
Acid Catalyzed Formation of C–C and C–S Bonds via Excited State Proton Transfer


1
Department of Chemistry, University of Pavia, Viale Taramelli 10, 27100 Pavia, Italy
2
PhotoGreen Lab, Department of Chemistry, University of Pavia, Viale Taramelli 10, 27100 Pavia, Italy
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(7), 1318; https://doi.org/10.3390/molecules24071318
Submission received: 11 February 2019
/
Revised: 28 March 2019
/
Accepted: 28 March 2019
/
Published: 3 April 2019
(This article belongs to the Special Issue Photocatalytic Strategies in Organic Synthesis)
Abstract
:The behavior of 2-naphthol and 7-bromo-2-naphthol as organic photoacids are exploited in organic synthesis for the preparation of benzyl sulfides (using a trichloroacetimidate derivative as the starting substrate) and polycyclic amines via acid catalyzed condensation of 1,2,3,4-tetrahydroisoquinoline with aldehydes.
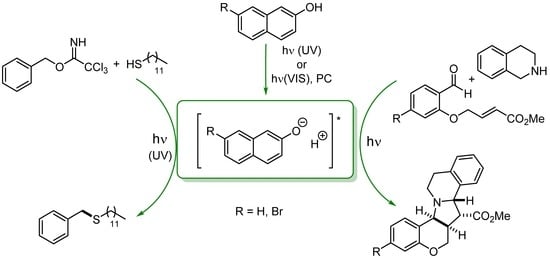
1. Introduction
Photocatalysis emerged in the last decade as an established strategy for organic synthesis [1,2,3,4,5], in view of the versatility of the approach and the mild conditions adopted, including working at room temperature, the adoption of aqueous solvents and the use of photons as a traceless reagent [6,7]. In the photocatalytic approach, the absorption of a photon by the compound that acts as the photocatalyst (PC in Scheme 1) leads to dramatic changes that can impact different aspects of its reactivity. As an example, irradiation of a molecule can affect its redox properties by increasing the oxidation or the reduction potential, in turn opening the way to an electron transfer (ET) step. Such behavior is the pillar of the visible-light photoredox catalytic approach, the synthetic potential of which has been widely demonstrated, starting from the seminal works of Mac Millan [8,9] and Yoon [10].
In some cases, the excited photocatalyst is able to activate selectively a C–H bond of a reactant via a direct Hydrogen Atom Transfer (HAT) path, to generate the corresponding carbon centered radical that can be trapped in turn by a suitable olefin, leading to the formation of a new C–C bond [11]. However, direct HAT processes are currently limited to few (photo)catalysts, namely aromatic ketones [12] and some polyoxometalate derivatives (e.g., decatungstate salts) [13]. In some cases, activation of the substrate towards the desired reaction occurs via a simple energy transfer (also known as sensitization) from the excited catalyst [14,15].
A further approach relies on the electron density shift occurring when a compound is promoted to the corresponding excited state, which can result in a dramatic increase of the acidity, by down to 13 pKa units [16,17,18,19]. A wide range of organic molecules (including substituted phenols and naphthols) exhibit such a property, and are therefore defined as “organophotoacids”.
Whereas HAT, sensitization and especially photoredox catalysis found an impressive range of applications in synthesis, little attention has been given to the use of organophotoacids, which, in in most cases, were mainly used in deprotection reactions [20] as well as in the protection of alcohols as tetrahydropyran ethers [21]. However, interest in such an approach recently emerged. Lei et al. described the potentialities of the xanthene dye Eosin Y [22,23] in the visible light driven acetalization of aldehydes; the same organophotoacid was recently employed in the stereoselective preparation of 2-deoxyglycosides [24]. Toshima et al. exploited the decrease of the pKa value (down to five magnitude orders) of a chiral thiourea derivative in photoinduced stereo-controlled glycosylation reactions [25]. A significant contribution to the use of organophotoacids in synthesis has been offered by Hanson et al., who optimized the visible-light enantioselective protonation of silyl enol ethers, using an Ir(III) complex as the photosensitizer, 7-bromo-2-naphthol as the photocatalyst and an excess of phenol as the proton source [26]. The approach was recently implemented, and the use of a chiral organophotoacid was able to induce some degree of enantioselectivity [27].
2. Results and Discussion
In view of these recent results, we reasoned that organophotoacids could potentially play a role in a wide range of synthetic processes, including C–S and C–C bond formation. With this target in mind, we embarked on the exploratory study of a model reaction, namely the acid catalyzed nucleophilic substitution occurring on trichloroacetimidate derivative 1 [28,29]. The process was investigated by using different light sources and, on the basis of the available literature, 2-naphthol (PA1) and 7-bromo-2-naphthol (PA2) were considered in the role of organophotoacid candidates. The choice of the solvent was carried out on the basis of their compatibility with the reaction conditions, which precluded the use of alcoholic solvents. The reaction was monitored by GC and the product yield was measured by calibration curve obtained by comparison with an authentic sample. The obtained results are summarized in Table 1.
Irradiation of 1 (0.10 M) at 366 nm, in the presence of an excess of 2 (0.50 M) and 2-naphthol (PA1, 30 mol%) as the photoacid in DMSO did not afford the desired thiolation, and 1 underwent exclusively thermal decomposition to the corresponding benzyl 2,2,2-trichloroacetate (entry 1). However, small amounts of 3 (up to 21% yield) were observed when moving from apolar (n-hexane and toluene, entries 2,3) to polar media, such as dichloroethane, diethyl ether and acetonitrile (entries 4–6). Better results were obtained when moving to dichloromethane, where 3 was formed in 28% yield (entry 7). The efficiency of the process was further improved by increasing either the equivalent of 2 (up to 1.0 M, entry 8) or of photoacid (up to 40% yield when PA1 was used in 50 mol%, entry 10). Irradiation with different wavelength (310 nm, 15 W, Hg lamp), afforded expected thiolation product with a comparable yield (entry 11). Similar results were obtained when using 7-bromo-2-naphthol (PA2) as the organophotoacid, with the benzyl sulfide 3 obtained in up to 47% yield (entries 12, 13). As in the case of PA1, the reaction yield dropped when acetonitrile was used as the solvent (entry 14). Finally, the reaction took place only to a minor extent in the absence of the photoacid and of light, respectively (entries 15,16).
With this promising results in hand, we decided to investigate the potentialities of organophotoacid catalysis in the preparation of alkaloids 6a,b, from tetrahydroisoquinoline 5 and salicylaldehydes 4a,b (in turn obtained by adapting a known synthesis procedure; see Supplementary Materials for further details) [30]. Unfortunately, the conditions optimized for the synthesis of 3 were found inefficient to achieve this target (Table 2, entry 1). Taking inspiration from the work of Hanson et al. [26], we decided to move to visible light irradiation by adopting Ru(bpy)32+ as the photosensitizer. In this case, due to the poor solubility of the complex in DCM, experiments were performed in MeCN, and light emitting diodes (LEDs) were used as the light source. Gratifyingly, a discrete amount of 6a (19% yield, entry 2) was obtained when irradiating a solution of 4a and a slight excess of tetrahydroisoquinoline 5 (1.2 equiv) in the presence of 2-naphthol (50 mol%) and Ru(bpy)32+ (5 mol%) as the photoacid and the photocatalyst, respectively. A change of the 4a:5 stoichiometric ratio from 1:1.2 to 1.5:1 (entries 3,4) led to a significant improvement of the reaction efficiency, and the desired alkaloid 6a was isolated in up to 65% yield. Further increase in the amount of 4a did not significantly influence the process (entry 5), whereas in the absence of PA1 the product was obtained in low yield, due to the unsatisfactory consumption of 5 (entry 6). Good results were also obtained with PA2. In this case, irradiation of a RuII-sensitized solution of 4a and 5 in dry acetonitrile in the presence of 7-bromo-2-naphthol as the photoacid and 1 equiv of phenol afforded 6a a 62% yield (entry 7). Finally, the protocol was extended to the preparation of methoxy substituted derivative 6b. In this case, however, the desired product was obtained only in modest yield (25%, entry 8).
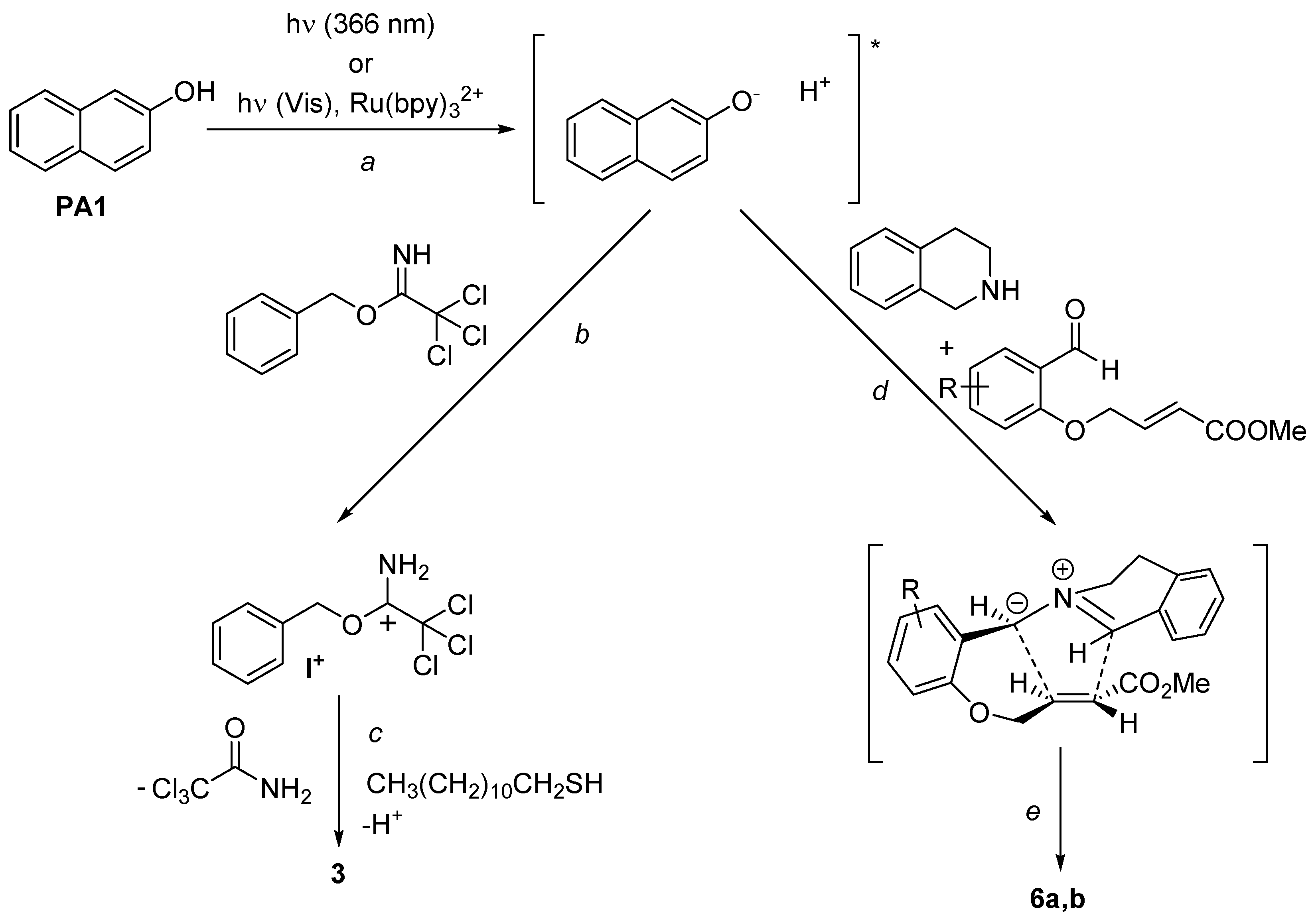
As stated in the Introduction, the behavior of naphthol derivatives as organophotoacids has been widely investigated [31,32,33]. In particular, the pKa* value for the photoexcited 2-naphthol (PA1) in water has been calculated as 2.8, with an enhancement of more than six orders of magnitude with respect to the value for the ground state (pKa = 9.45) [34]. Analogous results have been described in the literature for 7-bromo-2-naphthol, for which a ΔpKa > 10 was observed upon excitation [26]. In this case, intersystem crossing from the singlet excited state to the triplet of PA2 is favored by the presence of a bromine atom as substituent, thus the 3PA2 state is responsible for the photoacidity of the molecule [35]. Such excited states could also be populated by using RuII(bpy)3Cl2 as the photosensitizer under visible light irradiation. For the sake of simplicity, the mechanism of the reactions has been described in Scheme 2 for the case of 2-naphthol. Thus, irradiation of 2-naphthol results in the formation of a short-lived, photoexcited acid, that can interact with the proton sensitive moiety of the substrate. As depicted in Scheme 2, interaction of the released proton with trichloroacetimidates (path b) results in the formation of the cationic intermediate I+, which undergoes nucleophilic substitution to form the desired sulfide 3 [36]. On the other hand, the photostimulated acidity of PA1 and PA2 is also responsible for the formation of azomethine ylides 7 via proton-induced coupling (path d) of tetrahydroquinoline 5 and aldehydes 4a,b [30]. As also described in literature [26,27] phenol (1 equiv) could act as a sacrificial proton source to restore the starting photoacid. The intermediate then afforded the desired alkaloids 6a,b (path e).
In conclusion, we demonstrated that molecules exhibiting a behavior as organophotoacids can be efficiently used in organic synthesis, in both C–S and C–C bond formation processes, avoiding the use of aggressive acids as well as of harsh conditions.
3. Material and Methods
3.1. General
All the chemicals and solvents used for the synthesis of 3, 4a,b and 6a,b were commercially available and used as received. The solvents used for the irradiations (GC or HPLC grade purity), as well as 2-naphthol, 7-bromo-2-naphthol and tris(2,2’-bipyrydyl)dichlororuthenium(II) hexahydrate were likewise commercially available and used as received. Dichloromethane (GC grade purity) and Toluene (GC grade purity) were purified by distillation over the drying agent CaCl2. All sensitive reactions were carried out under a positive static atmosphere of Ar. Syringes, needles and the other glassware were dried at 140 °C for at least one night and allowed to cool in a desiccator over P2O5 before use. Routine monitoring of reactions was performed using silica gel 60 mesh (0.25 mm) aluminum-supported Thin Layer Chromatography (TLC) plates (purchased from Merck, Rome, Italy). Compounds were visualized by UV irradiation at a wavelength of 254 nm or stained by exposure to a 0.5% solution of vanillin in H2SO4/EtOH, followed by charring. NMR spectra were recorded at 200 or 300 (for 1H) and 75 MHz (for 13C), respectively, in the solvents indicated; chemical shifts (δ) are given in ppm relative to TMS, and coupling constants (J) are in hertz (Hz). The solvent signals were used as references, and the chemical shifts were converted to the TMS scale (CDCl3: δ-C 77.00; residual CHCl3 in CDCl3: 7.26 ppm). The number of H-atoms attached to each C-atom was determined by DEPT experiments. Photochemical reactions were carried out by using argon purged solutions in quartz tubes. Irradiations were performed in a multi-lamp reactor fitted with 10 × 15 W phosphor-coated Hg lamps (emission centered at 366 or 310 nm). In selected cases, the processes were also performed by irradiation in Pyrex vials in a reactor equipped with LED 1 W (emission centered at 410 nm). For compounds 3, 4a and 6a, GC/MS analyses were carried out using a single quadrupole GC/MS system. The GC oven temperature was held at 80 °C for 1 min, increased to 280 °C by a temperature ramp of 10 °C min-1 and held for 5 min. For compounds 4b and 6b, the GC oven temperature was held at 80 °C for 1 min, increased to 280 °C by a temperature ramp of 10 °C min−1 and held for 15 min. Mass spectral analysis was carried out in full scan mode.
3.2. Analytical Data for Compounds 3–6
3.2.1. Synthesis of Benzyl(dodecyl)sulfide (3)
Compound 3 was prepared by following a known procedure [37]. Spectroscopical data were in accordance with the literature [38]. 1H-NMR (200 MHz, CDCl3) δ(ppm); 7.40–7.15 (m, 5H), 3.72 (s, 2H), 2.42 (t, J = 7.3 Hz, 2H), 1.55 (d, J = 9.0 Hz, 4H), 1.26 (s, 24H), 0.99–0.77 (m, 4H). GC–MS (m/z): 90.9 (100), 92.0 (32).
3.2.2. Photochemical synthesis of 3
A nitrogen-saturated solution of benzyl trichloroacetimidate (1, 0.1 M), dodecylsulfide (2, 0.5 M, 5 equiv), and 7-bromo-2-naphthol (0.05 M, PA2, 50 mol%) and Ru[(bpy)3]2+ (5 mol%) in dry MeCN (1 mL) was irradiated for 12 h in a multi-lamp reactor equipped with 10 phosphor coated Hg lamps (15 W, λem = 366 nm). The product formed (3) was identified and quantified by comparison with calibration curves of authentic samples.
3.2.3. Synthesis of (E)-methyl 4-(2-formylphenoxy)but-2-enoate (4a)
Compound 4a was prepared by following a known procedure [30]. 1H-NMR (300 MHz, CDCl3) δ(ppm): 10.56 (s, 1H), 7.87 (ddd, J = 7.7, 1.8, 0.4 Hz, 1H), 7.56 (ddd, J = 8.5, 7.4, 1.9 Hz, 1H), 7.19–7.03 (m, 3H), 6.95 (d, J = 8.5 Hz, 0H), 6.24 (dt, J = 15.8, 2.1 Hz, 1H), 4.88–4.81 (m, 2H), 3.79 (s, 3H). 13C NMR (75 MHz, CDCl3) δ(ppm): 189.1, 166.1, 160.0, 141.4, 135.8, 128.7, 125.0, 121.9, 121.3, 112.4, 66.7, 51.7
3.2.4. Synthesis of Methyl-6a,7,7a,12,13,14a-hexahydro-6H chromeno[3′,4′:4,5]pyrrolo[2,1-a]isoquinoline-7-carboxylate (6a)
Compound 6a was prepared by following a known procedure [30]. 1H-NMR (300 MHz, CDCl3) δ(ppm): 7.51–7.42 (m, 1H), 7.25–7.13 (m, 1H), 7.15–7.04 (m, 3H), 7.06–6.91 (m, 2H), 6.88 (dd, J = 8.2, 1.3 Hz, 1H), 4.48 (dd, J = 8.1, 3.6 Hz, 2H), 4.34 (dd, J = 11.5, 3.9 Hz, 1H), 4.23 (dd, J = 11.5, 2.7 Hz, 1H), 3.59 (dd, J = 9.3, 6.2 Hz, 1H), 3.49 (ddd, J = 11.3, 8.6, 4.4 Hz, 1H), 3.30 (s, 1H), 3.08–2.88 (m, 3H). 13C-NMR (75 MHz, CDCl3) δ(ppm): 174.4, 155.5, 135.1, 133.4, 130.4, 128.4, 128.3, 127.4, 126.3, 125.1, 121.2, 116.6, 65.9, 62.4, 62.3, 51.5, 51.3, 46.0, 40.5, 28.6. GC-MS (m/z): 27.9 (78), 76.9 (100), 114.9 (90), 130.9 (78), 144.9 (90), 203.0 (54).
3.2.5. Synthesis of (E)-methyl 4-(2-formyl-5-methoxyphenoxy)but-2-enoate 4b
Compound 4b was prepared by following a known procedure [30]. 1H-NMR (300 MHz, CDCl3) δ(ppm) 10.37 (s, 1H), 7.84 (d, J = 8.7 Hz, 1H), 7.11 (dt, J = 15.7, 4.0 Hz, 1H), 6.59 (ddd, J = 8.6, 2.3, 0.8 Hz, 1H), 6.40 (d, J = 2.2 Hz, 1H), 6.23 (dt, J = 15.7, 2.0 Hz, 1H), 4.80 (dd, J = 4.1, 2.1 Hz, 2H), 3.87 (s, 3H), 3.78 (s, 3H). 13C-NMR (75 MHz, CDCl3) δ(ppm): 187.7, 166.1, 165.9, 161,7, 141.2, 130.8, 121.9, 119.1, 106.2, 98.9, 66.7, 55.6, 51.7.
3.2.6. Synthesis of Methyl-methoxy-6a,7,7a,12,13,14a-hexahydro-6H-chromeno[3′,4′:4,5]pyrrolo[2,1-a]isoquinoline-7-carboxylate 6b
Compound 6b was prepared by following a known procedure [30]. 1H-NMR (300 MHz, CDCl3) δ(ppm): 7.35 (d, J = 8.5 Hz, 1H), 7.13–7.05 (m, 3H), 7.04–6.98 (m, 1H), 6.56 (dd, J = 8.6, 2.6 Hz, 1H), 6.42 (d, J = 2.6 Hz, 1H), 4.45 (d, J = 7.1 Hz, 2H), 4.33 (dd, J = 11.5, 3.9 Hz, 1H), 4.21 (dd, J = 11.5, 2.8 Hz, 1H), 3.79 (s, 3H), 3.57 (dd, J = 9.3, 6.1 Hz, 1H), 3.45 (q, J = 8.0, 5.9 Hz, 1H), 3.29 (s, 3H), 2.96 (qd, J = 12.8, 11.4, 5.8 Hz, 4H). 13C-NMR (75 MHz, CDCl3) δ(ppm): 174.3, 159.8, 156.4, 135.1, 130.8, 128.3, 127.4, 126.3, 125.1, 108.4, 101.1, 66.1, 62.4, 61.9, 55.2, 51.5, 51.3, 45.9, 40.5, 28.5. GC-MS (m/z): 161.1 (100), 175.1 (90), 203.1 (62), 204.1 (48), 235.1 (100), 266.7 (30)
3.2.7. Photochemical synthesis of 6a,b
A nitrogen-saturated solution of 1,2,3,4-tetrahydroisoquinoline 5 (0.1 M), salicylaldehydes 4a,b (0.15 M, 1.5 equiv), 2-naphthol (0.075 M, PA1, 50 mol% with respect to the chosen salicylaldehyde) and Ru[(bpy)3]2+ (5 mol%) in dry MeCN (1 mL, the reaction was carried out in the presence of 4 Å MS) was poured in a Pyrex vial and irradiated at 410 nm for 24 h (LED, 1 W). The use of phenol (1 equiv) as the proton source in selected cases was also investigated. The product formed was identified and quantified by comparison with calibration curves of authentic samples.
Supplementary Materials
The following are available online at https://www.mdpi.com/1420-3049/24/7/1318/s1: Copy of the 1H and 13 NMR spectra of the synthesized compounds (Section S1), and UV-Visible spectra of compounds PA1, PA2, 1 and 4a Figures S1–S4.
Author Contributions
Conceptualization, S.P. and G.Z.; methodology, A.S. and M.F.; formal analysis, A.S.; investigation, A.S. and M.F. writing—original draft preparation, S.P.; writing—review and editing, G.Z.
Funding
This research received no external funding
Conflicts of Interest
The authors declare no conflict of interest.
References
- Oelgemöller, M. Solar Photochemical Synthesis: From the Beginnings of Organic Photochemistry to the Solar Manufacturing of Commodity Chemicals. Chem. Rev. 2016, 116, 9664–9682. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, D.; Protti, S.; Fagnoni, M. Carbon–Carbon Bond Forming Reactions via Photogenerated Intermediates. Chem. Rev. 2016, 116, 9850–9913. [Google Scholar] [CrossRef]
- Marzo, L.; Pagire, S.K.; Reiser, O.; König, B. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem. Int. Ed. 2018, 57, 10034–10072. [Google Scholar] [CrossRef]
- Yoon, T.P.; Ischay, M.A.; Du, J. Visible Light Photocatalysis As A Greener Approach To Photochemical Synthesis. Nat. Chem. 2010, 2, 527–532. [Google Scholar] [CrossRef]
- Albini, A.; Protti, S. Photochemistry; The Royal Society of Chemistry: London, UK, 2019. [Google Scholar]
- Hoffmann, N. Photochemical reactions of aromatic compounds and the concept of the photon as a traceless reagent. Photochem. Photobiol. Sci. 2012, 11, 1613–1641. [Google Scholar] [CrossRef]
- Albini, A.; Fagnoni, M. Green chemistry and photochemistry were born at the same time. Green Chem. 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Nagib, D.A.; MacMillan, D.W.C. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 480, 224–228. [Google Scholar] [CrossRef]
- Pham, P.V.; Nagib, D.A.; MacMillan, D.W.C. Photoredox Catalysis: A Mild, Operationally Simple Approach to the Synthesis of α-Trifluoromethyl Carbonyl Compounds. Angew. Chem. Int. Ed. 2011, 50, 6119–6122. [Google Scholar] [CrossRef]
- Ischay, M.A.; Anzovino, M.E.; Du, J.; Yoon, T.P. Efficient Visible Light Photocatalysis of 2 + 2 Enone Cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. [Google Scholar] [CrossRef]
- Protti, S.; Fagnoni, M.; Ravelli, D.D. Photocatalytic C-H Activation by Hydrogen-Atom Transfer in Synthesis. ChemCatChem 2015, 7, 1516–1523. [Google Scholar] [CrossRef]
- Yavorskyy, A.; Shvydkiv, O.; Hoffmann, N.; Nolan, K.; Oelgemoeller, M. Parallel Microflow Photochemistry: Process Optimization, Scale-up, and Library Synthesis. Org. Lett. 2012, 14, 4342–4345. [Google Scholar] [CrossRef]
- Ravelli, D.; Protti, S.; Fagnoni, M. Decatungstate Anion for Photocatalyzed “Window Ledge” Reactions. Acc. Chem. Res. 2016, 49, 2232–2242. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Shaikh, R.S.; König, B. Sensitization-Initiated Electron Transfer for Photoredox Catalysis. Angew. Chem. Int. Ed. 2017, 56, 8544–8549. [Google Scholar] [CrossRef] [PubMed]
- Marchini, M.; Bergamini, G.; Cozzi, P.G.; Ceroni, P.; Balzani, V. Photoredox Catalysis: The Need to Elucidate the Photochemical Mechanism. Angew. Chem. Int. Ed. 2017, 56, 12820–12821. [Google Scholar] [CrossRef]
- Tolbert, L.M.; Solntsev, K.M. Excited-State Proton Transfer: From Constrained Systems to “Super” Photoacids to Superfast Proton Transfer. Acc. Chem. Res. 2002, 35, 19–27. [Google Scholar] [CrossRef]
- Solntsev, K.M.; Huppert, D.; Tolbert, L.M.; Agmon, N. Solvatochromic Shifts of “Super” Photoacids. J. Am. Chem. Soc. 1998, 120, 7981–7982. [Google Scholar] [CrossRef]
- Solntsev, K.M.; Huppert, D.; Agmon, N.; Tolbert, L.M. Photochemistry of “Super” Photoacids. 2. Excited-State Proton Transfer in Methanol/Water Mixtures. J. Phys. Chem. A 2000, 104, 4658–4669. [Google Scholar] [CrossRef]
- Claya, A.; Krishnan, R.; Sibi, M.; Websterd, D.; Jockusch, S.; Sivaguru, J. Photoacidity of vanillin derivatives. J. Photochem. Photobiol. A Chem. 2018, 355, 38–41. [Google Scholar] [CrossRef]
- Nishikubo, Y.; Kanzaki, S.; Matsumura, S.; Toshima, K. The use of organophotoacids for deprotection reactions in organic synthesis. Tetrahedron Lett. 2006, 47, 8125–8128. [Google Scholar] [CrossRef]
- Oates, R.P.; Jones, P.B. Photosensitized Tetrahydropyran Transfer. J. Org. Chem. 2008, 73, 4743–4745. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Niu, L.; Wang, S.; Liu, T.; Singh, A.K.; Lei, A. Visible-Light-Induced Acetalization of Aldehydes with Alcohols. Org. Lett. 2017, 19, 122–125. [Google Scholar] [CrossRef]
- Yan, D.-M.; Chen, J.-R.; Xiao, W.-J. New Roles for Photoexcited Eosin Y in Photochemical Reactions. Angew. Chem. Int. Ed. 2019, 58, 378–380. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, T. Stereoselective Synthesis of 2-Deoxyglycosides from Glycals by Visible-Light-Induced Photoacid Catalysis. Angew. Chem. Int. Ed. 2018, 57, 6120–6124. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Eto, T.; Takahashi, D.; Toshima, K. Stereocontrolled Photoinduced Glycosylation Using an Aryl Thiourea as an Organo Photoacid. Org. Lett. 2016, 18, 3190–3193. [Google Scholar] [CrossRef]
- Das, A.; Banerjee, T.; Hanson, K. Protonation of silylenol ether via excited state proton transfer catalysis. Chem. Commun. 2016, 52, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Ayad, S.; Hanson, K. Enantioselective Protonation of Silyl Enol Ether Using Excited State Proton Transfer Dyes. Org. Lett. 2016, 18, 5416–5419. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.Y.; Roush, W.R. Concerning the Origin of the High β-Selectivity of Glycosidation Reactions of 2-Deoxy-2-iodo-glucopyranosyl Trichloroacetimidates. Org. Lett. 2002, 4, 4523–4526. [Google Scholar] [CrossRef]
- Iwata, R.; Uda, K.; Takahashi, D.; Toshima, K. Photo-induced glycosylation using reusable organophotoacids. Chem. Commun. 2014, 50, 10695–10698. [Google Scholar] [CrossRef] [PubMed]
- Mantelingu, K.; Lin, Y.; Seidel, D. Intramolecular [3 + 2]-Cycloadditions of Azomethine Ylides Derived from Secondary Amines via Redox-Neutral C–H Functionalization. Org. Lett. 2014, 16, 5910–5913. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Cheng, Y.-C. Molecular electrostatic potential on the proton-donating atom as a theoretical descriptor of excited state acidity. Phys. Chem. Chem. Phys. 2018, 20, 4351–4359. [Google Scholar] [CrossRef]
- Acharya, A.; Chaudhuri, S.; Batista, V.S. Can TDDFT Describe Excited Electronic States of Naphthol Photoacids? A Closer Look with EOM-CCSD. J. Chem. Theory Comput. 2018, 14, 867–876. [Google Scholar] [CrossRef]
- Pines, E. UV-Visible Spectra and Photoacidity of Phenols, Naphthols and Pyrenols. In The Chemistry of Phenols-PATAI’S Chemistry of Functional Groups; Rappoport, Z., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Tolbert, L.M.; Haubrich, J.E. Photoexcited Proton Transfer from Enhanced Photoacids. J. Am. Chem. Soc. 1994, 116, 10593–10600. [Google Scholar] [CrossRef]
- Mcclure, D.S.; Blake, N.W.; Hanst, P.L. Singlet-Triplet Absorption Bands in Some Halogen Substituted Aromatic Compounds. J. Chem. Phys. 1954, 22, 255–258. [Google Scholar] [CrossRef]
- Eckenberg, P.; Groth, U.; Huhn, T.; Richter, N.; Schmeck, C. A Useful Application of Benzyl Trichloroacetimidate for the Benzylation of Alcohols. Tetrahedron 1993, 49, 1619–1624. [Google Scholar] [CrossRef]
- Yin, J.; Pidgeon, C. A simple and efficient method for preparation of unsymmetrical sulfides. Tetrahedron Lett. 1997, 38, 5953–5954. [Google Scholar] [CrossRef]
- Tanaka, K.; Ajiki, K. Rhodium-Catalyzed Reaction of Thiols with Polychloroalkanes in the Presence of Triethylamine. Org. Lett. 2005, 7, 1537–1539. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
Scheme 1.
Photocatalytic Activation of Substrates.

Scheme 2.
Proposed mechanism for the acid induced formation of sulfides 3 and alkaloids 6a,b via photoexcited 2-napthol (* PA1).
Scheme 2.
Proposed mechanism for the acid induced formation of sulfides 3 and alkaloids 6a,b via photoexcited 2-napthol (* PA1).


{kind=link}
{kind=link}
{kind=link}
Table 1.
Acid catalyzed synthesis of benzylsulfide 3.
 | |||
|---|---|---|---|
| Entry | Conditions a | Light Source (nm) | Yield b (%) |
| 1 | DMSO, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | - |
| 2 | Hexane, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | 13 |
| 3 | PhMe, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | 15 |
| 4 | DCE, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | 21 |
| 5 | Et2O, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | 8 |
| 6 | MeCN, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | 4 |
| 7 | DCM, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | 28 |
| 8 | DCM, 1 (0.1 M), 2 (1.0 M), PA1 (30 mol%) | 366 | 40 |
| 9 | DCM, 1 (0.1 M), 2 (0.2 M), PA1 (30 mol%) | 366 | 12 |
| 10 | DCM, 1 (0.1 M), 2 (0.5 M), PA1 (50 mol%) | 366 | 40 |
| 11 | DCM, 1 (0.1 M), 2 (0.5 M), PA1 (50 mol%) | 310 | 21 |
| 12 | DCM, 1 (0.1 M), 2 (0.5 M), PA2 (30 mol%) | 366 | 32 |
| 13 | DCM, 1 (0.1 M), 2 (0.5 M), PA2 (50 mol%) | 366 | 47 |
| 14 | MeCN, 1 (0.1 M), 2 (0.5 M), PA2 (50 mol%) c | 366 | 9 |
| 15 | DCM, 1 (0.1 M), 2 (0.5 M) | 366 | 12 |
| 16 | DCM, 1 (0.1 M), 2 (0.5 M), PA2 (50 mol%) d | - d | - |
a mol% of 2-naphthol (PA1) and 7-bromo-2-naphthol (PA2) were calculated with respect to the amount of 1; b Determined by GC analyses; c 1.5 mol% of RuII was used; d No light.
Table 2.
Acid induced synthesis of alkaloids 6a,b.
 | |||
|---|---|---|---|
| Entry | Conditions a | Light Source (nm) | Product (% Yield) b |
| 1 | 4a (0.1 M) 5 (0.12 M), PA1 or PA2 (50 mol%) | 366 | 6a, < 5% |
| dry DCM | |||
| 2 | 4a (0.1 M), 5 (0.12 M), PA1 (50 mol%), Ru[(bpy)3]2+ | 410 | 6a, 19% |
| (5 mol%), dry MeCN | |||
| 3 | 4a (0.1 M), 5 (0.1 M), PA1 (50 mol%), Ru[(bpy)3]2+ | 410 | 6a, 43% |
| (5 mol%), dry MeCN | |||
| 4 | 4a (0.15 M), 5 (0.10 M), PA1 (50 mol%), Ru[(bpy)3]2+ | 410 | 6a, 65% |
| (5 mol%), dry MeCN | |||
| 5 | 4a (0.2 M), 5 (0.10 M), PA1 (50 mol%), Ru[(bpy)3]2+ | 410 | 6a, 63% |
| (5 mol%), dry MeCN c | |||
| 6 | 4a (0.15 M), 5 (0.10 M), Ru[(bpy)3]2+ | 410 | 6a, 20% |
| (5 mol%), dry MeCN | |||
| 7 | 4a (0.15 M), 5 (0.10 M), PA2 (30 mol%), Ru[(bpy)3]2+ | 410 | 6a, 62% |
| (5 mol%), dry MeCN | |||
| 8 | 4b (0.15 M), 5 (0.10 M), PA1 (50 mol%), Ru[(bpy)3]2+ | 410 | 6b, 25% |
| (5 mol%), dry MeCN | |||
a mol% of photocatalyst and PA1, PA2 were calculated with respect to the amount of 1; b Determined by GC analyses; c 1.0 equiv of phenol was used.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Strada, A.; Fredditori, M.; Zanoni, G.; Protti, S. Acid Catalyzed Formation of C–C and C–S Bonds via Excited State Proton Transfer. Molecules 2019, 24, 1318. https://doi.org/10.3390/molecules24071318
AMA Style
Strada A, Fredditori M, Zanoni G, Protti S. Acid Catalyzed Formation of C–C and C–S Bonds via Excited State Proton Transfer. Molecules. 2019; 24(7):1318. https://doi.org/10.3390/molecules24071318
Chicago/Turabian StyleStrada, Alessandro, Mattia Fredditori, Giuseppe Zanoni, and Stefano Protti. 2019. "Acid Catalyzed Formation of C–C and C–S Bonds via Excited State Proton Transfer" Molecules 24, no. 7: 1318. https://doi.org/10.3390/molecules24071318