
Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases


Beijing Key Laboratory of Bioprocess, College of Life Science and Technology, Beijing University of Chemical Technology, Beijing 100029, China
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(8), 1622; https://doi.org/10.3390/molecules24081622
Submission received: 30 January 2019
/
Revised: 12 April 2019
/
Accepted: 17 April 2019
/
Published: 24 April 2019
(This article belongs to the Special Issue Recent Advances in Self-Assembled Peptides)
Abstract
:Fused in sarcoma (FUS) is a DNA/RNA binding protein that is involved in RNA metabolism and DNA repair. Numerous reports have demonstrated by pathological and genetic analysis that FUS is associated with a variety of neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration (FTLD), and polyglutamine diseases. Traditionally, the fibrillar aggregation of FUS was considered to be the cause of those diseases, especially via its prion-like domains (PrLDs), which are rich in glutamine and asparagine residues. Lately, a nonfibrillar self-assembling phenomenon, liquid–liquid phase separation (LLPS), was observed in FUS, and studies of its functions, mechanism, and mutual transformation with pathogenic amyloid have been emerging. This review summarizes recent studies on FUS self-assembling, including both aggregation and LLPS as well as their relationship with the pathology of ALS, FTLD, and other neurodegenerative diseases.
1. Introduction
Fused in sarcoma (FUS) is a DNA/RNA binding protein containing 526 amino acids. The FUS gene was initially identified as a fusion oncogene on chromosome 16 in human liposarcoma [1], the translocation and fusion of which to transcription factors results in strong transcriptional activation of the proteins. FUS is one of the components of the heterogeneous nuclear ribonucleoprotein (hnRNP) complex. Increasing evidence suggests that FUS is involved in various cellular processes, including gene transcription and regulation, DNA repair, RNA shearing, RNA transport, translation, processing of microRNAs, and maintenance of genomic stability [2]. FUS can bind to RNA, ssDNA, and possibly to dsDNA [3]. The most important binding sequence of FUS is GUGGU, which is rich in 5′ untranslated regions (UTRs), and mutant FUS preferentially binds 3′-UTR and intron sequences [4,5,6]. As an abundant nuclear protein, FUS forms stable complexes with numerous members of the hnRNP family, and some components of the FUS–hnRNP complex have RNA-binding activity [7,8]. The physiological functions of FUS are apparently diverse. Here, we describe its role in RNA metabolism, DNA repair, and its relationship to disease.
1.1. Role of FUS in RNA Metabolism
FUS is well-studied for its transcriptional regulation function in cells. FUS binds to single-stranded DNA motifs in the promoter region of certain genes, which are transcribed by RNA polymerases II and III, and then accumulates near the transcription start site (TSS), resulting in changes of transcription levels [9,10]. Additionally, FUS also regulates transcription levels partly via the interaction with specific transcription factors (e.g., Spi-1/PU.1, NF-κB, and Runx2) and transcription initiation factor TFIID [11,12,13,14]. One recognized mechanism of this regulation is that FUS binds to and recruits RNA polymerase II, subsequently altering its phosphorylation status [15]. RNA can promote the oligomerization of FUS, allowing it to form high-order components near TSS, which interact with CTD of RNA polymerase II. This allows more RNA polymerase II to be recruited to TSS and also prevents premature phosphorylation of Ser2 in CTD, stimulating the transition of polymerase from initiation to activity extension [5]. FUS also interacts with the DNA binding domain of certain nuclear hormone receptors to initiate their transcription [16]. Immunofluorescence studies have found that FUS preferentially localizes on active chromatin to execute its role in transcription regulation [17]. Specifically, during meiosis, FUS binds to autosomes other than transcriptionally silent sex chromosomes [18]. Similarly, it does not bind to transcriptionally silenced chromatin during mitosis [19].
FUS modulates RNA splicing in conjunction with splicing regulators or precursor mRNAs [19]. FUS is a transporter of mRNA between the cytoplasm and the nucleus. For instance, the transfer of mRNA between neuronal dendrites and dendritic spines by FUS is essential for neuronal cell maturation, plasticity, and dendrite integrity [20]. Other relevant studies have indicated that FUS boosts the synthesis of microRNAs, as FUS is an integral part of the Drasha complex, which is required as a ribonuclease III enzyme for microRNA biosynthesis [21].
1.2. Role of FUS in DNA Damage Repair
During DNA damage repair, FUS is one of the proteins firstly recruited to the DNA damage site [22,23]. Loss of FUS results in impaired ATM/γH2AX (ataxia telangiectasia-mutated gene) signaling. The mechanism by which FUS regulates DNA damage repair also depends on interactions with histone deacetylase 1 (HADC1) and poly-ADP ribose, a by-product of DNA damage [22,24]. Knockdown of FUS during cell growth leads to defects in DNA damage recovery, and disease-associated mutations may limit FUS participation in DNA damage response (DDR) [25].
In 1999, Baechtold demonstrated that FUS participates in DDR, which promotes complementary ssDNA annealing and single-stranded oligonucleotide uptake of homologous supercoiled DNA to form a D-loop. The D-loop formation is a principal step in DNA double-strand break repair via recombination, and the oncogenic fusion form FUS–CHOP does not promote DNA pairing [26]. Wild-type FUS can be phosphorylated by ATM in response to DNA double-strand breaks (DSBs), as phosphorylated FUS binds to dsDNA cleavage and Holliday junctions [27]. When DNA damage occurs, FUS is recruited by sense and antisense noncoding RNAs transcribed from 5′ regulatory regions of the cyclin D1 (CCND1) gene [28]. FUS directly interacts with the CREB-binding protein/p300 histone acetyltransferase via its N-terminal domain, resulting in inhibition of CCND1 transcription [28]. Interestingly, FUS’s ability to respond to DNA damage relies on allosteric interactions with single-stranded, low-copy-number long noncoding RNA transcripts [28]. Although the detailed processes of FUS in DDR have not been fully illustrated, its important function in DDR is without question.
1.3. The Link between FUS and Neurodegenerative Disease
Amyotrophic lateral sclerosis (ALS or Lou Gehrig’s disease) is a well-known neurodegenerative disease caused by the loss of both upper and lower motor neurons, first described by the French neurologist Jean-Martin Charcot in 1869 [29]. The first mutation of FUS associated with ALS was discovered in 2009 and later on FUS-immunoreactive cytoplasmic inclusions were found in ALS–FUS patients [30]. Shortly thereafter, FUS aggregation has been observed in various neurodegenerative diseases, such as frontotemporal lobar degeneration (FTLD), the polyglutamine diseases (Huntington’s disease, spinocerebellar ataxia, and dentatorubral–pallidoluysian atrophy). Further studies showed that normal FUS proteins are mainly located in the nucleus, whereas the mutants are primarily found in the cytoplasm [31,32,33]. FUS mutations have also been detected in patients with essential tremor (ET) and Parkinson’s disease. For example, a mutation in its exon (c.868C > T) leads to a stop mutation during FUS expression (p.Q290X), resulting in a potential cause of ET [34,35]. Interestingly, FUS usually coaggregates with other FET proteins in the pathologic inclusions of FTLD, while in all cases of ALS with FUS inclusions, the FUS mutant is dominant in the composition of ALS FUS inclusion [36,37]. Further studies showed that normal FUS proteins are mainly located in the nucleus, whereas the mutants are primarily found in the cytoplasm. Most mutations alter the C-terminal nuclear localization signal, resulting in FUS redistribution from the nucleus to the cytoplasm. Excess FUS in the cytoplasm is involved in the formation of stress granules (SGs) during stress, which are membraneless granules composed of mRNAs, ribosome translation initiation factors, and other RNA binding proteins [38,39,40,41]. These granules can be induced by various cellular stresses such as oxidative stress, mitochondrial dysfunction, and viral infection that inhibit translation initiation [42].
FUS has two possible models for cytoplasmic aggregation leading to neurological diseases. One is the gain-of-function model, in which FUS gains a toxic function in the cytoplasm. FUS may sequester important regulators or trigger abnormal signaling pathways to alter cell physiology [43]. The alternative model is that FUS aggregation depletes functional FUS, especially in the dendritic spines of neurons, which can cause instability of mRNA and trigger downstream immune responses such as inflammation, resulting in further neuronal damage [44]. Loss of FUS in the nucleus affects transcription, alternative splicing, and also DNA repair [45]. FUS cytoplasmic aggregation may spread across the anatomical network in a prion-like manner, as observed by biophysical and histological analysis, ultimately leading to neurodegenerative disease [46].
2. Domains of the FUS Protein
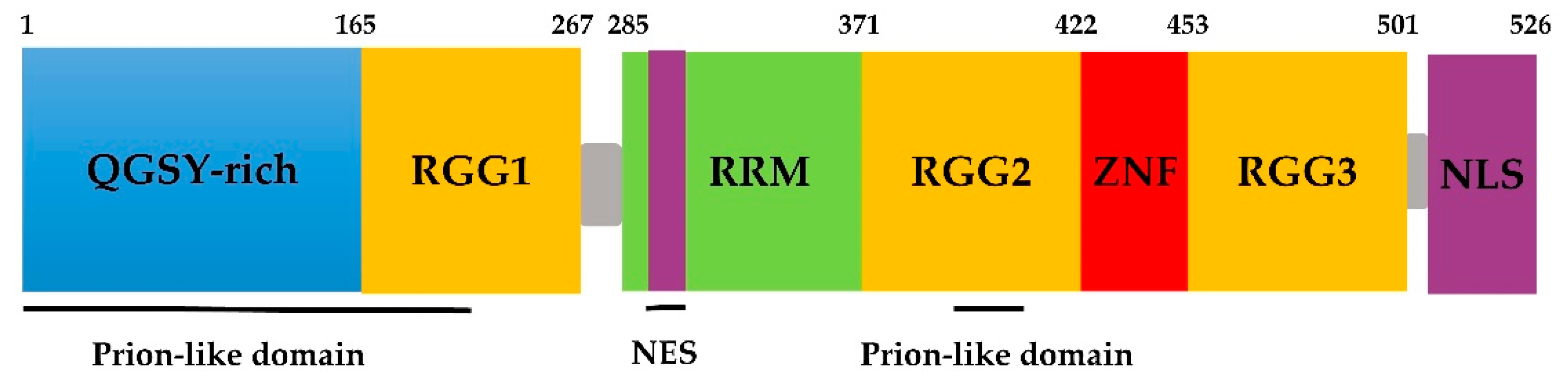
FUS belongs to the FET/TET family, in which proteins share domains similar in both structure and function [47]. FUS contains an N-terminal transcriptional activation domain, which is a glutamine-glycine-serine-tyrosine-rich domain (QGSY-rich domain, amino acids 1–165); three arginine-glycine-glycine repetitive region (RGG, named RGG1–RGG3); an RNA recognition motif (RMM, amino acids 285–371); a Cys2–Cys2 zinc finger (ZnF) motif; a nuclear export signal (NES); and a C-terminus nonclassical nuclear positioning signal (NLS) (Figure 1) [48,49]. In addition, according to bioinformatics analysis, FUS has two prion-like domains (PrLDs) predicted to be amino acids 1–239 and 391–407 [50].
The binding of FUS to RNA may be related to the ZnF motif. The RGG–ZnF–RGG domain is thought to be the major RNA-binding sequence [7,51]. Furthermore, Lerga et al. identified that RNA oligoribonucleotides bind to the FUS protein via the GGUG motif [52]. Survival motor neuron (SMN) proteins are part of a large multiprotein complex that plays a vital role in the biogenesis of small nuclear ribonucleoprotein (snRNP) granules, which results in the fatal childhood motor neuron disease spinal muscular atrophy (SMA) [53]. Sun et al. further demonstrated that the RGG domain in FUS and the Tudor domain in SMN are necessary for the protein–protein interaction [32]. The RGG2 domain recruits FUS to DNA damage sites, which is enhanced by PrLDs [23]. The C-terminal NLS of FUS is recognized by the nuclear input receptor, facilitating the transportation of the protein from the cytoplasm to the nucleus. The majority of mutations discovered in familial ALS is in the NLS. Loss-of-function mutations or deletion in this signaling motif prevents FUS from binding to transport proteins and being transported into the nucleus, and eventually, FUS accumulates in the cytoplasm [54,55].
3. FUS Self-Assembly
3.1. Prion-Like Domains and Self-Assembly of FUS
Prions are self-replicating proteins with misfolded conformations and can lead to diseases, also known as infectious proteins. A single prion can act as a template to fold soluble proteins containing the same amino acid sequence into the prion conformation, resulting in self-replication [56]. Prions typically form stable amyloid fibers with a hallmark “cross-β” structure [57,58]. Yeast prions such as Sup35, Ure2, and Rnq1 are rich in uncharged polar amino acids (glutamine, asparagine, tyrosine, and serine) and glycines, which are essential for prion-like propagation and amyloid formation, and those areas are referred to as the “prion domain”. Adding this region to other innocuous proteins is sufficient to confer prion behavior [59]. Proteins containing prion-related Q/N-rich domains could induce conformational changes and finally lead to self-assembling, which alters protein functions such as SG formation and synaptic translation [60]. The most neurodegenerative diseases share similar cellular and biochemical mechanisms, which is the accumulation of misfolded proteins in brains. Thus, numerous reports suggest that aggregation-prone RNA-/DNA-binding proteins with PrLDs cause neurodegenerative disease by seed formation in a prion-like manner [41,61]. In other words, misfolded proteins act as seeds of aggregation, folding their primary subtypes and converting them into pathological aggregates, which ultimately generate neurological diseases through further recruitment and transformation [62]. A typical example of a pathogenic PrLD is that the trinucleotide is repeatedly amplified in the gene encoding ataxin 1 (ATXN1) in PrLDs and promotes aggregation of ATXN1, leading to polyglutamine protein production and, ultimately, to spinocerebellar ataxia type 1 [61,63,64,65].
The experimental results from the interactions between FUS and polyglutamine inclusions and the predictions suggested that the FUS prion-like domains are located in the N-terminal QGSY domain and are part of the first RGG domain (amino acids 1–239), with an additional region in the RGG3 domain (amino acids 391–405) [66,67]. By using a prediction algorithm based on a yeast prion domain to score 27,879 human proteins, FUS ranked 15th for its prion-like property and 1st in RNA binding proteins [45]. Prion-like domains in RNA-binding proteins are essential for neuronal proteins to enter ribonucleoprotein granules, forming a functional assembling state, which also drives pathological protein aggregation in neurons. It can therefore be speculated that the prion-like domain in FUS is essential for its aggregation in neurons [68]. It was reported that the PrLD of FUS can be assembled into amyloid-like fibrils in a cell-free system and the morphology of aggregates is similar to the general pathogenic amyloid fibers. Reports showed that the process is reversible and highly regulated. Amyloid fibrils formed by FUS are susceptible to depolymerization due to a variety of factors, including FUS concentration, DNA or RNA levels, and SYGQ-rich domain phosphorylation [50,69,70,71,72,73,74].
Phase separation is a physical process in which a supersaturated solution of components spontaneously separates into two phases with different densities, both stable and coexisting [75]. This phenomenon is common in polymer chemistry and has recently been found in biomacromolecules [76]. For example, FUS undergoes liquid–liquid phase separation (LLPS) both in vivo and in vitro at physiological concentrations [77]. It is progressively recognized that phase separation controls the formation of membrane-free organelles, regulating biological functions and activities [78]. FUS is rapidly recruited to the site of DNA damage, then phase separation allows FUS to locally form droplet-like compartments, increasing the concentration of FUS and other substances and possibly promoting DNA repair [79,80].
Studies have shown that FUS has three forms of existence—dispersion, droplets, and hydrogels—and the reversible dynamic phase transition is driven by the low-complexity (LC) domain at the N-terminus of FUS [81]. According to the results of Patel et al., FUS closed compartments have two states of droplets and hydrogels which are reversible and dynamic [82]. Their experiments showed that increasing the concentration of FUS in vivo or in vitro causes its transformation from liquid to an aggregated state. Under stress conditions, FUS rapidly passes through the nucleus to the cytoplasm, then forms SGs [83]. SGs act as condensation sites, where the FUS concentration is locally increased and eventually may aggregate [3].
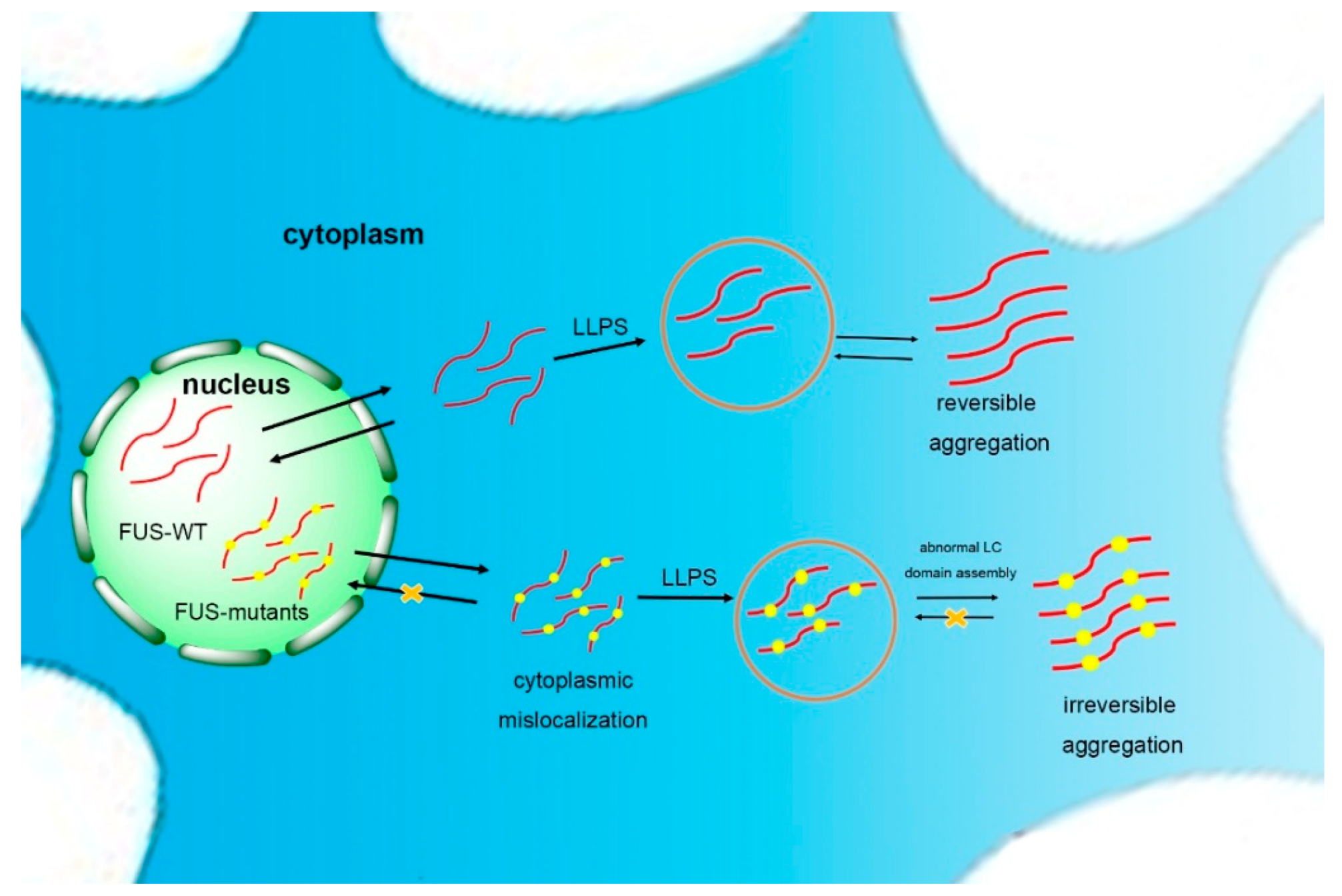
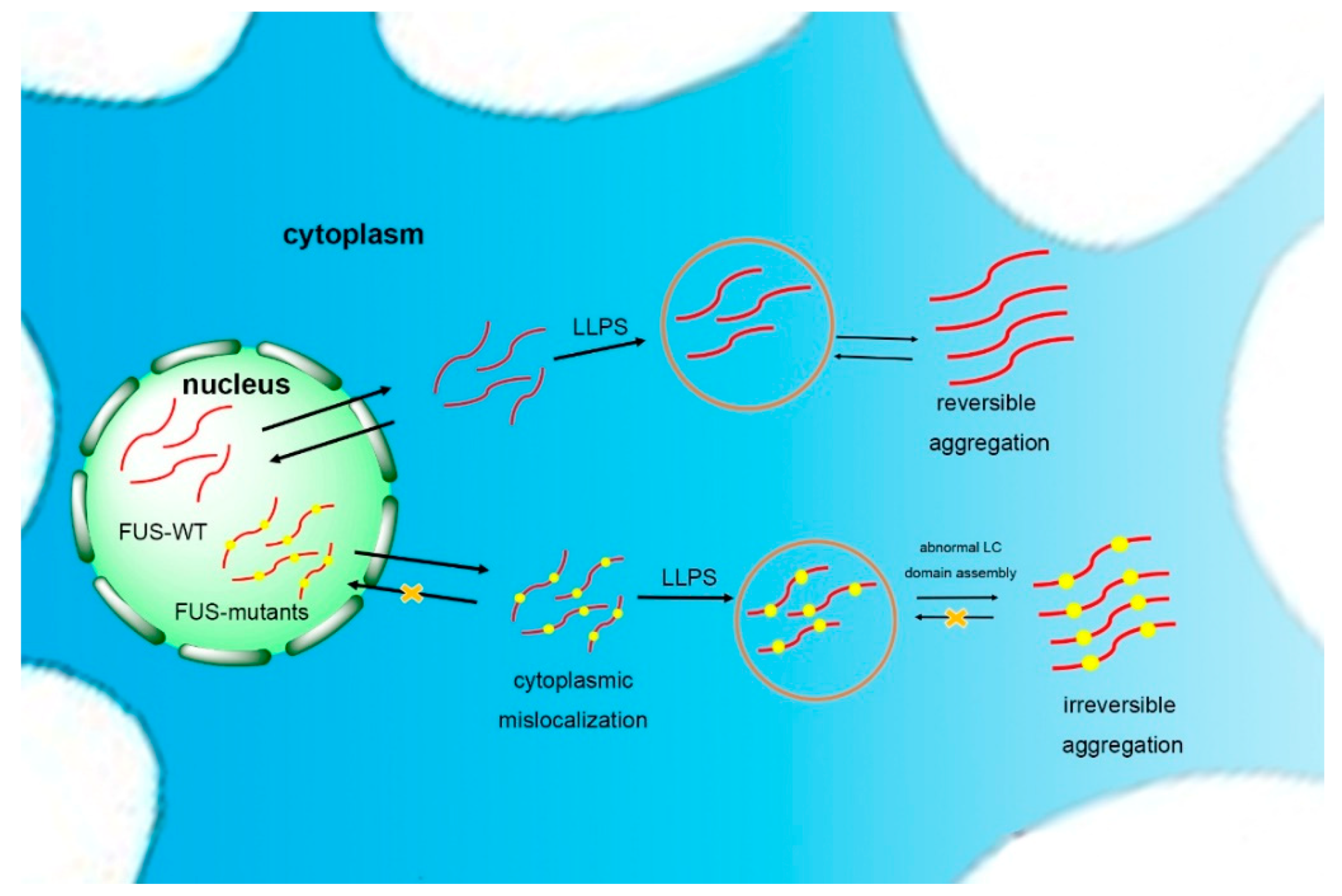
The basis of the reversible phase separation of FUS is the self-assembly of the LC domain [69]. Once the self-assembly of the LC domain goes beyond normality, usually caused by mutations in ALS, FUS is driven to form a poorly soluble and stable hydrogel (Figure 2) [45,84]. Murakami et al. demonstrated that pathogenic FUS mutations reduce the ability for reversible phase transitions. More specifically, the liquid-to-solid transition is accelerated by FUS mutants, indicating that FUS phase transitions are closely related to disease. In addition, the repeated cycles of gelation and degelation of FUS promote it to form an irreversible hydrogel [85]. The droplets and hydrogels formed by the LC domains spontaneously mature into fibrous solid aggregates as the cell ages and the ability of the quality control mechanism diminishes [82]. However, the rest of the FUS domains will reduce the tendency of FUS assembly to hinder the formation of high-grade assemblies of the full-length protein. Meanwhile, specific cellular proteins, such as the receptors transportin (TNPO1) and karyopherin-b2 (Kapb2), inhibit the solidification of FUS [86,87]. Structural analysis has shown that the LC domain noncovalently aggregates into morphologically uniform amyloid fibrils in FUS protein hydrogels [75], with a structure similar to FUS aggregates in the cytoplasm of spinal motor neurons of FUS mutant patients (with amyloid protein-like prominent filamentous cross-β structure) [88].
3.2. Amyloid Core in LC Domains of FUS Regulates Its Self-Assembly
Recent solid-state nuclear magnetic resonance (NMR) studies have indicated a structurally ordered amyloid core in the LC domain of FUS, also known as the prion-like domain. At the same time, it was discovered that the residues 39–95 constitute the structurally ordered core of the prion-like domain fibers in combination with the computational assistance method based on the MCASSIGN algorithm [89,90]. The droplets and hydrogels formed by FUS are stabilized by hydrogen bonds between the antiparallel β-sheet conformations [90]. The hallmark structure of pathogenic amyloid fibers is an in-register parallel cross-β structure in which the β-strands are perpendicular to the fiber axis, generally formed from 2 to 9 repetitive residues [91]. β-strands, irregular crimps, and loops constitute the backbone conformation of the fibers and the β-sheet layer interacts by van der Waals forces [92,93]. Sayawa et al. named this structure a “stereo zipper” that is highly stable and generally resistant to high temperatures, proteases, and detergents [94]. The FUS-LC fibrils have a single cross-β unit, with all molecules having the same structural environments and conformations and a cross-β-core-aligned parallel intermolecular alignment.
The overall bone architecture of residues 43–95 and the orientation of the side chains of residues 44–52 and 65–95 have been determined, while residues 55–62 form a dynamic loop. The fragments of the β-strands forming the cross-β contain residues 44–46, 52–54, 62–64, 67–70, 85–90, and 93–95, determined from the characteristic arrangement of the backbone carbonyl and the fiber growth axis [90]. This also indicates that many of the structurally ordered cores of the FUS-LC fibers do not participate in the formation of hydrogen bonds, which makes the cross-β structure more stable [90]. Probably because of the more rational arrangements, residues 39–95 dynamically convert into stable cross-β structures, driven by entropy cost or desolvation energy [95]. From the structural model, it is known that S84 forms a hydrogen bond network with Y75 and T78, and the stability of the loop between 74 and 87 is related to this hydrogen bond network. Phosphorylation sites S84 and S87 are located between the loops 74 and 87; therefore, the disruption of the loop conformation may be critical to the formation of FUS-LC fibers [90]. In addition, the folding of residues 44–50 against residues 64–80 is stabilized via the interaction of S48 with Q69 and T71 and the interaction of T45 and T47 with S77. The folding of residues 69–73 to 90–95 may be stabilized by the interaction of S70, Q73, S90, Q93, and S95 with each other. Y50 and Y66 may participate in π-stacking interactions (attractive, noncovalent interactions between aromatic rings) which help in stabilizing the core structure [90]. In conclusion, hydrogen bonding and electrostatic dipole–dipole interactions between polar side chains may improve the stability of the PrLDs of the FUS structure. The prion-like domain fibers of FUS lack pure hydrophobic side chains, which are conducive to disassembly [74].
Liu et al. found that 37SYSGYS42 and 54SYSSYG59 in the amyloid core region 39–95 can form reversible fibers that are sensitive to temperature and phosphorylation, termed the reversible amyloid nucleus [96]. Reversible amyloid cores (RACs) are assembled at low temperatures and disassembled as the temperature increases. The absence of either 37SYSGYS42 (RAC1) or 54SYSSYG59 (RAC2) significantly reduces the ability of self-assembling for FUS-LC; thus, it is considered the core area for FUS self-assembly [96]. Using microelectronics and X-ray diffraction techniques, Liu et al. determined that RAC1 is not a fiber spine of β-strands but a coil arranged along the fiber axis with Gly40 as a kink point. The hydroxyl group of the same layer of Tyr38 forms a hydrogen bond with the carbonyl group of Tyr41 to block the coil, and the adjacent sheets of Tyr38 and Ser42 also form hydrogen bonds, which facilitates the formation of a hydrogen bond network [96]. When the formation of these hydrogen bonds is disrupted, such as by Ser42 phosphorylation, the RAC1 fiber disassembles [24]. The π-stacking of Tyr38 enhances the interaction of adjacent layers. The RAC1 fibril spine uses polar residues to form the interface between the mating layers, which is different from the hydrophobic interface composed of nonpolar residues of pathogenic fibers [97]. Additionally, the interaction between the pathogenic fibrous sheets also has hydrogen bonds and van der Waals forces, resulting in a relatively stable fiber structure [96,98]. Unlike RAC1, in the crystal structure, the RAC2 fiber contains a cross-β structure, the marker of the pathogenic fiber. However, in the RAC2 fibril spine, water molecules, together with the hydroxyl groups of Tyr58, form a hydrogen bond, so that the distance between the sheets increases to 13 Å, while the spacing of the layers of normal pathogenic fibers is ~10 Å. The interlayer bonding is not tight and the fiber is remarkably unstable [94,96]. The unique structure of the fibers formed by RAC1 and RAC2 makes them reversible, facilitating FUS reversible self-assembly [69,96].
4. Regulation Factors of FUS Self-Assembly
The physiological function of FUS is performed while in a droplet state. In vitro, FUS droplets are amorphous, and it is speculated that, in vivo, FUS may have no ordered structure, and the oligomers formed may be gradually decomposed [99,100]. Currently, the confirmed mechanism for driving droplet formation is through its prion-like LC domains [82,101]. It can therefore be hypothesized that increasing the interaction of the LC domains accelerates the formation of aggregates. Conversely, weakening the interaction of the LC domain can be detrimental to liquid–liquid phase separation [77,101]. Irreversible FUS aggregates selectively capture ribonucleoproteins, thereby disrupting RNP granule function and impeding the synthesis of new proteins at the axon terminals of neuronal cells [85,102]. DNA-PK phosphorylates the LC domain of FUS [27]. Tycko et al. identified 14 phosphorylation sites, including 30, 42, 54, 61, 84, 87, 112, 117, 131, and 142 as well as Thr residues 7, 11, 19, and 68. In the fibril core formation segment, the phosphorylation of Ser and Thr residues disrupts the interaction of the cross-β, inhibiting hydrogel binding and droplet formation [69]. RAC1 does not form amyloid fibrils when Ser42 is phosphorylated, and S42D mutation significantly inhibits phase separation of FUS-LC [96].
Numerous studies have shown that the regions of FUS other than the LC domains, especially the C-terminal domain, are important in the phase transition behavior of FUS. Qamar et al. demonstrated that the synergistic cation–π interaction between the tyrosine of the LC domain and arginine of the structured C-terminal is related to phase separation [103]. The electrons of the tyrosine benzene ring form a cation–π interaction with the protons in the guanidino moiety of arginine, resulting in a local high concentration of the LC domains. Methylation of arginine is a common post-translational modification of RNA-binding proteins, and the C-terminal arginine rich in FUS is modified by methyltransferase with asymmetric dimethyl groups [104]. This changes the local hydrophobicity and hydrogen bonding of arginine, thereby affecting the cation–π interaction [105]. The degree of methylation of arginine in the C-terminus regulates FUS assembly. Arginine hypomethylation at the C-terminus domain greatly accelerates phase separation and gelation, allowing FUS to form a stable hydrogel containing β-sheets. This is consistent with hypomethylation in FUS-related patients with frontotemporal degeneration [75,106]. Experiments indicated that unmethylated FUS accounts for more than 1% of the total amount of FUS, which can induce the dispersed FUS to assemble into droplets and, in turn, become a stable fibrous gel [103].
The nuclear import receptor TNPO1 and Kapb2 regulate FUS phase separation by binding to FUS via its C-terminal domain [54]. They exist in the axonal terminal compartment of neurons, mediating nuclear import of FUS. TNPO1 acts as a molecular chaperone to reduce the tendency of FUS to form stress granules, suppressing FUS phase separation and liquid–solid phase transition. TNPO1 plays a central role in the FUS quality control mechanism. The highly stable combination of Kapb2 and FUS NLS weakens and dynamically interacts with the self-assembly-related regions, hindering the self-assembly of FUS. In vivo, Kapb2 dissociates FUS and RNA, regulating cytoplasmic RNA particles [107]. In vitro, Kapb2 dissolves the FUS phase separated liquid and aberrant fibrous hydrogels [108].
It is currently known that over 50 mutations/deletions in FUS are associated with ALS, accounting for about 5% of familial cases (FALS) and 0.7–1.8% of sporadic cases (SALS) [36]. Mutations in FUS mainly occur in exons 3–6 and exons 12–15. According to statistics, approximately two-thirds of FUS mutations are in exons 12–15, which encode ZnF motif, RGG2 and RGG3, and nuclear localization signaling domains. In particular, more than 50% of ALS-associated FUS mutations were found in the C-terminal NLS domain. Exons 3–6 encode the QGSY-rich and RGG1 regions, and about one-third of the mutations are in them [30,88,109]. The known mutations of FUS are shown in Table 1. A series of mutations regulate the self-assembling of FUS. NLS mutations, on the one hand, can reduce the nuclear import, increasing the concentration of cytoplasmic FUS and promoting the formation of a more durable and mature fiber structure; on the other hand, they can change the kinetics of phase transition [82,85]. It has been shown that the NLS mutant of FUS reduces the binding of TNPO1, such as P525L and R495X, affecting the nuclear import of FUS [75]. The NLS mutations of FUS cause the compulsive accumulation of FUS in the cytoplasm, resulting in aggregates that may produce toxic effects and trigger neuronal degeneration [109]. In 2011, Suzuki and colleagues constructed a series of FUS-deleted domains and explored the SG formation of them. The results showed that C-terminal deletion formed a large number of stress granules, indicating that the C-terminus is important in stress granule formation. Later studies showed that mutation R514S or P525L formed SGs in HeLa cells, and double mutations of R514S and P525L or triple mutations of R514S, R521C, and P525L promoted the formation of stress granules [82]. Mutations in PrLDs accelerate the conversion of liquid to hydrogels. The G156E mutant tended to increase the aggregation of FUS in a manner of seed aggregation, causing rapid progression of ALS by using aggregated protein fibrils as a structural template for promoting nonaggregated protein fibrosis [85,110].
Unlike the mechanism by which the mutated FUS locates in the cytoplasm, the FALS-associated FUS mutation enhances the interaction between FUS and SMN and reduces its levels at the normal expression sites, thereby impairing motor neurons [53]. Primarily mutant FUS impairs neuronal survival and causes defects in dendritic growth and synaptic connectivity by interfering with the ability of FUS in DNA damage repair and RNA splicing, leading to neurological disorders [12]. Mutations of FUS in the glycine-rich domain (amino acids 156–262) and the C-terminal domain (amino acids 450–526) affect its interaction with the chromatin remodeling factor histone deacetylase 1, which plays a fundamental role in DNA repair and genomic stability maintenance via interaction with FUS. Experiments by Qiu et al. showed that there is no wild-type FUS and interaction in FUS-R521C transgenic mice due to the mutated FUS-R521C protein in those mice, with HDAC1 forming a more stable complex with the wild-type FUS protein, implying that the mutated FUS acquired abnormal functional properties [4]. The study of Nishimoto et al. indicated that FUS affects the integrity of paraspeckles (subnuclear structures that regulate gene expression by nuclear retention of RNA) by interacting with a long noncoding RNA nuclear-enriched abundant transcript (NEAT1), and mutations of FUS may disrupt integrity and maintenance of paraspeckles [35,149]. In the same year, cytoplasmic aggregates of P45NRB/NONO, which are core proteins of paraspeckles, were found in spinal motoneurons in transgenic mice expressing truncated FUS mutant proteins (amino acids 1–359) [150]. This verified the FUS-mutation-disrupting integrity of paraspeckles.
5. Expectations
Most neurodegenerative diseases are associated with the misfolding of proteins. A growing number of studies have shown that FUS mutations in prion-like domains mediate the aggregation of FUS, which is an important reason for the development and progression of most neurodegenerative diseases. The mechanism of action of prion-associated regions of FUS needs to be further investigated, such as under what conditions the pathological aggregation of FUS occurs and whether they can play a role in disease progression by allowing the cell–cell metastasis of pathological protein aggregates. Although emergency particle assembly and prion proliferation has been confirmed, whether FUS mutations associated with neurodegenerative diseases cause normal function loss, a gain of toxic properties of FUS aggregates, or a combination of the both requires clarification and further studies. In addition, based on known mechanisms, the research of small-molecule-targeting inhibitors of FUS is also a topic of particular interest regarding finding a cure for related diseases. We are optimistic that with the deepening of FUS research, this field will make a major breakthrough.
Author Contributions
C.C. wrote the manuscript. S.X. contributed to discussions and revised the manuscript. X.D. and N.A. revised the manuscript. This review was written based on the design of S.-Z.L.
Funding
This work was supported by the National Natural Science Foundation of China (91853116, 21672019), the Fundamental Research Funds for the Central Universities and Research projects on biomedical transformation of China-Japan Friendship Hospital (No. PYBZ1812PYBZ1815).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Crozat, A.; Aman, P.; Mandahl, N.; Ron, D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993, 363, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Lagiertourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416. [Google Scholar] [CrossRef]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Invest. 2014, 124, 981–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, J.C.; Cech, T.R.; Parker, R.R. Biochemical Properties and Biological Functions of FET Proteins. Annu. Rev. Biochem. 2015, 84, 355. [Google Scholar] [CrossRef]
- Tan, A.Y.; Manley, J.L. The TET Family of Proteins: Functions and Roles in Disease. J. Mol. Cell Biol. 2009, 1, 82. [Google Scholar] [CrossRef]
- Zinszner, H.; Sok, J.; Immanuel, D.; Yin, Y.; Ron, D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci. 1997, 110, 1741–1750. [Google Scholar] [PubMed]
- Calvio, C.; Neubauer, G.; Mann, M.; Lamond, A.I. Identification of hnRNP P2 as TLS/FUS using electrospray mass spectrometry. Rna—Publ. Rna Soc. 1995, 1, 724–733. [Google Scholar]
- Tan, A.Y.; Manley, J.L. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc. Natl Acad. Sci. USA 2012, 109, 6030–6035. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.Y.; Manley, J.L. TLS inhibits RNA polymerase III transcription. Mol. Cell. Biol. 2010, 30, 186–196. [Google Scholar] [CrossRef]
- Hallier, M.; Lerga, A.; Barnache, S.; Tavitian, A.; Moreau-Gachelin, F. The transcription factor Spi-1/PU.1 interacts with the potential splicing factor TLS. J. Biol. Chem. 1998, 273, 4838–4842. [Google Scholar] [CrossRef]
- Uranishi, H.; Tetsuka, T.; Yamashita, M.; Asamitsu, K.; Shimizu, M.; Itoh, M.; Okamoto, T. Involvement of the pro-oncoprotein TLS (translocated in liposarcoma) in nuclear factor-kappa B p65-mediated transcription as a coactivator. J. Biol. Chem. 2001, 276, 13395–13401. [Google Scholar] [CrossRef]
- Li, X.; Decker, M.; Westendorf, J.J. TEThered to Runx: Novel binding partners for runx factors. Blood Cells Mol. Dis. 2010, 45, 82–85. [Google Scholar] [CrossRef]
- Bertolotti, A.; Lutz, Y.; Heard, D.J.; Chambon, P.; Tora, L. hTAF(II)68, a novel RNA/ssDNA-binding protein with homology to the pro-oncoproteins TLS/FUS and EWS is associated with both TFIID and RNA polymerase II. Embo J. 1996, 15, 5022–5031. [Google Scholar] [CrossRef]
- Schwartz, J.; R Cech, T.; R Parker, R. Biochemical Properties and Biological Functions of FET Proteins. Ann. Rev. Biochem. 2015, 84, 355–379. [Google Scholar] [CrossRef]
- Fay, R.M.; Walker, C.S.; Powers, J.M. Discoloration of a compomer by stains. J. Gt Houst Dent. Soc. 1998, 69, 12–13. [Google Scholar]
- Immanuel, D.; Zinszner, H.; Ron, D. Association of SARFH (sarcoma-associated RNA-binding fly homolog) with regions of chromatin transcribed by RNA polymerase II. Mol. Cell. Biol. 1995, 15, 4562–4571. [Google Scholar] [CrossRef]
- Kuroda, M.; Sok, J.; Webb, L.; Baechtold, H.; Urano, F.; Yin, Y.; Chung, P.; Rooij, D.G.D.; Akhmedov, A.; Ashley, T. Male sterility and enhanced radiation sensitivity in TLS−/−mice. Embo J. 2014, 19, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Alliegro, M.C.; Alliegro, M.A. A nuclear protein regulated during the transition from active to quiescent phenotype in cultured endothelial cells. Dev. Biol. 1996, 174, 288–297. [Google Scholar] [CrossRef]
- Fujii, R.; Okabe, S.; Urushido, T.; Inoue, K.; Yoshimura, A.; Tachibana, T.; Nishikawa, T.; Hicks, G.G.; Takumi, T. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr. Biol. 2005, 15, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.I.; Yan, K.P. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Wen-Yuan, W.; Ling, P.; Su, S.C.; Quinn, E.J.; Megumi, S.; Jimenez, J.C.; Mackenzie, I.R.A.; Huang, E.J.; Li-Huei, T. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurol. 2013, 16, 1383. [Google Scholar]
- Mastrocola, A.S.; Sang, H.K.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding Protein Fused in Sarcoma (FUS) Functions Downstream of Poly(ADP-ribose) Polymerase (PARP) in Response to DNA Damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef] [PubMed]
- Qiudong, D.; Holler, C.J.; Georgia, T.; Hudson, K.F.; William, W.; Marla, G.; Daisuke, I.; Murray, M.E.; Dickson, D.W.; Seyfried, N.T. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J. Neurosci. 2014, 34, 7802–7813. [Google Scholar]
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.; Gómez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014, 42, 307–314. [Google Scholar] [CrossRef]
- Baechtold, H.; Kuroda, M.; Sok, J.; Ron, D.; Lopez, B.S.; Akhmedov, A.T. Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J. Biol. Chem. 1999, 274, 34337–34342. [Google Scholar] [CrossRef] [PubMed]
- Mary, G.; Rachel, T.; Franck, V.; Nicholas, A.M.; John, R. Identification and characterization of FUS/TLS as a new target of ATM. Biochem. J. 2008, 415, 297–307. [Google Scholar] [Green Version]
- Xiangting, W.; Shigeki, A.; Xiaoyuan, S.; Donna, R.; Kun, D.; Gabriel, P.; Paul, T.; Rosenfeld, M.G.; Glass, C.K.; Riki, K. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126. [Google Scholar]
- Charcot, J.M. Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux anterolateraux de la moelle epiniere. Arch. Pathol. Norm. Pathol. 1869, 2, 744–760. [Google Scholar]
- Kwiatkowski, T.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Neumann, M.; Roeber, S.; Kretzschmar, H.A.; Rademakers, R.; Baker, M.; Mackenzie, I.R. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009, 118, 605–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, H.; Koyano, S.; Suzuki, Y.; Nukina, N.; Kuroiwa, Y. The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci. Res. 2010, 66, 131–133. [Google Scholar] [CrossRef]
- Woulfe, J.; Gray, D.A.; Mackenzie, I.R. FUS-immunoreactive intranuclear inclusions in neurodegenerative disease. Brain Pathol. 2010, 20, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Girard, S.L.; Catoire, H.; Bourassa, C.V.; Belzil, V.V.; Rivière, J.B.; Hince, P.; Levert, A.; Dionnelaporte, A.; Spiegelman, D. Exome sequencing identifies FUS mutations as a cause of essential tremor. Am. J. Hum. Genet. 2012, 91, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Labbé, C.; Rayaprolu, S.; Soto-Ortolaza, A.; Ogaki, K.; Uitti, R.J.; Wszolek, Z.K.; Ross, O.A. Investigating FUS variation in Parkinson’s disease. Parkinsonism Relat. Disord. 2014, 20, S147. [Google Scholar] [CrossRef]
- Svetoni, F.; Frisone, P.; Paronetto, M.P. Role of FET proteins in neurodegenerative disorders. Rna Biol. 2016, 13, 1089–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.; Bentmann, E.; Dormann, D.; Jawaid, A.; DeJesus-Hernandez, M.; Ansorge, O.; Roeber, S.; Kretzschmar, H.A.; Munoz, D.G.; Kusaka, H.; et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain J. Neurol. 2011, 134, 2595–2609. [Google Scholar] [CrossRef] [Green Version]
- Caroline, V.; Emma, L.S.; Agnes, L.N.; Claire, T.; Jacqueline, C.M.; Claudia, K.; Hazel, U.; Catherine, M.; Christopher, C.M.; Tibor, H.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [Green Version]
- Bosco, D.A.; Nathan, L.; Hae Kyung, K.; Hongru, Z.; Chris, B.; Kwiatkowski, T.J.; Peter, S.; Diane, M.K.Y.; Brown, R.H.; Hayward, L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010, 19, 4160–4175. [Google Scholar] [CrossRef]
- Sama, R.R.; Ward, C.L.; Kaushansky, L.J.; Lemay, N.; Ishigaki, S.; Urano, F.; Bosco, D.A. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J. Cell. Phys. 2013, 228, 2222–2231. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell. Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.B.; Almeida, S.; Lopez-Gonzalez, R. Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J. 2017, 36, e201797342. [Google Scholar] [CrossRef]
- Ramaswami, M.; Taylor, J.P.; Parker, R. Altered Ribostasis: RNA-Protein Granules in Degenerative Disorders. Cell 2013, 154, 727–736. [Google Scholar] [CrossRef]
- Neumann, M.; Rademakers, R.; Roeber, S.; Baker, M.; Kretzschmar, H.A.; Mackenzie, I.R.A. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 2009, 132, 2922–2931. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.; Huang, E.J. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 2016, 1647, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, A.A. Neuronal cytoplasmic inclusions in tau, TDP-43, and FUS molecular subtypes of frontotemporal lobar degeneration share similar spatial patterns. Folia Neuropathol. 2017, 55, 185–192. [Google Scholar]
- Morohoshi, F.; Ootsuka, Y.; Arai, K.; Ichikawa, H.; Mitani, S.; Munakata, N.; Ohki, M. Genomic structure of the human RBP56/hTAFII68 and FUS/TLS genes. Gene 1998, 221, 191–198. [Google Scholar] [CrossRef]
- Lanson, N.A., Jr.; Pandey, U.B. FUS-related proteinopathies: lessons from animal models. Brain Res. 2012, 1462, 44–60. [Google Scholar]
- Guerrero, E.N.; Wang, H.; Mitra, J.; Hegde, P.M.; Stowell, S.E.; Liachko, N.F.; Kraemer, B.C.; Garruto, R.M.; Rao, K.S.; Hegde, M.L. TDP-43/FUS in motor neuron disease: Complexity and challenges. Prog. Neurobiol. 2016, 145, 78–97. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular Determinants and Genetic Modifiers of Aggregation and Toxicity for the ALS Disease Protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef]
- Iko, Y.; Kodama, T.S.; Kasai, N.; Oyama, T.; Morita, E.H.; Muto, T.; Okumura, M.; Fujii, R.; Takumi, T.; Tate, S. Domain Architectures and Characterization of an RNA-binding Protein, TLS. J. Biol. Chem. 2004, 279, 44834. [Google Scholar] [CrossRef] [PubMed]
- Lerga, A.; Hallier, M.; Delva, L.; Orvain, C.; Gallais, I.; Marie, J.; Moreaugachelin, F. Identification of an RNA binding specificity for the potential splicing factor TLS. J. Biol. Chem. 2001, 276, 6807–6816. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y.; Ling, S.C.; Qiu, J.S.; Albuquerque, C.P.; Zhou, Y.; Tokunaga, S.; Li, H.R.; Qiu, H.Y.; Bui, A.; Yeo, G.W.; et al. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat. Commun. 2015, 6, 6171. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.J.; Cansizoglu, A.E.; Süel, K.E.; Louis, T.H.; Zhang, Z.; Chook, Y.M. Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell 2006, 126, 543–558. [Google Scholar] [CrossRef]
- Zhang, Z.C.; Chook, Y.M. Structural and energetic basis of ALS-causing mutations in the atypical proline-tyrosine nuclear localization signal of the Fused in Sarcoma protein (FUS). Proc. Natl. Acad. Sci. USA 2012, 109, 12017–12021. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J.; Lindquist, S. Prions as adaptive conduits of memory and inheritance. Nat. Rev. Genet. 2005, 6, 435–450. [Google Scholar] [CrossRef]
- Shorter, J. Emergence and natural selection of drug-resistant prions. Mol. Biosyst. 2010, 6, 1115–1130. [Google Scholar] [CrossRef] [Green Version]
- Wiltzius, J.J.; Landau, M.; Nelson, R.; Sawaya, M.R.; Apostol, M.I.; Goldschmidt, L.; Soriaga, A.B.; Cascio, D.; Rajashankar, K.; Eisenberg, D. Molecular mechanisms for protein-encoded inheritance. Nat. Struct. Mol. Biol. 2009, 16, 973–978. [Google Scholar] [CrossRef] [Green Version]
- Harrison, A.F.; Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef] [Green Version]
- Udan, M.; Baloh, R.H. Implications of the prion-related Q/N domains in TDP-43 and FUS. Prion 2011, 5, 1–5. [Google Scholar] [CrossRef] [Green Version]
- March, Z.M.; King, O.D.; Shorter, J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016, 1647, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniecka, Z.; Polymenidou, M. From nucleation to widespread propagation: A prion-like concept for ALS. Virus Res. 2015, 207, 94–105. [Google Scholar] [CrossRef] [Green Version]
- Banfi, S.; Servadio, A.; Chung, M.Y.; Kwiatkowski, T.J., Jr.; Mccall, A.E.; Duvick, L.A.; Shen, Y.; Roth, E.J.; Orr, H.T.; Zoghbi, H.Y. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat. Genet. 1994, 7, 513–520. [Google Scholar] [CrossRef]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; Defranco, D.B.; Orr, H.T.; Zoghbi, H.Y. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998, 19, 148. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Shorter, J. RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion 2011, 5, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Cushman, M.; Johnson, B.S.; King, O.D.; Gitler, A.D.; Shorter, J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J. Cell. Sci. 2010, 123, 1191–1201. [Google Scholar] [CrossRef] [Green Version]
- King, O.D.; Gitler, A.D.; Shorter, J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012, 1462, 61–80. [Google Scholar] [CrossRef] [Green Version]
- Han, T.; Kato, M.; Xie, S.; Wu, L.; Mirzaei, H.; Pei, J.; Chen, M.; Xie, Y.; Allen, J.; Xiao, G. Cell-free Formation of RNA Granules: Bound RNAs Identify Features and Components of Cellular Assemblies. Cell 2012, 149, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryndushkin, D.; Wickner, R.B.; Shewmaker, F. FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein Cell 2011, 2, 223–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, S.; Tardiff, D.F.; Han, H.; Divya, K.; Quan, Z.; Maquat, L.E.; Bosco, D.A.; Hayward, L.J.; Brown, R.H., Jr.; Lindquist, S. A Yeast Model of FUS/TLS-Dependent Cytotoxicity. PLoS Biol. 2011, 9, e1001052. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Wang, X.; Podell, E.R.; Cech, T.R. RNA seeds higher-order assembly of FUS protein. Cell Rep. 2013, 5, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Kato, M.; Xiang, S.; Wu, L.; Theodoropoulos, P.; Mirzaei, H.; Han, T.; Xie, S.; Corden, J.; Mcknight, S. Phosphorylation-Regulated Binding of RNA Polymerase II to Fibrous Polymers of Low-Complexity Domains. Cell 2013, 155, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Han, T.N.W.; Xie, S.H.; Shi, K.; Du, X.L.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.M.; et al. Cell-free Formation of RNA Granules: Low Complexity Sequence Domains Form Dynamic Fibers within Hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.D.; Simons, M.; Niessing, D.; Madl, T. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 2018, 173, 706–719. [Google Scholar] [CrossRef]
- Broide, M.L.; Berland, C.R.; Pande, J.; Ogun, O.O.; Benedek, G.B. Binary-liquid phase separation of lens protein solutions. Proc. Natl. Acad. Sci. USA 1991, 88, 5660–5664. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.A.; Janke, A.M.; Rhine, C.L.; Fawzi, N.L. Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol. Cell. 2015, 60, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Sign. 2016, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutertre, M.; Lambert, S.; Carreira, A.; Amor-Guéret, M.; Vagner, S. DNA damage: RNA-binding proteins protect from near and far. Trends Biochem. Sci. 2014, 39, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Hyman, A.A.; Brangwynne, C.P. Beyond Stereospecificity: Liquids and Mesoscale Organization of Cytoplasm. Dev. Cell 2011, 21, 14–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Banjade, S.; Cheng, H.C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F. Phase Transitions in the Assembly of Multi-Valent Signaling Proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dormann, D.; Haass, C. TDP-43 and FUS: a nuclear affair. Trends Neurosci. 2011, 34, 339–348. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: from genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.K.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.S.; Michel, C.H. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Hyman, A.A. Are aberrant phase transitions a driver of cellular aging? Bioessays 2016, 38, 959–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, P.R.; Milin, A.N.; Moosa, M.M.; Onuchic, P.L.; Deniz, A.A. Reentrant Phase Transition Drives Dynamic Substructure Formation in Ribonucleoprotein Droplets. Angew. Chem. Int. Ed. 2017, 56, 11354–11359. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Hu, K.N.; Qiang, W.; Tycko, R. A general Monte Carlo/simulated annealing algorithm for resonance assignment in NMR of uniformly labeled biopolymers. J. Biomol. NMR 2011, 50, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; Mcknight, S.L.; Tycko, R. Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell 2017, 171, 615–627. [Google Scholar] [CrossRef]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.Ø.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The Amyloid State of Proteins in Human Diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef]
- Rambaran, R.N.; Serpell, L.C. Amyloid fibrils Abnormal protein assembly. Prion 2008, 2, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.W.; McFarlane, H.T. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef]
- Thirumalai, D.; Reddy, G.; Straub, J.E. Role of water in Protein Aggregation and Amyloid Polymorphism. Acc. Chem. Res. 2011, 45, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Gui, X.; Zhou, H.; Gu, J.; Li, Y.; Liu, X.; Zhao, M.; Li, D.; Li, X.; Liu, C. Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation. Nat. Struct. Mol. Biol. 2018, 25, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Eisenberg, D. Recent atomic models of amyloid fibril structure. Curr. Opin. Struct. Biol. 2006, 16, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Eisenberg, D.S. The activities of amyloids from a structural perspective. Nature 2016, 539, 227–235. [Google Scholar] [CrossRef]
- Amen, T.; Kaganovich, D. Dynamic droplets: the role of cytoplasmic inclusions in stress, function, and disease. Cell. Mol. Life Sci. 2015, 72, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, S.N.; Monahan, Z.T.; Yee, D.S.; Shewmaker, F.P. The Role of Post-Translational Modifications on Prion-Like Aggregation and Liquid-Phase Separation of FUS. Int. J. Mol. Sci. 2018, 19, 886–905. [Google Scholar] [CrossRef]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef]
- Takanashi, K.; Yamaguchi, A. Aggregation of ALS-linked FUS mutant sequesters RNA binding proteins and impairs RNA granules formation. Biochem. Biophys. Res. Commun. 2014, 452, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Qamar, S.; Wang, G.; Randle, S.J.; Ruggeri, F.S.; Varela, J.A.; Lin, J.Q.; Phillips, E.C.; Miyashita, A.; Williams, D.; Ströhl, F. FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-π Interactions. Cell 2018, 173, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Chiara, S.; John, M.; Carmelo, M.; Lanson, N.A.; Astha, M.; Tanya, A.; Ian, C.; Fackelmayer, F.O.; Maria, P.; Udai Bhan, P. Protein arginine methyltransferase 1 and 8 interact with FUS to modify its sub-cellular distribution and toxicity in vitro and in vivo. PLoS ONE 2013, 8, e61576. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Clancy, K.W.; Thompson, P.R. Chemical biology of protein arginine modifications in epigenetic regulation. Chem. Rev. 2015, 115, 5413–5461. [Google Scholar] [CrossRef]
- Dormann, D.; Madl, T.; Valori, C.F.; Bentmann, E.; Tahirovic, S.; Abou-Ajram, C.; Kremmer, E.; Ansorge, O.; Mackenzie, I.R.; Neumann, M.; Haass, C. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012, 31, 4258–4275. [Google Scholar] [CrossRef] [Green Version]
- Yoshizawa, T.; Ali, R.; Jiou, J.; Fung, H.; Burke, K.A.; Kim, S.J.; Lin, Y.; Peeples, W.B.; Saltzberg, D.; Soniat, M. Nuclear Import Receptor Inhibits Phase Separation of FUS through Binding to Multiple Sites. Cell 2018, 173, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Hong, J.K.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.M.; Diaz, Z.; Singh, N. Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 2018, 173, 677–692. [Google Scholar] [CrossRef]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Watanabe, S.; Kaneko, K.; Yamanaka, K.; Nukina, N.; Furukawa, Y. Intranuclear Aggregation of Mutant FUS/TLS as a Molecular Pathomechanism of Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2014, 289, 1192–1202. [Google Scholar] [CrossRef]
- Belzil, V.V.; St-Onge, J.; Daoud, H.; Desjarlais, A.; Bouchard, J.P.; Dupré, N.; Camu, W.; Dion, P.A.; Rouleau, G.A. Identification of a FUS splicing mutation in a large family with amyotrophic lateral sclerosis. J. Hum. Genet. 2011, 56, 247. [Google Scholar] [CrossRef]
- Belzil, V.V.; Valdmanis, P.N.; Dion, P.A.; Daoud, H.; Kabashi, E.; Noreau, A.; Gauthier, J.; Hince, P.; Desjarlais, A.; Desjarlais, J.-P.; et al. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology 2009, 73, 1176–1179. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Deng, H.X.; Siddique, N.; Fecto, F.; Chen, W.; Yang, Y.; Liu, E.; Donkervoort, S.; Zheng, J.G.; Shi, Y. Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 2010, 75, 807–814. [Google Scholar] [CrossRef]
- Marka, V.B.; Van Es, M.A.; Hennekam, E.A.M.; Dennis, D.; Wouter, V.R.; Jelena, M.; Bourque, P.R.; Schelhaas, H.J.; Van Der Kooi, A.J.; Marianne, D.V. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 3776–3784. [Google Scholar]
- Brown, J.A.; Min, J.; Staropoli, J.F.; Collin, E.; Bi, S.; Feng, X.; Barone, R.; Cao, Y.; O’Malley, L.; Xin, W. SOD1, ANG, TARDBP and FUS mutations in amyotrophic lateral sclerosis: a United States clinical testing lab experience. Amyotroph. Lateral Scler. 2012, 13, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Ticozzi, N.; Silani, V.; Leclerc, A.L.; Keagle, P.; Gellera, C.; Ratti, A.; Taroni, F.; Kwiatkowski, T.J.; Mckenna-Yasek, D.M.; Sapp, P.C. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 2009, 73, 1180–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucia, C.; Roberto, D.B.; Barbara, C.; Antonia, R.; Cristina, C.; Silvana, P.; Gianni, S.; Yari, C.; Serena, G.; Viviana, P. Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J. Med. Genetics 2010, 47, 190–194. [Google Scholar]
- Kwon, M.J.; Baek, W.; Ki, C.S.; Kim, H.Y.; Koh, S.H.; Kim, J.W.; Kim, S.H. Screening of the SOD1, FUS, TARDBP, ANG, and OPTN mutations in Korean patients with familial and sporadic ALS. Neurobio. Aging 2012, 33, 1017.e17–1017.e23. [Google Scholar] [CrossRef] [PubMed]
- Tarlarini, C.; Lunetta, C.; Mosca, L.; Avemaria, F.; Riva, N.; Mantero, V.; Maestri, E.; Quattrini, A.; Corbo, M.; Melazzini, M.G. Novel FUS mutations identified through molecular screening in a large cohort of familial and sporadic amyotrophic lateral sclerosis. Eur. J. Neurol. 2015, 22, 1474–1481. [Google Scholar] [CrossRef]
- Murni, T.; Wen, R.; Lin, L.Y.; Wang, H.; Ling, S.C.; Yi, Z.; Tan, E.K. FUS-linked essential tremor associated with motor dysfunction inDrosophila. Hum. Genet. 2016, 135, 1223–1232. [Google Scholar]
- Rajput, A.; Rajput, A.H.; Rajput, M.L.; Encarnacion, M.; Bernales, C.Q.; Ross, J.P.; Farrer, M.J.; Vilariñogüell, C. Identification of FUS p.R377W in essential tremor. Eur. J. Neurol. 2014, 21, 361–363. [Google Scholar] [CrossRef]
- Gao, K.; Zheng, W.; Deng, X.; Xiong, W.; Song, Z.; Yang, Y.; Deng, H. Genetic analysis of the fused in sarcoma gene in Chinese Han patients with Parkinson’s disease. Parkinsonism Relat. Disord. 2014, 20, 119–121. [Google Scholar] [CrossRef]
- Mariely, D.J.H.; Jannet, K.; Nicole, F.; Richard, C.; Matt, B.; Pamela, D.; Amelia, J.; Nicola, R.; Aleksandra, W.; Kathleen, K. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum. Mutat. 2010, 31, E1377–E1389. [Google Scholar]
- Labbé, C.; Sotoortolaza, A.I.; Rayaprolu, S.; Harriott, A.M.; Strongosky, A.J.; Uitti, R.J.; Gerpen, J.A.V.; Wszolek, Z.K.; Ross, O.A. Investigating the role of FUS exonic variants in Essential Tremor. Parkinsonism Relat. Disord. 2013, 19, 755–757. [Google Scholar] [CrossRef] [Green Version]
- Groen, E.J.; Van, E.M.V.P. FUS mutations in familial amyotrophic lateral sclerosis in the Netherlands. Arch. Neurol. 2010, 67, 224–230. [Google Scholar] [CrossRef]
- Nagayama, S.; Minato-Hashiba, N.; Nakata, M.; Kaito, M.; Nakanishi, M.; Tanaka, K.; Arai, M.; Akiyama, H.; Matsui, M. Novel FUS mutation in patients with sporadic amyotrophic lateral sclerosis and corticobasal degeneration. J. Clin. Neurol. 2012, 19, 1738–1739. [Google Scholar] [CrossRef]
- Hara, M.; Minami, M.; Suzuki, N.; Kato, M.; Aoki, M. Lower motor neuron disease caused by a novel FUS/TLS gene frameshift mutation. J. Neurol. 2012, 259, 2237–2239. [Google Scholar] [CrossRef]
- Liu, Z.J.; Lin, H.X.; Liu, G.L.; Tao, Q.Q.; Ni, W.; Xiao, B.G.; Wu, Z.Y. The investigation of genetic and clinical features in Chinese patients with juvenile amyotrophic lateral sclerosis. Clin. Genet. 2017, 92, 267–273. [Google Scholar] [CrossRef]
- Satoshi, Y.; Akira, M.; Hideya, S.; Tomohiro, S.; Daijirou, I.; Akihiko, U.; Taro, Y.; Yasushi, M.; Makoto, U.; Teruyuki, H. Sporadic juvenile amyotrophic lateral sclerosis caused by mutant FUS/TLS: possible association of mental retardation with this mutation. J. Neurol. 2012, 259, 1039–1044. [Google Scholar]
- Zou, Z.Y.; Cui, L.Y.; Sun, Q.; Li, X.G.; Liu, M.S.; Xu, Y.; Zhou, Y.; Yang, X.Z. De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. Neurol. Aging 2013, 34, 1312.e1–1312.e8. [Google Scholar] [CrossRef]
- Waibel, S.; Neumann, M.; Rabe, M.; Meyer, T.; Ludolph, A.C. Novel missense and truncating mutations in FUS/TLS in familial ALS. Neurology 2010, 75, 815–817. [Google Scholar] [CrossRef]
- Belzil, V.V.; Jean-Sébastien, L.; Hussein, D.; Dion, P.A.; Bernard, B.; Rouleau, G.A. Novel FUS deletion in a patient with juvenile amyotrophic lateral sclerosis. Arch. Neurol. 2012, 69, 653–656. [Google Scholar]
- Sproviero, W.; Bella, V.L.; Mazzei, R.; Valentino, P.; Rodolico, C.; Simone, I.L.; Logroscino, G.; Ungaro, C.; Magariello, A.; Patitucci, A. FUS mutations in sporadic amyotrophic lateral sclerosis: Clinical and genetic analysis. Neurobiol. Aging 2012, 33, 837.e1–837.e5. [Google Scholar] [CrossRef] [Green Version]
- Christopher, H.; Janine, K.; Highley, J.R.; Hartley, J.A.; Rachael, H.; Hollinger, H.C.; Williams, T.L.; Ince, P.G.; Mcdermott, C.J.; Shaw, P.J. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2010, 67, 455–461. [Google Scholar]
- Suzuki, M.; Mikami, H.; Watanabe, T.; Yamano, T.; Yamazaki, T.; Nomura, M.; Yasui, K.; Ishikawa, H.; Ono, S. Increased expression of TDP-43 in the skin of amyotrophic lateral sclerosis. Acta Neurol. Scand. 2010, 122, 367–372. [Google Scholar] [CrossRef]
- Syriani, E.; Morales, M.; Gamez, J. FUS/TLS gene mutations are the second most frequent cause of familial ALS in the Spanish population. Amyotroph. Lateral Scler. 2011, 12, 118–123. [Google Scholar] [CrossRef]
- Chiò, A.; Mora, G.; Calvo, A.; Mazzini, L.; Bottacchi, E.; Mutani, R. Epidemiology of ALS in Italy: A 10-year prospective population-based study. Neurology 2009, 72, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chio, A.; Restagno, G.M. Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol. Aging 2009, 30, 1272–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stéphanie, M.; Francois, S.; Cécile, C.; Paul, G.; Bernard, B.; Agnès, C.; Léna, G.N.L.; Odile, R.; Gaelle, B.; Pierre-Francois, P.; et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: Genotype-phenotype correlations. J. Med. Genet. 2010, 47, 554–560. [Google Scholar]
- Robertson, J.; Bilbao, J.; Zinman, L.; Hazrati, L.-N.; Tokuhiro, S.; Sato, C.; Moreno, D.; Strome, R.; Mackenzie, I.R.; Rogaeva, E. A novel double mutation in FUS gene causing sporadic ALS. Neurobiol. Aging 2011, 32, 553.e27–553.e30. [Google Scholar] [CrossRef] [PubMed]
- Ching-Paio, T.; Bing-Wen, S.; Kon-Ping, L.; Pang-Hsien, T.; Jer-Li, L.; Yi-Chung, L. FUS, TARDBP, and SOD1 mutations in a Taiwanese cohort with familial ALS. Neurobiol. Aging 2011, 32, 553.e13–553.e21. [Google Scholar]
- BaUmer, D.; Hilton, D.; Paine, S.M.L.; Turner, M.R.; Lowe, J.; Talbot, K.; Ansorge, O. Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology 2010, 75, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Blair, I.P.; Williams, K.L.; Warraich, S.T.; Durnall, J.C.; Thoeng, A.D.; Jim, M.; Blumbergs, P.C.; Steve, V.; Kiernan, M.C.; Nicholson, G.A. FUS mutations in amyotrophic lateral sclerosis: Clinical, pathological, neurophysiological and genetic analysis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 639–645. [Google Scholar] [CrossRef]
- Yamamoto-Watanabe, Y.; Watanabe, M.; Okamoto, K.; Fujita, Y.; Jackson, M.; Ikeda, M.; Nakazato, Y.; Ikeda, Y.; Matsubara, E.; Kawarabayashi, T. A Japanese ALS6 family with mutation R521C in the FUS/TLS gene: A clinical, pathological and genetic report. J. Neurol. Sci. 2010, 296, 59–63. [Google Scholar] [CrossRef]
- Broustal, O.; Camuzat, A.; Guillot-Noël, L.; Guy, N.; Millecamps, S.; Deffond, D.; Lacomblez, L.; Golfier, V.; Hannequin, D.; Salachas, F. FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J. Alzheimers Dis. 2010, 22, 765–769. [Google Scholar] [PubMed]
- Damme, P.V.; Goris, A.V.; Hersmus, N.; Dubois, B.; Bosch, L.V.; Matthijs, G.; Robberecht, W. The occurrence of mutations in FUS in a Belgian cohort of patients with familial ALS. Eur. J. Neurol. 2010, 17, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Eysel, K. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology 2012, 79, 66–72. [Google Scholar]
- Hou, L.; Jiao, B.; Xiao, T.; Zhou, L.; Zhou, Z.; Du, J.; Yan, X.; Wang, J.; Tang, B.; Shen, L. Screening of SOD1, FUS and TARDBP genes in patients with amyotrophic lateral sclerosis in central-southern China. Sci. Rep. 2016, 6, 32478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimoto, Y.; Nakagawa, S.; Hirose, T.; Okano, H.J.; Takao, M.; Shibata, S.; Suyama, S.; Kuwako, K.; Imai, T.; Murayama, S. The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 2013, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Shelkovnikova, T.A.; Peters, O.M.; Deykin, A.V.; Connorrobson, N.; Robinson, H.; Ustyugov, A.A.; Bachurin, S.O.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R. Fused in Sarcoma (FUS) Protein Lacking Nuclear Localization Signal (NLS) and Major RNA Binding Motifs Triggers Proteinopathy and Severe Motor Phenotype in Transgenic Mice. J. Biol. Chem. 2013, 288, 25266–25274. [Google Scholar] [CrossRef]
Figure 1.
Schematic of fused in sarcoma (FUS) protein domains.
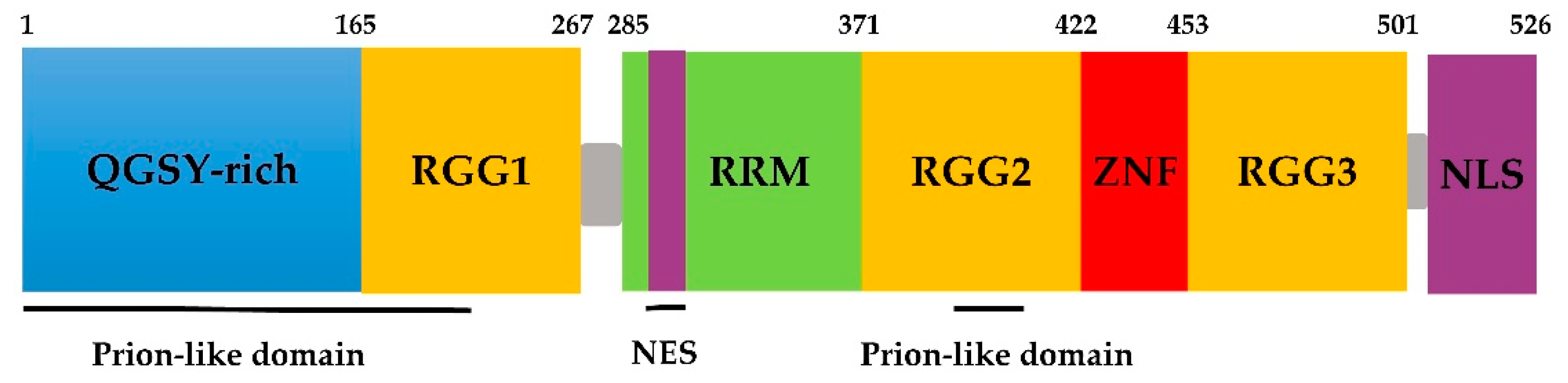
Figure 2.
Self-assembling model of FUS. FUS can phase separate to form granules and reversible aggregates. Once the self-assembly of the low-complexity (LC) domain goes beyond normality, usually caused by mutations in neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), FUS will form irreversible aggregation, possibly via liquid–liquid phase separation (LLPS).
Figure 2.
Self-assembling model of FUS. FUS can phase separate to form granules and reversible aggregates. Once the self-assembly of the low-complexity (LC) domain goes beyond normality, usually caused by mutations in neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), FUS will form irreversible aggregation, possibly via liquid–liquid phase separation (LLPS).

{kind=link}
{kind=link}
Table 1.
Summarizes of the mutations in the FUS gene. ALS: amyotrophic lateral sclerosis; FTLD, frontotemporal lobar degeneration; ET, essential tremor.
Table 1.
Summarizes of the mutations in the FUS gene. ALS: amyotrophic lateral sclerosis; FTLD, frontotemporal lobar degeneration; ET, essential tremor.
| NO. | Exon | Domain/Region | Nucleotide Change | Amino Acid Mutation | ALS | References |
|---|---|---|---|---|---|---|
| 1 | 3 | PrLDs | c.52 C > T | p.P18S | ALS | [111] |
| 2 | 3 | PrLDs | c.170_172delCTT | p.S57del | ALS | [112] |
| 3 | 4 | PrLDs | c.287_291delCCTACinsAT | p.S96del | ALS | [113] |
| 4 | 4 | PrLDs | c.344 G > A | p.S115N | ALS | [114] |
| 5 | 5 | PrLDs | c.430_447delGG | p.G144Y149del | ALS | [111,115] |
| 6 | 5 | PrLDs | c.467 G > A | p.G156E | ALS | [116] |
| 7 | 6 | PrLDs | c.518_523del GAGGTG | p.G174_G175del | AlS/FTLD | [30,113] |
| 8 | 6 | RGG1 | c.616 G > A | p.G206S | AlS/FTLD | [113] |
| 9 | 6 | RGG1 | c.646 C > T | p.R216C | ALS | [117] |
| 10 | 6 | RGG1 | c.674 G > T | p.G225V | ALS | [117] |
| 11 | 6 | RGG1 | c.681_684delGGC | p.G230delG | ALS | [118] |
| 12 | 6 | RGG1 | c.688 G > T | p.G230C | ALS | [117] |
| 13 | 6 | RGG1 | c.700 C > T | p.R234C | ALS | [117] |
| 14 | 6 | RGG1 | c.701 G > T | p.R234L | ALS | [116] |
| 15 | 6 | RGG1 | c.730 C > T | p.R244C | ALS | [30] |
| 16 | 6 | RGG1 | c.734 G > T | p.G245V | ALS | [119] |
| 17 | 9 | RRM | c.868 C > T | p.Q290X | ET | [120] |
| 18 | 11 | RGG2 | c.1129 C > T | p.R377W | ET | [121] |
| 19 | 12 | PrLDs | c.1176 G > A | p.M392I | ET | [122] |
| 20 | 12 | PrLDs | c.1196 G > T | p.G399V | ALS | [118] |
| 21 | 12 | RGG2 | c.1204 1232 delinsGGAGGTGGAGG | p.S402P | ALS | [123] |
| 22 | 13 | ZNF | c.1292 C > T | p.P431L | ET | [124] |
| 23 | 13 | RGG3 | c.1385 C > T | p.S462F | ALS | [125] |
| 24 | 13 | RGG3 | c.1392 G > T | M464I | ALS | [126] |
| 25 | 14 | RGG3 | c.1394_1541del | p.G466VfsX14 | ALS | [123] |
| 26 | 14 | RGG3 | c.1420_1421insGT | p.G472VfsX57 | ALS | [127] |
| 27 | 14 | RGG3 | c.1449_1488del | p.Y485AfsX514 | ALS | [113] |
| 28 | 14 | RGG3 | c.1456_1457delGG | p.G486PfsX30 | ALS | [128] |
| 29 | 14 | RGG3 | c.1459 C > T | p.R487C | ALS | [114] |
| 30 | 14 | RGG3 | c.1464 C > T | p.G488G | ALS | [119] |
| 31 | 14 | RGG3 | c.1475delG | p.G492EfsX527 | ALS | [129] |
| 32 | 14 | RGG3 | c.1483delC | p.R495EfsX527 | ALS | [113] |
| 33 | 14 | RGG3 | c.1483 C > T | p.R495X | ALS | [39,130,131] |
| 34 | 14 | RGG3 | c.1484delG | p.R495QfsX527 | ALS | [132] |
| 35 | 14 | RGG3 | c.1485delA | p.G497AfsX527 | ALS | [113] |
| 36 | 14 | RGG3 | c.1489_1490dupGG | p.R498AfsX32 | ALS | [128] |
| 37 | 14 | - | c.1506dupA | p.R502fsX15 | ALS | [111] |
| 38 | 14 | - | c.1507_1508delAG | p.G503WfsX12 | ALS | [118] |
| 39 | 14 | - | c.1509_1510delAG | p.G504WfsX12 | ALS | [130] |
| 40 | 14 | - | c.1520 G > A | p.G507D | ALS | [117,133,134] |
| 41 | 14 | NLS | c.1526 G > A | p.G509D | ALS | [119] |
| 42 | 14 | NLS | c.1527_1528insTGCC | p.K510WfsX517 | ALS | [113] |
| 43 | 14 | NLS | c.1528 A > G | p.K510E | ALS | [135,136] |
| 44 | 14 | NLS | c.1529 A > G | p.K510R | ALS | [131] |
| 45 | 14 | NLS | c.1537 T > C | p.S513P | ALS | [135] |
| 46 | 14 | NLS | c.1540 A > G | p.R514G | ALS | [88] |
| 47 | 14 | NLS | c.1542 G > C | p.R514S | ALS | [30,137,138,139,140] |
| 48 | 15 | NLS | c.1543 G > T | p.G515C | ALS | [30] |
| 49 | 15 | NLS | c.1544delG | p.G515VfsX14 | ALS | [128] |
| 50 | 15 | NLS | c.1547 A > T | p.E516V | ALS | [140] |
| 51 | 15 | NLS | c.1549 C > G | p.H517D | ALS | [141] |
| 52 | 15 | NLS | c.1550 A > C | p.H517P | ALS | [135] |
| 53 | 15 | NLS | c.1551 C > G | p.H517Q | ALS | [30] |
| 54 | 15 | NLS | c.1552 A > G | p.R518G | ALS | [104] |
| 55 | 15 | NLS | c.1553 G > A | p.R518K | ALS | [30] |
| 56 | 15 | NLS | c.1554_1557delACAG | p.R518del | ALS | [142] |
| 57 | 15 | NLS | c.1555 C > T | p.Q519X | ALS | [112] |
| 58 | 15 | NLS | c.1561C > T | p.R521C | ALS | [112,143,144] |
| 59 | 15 | NLS | c.1561 C > G | p.R521G | ALS | [115,133] |
| 60 | 15 | NLS | c.1561 C > A | p.R521S | ALS | [139] |
| 61 | 15 | NLS | c.1562 G > A | p.R521H | ALS | [112,139,145,146] |
| 62 | 15 | NLS | c.1562 G > T | p.R521L | ALS | [113,139,147] |
| 63 | 15 | NLS | c.1564 C > T | p.R522G | ALS | [30] |
| 64 | 15 | NLS | c.1570 A > T | p.R524W | ALS | [134] |
| 65 | 15 | NLS | c.1571 G > C | p.R524T | ALS | [30] |
| 66 | 15 | NLS | c.1572 G > C | p.R524S | ALS | [30,113] |
| 67 | 15 | NLS | c.1574 C > T | p.525L | ALS | [142] |
| 68 | 15 | NLS | c.1574 C > G | p.525R | ALS | [142] |
| 69 | 15 | NLS | c.1575_1576insTAT | p.P525_Y526ins | ALS | [148] |
| 70 | 15 | NLS | c.1581delA | p.X527YextX | ALS | [118] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, C.; Ding, X.; Akram, N.; Xue, S.; Luo, S.-Z. Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases. Molecules 2019, 24, 1622. https://doi.org/10.3390/molecules24081622
AMA Style
Chen C, Ding X, Akram N, Xue S, Luo S-Z. Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases. Molecules. 2019; 24(8):1622. https://doi.org/10.3390/molecules24081622
Chicago/Turabian StyleChen, Chen, Xiufang Ding, Nimrah Akram, Song Xue, and Shi-Zhong Luo. 2019. "Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases" Molecules 24, no. 8: 1622. https://doi.org/10.3390/molecules24081622