, R.; Zia-ul-Haq, M.; Duddeck, H.; Stefaniak, L.; Sitkowski, J. Study of the conformational equilibria of some 2-(2′-hydroxyphenyl)-4-aryl-3H-1,5-benzodiazepines using 1H, 13C, and 15N NMR spectroscopy. Mon. Chem. 1997, 128, 633–640. [Google Scholar] [CrossRef]
- Koster, R.; Anderson, M.; De Beer, E.J. Acetic acid for analgesic screening. Fed. Proc. 1959, 18, 412–415. [Google Scholar]
- Eddy, N.B.; Leimbach, D. Synthetic analgesics. II. Dithienylbutenyl- and dithienylbutylamines. J. Pharmacol. Exp. Ther. 1953, 107, 385–393. [Google Scholar] [PubMed]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharm. Rev. 2001, 53, 597–652. [Google Scholar] [PubMed]
- Sheets, M.F.; Fozzard, H.A.; Lipkind, G.M.; Hanck, D.A. Sodium channel molecular conformations and antiarrhythmic drug affinity. Trends Cardiovas. Med. 2010, 20, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Tosaki, A.; Szerdahelyi, P.; Engelman, R.M.; Das, D.K. Potassium channel openers and blockers: Do they possess proarrhythmic or antiarrhythmic activity in ischemic and reperfused rat hearts? J. Pharmacol. Exp. Ther. 1993, 267, 1355–1362. [Google Scholar]
- Zhang, X.; Gao, Y.; Zhou, Y.; Liu, Z.; Liu, R. Pharmacological mechanism of natural drugs and their active ingredients in the treatment of arrhythmia via calcium channel regulation. Biomed. Pharmacother. 2023, 160, 114413. [Google Scholar] [CrossRef]
- Loussouarn, G.; Sternberg, D.; Nicole, S.; Marionneau, C.; Le Bouffant, F.; Toumaniantz, G.; Barc, J.; Malak, O.A.; Fressart, V.; Péréon, Y.; et al. Physiological and pathophysiological insights of Nav1.4 and Nav1.5 comparison. Front. Pharmacol. 2015, 6, 314. [Google Scholar] [CrossRef]
- Chew, L.A.; Bellampalli, S.S.; Dustrude, E.T.; Khanna, R. Mining the Nav1.7 interactome: Opportunities for chronic pain therapeutics. Biochem. Pharmacol. 2019, 163, 9–20. [Google Scholar] [CrossRef]
- Tavares-Ferreira, D.; Ray, P.R.; Sankaranarayanan, I.; Mejia, G.L.; Wangzhout, A.; Shiers, S.; Uttarkar, R.; Magat, S.; Barragan-Iglesias, P.; Dussor, G.; et al. Sex differences in nociceptor translatomes contribute to divergent prostaglandin signaling in male and female mice. Biol. Psychiatry 2022, 91, 129–140. [Google Scholar] [CrossRef]
- Jiang, D.; Shi, H.; Tonggu, L.; El-Din, T.M.G.; Lenaeus, M.J.; Zhao, Y.; Yoshioka, C.; Zheng, N.; Catterall, W.A. Structure of the cardiac sodium channel. Cell 2020, 180, 122–134. [Google Scholar] [CrossRef]
- Sampson, M. Antiarrhythmic drugs. Part 1: An overview. Br. J. Cardiac. Nurs. 2019, 14, 1–10. [Google Scholar] [CrossRef]
- Dvorak, N.M.; Wadsworth, P.A.; Wang, P.; Zhou, J.; Laezza, F. Development of allosteric modulators of voltage-gated Na+ channels: A novel approach for an old target. Curr. Top. Med. Chem. 2021, 21, 841–848. [Google Scholar] [CrossRef]
- Li, Z.; Jin, X.; Wu, T.; Huang, G.; Wu, K.; Lei, J.; Pan, X.; Yan, N. Structural basis for pore blockade of the human cardiac sodium channel Nav1.5 by the antiarrhythmic drug quinidine. Angew. Chem. Int. Ed. 2021, 60, 11474–11480. [Google Scholar] [CrossRef] [PubMed]
- Mazola, Y.; Márquez Montesinos, J.C.E.; Ramírez, D.; Zúñiga, L.; Decher, N.; Ravens, U.; Yarov-Yarovoy, V.; González, W. Common structural pattern for flecainide binding in atrial-selective Kv1.5 and Nav1.5 channels: A computational approach. Pharmaceutics 2022, 14, 1356. [Google Scholar] [CrossRef] [PubMed]
- Sultankhodzhaev, M.N.; Yunusov, M.S.; Yunusov, S.Y. Karacoline—A new diterpene alkaloid from Aconitum karacolicum. Chem. Nat. Compd. 1972, 8, 399–400. [Google Scholar] [CrossRef]
- Nie, J.; Wang, F.; Ji, T.; Zhao, J.; Zhao, F. Assessment of in vitro cardiotoxicity of extract fractions and diterpene alkaloids from Aconitum leucostomum Worosch: A short communication. J. Pharm. Biomed. Anal. 2017, 137, 84–89. [Google Scholar] [CrossRef]
- Pankrushina, N.A.; Nikitina, I.A.; Anferova, N.V.; Osadchii, S.A.; Shakirov, M.M.; Shults, E.E.; Tolstikov, G.A. Study of alkaloids of the Siberian and Altai flora. 10. Synthesis of N(20)-deethyllappaconitine derivatives. Russ. Chem. Bull. Int. Ed. 2003, 52, 2490–2499. [Google Scholar] [CrossRef]
- Schrodinger Small Molecule Drug Discovery Suite; Schrödinger, LLC: New York, NY, USA, 2020.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Genet. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Dose, mg/kg | Acetic Acid-Induced Writhing Test | Dose, mg/kg | Hot Plate Test |
|---|---|---|---|---|
| Mean ± SD (MPE, %) a | Mean ± SD (MPE, %) b | |||
| 7 | 5 | 9.3 ± 0.94 * (21) | 5 | 6.8 ± 0.37 |
| 8 | 5 | 5.3 ± 0.62 * (54) | 5 | 12.8 ± 1.21 * (62) |
| 9 | 5 | 10.9 ± 1.01 | 5 | 10.0 ± 1.16 |
| 10 | 5 | 10.8 ± 1.66 | 5 | 8.4 ± 0.78 |
| 11 | 5 | 12.0 ± 0.98 | 5 | 8.9 ± 0.74 |
| 12 | 5 | 12.0 ± 1.00 | 5 | 8.9 ± 0.64 |
| 1 | 5 | 5.7 ± 1.25 * (49) | 5 | 12.3 ± 1.13 * (58) |
| Diclofenac sodium | 10 | 3.1 ± 0.47 * (72) | 10 | 11.3 ± 0.49 * (43) |
| Control | - | 11.3 ± 0.62 | - | 7.9 ± 0.51 |
| Compound | Dose, mg/kg | Acetic Acid-Induced Writhing Test | Dose, mg/kg | Hot Plate Test |
|---|---|---|---|---|
| Mean ± SD (MPE, %) a | Mean ± SD (MPE, %) b | |||
| 8 | 5 | 4.9 ± 0.59 * (59) | 5 | 11.8 ± 0.65 * (61) |
| 8 | 1 | 6.6 ± 1.07 * (44) | 1 | 9.6 ± 0.94 * (35) |
| Diclofenac sodium | 10 | 4.3 ± 0.65 * (64) | 10 | 11.4 ± 0.68 * (56) |
| Control | - | 11.9 ± 0.51 | - | 7.3 ± 0.62 |
| Compound | Dose, mg/kg | Surviving Animals (%) | Compound | Dose, mg/kg | Surviving Animals (%) | Compound | Dose, mg/kg | Surviving Animals (%) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Calcium Chloride Arrhythmia (250 mg/kg) | Epinephrine Arrhythmia (0.3 mg/kg) | Calcium Chloride Arrhythmia (250 mg/kg) | Epinephrine Arrhythmia (0.3 mg/kg) | Calcium Chloride Arrhythmia (250 mg/kg) | Epinephrine Arrhythmia (0.3 mg/kg) | ||||||
| 8 | 10 | 0 | 100 | 10 | 10 | 0 | 100 | Allapinine | 2.9 | 100 | 100 |
| 5.0 | 100 | 100 | 5.0 | 100 | 100 | 0.29 | 50 | 50 | |||
| 2.5 | NT | 50 | 0.5 | NT | 100 | 0.029 | 0 | 0 | |||
| 1.0 | NT | 0 | 0.05 | NT | 50 | 0.0029 | 0 | 0 | |||
| Ligand | IFD Calculation Parameters, kcal/mol | |||
|---|---|---|---|---|
| Docking Score | Ligand Efficiency | Emodel | IFD Score | |
| 1 | −9.903 | −0.236 | −74.681 | −2133.29 |
| 8 | −17.711 | −0.295 | −108.167 | −2241.11 |
| 10 | −16.595 | −0.277 | −137.495 | −2239.37 |
| 15 | −9.529 | −0.222 | −79.817 | −2133.29 |
| 18 | −9.547 | −0.354 | −43.472 | −2223.73 |
| 19 | −11.250 | −0.268 | −100.692 | −2227.94 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheremnykh, K.P.; Bryzgalov, A.O.; Baev, D.S.; Borisov, S.A.; Sotnikova, Y.S.; Savelyev, V.A.; Tolstikova, T.G.; Sagdullaev, S.S.; Shults, E.E. Synthesis, Pharmacological Evaluation, and Molecular Modeling of Lappaconitine–1,5-Benzodiazepine Hybrids. Molecules 2023, 28, 4234. https://doi.org/10.3390/molecules28104234
Cheremnykh KP, Bryzgalov AO, Baev DS, Borisov SA, Sotnikova YS, Savelyev VA, Tolstikova TG, Sagdullaev SS, Shults EE. Synthesis, Pharmacological Evaluation, and Molecular Modeling of Lappaconitine–1,5-Benzodiazepine Hybrids. Molecules. 2023; 28(10):4234. https://doi.org/10.3390/molecules28104234
Chicago/Turabian StyleCheremnykh, Kirill P., Arkadiy O. Bryzgalov, Dmitry S. Baev, Sergey A. Borisov, Yulia S. Sotnikova, Victor A. Savelyev, Tatyana G. Tolstikova, Shamansur S. Sagdullaev, and Elvira E. Shults. 2023. "Synthesis, Pharmacological Evaluation, and Molecular Modeling of Lappaconitine–1,5-Benzodiazepine Hybrids" Molecules 28, no. 10: 4234. https://doi.org/10.3390/molecules28104234