
Reversal of Enantioselectivity in the Conjugate Addition Reaction of Cyclic Enones with the CuOTf/Azolium Catalytic System


Abstract
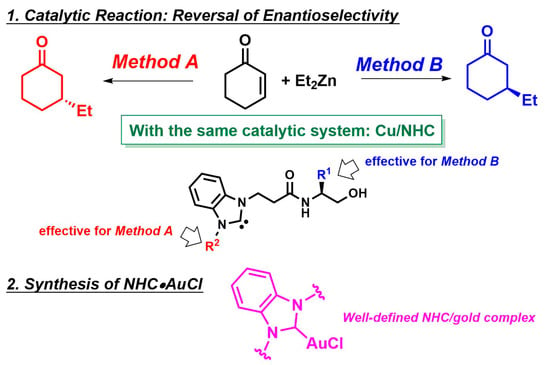
:
1. Introduction
2. Results and Discussion
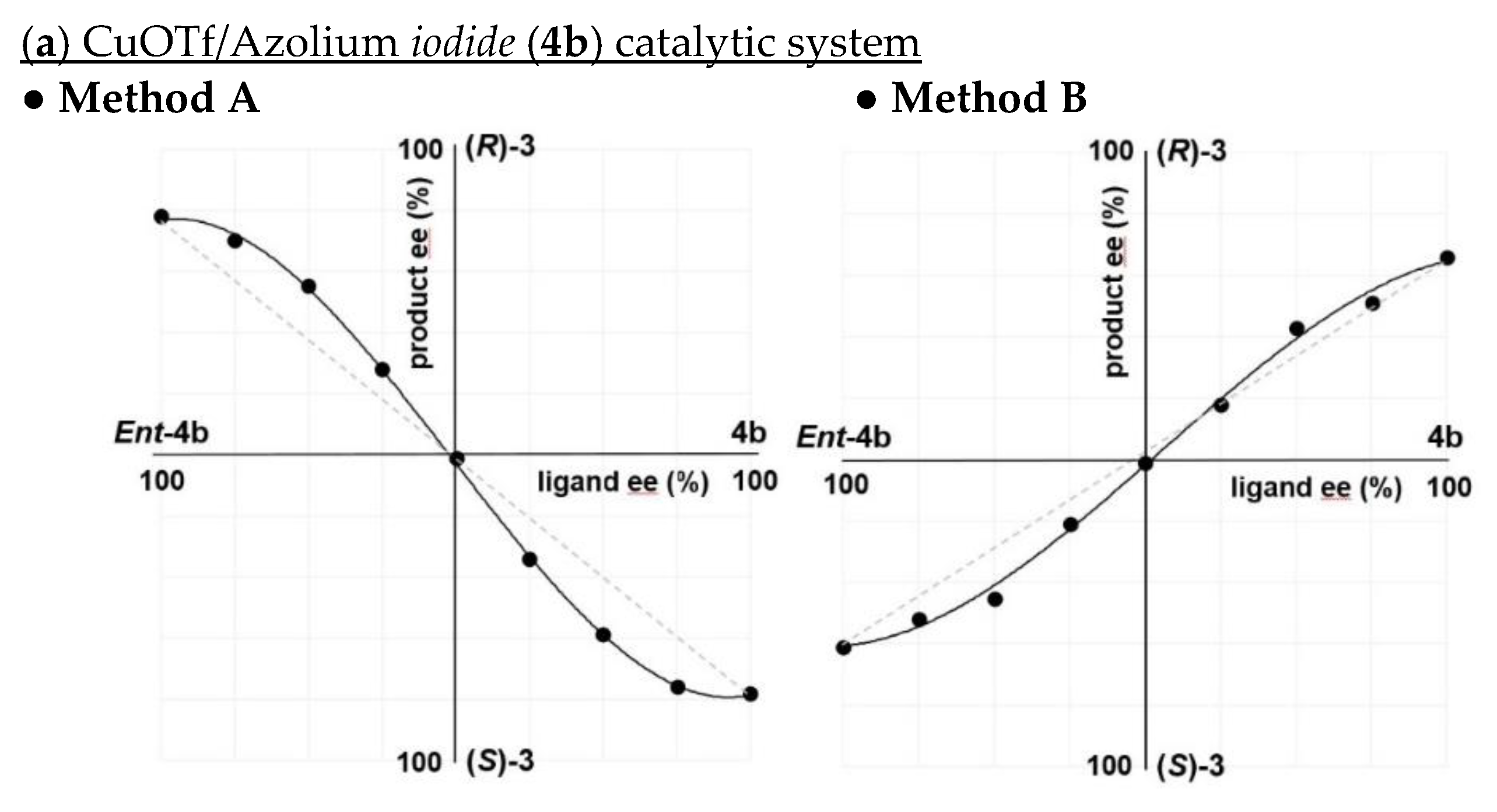
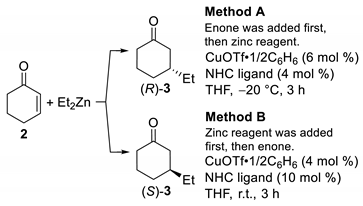
2.1. Switching of Enantioselectivity in the Cu-Catalyzed ACA Reaction Using Azolium Iodide
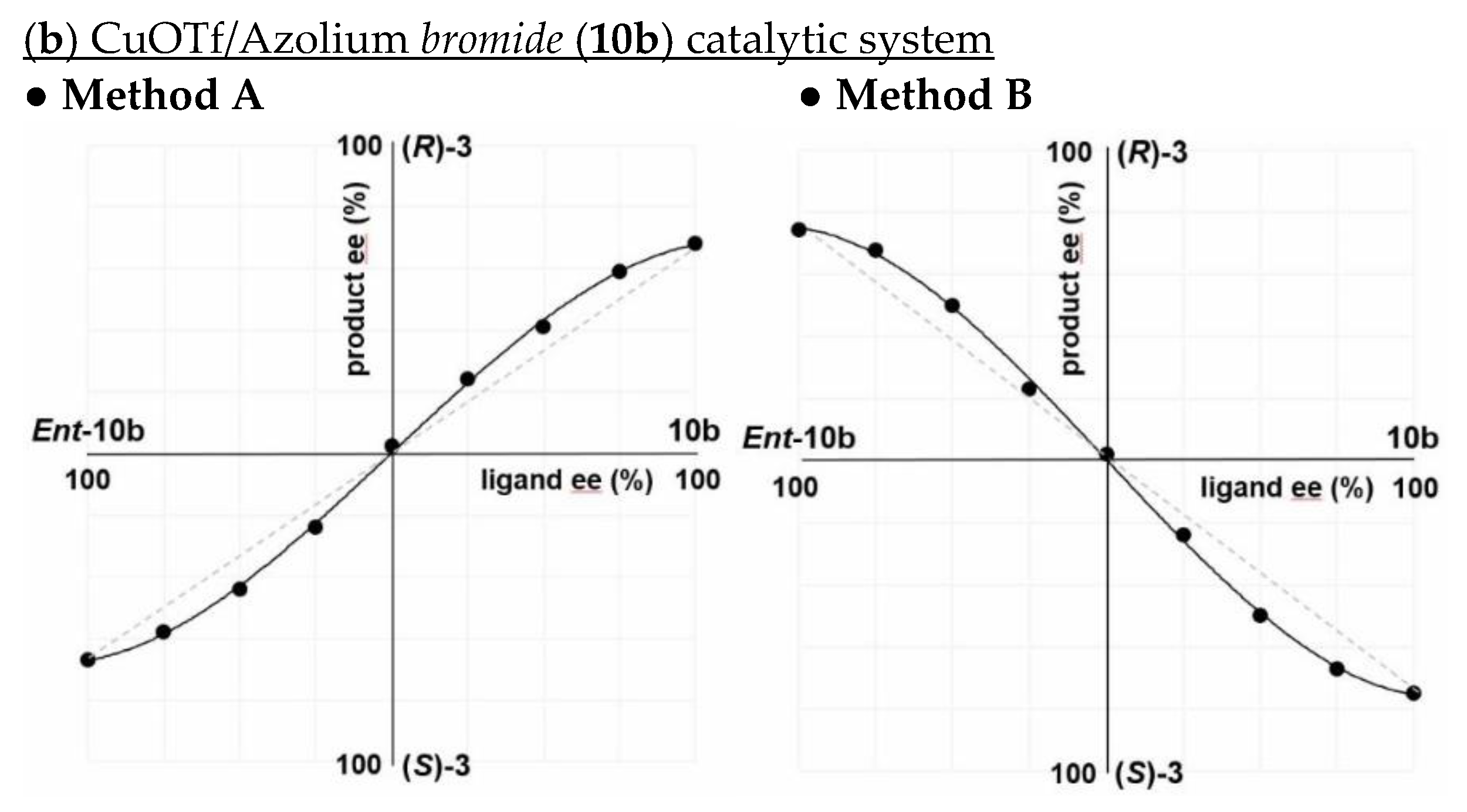
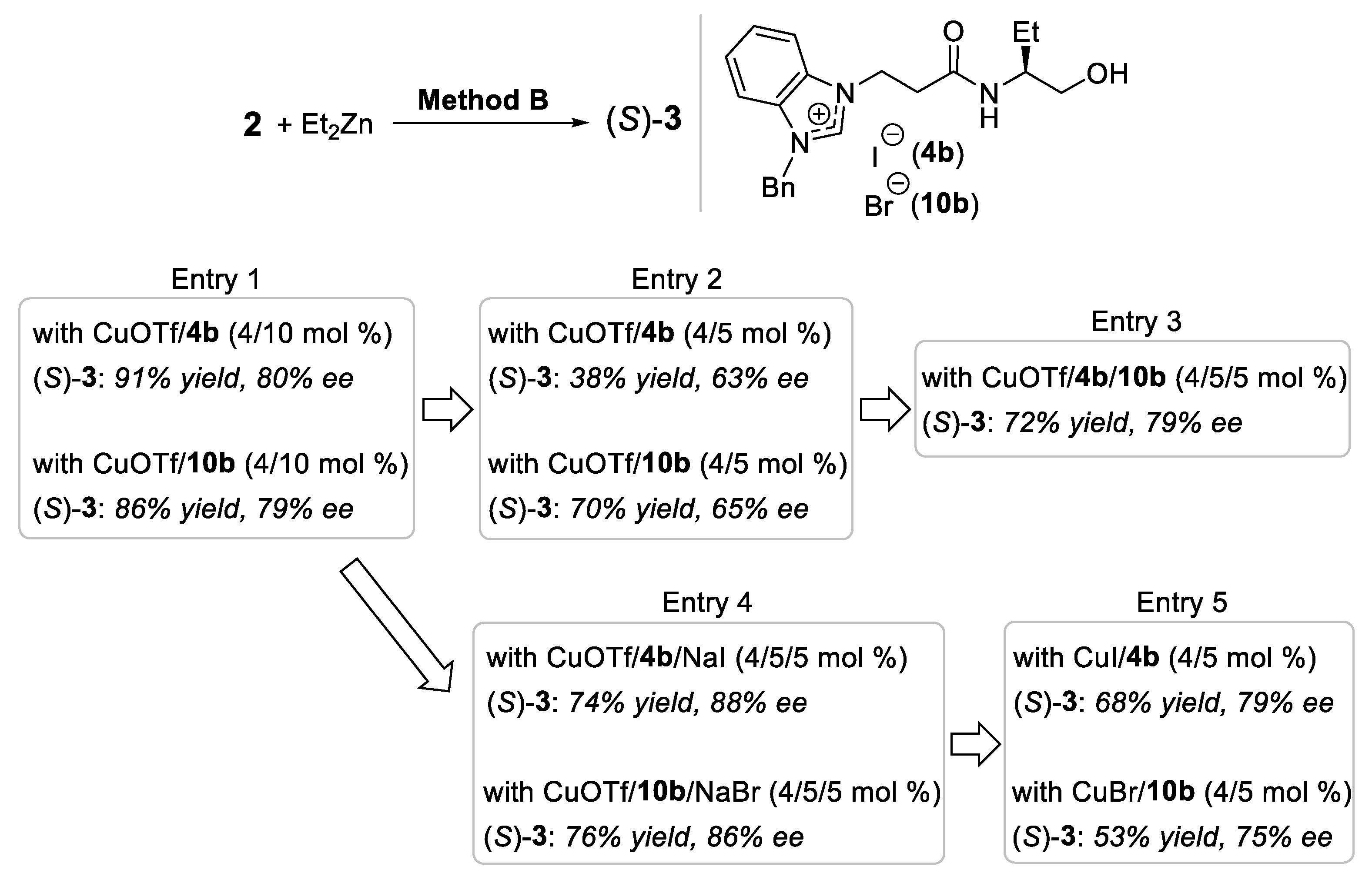
2.2. Influence of Counter Anion on Azolium Salt: The Effect of Halide Ion
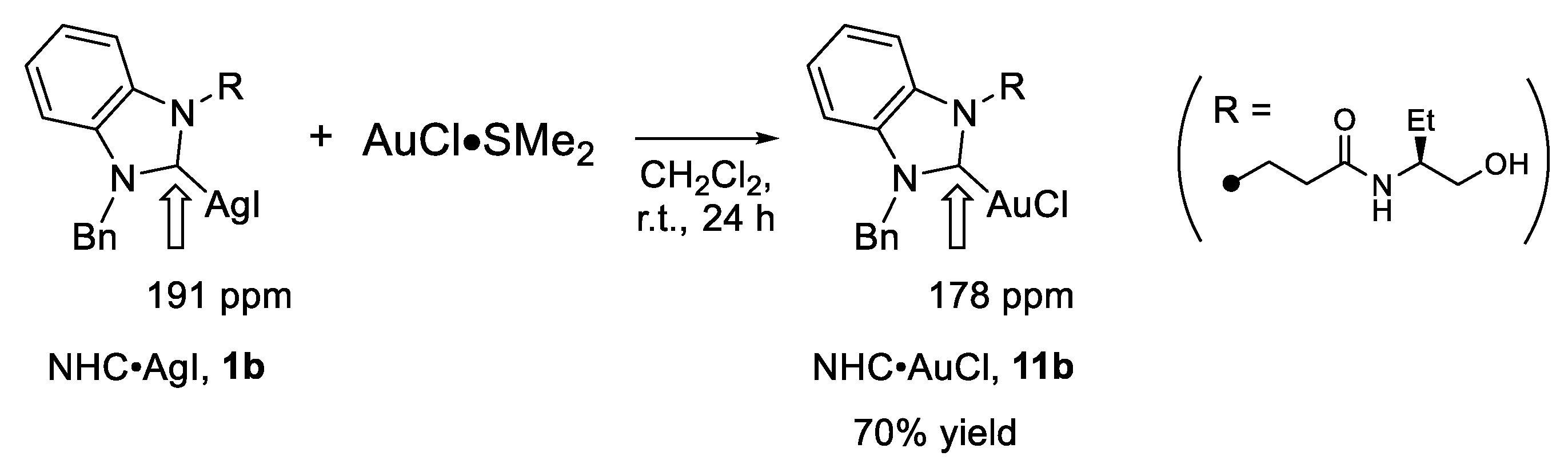
2.3. Investigation of the Reaction of NHC•AgI 1b with CuOTf
3. Materials and Methods
3.1. General Procedures
3.2. General Procedure for Method A
3.3. General Procedure for Method B
3.4. Procedure for Reaction of NHC•AgI Complex 1b with CuOTf•1/2C6H6
3.5. Procedure for Synthesis of NHC•AuCl Complex 1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hayashi, M.; Matsubara, R. Recent topics on catalytic asymmetric 1,4-addition. Tetrahedron Lett. 2017, 58, 1793–1805. [Google Scholar] [CrossRef]
- Alexakis, A.; Krause, N.; Woodward, S. (Eds.) Copper-Catalyzed Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Jerphagnon, T.; Pizzuti, M.G.; Minnaard, A.J.; Feringa, B.L. Recent advances in enantioselective copper-catalyzed 1,4-addition. Chem. Soc. Rev. 2009, 38, 1039–1075. [Google Scholar] [CrossRef] [Green Version]
- Alexakis, A.; Bäckvall, J.E.; Krause, N.; Pàmies, O.; Diéguez, M. Enantioselective copper-catalyzed conjugate addition and allylic substitution reactions. Chem. Rev. 2008, 108, 2796–2823. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhang, W. Renaissance of pyridine-oxazolines as chiral ligands for asymmetric catalysis. Chem. Soc. Rev. 2018, 47, 1783–1810. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Müller, D.; Schlepphorst, C.; Glorius, F. Privileged chiral N-heterocyclic carbene ligands for asymmetric transition-metal catalysis. Chem. Soc. Rev. 2017, 46, 4845–4854. [Google Scholar] [CrossRef]
- Zhou, Q.-L. (Ed.) Privileged Chiral Ligands and Catalysts; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Yoon, T.P.; Jacobsen, E.N. Privileged chiral catalysis. Science 2003, 299, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.P.; Nájera, C.; Yus, M. Stereodivergent catalysis. Chem. Rev. 2018, 118, 5080–5200. [Google Scholar] [CrossRef]
- Blanco, V.; Leigh, D.A.; Marcos, V. Artificial switchable catalysts. Chem. Soc. Rev. 2015, 44, 5341–5370. [Google Scholar] [CrossRef] [Green Version]
- Escorihuela, J.; Isabel Burguete, M.; Luis, S.V. New advances in dual stereocontrol for asymmetric reactions. Chem. Soc. Rev. 2013, 42, 5595–5617. [Google Scholar] [CrossRef]
- Bartók, M. Unexpected inversions in asymmetric reactions: Reactions with chiral metal complexes. Chem. Rev. 2010, 110, 1633–1705. [Google Scholar] [CrossRef]
- Tanaka, T.; Hayashi, M. Approach for complete reversal of enantioselectivity using a single chiral source. Synthesis 2008, 2008, 3361–3376. [Google Scholar]
- Zanoni, G.; Castronovo, F.; Franzini, M.; Vidari, G.; Giannini, E. Toggling enantioselective catalysis-a promising paradigm in the development of more efficient and versatile enantioselective synthetic methodologies. Chem. Soc. Rev. 2003, 32, 115–129. [Google Scholar] [CrossRef]
- Nakano, Y.; Sakaguchi, S. Inversions in asymmetric conjugate addition reaction of cyclic enones catalyzed by the Cu/NHC-AgX system: Factors affecting the stereoselective formation of both enantiomers. J. Organomet. Chem. 2017, 846, 407–416. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nakano, Y.; Shibata, N.; Sakaguchi, S. Enantioselectivity switch in copper-catalyzed conjugate addition reactions under the influence of a chiral N-heterocyclic carbene-silver complex. RSC Adv. 2016, 6, 7755–7759. [Google Scholar] [CrossRef]
- Lin, J.C.Y.; Huang, R.T.W.; Lee, C.S.; Bhattacharyya, A.; Hwang, W.S.; Lin, I.J.B. Coinage metal-N-heterocyclic carbene complexes. Chem. Rev. 2009, 109, 3561–3598. [Google Scholar] [CrossRef]
- Hahn, F.E.; Jahnke, M.C. Heterocyclic carbenes: Synthesis and coordination chemistry. Angew. Chem. Int. Ed. 2008, 47, 3122–3172. [Google Scholar] [CrossRef]
- Lin, I.J.B.; Vasam, C.S. Preparation and application of N-heterocyclic carbene complexes of Ag(I). Coord. Chem. Rev. 2007, 251, 642–670. [Google Scholar] [CrossRef]
- De Frémont, P.; Scott, N.M.; Stevens, E.D.; Ramnial, T.; Lightbody, O.C.; Macdonald, C.L.B.; Clyburne, J.A.C.; Abernethy, C.D.; Nolan, S.P. Synthesis of well-defined N-heterocyclic carbene silver(I) complexes. Organometallics 2005, 24, 6301–6309. [Google Scholar] [CrossRef]
- Garrison, J.C.; Youngs, W.J. Ag(I) N-heterocyclic carbene complexes: Synthesis, structure, and application. Chem. Rev. 2005, 105, 3978–4008. [Google Scholar] [CrossRef]
- Budagumpi, S.; Keri, R.S.; Achar, G.; Brinda, K.N. Coinage metal complexes of chiral N-heterocyclic carbene ligands: Syntheses and applications in asymmetric catalysis. Adv. Synth. Catal. 2020, 362, 970–997. [Google Scholar] [CrossRef]
- Khose, V.N.; John, M.E.; Pandey, A.D.; Karnik, A.V. Chiral benzimidazoles and their applications in stereodiscrimination processes. Tetrahedron Asymmetry 2017, 28, 1233–1289. [Google Scholar] [CrossRef]
- Wang, F.; Liu, L.-j.; Wang, W.; Li, S.; Shi, M. Chiral NHC–metal-based asymmetric catalysis. Coord. Chem. Rev. 2012, 256, 804–853. [Google Scholar] [CrossRef]
- Gade, L.H.; Bellemin-Laponnaz, S. Mixed oxazoline-carbenes as stereodirecting ligands for asymmetric catalysis. Coord. Chem. Rev. 2007, 251, 718–725. [Google Scholar] [CrossRef]
- César, V.; Bellemin-Laponnaz, S.; Gade, L.H. Chiral N-heterocyclic carbenes as stereodirecting ligands in asymmetric catalysis. Chem. Soc. Rev. 2004, 33, 619–636. [Google Scholar] [CrossRef]
- Perry, M.C.; Burgess, K. Chiral N-heterocyclic carbene-transition metal complexes in asymmetric catalysis. Tetrahedron Asymmetry 2003, 14, 951–961. [Google Scholar] [CrossRef]
- Herrmann, W.A. N-Heterocyclic carbenes: A new concept in organometallic catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Magrez, M.; Wencel-Delord, J.; Alexakis, A.; Crévisy, C.; Mauduit, M. Significant asymmetric amplification in enantioselective Cu/DiPPAM-catalyzed 1,6- and 1,4-conjugate additions of diethylzinc to (di)enone. Org. Lett. 2012, 14, 3576–3579. [Google Scholar] [CrossRef]
- Satyanarayana, T.; Abraham, S.; Kagan, H.B. Nonlinear effects in asymmetric catalysis. Angew. Chem. Int. Ed. 2009, 48, 456–494. [Google Scholar] [CrossRef]
- Luukas, T.O.; Fenwick, D.R.; Kagan, H.B. Presence or absence of a nonlinear effect according to the asymmetric catalyst preparation in the alkylation of benzaldehyde. C. R. Chim. 2002, 5, 487–491. [Google Scholar] [CrossRef]
- Walsh, P.J. Titanium-Catalyzed Enantioselective Additions of Alkyl Groups to Aldehydes: Mechanistic Studies and New Concepts in Asymmetric Catalysis. Acc. Chem. Res. 2003, 36, 739–749. [Google Scholar] [CrossRef]
- Li, D.; Ollevier, T. Mechanism studies of oxidation and hydrolysis of Cu(I)-NHC and Ag-NHC in solution under air. J. Organomet. Chem. 2020, 906, 121025. [Google Scholar] [CrossRef]
- Li, D.; Ollevier, T. Synthesis of imidazolidinone, imidazolone, and benzimidazolone derivatives through oxidation using copper and air. Org. Lett. 2019, 21, 3572–3575. [Google Scholar] [CrossRef]
- Cervantes-Reyes, A.; Rominger, F.; Hashmi, A.S.K. Sterically demanding Ag(I) and Cu(I) N-heterocyclic carbine complexes: Synthesis, structures, steric parameters and catalytic activity. Chem. Eur. J. 2020, 26, 5530–5540. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.; Gimeno, M.C. An efficient and sustainable synthesis of NHC gold complexes. Chem. Commun. 2016, 52, 9664–9667. [Google Scholar] [CrossRef] [PubMed]
- Marion, N.; Nolan, S.P. N-Heterocyclic carbenes in gold catalysis. Chem. Soc. Rev. 2008, 37, 1776–1782. [Google Scholar] [CrossRef]
- Shibata, N.; Yoshimura, M.; Yamada, H.; Arakawa, R.; Sakaguchi, S. Hydroxy-amide functionalized azolium salts for Cu-catalyzed asymmetric conjugate addition: Stereocontrol based on ligand structure and copper precatalyst. J. Org. Chem. 2012, 77, 4079–4086. [Google Scholar] [CrossRef]


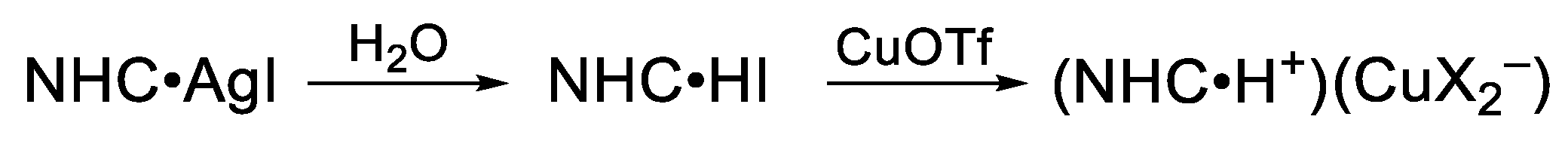

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | NHC Ligand | Method A 1 | Method B 2 |
| entry 1 3 |  | (R)-3 84% yield 74% ee | (S)-3 84% yield 87% ee |
| entry 2 | 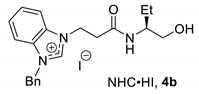 | (R)-3 91% yield 60% ee | (S)-3 91% yield 80% ee |
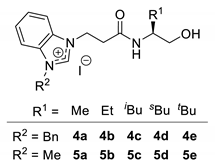 | |||||
|---|---|---|---|---|---|
| Entry | Azolium Salt (R1) | Method A 1 | Method B 2 | ||
| Product: (R)-3 | Product: (S)-3 | ||||
| Yield [%] | Ee [%] | Yield [%] | Ee [%] | ||
| entry 1 | 4a (Me) | 72 | 63 | 77 | 55 |
| entry 2 | 4b (Et) | 91 | 60 | 91 | 80 |
| entry 3 | 4c (iBu) | 57 | 65 | 85 | 87 |
| entry 4 | 4d (sBu) | 88 | 71 | 78 | 50 |
| entry 5 | 4e (tBu) | 90 | 75 | 46 | −42 3 |
| entry 6 | 5a (Me) | 89 | 50 | 77 | 74 |
| entry 7 | 5b (Et) | 86 | 53 | 80 | 89 |
| entry 8 | 5c (iBu) | 88 | 56 | 89 | 86 |
| entry 9 | 5d (sBu) | 83 | 60 | 77 | 56 |
| entry 10 | 5e (tBu) | 82 | 61 | 80 | −36 3 |
| Entry | Method A 1 | Method B 2 |
|---|---|---|
| entry 1 | 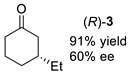 | 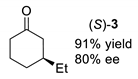 |
| entry 2 3 | 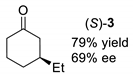 |  |
| entry 3 4 | 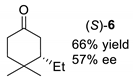 |  |
| entry 4 |  | 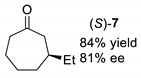 |
| entry 5 5 | n.d. 6 |  |
| entry 6 5 | n.d. 6 |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakano, Y.; Shimizu, S.; Takeda, C.; Sakaguchi, S. Reversal of Enantioselectivity in the Conjugate Addition Reaction of Cyclic Enones with the CuOTf/Azolium Catalytic System. Molecules 2021, 26, 3404. https://doi.org/10.3390/molecules26113404
Nakano Y, Shimizu S, Takeda C, Sakaguchi S. Reversal of Enantioselectivity in the Conjugate Addition Reaction of Cyclic Enones with the CuOTf/Azolium Catalytic System. Molecules. 2021; 26(11):3404. https://doi.org/10.3390/molecules26113404
Chicago/Turabian StyleNakano, Yuki, Satoki Shimizu, Chihiro Takeda, and Satoshi Sakaguchi. 2021. "Reversal of Enantioselectivity in the Conjugate Addition Reaction of Cyclic Enones with the CuOTf/Azolium Catalytic System" Molecules 26, no. 11: 3404. https://doi.org/10.3390/molecules26113404