Enhancing the Yield of Bioactive Compounds from Sclerocarya birrea Bark by Green Extraction Approaches

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
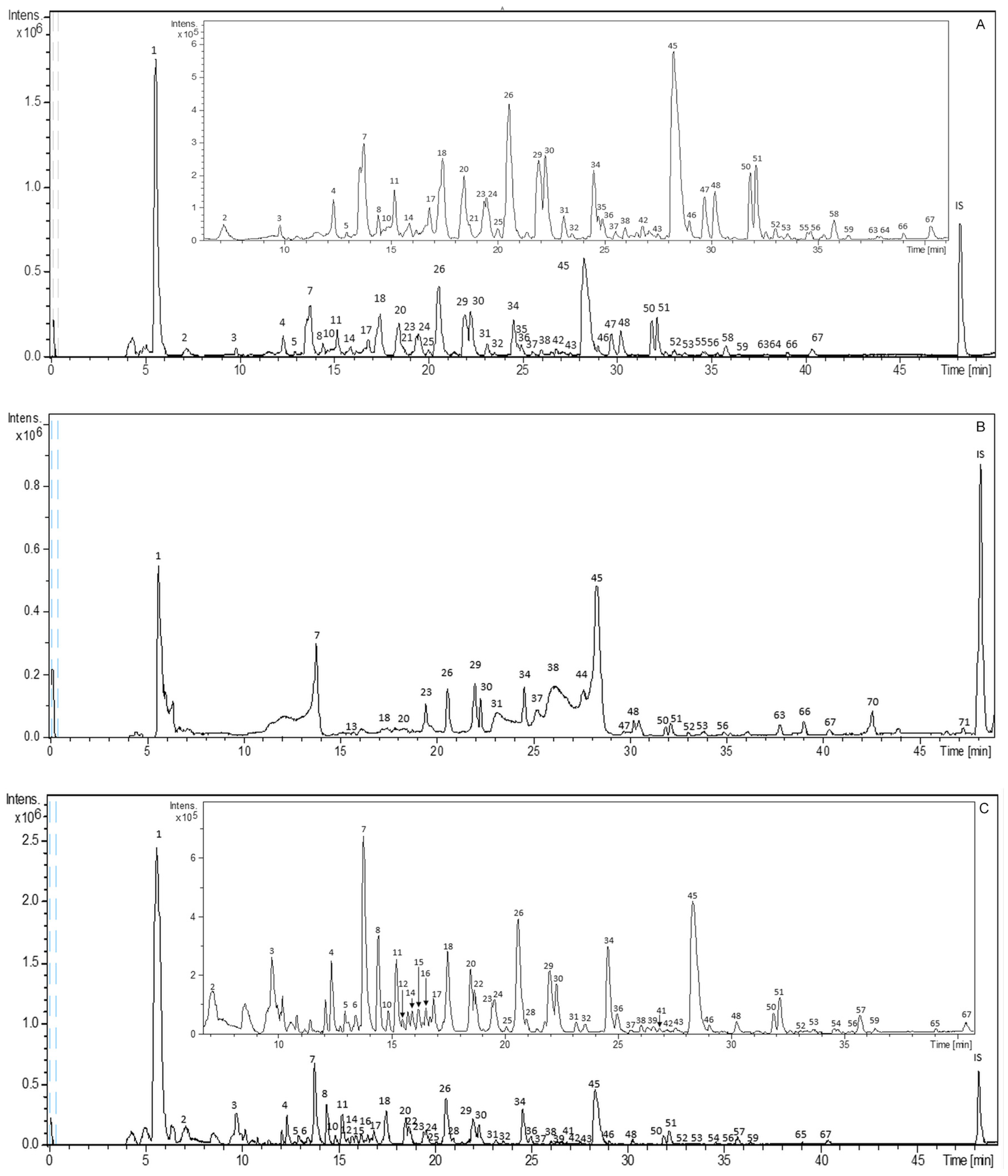
2.1. Characterization of Bioactive Compounds from S. birrea by HPLC–ESI–TOF–MS
2.1.1. Gallic Acid and Derivatives
2.1.2. Monomers and Dimers from (Epi)Catechin and Derivatives
2.1.3. Flavonoids
2.1.4. Other Compounds
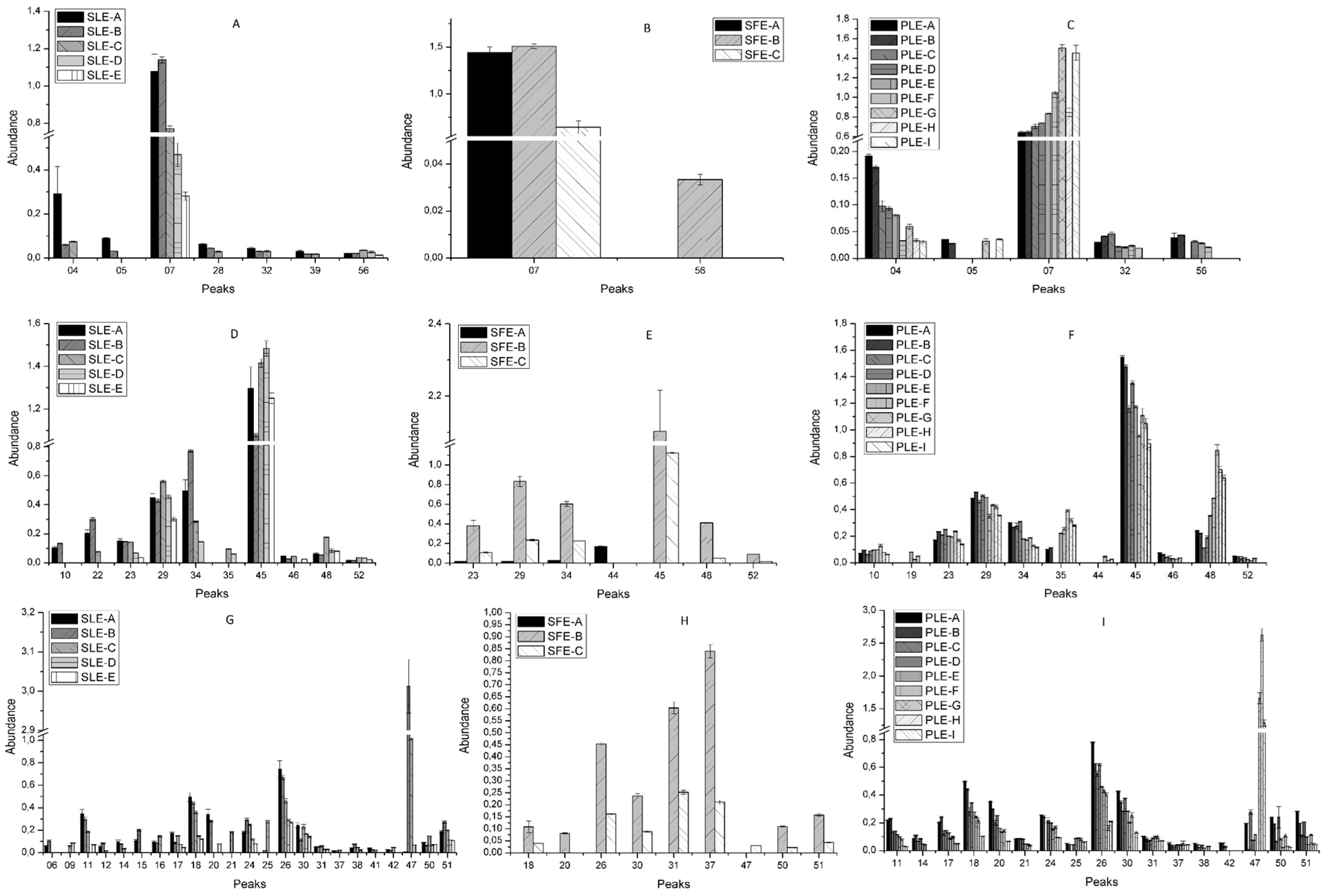
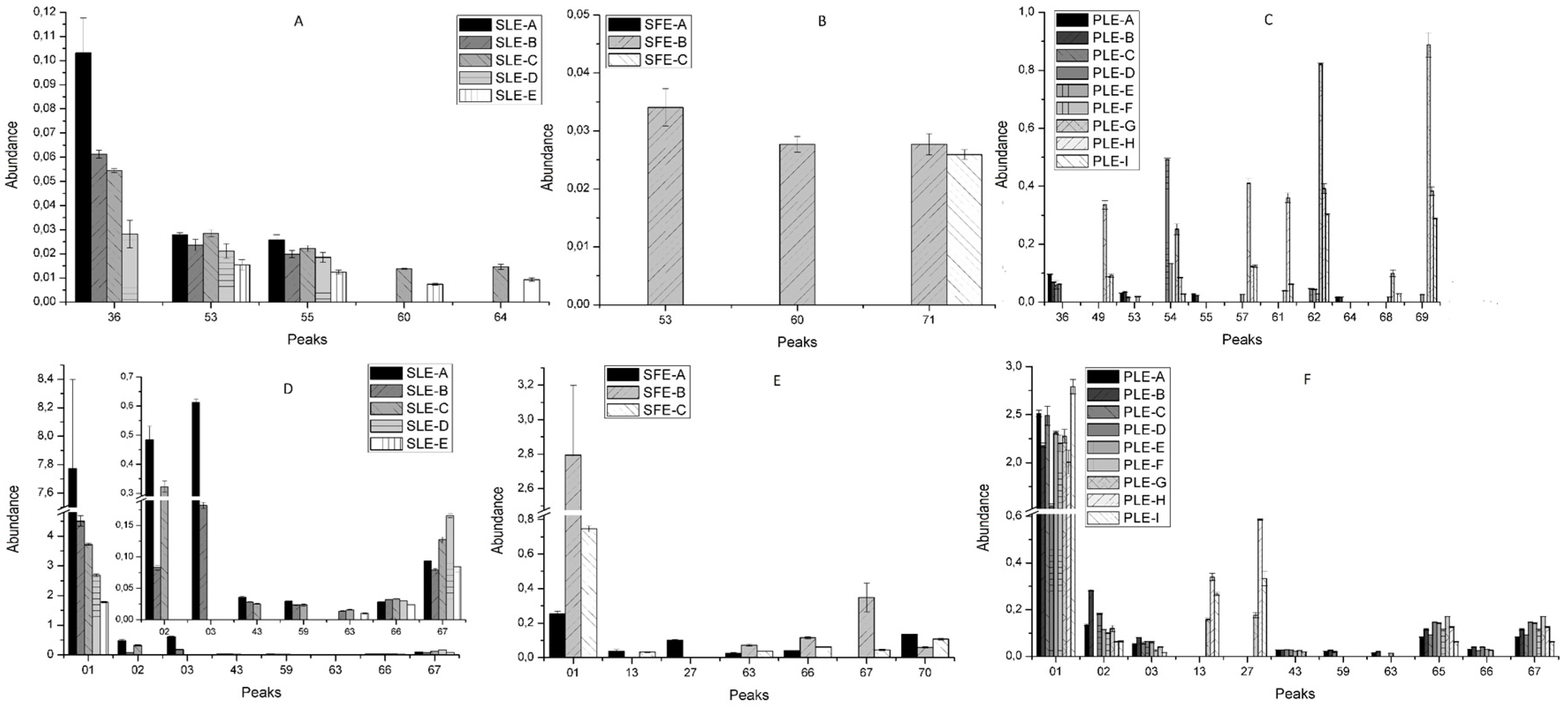
2.2. Effect of Conventional and New Extraction Techniques on Bioactive Compounds Recovery
Extraction Yield
2.3. Comparative Study of the Phytochemical Composition of S. Birrea Extracts Obtained with different Extraction Methodologies
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Instrumentation
3.3. Sample Preparation
3.4. Extraction Methods and Conditions
3.4.1. Conventional Solid–Liquid Extraction (SLE)
3.4.2. Supercritical Fluid Extraction (SFE)
3.4.3. Pressurized Liquid Extraction (PLE)
3.5. HPLC–ESI–TOF–MS Analysis
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Helm, C.V.; Scott, S.L.; Witkowski, E.T.F. Reproductive potential and seed fate of Sclerocarya birrea subsp. caffra (marula) in the low altitude savannas of South Africa. S. Afr. J. Bot. 2011, 77, 650–664. [Google Scholar] [CrossRef] [Green Version]
- Council, N.R. Lost Crops of Africa: Volume III: Fruits; The National Academies Press: Washington, DC, USA, 2008; ISBN 978-0-309-10596-5. [Google Scholar]
- Kutama, A.S.; Auyo, M.I.; Umar, M.L.; Hadiza, M.S. Assessing the antibacterial activity of morula (Sclerocarya birrea) stem bark and leaf extracts against some selected bacterial isolates in Kano, Nigeria. World J. Agric. Sci. 2013, 1, 209–214. [Google Scholar]
- Kpoviessi, D.S.S.; Gbaguidi, F.A.; Kossouoh, C.; Agbani, P.; Yayi-Ladekan, E.; Sinsin, B.; Moudachirou, M.; Accrombessi, G.C.; Quentin-Leclercq, J. Chemical composition and seasonal variation of essential oil of Sclerocarya birrea (A. Rich.) Hochst subsp birrea leaves from Benin. J. Med. Plants Res. 2011, 5, 4640–4646. [Google Scholar]
- Ojewole, J.A.O. Evaluation of the analgesic, anti-inflammatory and anti-diabetic properties of Sclerocarya birrea (A. Rich.) Hochst. stem-bark aqueous extract in mice and rats. Phyther. Res. 2004, 18, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Gondwe, M.; Kamadyaapa, D.R.; Tufts, M.; Chuturgoon, A.A.; Musabayane, C.T. Sclerocarya birrea [(A. Rich.) Hochst.] [Anacardiaceae] stem-bark ethanolic extract (SBE) modulates blood glucose, glomerular filtration rate (GFR) and mean arterial blood pressure (MAP) of STZ-induced diabetic rats. Phytomedicine 2008, 15, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Fotio, A.L.; Dimo, T.; Nguelefack, T.B.; Dzeufiet, P.D.D.; Ngo Lemba, E.; Temdie, R.J.; Ngueguim, F.; Olleros, M.L.; Vesin, D.; Dongo, E.; et al. Acute and chronic anti-inflammatory properties of the stem bark aqueous and methanol extracts of Sclerocarya birrea (Anacardiaceae). Inflammopharmacology 2009, 17, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Ojewole, J.A.O. Evaluation of the anti-inflammatory properties of Sclerocarya birrea (A. Rich.) Hochst. (family: Anacardiaceae) stem-bark extracts in rats. J. Ethnopharmacol. 2003, 85, 217–220. [Google Scholar] [CrossRef]
- Masoko, P.; Mmushi, T.J.; Mogashoa, M.M.; Mokgotho, M.P.; Mampuru, L.J.; Howard, R.L. In vitro evaluation of the antifungal activity of Sclerocarya birrea extracts against pathogenic yeasts. Afr. J. Biotechnol. 2008, 7, 3521–3526. [Google Scholar]
- Eloff, J.N. Antibacterial activity of Marula (Sclerocarya birrea (A. rich.) Hochst. subsp. caffra (Sond.) Kokwaro) (Anacardiaceae) bark and leaves. J. Ethnopharmacol. 2001, 76, 305–308. [Google Scholar] [CrossRef]
- Borochov-Neori, H.; Judeinstein, S.; Greenberg, A.; Fuhrman, B.; Attias, J.; Volkova, N.; Hayek, T.; Aviram, M. Phenolic Antioxidants and Antiatherogenic Effects of Marula (Sclerocarrya birrea Subsp. caffra) Fruit Juice in Healthy Humans. J. Agric. Food Chem. 2008, 56, 9884–9891. [Google Scholar] [CrossRef]
- Ojewole, J.A.O. Vasorelaxant and hypotensive effects of Sclerocarya birrea (A Rich) Hochst (Anacardiaceae) stem bark aqueous extract in rats. Cardiovasc. J. S. Afr. 2006, 17, 117–123. [Google Scholar] [PubMed]
- Mariod, A.A.; Matthäus, B.; Hussein, I.H. Antioxidant properties of methanolic extracts from different parts of Sclerocarya birrea. Int. J. Food Sci. Technol. 2008, 43, 921–926. [Google Scholar] [CrossRef]
- Mariod, A.A.; Matthäus, B.; Eichner, K.; Hussein, I.H. Antioxidant activity of extracts from Sclerocarya birrea kernel oil cake. Grasas y Aceites 2006, 57, 361–366. [Google Scholar] [CrossRef]
- Jiménez-Sánchez, C.; Lozano-Sánchez, J.; Gabaldón-Hernández, J.A.; Segura-Carretero, A.; Fernández-Gutiérrez, A. RP-HPLC–ESI–QTOF/MS2 based strategy for the comprehensive metabolite profiling of Sclerocarya birrea (marula) bark. Ind. Crops Prod. 2015, 71, 214–234. [Google Scholar] [CrossRef]
- Cádiz-Gurrea, M.d.l.L.; Fernández-Arroyo, S.; Joven, J.; Menéndez, J.A.; Micol, V.; Segura-Carretero, A. Proanthocyanidins in Agro-Industrial By-Products: Health Benefits. In Occurrences, Structure, Biosynthesis, and Health Benefits Based on Their Evidences of Medicinal Phytochemicals in Vegetables and Fruits; Motohashi, N., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2015; Volume 3, pp. 63–114. ISBN 978-1-63482-710-2. [Google Scholar]
- Kim, N.Y.; Jang, M.K.; Lee, D.G.; Yu, K.H.; Jang, H.J.; Kim, M.; Kim, S.G.; Yoo, B.H.; Lee, S.H. Comparison of methods for proanthocyanidin extraction from pine (Pinus densiflora) needles and biological activities of the extracts. Nutr. Res. Pract. 2010, 4, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Ojewole, J.A.O. Anticonvulsant effect of Sclerocarya birrea (A. Rich.) Hochst. subsp. caffra (Sond.) Kokwaro (Anacardiaceae) stem-bark aqueous extract in mice. J. Nat. Med. 2007, 61, 67–72. [Google Scholar] [CrossRef]
- Taamalli, A.; Arráez-Román, D.; Barrajón-Catalán, E.; Ruiz-Torres, V.; Pérez-Sánchez, A.; Herrero, M.; Ibañez, E.; Micol, V.; Zarrouk, M.; Segura-Carretero, A.; et al. Use of advanced techniques for the extraction of phenolic compounds from Tunisian olive leaves: Phenolic composition and cytotoxicity against human breast cancer cells. Food Chem. Toxicol. 2012, 50, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Herrero, M.; Cifuentes, A.; Ibañez, E. Sub- and supercritical fluid extraction of functional ingredients from different natural sources: Plants, food-by-products, algae and microalgae—A review. Food Chem. 2006, 98, 136–148. [Google Scholar] [CrossRef]
- Herrero, M.; Plaza, M.; Cifuentes, A.; Ibáñez, E. Green processes for the extraction of bioactives from Rosemary: Chemical and functional characterization via ultra-performance liquid chromatography-tandem mass spectrometry and in-vitro assays. J. Chromatogr. A 2010, 1217, 2512–2520. [Google Scholar] [CrossRef] [Green Version]
- Chemat, F.; Vian, M.A.; Cravotto, G. Green extraction of natural products: Concept and principles. Int. J. Mol. Sci. 2012, 13, 8615–8627. [Google Scholar] [CrossRef]
- Santos, C.C.d.S.; Masullo, M.; Cerulli, A.; Mari, A.; Estevam, C.D.S.; Pizza, C.; Piacente, S. Isolation of antioxidant phenolics from Schinopsis brasiliensis based on a preliminary LC-MS profiling. Phytochemistry 2017, 140, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Lampire, O.; Mila, I.; Raminosoa, M.; Michon, V.; Penhoat, C.H.D.; Faucheur, N.; Laprevote, O.; Scalbert, A. Polyphenols isolated from the bark of castanea sativa Mill. chemical structures and auto-association in honour of professor G. H. Neil Towers 75th birthday. Phytochemistry 1998, 49, 623–631. [Google Scholar] [CrossRef]
- Shi, C.; Xu, M.J.; Bayer, M.; Deng, Z.W.; Kubbutat, M.H.G.; Waejen, W.; Proksch, P.; Lin, W.H. Phenolic compounds and their anti-oxidative properties and protein kinase inhibition from the Chinese mangrove plant Laguncularia racemosa. Phytochemistry 2010, 71, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Jerez, M.; Touriño, S.; Sineiro, J.; Torres, J.J.L.; Núñez, M.J.M. Procyanidins from pine bark: Relationships between structure, composition and antiradical activity. Food Chem. 2007, 104, 518–527. [Google Scholar] [CrossRef]
- Barreca, D.; Bellocco, E.; Laganà, G.; Ginestra, G.; Bisignano, C. Biochemical and antimicrobial activity of phloretin and its glycosilated derivatives present in apple and kumquat. Food Chem. 2014, 160, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.-H.; Lin, C.-L.; Tsai, J.-H.; Ho, C.-T.; Chen, W.-J. 3,5,3′,4′,5′-Pentamethoxystilbene (MR-5), a Synthetically Methoxylated Analogue of Resveratrol, Inhibits Growth and Induces G1 Cell Cycle Arrest of Human Breast Carcinoma MCF-7 Cells. J. Agric. Food Chem. 2010, 58, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Horvath, Z.; Marihart-Fazekas, S.; Saiko, P.; Grusch, M.; Özsüy, M.; Harik, M.; Handler, N.; Erker, T.; Jaeger, W.; Fritzer-Szekeres, M.; et al. Novel resveratrol derivatives induce apoptosis and cause cell cycle arrest in prostate cancer cell lines. Anticancer Res. 2007, 27, 3459–3464. [Google Scholar] [PubMed]
- Chimento, A.; Sirianni, R.; Saturnino, C.; Caruso, A.; Sinicropi, M.S.; Pezzi, V. Resveratrol and Its Analogs As Antitumoral Agents For Breast Cancer Treatment. Mini Rev. Med. Chem. 2016, 16, 699–709. [Google Scholar] [CrossRef]
- Mustafa, A.; Turner, C. Pressurized liquid extraction as a green approach in food and herbal plants extraction: A review. Anal. Chim. Acta 2011, 703, 8–18. [Google Scholar] [CrossRef]
- Pinelo, M.; Zornoza, B.; Meyer, A.S. Selective release of phenols from apple skin: Mass transfer kinetics during solvent and enzyme-assisted extraction. Sep. Purif. Technol. 2008, 63, 620–627. [Google Scholar] [CrossRef]
- Hayatsu, H.; Inada, N.; Kakutani, T.; Arimoto, S.; Negishi, T.; Mori, K.; Okuda, T.; Sakata, I. Suppression of genotoxicity of carcinogens by (−)-epigallocatechin gallate. Prev. Med. (Baltim) 1992, 21, 370–376. [Google Scholar] [CrossRef]
- Gan, R.-Y.; Li, H.-B.; Sui, Z.-Q.; Corke, H. Absorption, metabolism, anti-cancer effect and molecular targets of epigallocatechin gallate (EGCG): An updated review. Crit. Rev. Food Sci. Nutr. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Della-Fera, M.A.; Baile, C.A. Green Tea Polyphenol Epigallocatechin Gallate Inhibits Adipogenesis and Induces Apoptosis in 3T3-L1 Adipocytes. Obes. Res. 2005, 13, 982–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lao, W.; Tan, Y.; Jin, X.; Xiao, L.; Kim, J.J.Y.; Qu, X. Comparison of Cytotoxicity and the Anti-Adipogenic Effect of Green Tea Polyphenols with Epigallocatechin-3-Gallate in 3T3-L1 Preadipocytes. Am. J. Chin. Med. 2015, 43, 1177–1190. [Google Scholar] [CrossRef] [PubMed]
- Ghoreishi, S.M.; Heidari, E. Extraction of epigallocatechin gallate from green tea via modified supercritical CO2: Experimental, modeling and optimization. J. Supercrit. Fluids 2012, 72, 36–45. [Google Scholar] [CrossRef]
- Cádiz-Gurrea, M.d.l.L.; Lozano-Sánchez, J.; Contreras-Gámez, M.d.M.; Legeai-Mallet, L.; Fernández-Arroyo, S.; Segura-Carretero, A. Isolation, comprehensive characterization and antioxidant activities of Theobroma cacao extract. J. Funct. Foods 2014, 10, 485–498. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak | Proposed Compound | RT | m/z Calc. | m/z Meas. | Err [ppm] | mSigma | Mol. Formula | SLE | SFE | PLE |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Quinic acid | 5.6 | 191.0561 | 191.0565 | 2.1 | 1.6 | C7H12O6 | A–E | A–C | A *–I |
| 2 | Sucrose | 7.2 | 341.1089 | 341.1095 | 1.6 | 0.8 | C12H22O11 | A–C | nd | A *,B,D–I |
| 3 | D-Raffinose | 9.9 | 503.1618 | 503.1607 | 2.1 | 2.7 | C18H32O16 | A, B | nd | A *–H |
| 4 | Galloyl glucose isomer 1 | 12.4 | 331.0671 | 331.0678 | 2.1 | 1.5 | C13H16O10 | A–C | nd | A *–I |
| 5 | Galloyl glucose isomer 2 | 13.1 | 331.0671 | 331.0677 | 1.9 | 12.6 | C13H16O10 | A,B | nd | A *,B,G,I |
| 6 | Gallo(epi)catechin dimer | 13.4 | 609.1250 | 609.1228 | 3.6 | 3.9 | C30H26O14 | A *, B | nd | nd |
| 7 | Gallic acid | 13.7 | 169.0142 | 169.0149 | 3.7 | 1.8 | C7H6O5 | A–E | A–C | A *–I |
| 8 | UK1 | 14.4 | 411.0259 | 411.0239 | −5.0 | 37.5 | C17H8N4O9 | A *–C | nd | A, B, I |
| 9 | Bis(epi)gallocatechin monogallate 1 | 14.8 | 761.1359 | 761.1359 | 0.1 | 5.3 | C37H30O18 | C *, D | nd | nd |
| 10 | (Epi)gallocatechin isomer 1 | 14.8 | 305.0667 | 305.0670 | 1.0 | 0.9 | C15H14O7 | A, B | nd | A *–I |
| 11 | Bis(epi)gallocatechin monogallate 2 | 15.2 | 761.1359 | 761.1365 | 0.7 | 4.6 | C37H30O18 | A–C, E | nd | A *–I |
| 12 | (Epi)gallocatechin gallate (epi)catechin isomer 1 | 15.5 | 745.1410 | 745.1414 | −0.5 | 8.7 | C37H30O17 | A *–C | nd | nd |
| 13 | Protocatechuic acid | 15.7 | 153.0193 | 153.0192 | 1.0 | 5.9 | C7H6O4 | nd | A, C | G *–I |
| 14 | Procyanidin B dimer isomer 1 | 15.9 | 577.1351 | 577.1335 | 2.8 | 2.6 | C30H26O12 | A *–C | nd | A–F |
| 15 | (Epi)catechin-(epi)gallocatechin | 16.5 | 593.1301 | 593.1311 | −1.8 | 2.7 | C30H26O13 | A *, B | nd | nd |
| 16 | (Epi)gallocatechin gallate (epi)catechin isomer 2 | 16.7 | 745.1410 | 745.1402 | 1.2 | 6.8 | C37H30O17 | A *–C | nd | nd |
| 17 | Bis(epi)gallocatechin digallate | 16.8 | 913.1469 | 913.1493 | 2.6 | 2.7 | C44H34O22 | A-E | nd | A *–I |
| 18 | (Epi)gallocatechin gallate (epi)catechin isomer 3 | 17.4 | 745.1410 | 745.1402 | 1.1 | 2.7 | C37H30O17 | A-–E | B, C | A *–G, I |
| 19 | (Epi)gallocatechin gallate isomer 1 | 17.8 | 457.0776 | 457.0769 | 1.7 | 1.7 | C22H18O11 | nd | nd | E *–G |
| 20 | (Epi)gallocatechin gallate (epi)catechin isomer 4 | 18.4 | 745.1410 | 745.1418 | 1.0 | 0.7 | C37H30O17 | A, E | B | A *–G, I |
| 21 | (Epi)gallocatechin gallate (epi)catechin isomer 5 | 18.6 | 745.1410 | 745.1414 | 0.5 | 3.7 | C37H30O17 | C | nd | A *–G |
| 22 | (Epi)gallocatechin isomer 2 | 18.7 | 305.0667 | 305.0669 | 0.8 | 1.7 | C15H14O7 | A*–C | nd | nd |
| 23 | Catechin | 19.4 | 289.0718 | 289.0725 | 2.7 | 3.2 | C15H14O6 | A–E | A–C | A*–I |
| 24 | (Epi)gallocatechin gallate (epi)catechin gallate isomer 1 | 19.5 | 897.1520 | 897.1543 | 2.6 | 7.2 | C44H34O21 | A–E | nd | A *, B,D–I |
| 25 | (Epi)catechin gallate (epi)catechin isomer 1 | 20 | 729.1461 | 729.1471 | −1.0 | 9.8 | C37H30O16 | B, C | nd | A *, B,D–I |
| 26 | (Epi)catechin gallate (epi)catechin isomer 2 | 20.5 | 729.1461 | 729.1476 | 2.0 | 3.1 | C37H30O16 | A–E | B, C | A *–I |
| 27 | Protocatechuic acid aldehide | 20.5 | 137.0244 | 137.0245 | −0.5 | 4.2 | C7H6O3 | nd | A * | G–I |
| 28 | Dimethoxy-hydroxyphenyl-O-galloyl-glucopyranoside | 20.8 | 483.1144 | 483.1134 | 2.2 | 4.1 | C21H24O13 | A *–C | nd | nd |
| 29 | (Epi)gallocatechin gallate isomer 2 | 21.9 | 457.0776 | 457.0783 | 1.5 | 5.7 | C22H18O11 | A–E | A–C | A *–I |
| 30 | (Epi)catechin gallate (epi)catechin gallate isomer 1 | 22.2 | 881.1571 | 881.1593 | −2.6 | 3.4 | C44H33O20 | A–E | B, C | A *–G, I |
| 31 | (Epi)catechin gallate (epi)catechin gallate isomer 2 | 23.1 | 881.1571 | 881.1586 | −1.7 | 11.8 | C44H33O20 | A–E | B, C | A *–I |
| 32 | Hydroxy-methoxyphenyl-O-galloyl-glucopyranoside | 23.5 | 453.1038 | 453.1042 | 0.8 | 2.3 | C20H22O12 | A–C | nd | A *–G |
| 33 | UK2 isomer 1 | 23.6 | 439.0671 | 439.0668 | 0.5 | 7.6 | C22H16O10 | nd | nd | G *–I |
| 34 | Epicatechin | 24.5 | 289.0718 | 289.0722 | 1.7 | 2.0 | C15H14O6 | A–D | A–C | A *–I |
| 35 | (Epi)gallocatechin gallate isomer 3 | 24.7 | 457.0776 | 457.0779 | 0.6 | 6.0 | C22H18O11 | C, D | nd | A *, B, E–I |
| 36 | Eriodictyol-O-glucoside | 24.9 | 449.1089 | 449.1069 | 4.6 | 2.3 | C21H22O11 | A *–D | nd | A–D |
| 37 | (Epi)catechin gallate (epi)catechin gallate isomer 3 | 25.5 | 881.1571 | 881.1586 | −1.7 | 11.8 | C44H34O20 | A–C | B, C | A *–G, I |
| 38 | (Epi)gallocatechin gallate (epi)catechin isomer 6 | 25.9 | 745.1410 | 745.1416 | 0.8 | 6.9 | C37H30O17 | A–D | nd | A *–E, G |
| 39 | Galloyl glucosyl dihydroxy methoxyacetophenone | 26.2 | 495.1144 | 495.1131 | 2.6 | 20.4 | C22H24O13 | A *–C | nd | nd |
| 40 | UK2 isomer 2 | 26.5 | 439.0671 | 439.0664 | 1.5 | 43.6 | C22H16O10 | nd | nd | F *–I |
| 41 | (Epi)gallocatechin-(epi)catechin-gallate | 26.6 | 743.1254 | 743.1282 | −2.8 | 50.5 | C37H28O17 | A *–C | nd | nd |
| 42 | (Epi)gallocatechin gallate (epi)catechin gallate isomer 2 | 26.7 | 897.1520 | 897.1516 | 0.4 | 6.1 | C44H34O21 | A *–C | nd | A–C |
| 43 | Lyoniside | 27.5 | 551.2134 | 551.2136 | 0.4 | 4.2 | C27H36O12 | A *–C | nd | A–G |
| 44 | (Epi)catechin gallate isomer 1 | 27.9 | 441.0827 | 441.0836 | 2.1 | 2.3 | C22H18O10 | nd | A * | G–I |
| 45 | (Epi)catechin gallate isomer 2 | 28.2 | 441.0827 | 441.0836 | 2.0 | 2.0 | C22H18O10 | A–E | B, C | A *–I |
| 46 | (Epi)catechin-3-O-glucoside-gallate | 28.9 | 603.1355 | 603.1355 | 0.1 | 15.1 | C28H28O15 | A *–C, E | nd | A–G |
| 47 | Procyanidin B dimer isomer 2 | 29.6 | 577.1351 | 577.1346 | 0.9 | 12.1 | C30H26O12 | A–D | C | A *–E, G–I |
| 48 | (Epi)catechin gallate isomer 3 | 30.1 | 441.0827 | 441.0814 | 2.9 | 2.4 | C22H18O10 | A–E | B, C | A *–I |
| 49 | Dihydromyricetin isomer 1 | 31 | 319.0459 | 319.0465 | 1.7 | 0.3 | C15H12O8 | nd | nd | G *–I |
| 50 | (Epi)catechin gallate (epi)catechin gallate isomer 4 | 31.8 | 881.1571 | 881.1602 | −3.6 | 5.0 | C44H34O20 | A–E | B, C | A *–I |
| 51 | (Epi)catechin gallate (epi)catechin isomer 3 | 32.1 | 729.1461 | 729.1488 | 3.8 | 2.8 | C37H30O16 | A–E | B, C | A *–I |
| 52 | (Epi)afzelechin gallate | 33 | 425.0878 | 425.0892 | 3.2 | 9.5 | C22H18O9 | A–E | B, C | A *–G |
| 53 | Myricetin glucoside | 33.5 | 479.0831 | 479.0849 | −3.8 | 14.0 | C21H20O13 | A *–E | B | A–C, F |
| 54 | Jaceidin triacetate | 33.6 | 485.1089 | 485.1093 | 0.8 | 8.3 | C24H22O11 | nd | nd | D *, E, G–I |
| 55 | Phloretin-di-C-glucoside | 34.6 | 597.1825 | 597.1807 | 3.0 | 13.1 | C27H34O15 | A *–E | nd | A, B |
| 56 | Trihydroxystilbene glucosyl-O-gallate | 34.7 | 541.1351 | 541.1362 | −1.9 | 14.9 | C27H26O12 | A *–E | B | A, B, D–F |
| 57 | Dihydromyricetin isomer 2 | 35.6 | 319.0459 | 319.0465 | 1.7 | 0.3 | C15H12O8 | nd | nd | E, G *–I |
| 58 | UK3 | 35.7 | 439.1093 | 439.1073 | 4.5 | 36.7 | C16H24O14 | A *–C, E | B | A, B |
| 59 | Homaloside D | 36.4 | 543.1508 | 543.1529 | −3.8 | 19.2 | C27H28O12 | A *–C | nd | A–C |
| 60 | Phloretin-C-glucoside (nothofagin) | 36.4 | 435.1297 | 435.1281 | 3.6 | 8.8 | C21H24O10 | C *, E | B | nd |
| 61 | Rhamnetin | 36.6 | 315.0510 | 315.0510 | −0.1 | 46.1 | C16H12O7 | nd | nd | G *–I |
| 62 | Dihydroquercetin | 37.6 | 303.0510 | 303.0514 | 1.2 | 17.9 | C15H12O7 | nd | nd | D *–I |
| 63 | Pentamethoxystilbene isomer 1 | 37.7 | 329.1394 | 329.1402 | 2.3 | 6.8 | C19H22O5 | B *, C, E | A–C | A, B, E |
| 64 | Quercetin glucoside | 37.9 | 463.0882 | 463.0883 | −0.3 | 0.7 | C21H20O12 | C *, E | nd | A, B |
| 65 | Syringic aldehyde | 38.7 | 181.0506 | 181.0503 | 1.7 | 0.8 | C9H10O4 | nd | nd | G *–I |
| 66 | Pentamethoxystilbene isomer 2 | 38.9 | 329.1394 | 329.1387 | 2.4 | 5.5 | C19H22O5 | A *–E | A-C | A–F |
| 67 | Ellagic acid | 40.2 | 300.9990 | 301.0004 | 4.6 | 15.7 | C14H6O8 | A–E | B, C | A *–I |
| 68 | Naringenin | 40.8 | 271.0612 | 271.0609 | 1.3 | 7.8 | C15H12O5 | nd | nd | F, G *, I |
| 69 | Taxifolin | 41.6 | 303.0510 | 303.0519 | 2.8 | 0.9 | C15H12O7 | nd | nd | E, G *–I |
| 70 | Nonanedioic acid (azelaic acid) | 42.3 | 187.0981 | 187.0976 | −2.8 | 5.1 | C9H16O4 | nd | A *–C | nd |
| 71 | Flavanone | 47.2 | 271.0612 | 271.0622 | −3.5 | 8.3 | C15H12O5 | nd | B *, C | nd |
| SLE | SFE | PLE | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | A | B | C | A | B | C | D | E | F | G | H | I |
| 6.2 ± 0.2 | 12 ± 1 | 12.1 ± 0.2 | 9.3 ± 0.5 | 7 ± 1 | 0.6 ± 0.2 | 2.2 ± 0.4 | 0.6 ± 0.1 | 23.0 ± 0.3 | 20 ± 3 | 25 ± 1 | 20 ± 1 | 29 ± 1 | 21 ± 2 | 31 ± 3 | 21 ± 2 | 42 ± 1 |
| Conditions | P (psi) | T (min) | T (°C) | EtOH (%) | H2O (%) | Dielectric Constant |
|---|---|---|---|---|---|---|
| A | 1500 | 20 | 40 | 50 | 50 | 48.02 |
| B | 63 | 85 | 15 | 31.02 | ||
| C | 63 | 15 | 85 | 59.09 | ||
| D | 120 | 100 | 0 | 19.00 | ||
| E | 120 | 50 | 50 | 34.71 | ||
| F | 120 | 0 | 100 | 50.41 | ||
| G | 176 | 85 | 15 | 21.55 | ||
| H | 176 | 15 | 85 | 33.43 | ||
| I | 200 | 50 | 50 | 26.00 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cádiz-Gurrea, M.d.l.L.; Lozano-Sánchez, J.; Fernández-Ochoa, Á.; Segura-Carretero, A. Enhancing the Yield of Bioactive Compounds from Sclerocarya birrea Bark by Green Extraction Approaches. Molecules 2019, 24, 966. https://doi.org/10.3390/molecules24050966
Cádiz-Gurrea MdlL, Lozano-Sánchez J, Fernández-Ochoa Á, Segura-Carretero A. Enhancing the Yield of Bioactive Compounds from Sclerocarya birrea Bark by Green Extraction Approaches. Molecules. 2019; 24(5):966. https://doi.org/10.3390/molecules24050966
Chicago/Turabian StyleCádiz-Gurrea, María de la Luz, Jesús Lozano-Sánchez, Álvaro Fernández-Ochoa, and Antonio Segura-Carretero. 2019. "Enhancing the Yield of Bioactive Compounds from Sclerocarya birrea Bark by Green Extraction Approaches" Molecules 24, no. 5: 966. https://doi.org/10.3390/molecules24050966