A Critical Review of Solid Materials for Low-Temperature Thermochemical Storage of Solar Energy Based on Solid-Vapour Adsorption in View of Space Heating Uses


Abstract
:
1. Introduction
2. Adequacy of Adsorption Phenomena in Heat Storage
3. Practical Aspects of Heat Storage by Adsorption in Reference to Adsorption Mechanisms
4. Comments on the Criteria for Selection of Working Materials as Adsorbents
- (1)
- Specific surface area and pore volume for high adsorption capacity towards the selected adsorbate,
- (2)
- Affinity between the active surface of the adsorbent and the adsorbate for high heat of adsorption,
- (3)
- Selectivity towards a given adsorbate when adsorbed from a gas mixture,
- (4)
- Ability to provide appropriate mass transport and kinetics of adsorption,
- (5)
- Thermal conductivity and specific heat capacity for good heat transfer from/to the adsorbent bed,
- (6)
- Ease of regeneration, thermal and chemical stability, and usable lifetime under operating conditions for long-term resistance to repeated cycles of charging and discharging,
- (7)
- Toxicity, environmental impact, corrosiveness, flammability, and compatibility with materials of construction,
- (8)
- Cost, availability, ease of handling, ease of shaping and up-scaling.
5. Presentation of Adsorbent Candidates for Adsorption-Based Thermochemical Storage of Energy
5.1. Amorphous Silicas and Aluminoslicates
5.1.1. Surface Reactivity and Hydrothermal Stability of Amorphous Silica Materials
5.1.2. Surface Reactivity and Hydrothermal Stability of Ordered Mesoporous Silicas and Alumino-silicates
5.1.3. Adsorbents for Heat Storage by Gas-Solid Adsorption
5.2. Zeolitic Materials
5.2.1. Surface Reactivity and Hydrothermal Stability of Zeolites
5.2.2. Zeolitic Adsorbents for Heat Storage by Gas-Solid Adsorption
5.3. Other Zeotype Materials
5.4. Metal-Organic Framework Structures (MOFs)
Surface Reactivity and Hydrothermal Stability of MOFs in View of Their Uses as Water Vapour Adsorbents
5.5. Other Adsorbents and Adsorbates
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- European Commission. Europe 2020: Europe’s Growth Strategy; European Commission: Brussels, Belgium, 2012; ISBN 9789279239724. [Google Scholar] [CrossRef]
- Pérez-Lombard, L.; Ortiz, J.; Pout, C. A review on buildings energy consumption information. Energy Build. 2008, 40, 394–398. [Google Scholar] [CrossRef]
- Lin, H.-W.; Hong, T. On variations of space-heating energy use in office buildings. Appl. Energy 2013, 111, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Lizana, J.; Chacartegui, R.; Barrios-Padura, A.; Valverde, J.M. Advances in thermal energy storage materials and their applications towards zero energy buildings: A critical review. Appl. Energy 2017, 203, 219–239. [Google Scholar] [CrossRef]
- Sajn, N. European Parliamentary Research Service: Energy Efficiency of Buildings: A Nearly Zero-Energy Future? European Parliament: Brussels, Belgium, 2016. [Google Scholar]
- Agence de l’Environnement et de la Maîtrise de l’Energie (ADEME). Climat, air et énergie: Chiffres-clés; ADEME éditions: Angers, France, 2017. [Google Scholar]
- Roeb, M.; Neises, M.; Monnerie, N.; Sattler, C.; Pitz-Paal, R. Technologies and trends in solar power and fuels. Energy Environ. Sci. 2011, 4, 2503–2511. [Google Scholar] [CrossRef]
- Aydin, D.; Casey, S.P.; Riffat, S. Theoretical analysis of the potential for thermochemical heat storage under Mediterranean climate conditions: Northern Cyprus Case. Future Cities Environ. 2015, 5, 231–295. [Google Scholar] [CrossRef]
- Renewable Energy Policy Network for the 21st Century (REN21). Renewables 2017: Global Status Report; REN21: Paris, France, 2017; Volume 72, ISBN 978-3-9818107-0-7. [Google Scholar] [CrossRef]
- International Energy Agency (IEA); Bahar, H. Renewables 2017; International Energy Agency: Paris, France, 2017; ISBN 978-92-64-28187-5. [Google Scholar] [CrossRef]
- Anisur, M.R.; Mahfuz, M.H.; Kibria, M.A.; Saidur, R.; Metselaar, I.H.S.C.; Mahlia, T.M.I. Curbing global warming with phase change materials for energy storage. Renew. Sustain. Energy Rev. 2013, 18, 23–30. [Google Scholar] [CrossRef]
- Moth-Poulsen, K.; Ćoso, D.; Börjesson, K.; Vinokurov, N.; Meier, S.K.; Majumdar, A.; Vollhardt, K.P.C.; Segalman, R.A. Molecular solar thermal (MOST) energy storage and release system. Energy Environ. Sci. 2012, 5, 8534–8537. [Google Scholar] [CrossRef]
- Haji Abedin, A.; Rosen, M.A. Energy and exergy analyses of an open thermochemical energy storage system: Methodology and illustrative application. Open Renew. Energy J. 2012, 5, 41–48. [Google Scholar] [CrossRef]
- Yu, N.; Wang, R.Z.; Wang, L.W. Sorption thermal storage for solar energy. Prog. Energy Combust. Sci. 2013, 39, 489–514. [Google Scholar] [CrossRef]
- N’Tsoukpoe, K.E.; Liu, H.; Le Pierrès, N.; Luo, L. A review on long-term sorption solar energy storage. Renew. Sustain. Energy Rev. 2009, 13, 2385–2396. [Google Scholar] [CrossRef]
- Shigeishi, R.A.; Langford, C.H.; Hollebone, B.R. Solar energy storage using chemical potential changes associated with drying of zeolites. Sol. Energy 1979, 23, 489–495. [Google Scholar] [CrossRef]
- Schmidt, T.; Mangold, D.; Müller-Steinhagen, H. Central solar heating plants with seasonal storage in Germany. Sol. Energy 2004, 76, 165–174. [Google Scholar] [CrossRef]
- Nielsen, K. Thermal Energy Storage: A State-Of-The-Art. A Report within the Research Program Smart Energy-Efficient Buildings at the Norwegian University of Science and Technology and SINTEF; Department of Geology and Mineral Resources Engineering NTNU: Trondheim, Norway, 2003. [Google Scholar]
- André, L.; Abanades, S.; Flamant, G. Screening of thermochemical systems based on solid-gas reversible reactions for high temperature solar thermal energy storage. Renew. Sustain. Energy Rev. 2016, 64, 703–715. [Google Scholar] [CrossRef]
- André, L.; Abanades, S. Evaluation and performances comparison of calcium, strontium and barium carbonates during calcination/carbonation reactions for solar thermochemical energy storage. J. Energy Storage 2017, 13, 193–205. [Google Scholar] [CrossRef]
- Prieto, C.; Cooper, P.; Fernández, A.I.; Cabeza, L.F. Review of technology: Thermochemical energy storage for concentrated solar power plants. Renew. Sustain. Energy Rev. 2016, 60, 909–929. [Google Scholar] [CrossRef]
- Aydin, D.; Casey, S.P.; Riffat, S. The latest advancements on thermochemical heat storage systems. Renew. Sustain. Energy Rev. 2015, 41, 356–367. [Google Scholar] [CrossRef]
- Bales, C.; Gantenbein, P.; Hauer, A.; Henning, H.-M.; Jaenig, D.; Kerskes, H.; Nuñez, T.; Visscher, K. Thermal Properties of Materials for Thermo-Chemical Storage of Solar Heat: Report B2 of Subtask B; The Solar Heating and Cooling Programme: Cedar, MI, USA, 2005. [Google Scholar]
- Singh, H.K.; Buddhi, D. Experimental investigation on CaCl2.6H2O for subcooling behavior and its correction for low temperature thermal energy storage. Int. J. Appl. Eng. Res. 2018, 13, 9858–9867. [Google Scholar]
- Henninger, S.K.; Ernst, S.J.; Gordeeva, L.; Bendix, P.; Fröhlich, D.; Grekova, A.D.; Bonaccorsi, L.; Aristov, Y.; Jaenchen, J. New materials for adsorption heat transformation and storage. Renew. Energy 2017, 110, 59–68. [Google Scholar] [CrossRef]
- Cabeza, L.F.; Solé, A.; Barreneche, C. Review on sorption materials and technologies for heat pumps and thermal energy storage. Renew. Energy 2017, 110, 3–39. [Google Scholar] [CrossRef]
- Frazzica, A.; Freni, A. Adsorbent working pairs for solar thermal energy storage in buildings. Renew. Energy 2017, 110, 87–94. [Google Scholar] [CrossRef]
- Scapino, L.; Zondag, H.A.; Van Bael, J.; Diriken, J.; Rindt, C.C.M. Sorption heat storage for long-term low-temperature applications: A review on the advancements at material and prototype scale. Appl. Energy 2017, 190, 920–948. [Google Scholar] [CrossRef]
- Steele, W.A. The Interaction of Gases with Solid Surfaces; Pergamon Press: Oxford, UK, 1974. [Google Scholar]
- Gregg, S.J.; Sing, K.S.W. Adsorption, Surface Area, and Porosity, 2nd ed.; Academic Press: London, UK, 1982. [Google Scholar]
- Bolis, V. Fundamentals in adsorption at the solid-gas interface. Concepts and thermodynamics. In Calorimetry and Thermal Methods in Catalysis; Auroux, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–50. ISBN 978-3-642-11954-5. [Google Scholar] [CrossRef]
- Lowell, S.; Shields, J.E.; Thomas, M.A.; Thommes, M. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density; Springer: Berlin/Heidelberg, Germany, 2004; Volume 16, ISBN 978-90-481-6633-6. [Google Scholar] [CrossRef]
- Sing, K.S.W. The use of gas adsorption for the characterization of porous solids. Colloids Surf. 1989, 38, 113–124. [Google Scholar] [CrossRef]
- Everett, D.H.; Powl, J.C. Adsorption in slit-like and cylindrical micropores in the Henry’s law region: A model for the microporosity of carbons. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 1976, 72, 619–636. [Google Scholar] [CrossRef]
- Healey, F.; Carter, R.N.; Worthy, G.; Hodgson, A. Endothermic dissociative chemisorption of molecular D2 on Ag(111). Chem. Phys. Lett. 1995, 243, 133–139. [Google Scholar] [CrossRef]
- De Boer, J.H. 50 endothermic chemisorption and catalysis. In Advances in Catalysis; Farkas, A., Ed.; Academic Press: Cambridge, MA, USA, 1957; Volume 9, pp. 472–480. ISBN 0360-0564. [Google Scholar] [CrossRef]
- Álvarez-Falcón, L.; Viñes, F.; Notario-Estévez, A.; Illas, F. On the hydrogen adsorption and dissociation on Cu surfaces and nanorows. Surf. Sci. 2016, 646, 221–229. [Google Scholar] [CrossRef]
- Zajac, J.; Dutartre, R.; Jones, D.J.; Roziere, J. Determination of surface acidity of powdered porous materials based on ammonia chemisorption: Comparison of flow-microcalorimetry with batch volumetric method and temperature-programmed desorption. Thermochim. Acta 2001, 379, 123–130. [Google Scholar] [CrossRef]
- Auroux, A. Acidity characterzation by microcalorimetry and relationship with reactivity. Top. Catal. 1997, 4, 71–89. [Google Scholar] [CrossRef]
- Stošić, D.; Bennici, S.; Sirotin, S.; Calais, C.; Couturier, J.L.; Dubois, J.L.; Travert, A.; Auroux, A. Glycerol dehydration over calcium phosphate catalysts: Effect of acidic-basic features on catalytic performance. Appl. Catal. A Gen. 2012, 447–448, 124–134. [Google Scholar] [CrossRef]
- Damjanović, L.; Auroux, A. Determination of acid/base properties by temperature programmed desorption (TPD) and adsorption calorimetry. In Zeolite Characterization and Catalysis: A Tutorial; Chester, A.W., Derouane, E.G., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 107–167. ISBN 978-1-4020-9678-5. [Google Scholar] [CrossRef]
- Ottiger, S.; Pini, R.; Storti, G.; Mazzotti, M. Measuring and modeling the competitive adsorption of CO2, CH4, and N2 on a dry coal. Langmuir 2008, 24, 9531–9540. [Google Scholar] [CrossRef] [PubMed]
- Bellat, J.-P. Study of selective adsorption of gases by calorimetry. In Calorimetry and Thermal Methods in Catalysis; Auroux, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 273–318. ISBN 978-3-642-11954-5. [Google Scholar] [CrossRef]
- Rouquerol, F.; Rouquerol, J.; Everett, D.H. Gas—Solid interactions. General derivation of reaction enthalpies from the data of isothermal microcalorimetry. Thermochim. Acta 1980, 41, 311–322. [Google Scholar] [CrossRef]
- Auroux, A. Calorimetry and Thermal Methods in Catalysis; Springer: New York, NY, USA, 2013; Volume 154, ISBN 978-3-642-11954-5. [Google Scholar] [CrossRef]
- Tian, Y.; Wu, J. Differential heat of adsorption and isosteres. Langmuir 2017, 33, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Mohr, R.; Rao, M.B. Isosteric Heat of Adsorption: Theory and Experiment. J. Phys. Chem. B 1999, 103, 6539–6546. [Google Scholar] [CrossRef]
- Builes, S.; Sandler, S.I.; Xiong, R. Isosteric heats of gas and liquid adsorption. Langmuir 2013, 29, 10416–10422. [Google Scholar] [CrossRef] [PubMed]
- Sircar, S. Excess properties and column dynamics of multicomponent gas adsorption. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 1985, 81, 1541–1545. [Google Scholar] [CrossRef]
- Rudzinski, W.; Everett, D.H. Adsorption of Gases on Heterogeneous Surfaces, 1st ed.; Press, A., Ed.; Academic Press: London, UK, 1992; ISBN 978-0-12-601690-1. [Google Scholar] [CrossRef]
- Knor, Z. Static volumetric methods for determination of absorbed amount of gases on clean solid surfaces. Catal. Rev. 1968, 1, 257–313. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Degas Options for Micropore Samples, Application Note N° 160, Micromeritics’ Application Notes and Technical Tips, Norcross, USA. September 2012. Available online: https://www.micromeritics.com/Library/ (accessed on 1 February 2019).
- Bejan, A.S.; Labihi, A.; Croitoru, C.; Catalina, T. Air solar collectors in building use—A review. In Proceedings of the E3s Web Conf., Bucharest, Romania, 24 November 2017. [Google Scholar] [CrossRef]
- D’Antoni, M.; Saro, O. Massive solar-thermal collectors: A critical literature review. Renew. Sustain. Energy Rev. 2012, 16, 3666–3679. [Google Scholar] [CrossRef]
- Fabian, F.; Eberhard, L. Multiple sample setup for testing the hydrothermal stability of adsorbents in thermal energy storage applications. Meas. Sci. Technol. 2015, 26, 65603. [Google Scholar] [CrossRef]
- Storch, G.; Reichenauer, G.; Scheffler, F.; Hauer, A. Hydrothermal stability of pelletized zeolite 13X for energy storage applications. Adsorption 2008, 14, 275–281. [Google Scholar] [CrossRef]
- Cruciani, G. Zeolites upon heating: Factors governing their thermal stability and structural changes. J. Phys. Chem. Solids 2006, 67, 1973–1994. [Google Scholar] [CrossRef]
- Fischer, U.R. What is the best possible heat storage density for a seasonal adsorptive thermal energy storage. In Proceedings of the Effstock 2009, Thermal Energy Storage for Efficiency and Sustainability: 11th International conference on Thermal Energy Storage, Stockholm, Sweden, 14–17 June 2009. [Google Scholar]
- Jänchen, J.; Ackermann, D.; Weiler, E.; Stach, H.; Brösicke, W. Calorimetric investigation on zeolites, AlPO4′s and CaCl2 impregnated attapulgite for thermochemical storage of heat. Thermochim. Acta 2005, 434, 37–41. [Google Scholar] [CrossRef]
- Zettl, B.; Englmair, G.; Steinmaurer, G. Development of a revolving drum reactor for open-sorption heat storage processes. Appl. Therm. Eng. 2014, 70, 42–49. [Google Scholar] [CrossRef]
- Energy-Hub for Residential and Commercial Districts and Transport (E-HUB), Report on a Combination of Thermal Storage Techniques and Components. Available online: https://www.e-hub.org/pdf/D3.2_Thermal_storage_techniques_components.pdf (accessed on 18 March 2014).
- Paksoy, H.Ö. (Ed.) Thermal Energy Storage for Sustainable Energy Consumption: Fundamentals, Case Studies and Design; Springer: Berlin/Heidelberg, Germany, 2007; ISBN 978-1-4020-5290-3. [Google Scholar] [CrossRef]
- Nir, S.; Adams, S.; Rein, R. Polarizability calculations on water, hydrogen, oxygen, and carbon dioxide. J. Chem. Phys. 1973, 59, 3341–3355. [Google Scholar] [CrossRef]
- Perry, R.H.; Green, D.W. Perry’s Chemical Engineers’ Handbook, 7th ed.; Perry, R.H., Green, D.W., Maloney, J.O., Eds.; McGraw-Hill: New York, NY, USA, 1997; ISBN 0070498415. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Brauman, J.I.; Biairlb, L.K. Gas-phase acidities of alcohols. J. Am. Chem. Soc. 1970, 2821, 5986–5992. [Google Scholar] [CrossRef]
- Mette, B.; Kerskes, H.; Drück, H. Concepts of long-term thermochemical energy storage for solar thermal applications—Selected examples. Energy Procedia 2012, 30, 321–330. [Google Scholar] [CrossRef]
- Jänchen, J.; Ackermann, D.; Stach, H.; Brösicke, W. Studies of the water adsorption on Zeolites and modified mesoporous materials for seasonal storage of solar heat. Sol. Energy 2004, 76, 339–344. [Google Scholar] [CrossRef]
- De Lange, M.F.; Verouden, K.J.F.M.; Vlugt, T.J.H.; Gascon, J.; Kapteijn, F. Adsorption-driven heat pumps: The potential of metal-organic frameworks. Chem. Rev. 2015, 115, 12205–12250. [Google Scholar] [CrossRef] [PubMed]
- Demir, H.; Mobedib, M.; Ülkü, S. A review on adsorption heat pump: Problems and solutions. Renew. Sustain. Energy Rev. 2008, 12, 2381–2403. [Google Scholar] [CrossRef] [Green Version]
- Fischer, F.; Lävemann, E.; Krönauer, A.; Hauer, A. BIOWKK Workshop Dortmund 2012 Open Adsorption Systems for Thermal Energy Storage Applications; Bavarian Centre for Applied Energy Research: Wuerzburg, Germany, 2012. [Google Scholar]
- Kerskes, H.; Mette, B.; Bertsch, F.; Asenbeck, S.; Drück, H. Chemical energy storage using reversible solid/gas-reactions (CWS)—Results of the research project. Energy Procedia 2012, 30, 294–304. [Google Scholar] [CrossRef]
- Hauer, A. Evaluation of adsorbent materials for heat pump and thermal energy storage applications in open systems. Adsorption 2007, 13, 399–405. [Google Scholar] [CrossRef]
- Hauer, A. Thermal energy storage with zeolite for heating and cooling applications. In Proceedings of the International Sorption Heat Pump Conference 2002, Shanghai, China, 24–27 September 2002; pp. 385–390. [Google Scholar]
- Stritih, U.; Koželj, R. Analysis of adsorption thermal storage device for solar energy storage. Int. J. Green Technol. 2017, 3, 23–34. [Google Scholar] [CrossRef]
- Mette, B.; Kerskes, H.; Drück, H.; Müller-Steinhagen, H. Experimental and numerical investigations on the water vapor adsorption isotherms and kinetics of binderless zeolite 13X. Int. J. Heat Mass Transf. 2014, 71, 555–561. [Google Scholar] [CrossRef]
- Poling, B.E.; Prausnitz, J.M.; O’Connell, J.P. Properties of Gases and Liquids, 5th ed.; McGraw-Hill Education: New York, NY, USA, 2001; ISBN 0071499997. [Google Scholar] [CrossRef]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics, 85th ed.; Taylor & Francis: Abingdon, UK, 2004; ISBN 0849304849. [Google Scholar] [CrossRef]
- Lias, S.G.; Liebman, J.F.; Levin, R.D. Evaluated gas phase basicities and proton affinities of molecules; Heats of formation of protonated molecules. J. Phys. Chem. Ref. Data 1984, 13, 695–808. [Google Scholar] [CrossRef]
- Nelson, J.R.D.; Lide, J.D.R.; Maryott, A.A. Selected Values of Electric Dipole Moments for Molecules in the Gas Phase; U.S. Department of Commerce: Washington, DC, USA, 1967. [Google Scholar]
- Michaels, R.; Satterfield, W.J.; Colonna, G. NFPA 325, 1994 Edition: Fire Hazard Properties of Flammable Liquids, Gases, and Volatile Solids. Fire Hazard Properties of Flammable Liquids, Gases, and Volatile Solids; NFPA: Quincy, MA, YSA, 1994. [Google Scholar]
- O’Kane, G.J. Inhalation of ammonia vapour. A report on the management of eight patients during the acute stages. Anaesthesia 1983, 38, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Guais, A.; Brand, G.; Jacquot, L.; Karrer, M.; Dukan, S.; Grévillot, G.; Molina, T.J.; Bonte, J.; Regnier, M.; Schwartz, L. Toxicity of carbon dioxide: A review. Chem. Res. Toxicol. 2011, 24, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Henninger, S.K.; Schmidt, F.P.; Henning, H.M. Water adsorption characteristics of novel materials for heat transformation applications. Appl. Therm. Eng. 2010, 30, 1692–1702. [Google Scholar] [CrossRef]
- Henninger, S.K.; Jeremias, F.; Kummer, H.; Schossig, P.; Henning, H.M. Novel sorption materials for solar heating and cooling. Energy Procedia 2012, 30, 279–288. [Google Scholar] [CrossRef]
- Henninger, S.K.; Jeremias, F.; Kummer, H.; Janiak, C. MOFs for use in adsorption heat pump processes. Eur. J. Inorg. Chem. 2012, 2625–2634. [Google Scholar] [CrossRef]
- Ehrenmann, J.; Henninger, S.K.; Janiak, C. Water adsorption characteristics of MIL-101 for heat-transformation applications of MOFs. Eur. J. Inorg. Chem. 2011, 471–474. [Google Scholar] [CrossRef]
- Jabbari-Hichri, A.; Bennici, S.; Auroux, A. Water sorption heats on silica-alumina-based composites for interseasonal heat storage. J. Therm. Anal. Calorim. 2014, 118, 1111–1118. [Google Scholar] [CrossRef]
- Jabbari-Hichri, A.; Bennici, S.; Auroux, A. Enhancing the heat storage density of silica–alumina by addition of hygroscopic salts (CaCl2, Ba(OH)2, and LiNO3). Sol. Energy Mater. Sol. Cells 2015, 140, 351–360. [Google Scholar] [CrossRef]
- Jabbari-Hichri, A.; Bennici, S.; Auroux, A. Effect of aluminum sulfate addition on the thermal storage performance of mesoporous SBA-15 and MCM-41 materials. Sol. Energy Mater. Sol. Cells 2016, 149, 232–241. [Google Scholar] [CrossRef]
- Whiting, G.; Grondin, D.; Bennici, S.; Auroux, A. Heats of water sorption studies on zeolite–MgSO4 composites as potential thermochemical heat storage materials. Sol. Energy Mater. Sol. Cells 2013, 112, 112–119. [Google Scholar] [CrossRef]
- Whiting, G.T.; Grondin, D.; Stosic, D.; Bennici, S.; Auroux, A. Zeolite–MgCl2 composites as potential long-term heat storage materials: Influence of zeolite properties on heats of water sorption. Sol. Energy Mater. Sol. Cells 2014, 128, 289–295. [Google Scholar] [CrossRef]
- Henninger, S.K.; Habib, H.A.; Janiak, C. MOFs as adsorbents for low temperature heating and cooling applications. J. Am. Chem. Soc. 2009, 131, 2776–2777. [Google Scholar] [CrossRef] [PubMed]
- Farha, O.K.; Eryazici, I.; Jeong, N.C.; Hauser, B.G.; Wilmer, C.E.; Sarjeant, A.A.; Snurr, R.Q.; Nguyen, S.T.; Yazaydın, A.Ö.; Hupp, J.T. Metal–Organic Framework materials with ultrahigh surface areas: Is the sky the limit? J. Am. Chem. Soc. 2012, 134, 15016–15021. [Google Scholar] [CrossRef] [PubMed]
- Titinchi, S.J.J.; Piet, M.; Abbo, H.S.; Bolland, O.; Schwieger, W. Chemically modified solid adsorbents for CO2 capture. Energy Procedia 2014, 63, 8153–8160. [Google Scholar] [CrossRef]
- Tarasevich, Y.I. The surface energy of hydrophilic and hydrophobic adsorbents. Colloid J. 2007, 69, 212–220. [Google Scholar] [CrossRef]
- Derouane, E.G.; Védrine, J.C.; Pinto, R.R.; Borges, P.M.; Costa, L.; Lemos, M.A.N.D.A.; Lemos, F.; Ribeiro, F.R. The acidity of zeolites: Concepts, measurements and relation to catalysis: A review on experimental and theoretical methods for the study of zeolite acidity. Catal. Rev. 2013, 55, 454–515. [Google Scholar] [CrossRef]
- Kawabuchi, Y.; Sotowa, C.; Kishino, M.; Kawano, S.; Whitehurst, D.D.; Mochida, I. Chemical vapor deposition of heterocyclic compounds over active carbon fiber to control its porosity and surface function. Langmuir 1997, 13, 2314–2317. [Google Scholar] [CrossRef]
- Alby, D.; Salles, F.; Fullenwarth, J.; Zajac, J. On the use of metal cation-exchanged zeolites in sorption thermochemical storage: Some practical aspects in reference to the mechanism of water vapor adsorption. Sol. Energy Mater. Sol. Cells 2018, 179, 223–230. [Google Scholar] [CrossRef]
- Medveď, I.; Černý, R. Surface diffusion in porous media: A critical review. Microporous Mesoporous Mater. 2011, 142, 405–422. [Google Scholar] [CrossRef]
- Fletcher, A.J.; Yüzak, Y.; Thomas, K.M. Adsorption and desorption kinetics for hydrophilic and hydrophobic vapors on activated carbon. Carbon 2006, 44, 989–1004. [Google Scholar] [CrossRef]
- Schwieger, W.; Machoke, A.G.; Weissenberger, T.; Inayat, A.; Selvam, T.; Klumpp, M.; Inayat, A. Hierarchy concepts: Classification and preparation strategies for zeolite containing materials with hierarchical porosity. Chem. Soc. Rev. 2016, 45, 3353–3376. [Google Scholar] [CrossRef] [PubMed]
- Colmenares, M.G.; Simon, U.; Cruz, O.; Thomas, A.; Goerke, O.; Gurlo, A. Batch and continuous synthesis upscaling of powder and monolithic ordered mesoporous silica COK-12. Microporous Mesoporous Mater. 2018, 256, 102–110. [Google Scholar] [CrossRef]
- Martens, J.A.; Jammaer, J.; Bajpe, S.; Aerts, A.; Lorgouilloux, Y.; Kirschhock, C.E.A. Simple synthesis recipes of porous materials. Microporous Mesoporous Mater. 2011, 140, 2–8. [Google Scholar] [CrossRef]
- John, V.B. The shaping of materials. In Introduction to Engineering Materials; Palgrave Macmillan UK: London, UK, 1983; pp. 137–177. ISBN 978-1-349-17190-3. [Google Scholar] [CrossRef]
- Hausner, H.H. New methods for the consolidation of metal powders. In Perspectives in Powder Metallurgy; Hausner, H.H., Roll, K.H., Johnson, P.K., Eds.; Springer: Boston, MA, USA; New York, NY, USA, 1967; p. 255. ISBN 978-1-4899-6209-6. [Google Scholar] [CrossRef]
- Akhtar, F.; Andersson, L.; Ogunwumi, S.; Hedin, N.; Bergström, L. Structuring adsorbents and catalysts by processing of porous powders. J. Eur. Ceram. Soc. 2014, 34, 1643–1666. [Google Scholar] [CrossRef] [Green Version]
- Nickel, W.; Oschatz, M.; Von Der Lehr, M.; Leistner, M.; Hao, G.P.; Adelhelm, P.; Müller, P.; Smarsly, B.M.; Kaskel, S. Direct synthesis of carbide-derived carbon monoliths with hierarchical pore design by hard-templating. J. Mater. Chem. A 2014, 2, 12703–12707. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Long, H.; Li, T.; Wang, Y.; Liu, S.; Ru, H. Hierarchical trimodal macro-mesoporous silica monoliths with co-continuous macrostructures and isotropic skeletons constructed by randomly oriented SBA-15-type primary particles. Microporous Mesoporous Mater. 2018, 258, 262–268. [Google Scholar] [CrossRef]
- Knez, Z.; Novak, Z. Adsorption of water vapor on silica, alumina, and their mixed oxide aerogels. J. Chem. Eng. Data 2001, 858–860. [Google Scholar] [CrossRef]
- Aristov, Y.I. Selective water sorbents, a new family of materials for adsorption cooling/heating: State of the art. In Proceedings of the Vminsk International Seminar “Heat Pipe, Heat Pumps, Refrigerators”, Minsk, Belarus, 8–11 September 2003; pp. 379–390. [Google Scholar]
- Dawoud, B.; Aristov, Y. Experimental study on the kinetics of water vapor sorption on selective water sorbents, silica gel and alumina under typical operating conditions of sorption heat pumps. Int. J. Heat Mass Transf. 2003, 46, 273–281. [Google Scholar] [CrossRef]
- Freni, A.; Russo, F.; Vasta, S.; Tokarev, M.; Aristov, Y.I.; Restuccia, G. An advanced solid sorption chiller using SWS-1L. Appl. Therm. Eng. 2007, 27, 2200–2204. [Google Scholar] [CrossRef]
- Simonova, I.A.; Freni, A.; Restuccia, G.; Aristov, Y.I. Water sorption on composite “silica modified by calcium nitrate”. Microporous Mesoporous Mater. 2009, 122, 223–228. [Google Scholar] [CrossRef]
- Gorbach, A.; Stegmaier, M.; Eigenberger, G. Measurement and modeling of water vapor adsorption on zeolite 4A–Equilibria and kinetics. Adsorption 2004, 10, 29–46. [Google Scholar] [CrossRef]
- Hongois, S.; Kuznik, F.; Stevens, P.; Roux, J.J. Development and characterisation of a new MgSO4-zeolite composite for long-term thermal energy storage. Sol. Energy Mater. Sol. Cells 2011, 95, 1831–1837. [Google Scholar] [CrossRef]
- Seo, Y.K.; Yoon, J.W.; Lee, J.S.; Hwang, Y.K.; Jun, C.H.; Chang, J.S.; Wuttke, S.; Bazin, P.; Vimont, A.; Daturi, M.; et al. Energy-efficient dehumidification over hierachically porous metal-organic frameworks as advanced water adsorbents. Adv. Mater. 2012, 24, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Barton, S.S.; Evans, M.J.B.; Holland, J.; Koresh, J.E. Water and cyclohexane vapour adsorption on oxidized porous carbon. Carbon 1984, 22, 265–272. [Google Scholar] [CrossRef]
- Pedram, E.O.; Hines, A.L. Pure vapor adsorption of water on Mobil Sorbead R silica gel. J. Chem. Eng. Data 1983, 28, 11–14. [Google Scholar] [CrossRef]
- Li, G.; Xiao, P.; Webley, P.A.; Zhang, J.; Singh, R. Competition of CO2/H2O in adsorption based CO2 capture. Energy Procedia 2009, 1, 1123–1130. [Google Scholar] [CrossRef]
- Serbezov, A. Adsorption equilibrium of water vapor on F-200 activated alumina. J. Chem. Eng. Data 2003, 48, 421–425. [Google Scholar] [CrossRef]
- Teo, H.W.B.; Chakraborty, A.; Fan, W. Improved adsorption characteristics data for AQSOA types zeolites and water systems under static and dynamic conditions. Microporous Mesoporous Mater. 2017, 242, 109–117. [Google Scholar] [CrossRef]
- Thach, U.D.; Trens, P.; Prelot, B.; Zajac, J.; Hesemann, P. Tuning the interfacial properties of mesoporous ionosilicas: Effect of cationic precursor and counter anion. J. Phys. Chem. C 2016, 120, 27412–27421. [Google Scholar] [CrossRef]
- Sing, K.S.W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Gregg, S.J.; Nashed, S.; Malik, M.T. The adsorption of water vapour on a microporous carbon black. Powder Technol. 1973, 7, 15–19. [Google Scholar] [CrossRef]
- Brinker, C.J.; Scherer, G.W. (Eds.) Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing; Elsevier Science: Boston, MA, USA, 2013; ISBN 978-0-08-057103-4. [Google Scholar] [CrossRef]
- Iler, R.K. The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry of Silica; Wiley Intersciences Publications: New York, NY, USA, 1979. [Google Scholar]
- Soleimani Dorcheh, A.; Abbasi, M.H. Silica aerogel; synthesis, properties and characterization. J. Mater. Process. Technol. 2008, 199, 10–26. [Google Scholar] [CrossRef]
- Kleitz, F. Ordered Microporous and Mesoporous Materials, 2nd ed.; Klabunde, K.J., Richards, R.M., Eds.; Wiley: Hoboken, NJ, USA, 2009; ISBN 9780470222706. [Google Scholar] [CrossRef]
- Van Der Voort, P.; Esquivel, D.; De Canck, E.; Goethals, F.; Van Driessche, I.; Romero-Salguero, F.J. Periodic mesoporous organosilicas: From simple to complex bridges; A comprehensive overview of functions, morphologies and applications. Chem. Soc. Rev. 2013, 42, 3913–3955. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-H.; Mou, C.-Y.; Lin, H.-P. Synthesis of mesoporous silica nanoparticles. Chem. Soc. Rev. 2013, 42, 3862. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-Y.; Su, B.-L. Insights into hierarchically meso–macroporous structured materials. J. Mater. Chem. 2006, 16, 663–677. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Alothman, Z.A. A review: Fundamental aspects of silicate mesoporous materials. Materials 2012, 5, 2874–2902. [Google Scholar] [CrossRef]
- Ying, J.Y.; Mehnert, C.P.; Wong, M.S. Synthesis and applications of supramolecular-templated mesoporous materials. Angew. Chem. Int. Ed. 1999, 38, 56–77. [Google Scholar] [CrossRef]
- Neimark, A.V.; Ravikovitch, P.I.; Grün, M.; Schüth, F.; Unger, K.K. Pore size analysis of MCM-41 type adsorbents by means of nitrogen and argon adsorption. J. Colloid Interface Sci. 1998, 207, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Tanev, P.T.; Pinnavaia, T.J. Mesoporous silica molecular sieves prepared by ionic and neutral surfactant templating: A comparison of physical properties. Chem. Mater. 1996, 8, 2068–2079. [Google Scholar] [CrossRef]
- Markowitz, M.A.; Schoen, P.E.; Kust, P.; Gaber, B.P. Surface acidity and basicity of functionalized silica particles. Colloids Surf. A Physicochem. Eng. Asp. 1999, 150, 85–94. [Google Scholar] [CrossRef]
- Tsyganenko, A.A.; Storozheva, E.N.; Manoilova, O.V.; Lesage, T.; Daturi, M.; Lavalley, J.C. Brønsted acidity of silica silanol groups induced by adsorption of acids. Catal. Lett. 2000, 70, 159–163. [Google Scholar] [CrossRef]
- Rouxhet, P.G.; Sempels, R.E. Hydrogen bond strengths and acidities of hydroxyl groups on silica-alumina surfaces and in molecules in solution. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 1974, 70, 2021–2032. [Google Scholar] [CrossRef]
- Cimas, Á.; Tielens, F.; Sulpizi, M.; Gaigeot, M.P.; Costa, D. The amorphous silica-liquid water interface studied by ab initio molecular dynamics (AIMD): Local organization in global disorder. J. Phys. Condens. Matter 2014, 26. [Google Scholar] [CrossRef]
- Zhuravlev, L.T. The surface chemistry of amorphous silica. Zhuravlev model. Colloids Surf. A Physicochem. Eng. Asp. 2000, 173, 1–38. [Google Scholar] [CrossRef] [Green Version]
- Snyder, L.R.; Ward, J.W. The surface structure of porous silicas. J. Phys. Chem. 1966, 70, 3941–3952. [Google Scholar] [CrossRef]
- Warring, S.L.; Beattie, D.A.; McQuillan, A.J. Surficial siloxane-to-silanol interconversion during room-temperature hydration/dehydration of amorphous silica films observed by ATR-IR and TIR-Raman spectroscopy. Langmuir 2016, 32, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Ramsey, C.; Baran, G. Thermal pretreatment of silica composite filler materials. J. Therm. Anal. Calorim. 2010, 99, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.S.D.; Pantano, C.G. Hydroxylation and dehydroxylation behavior of silica glass fracture surfaces. J. Am. Ceram. Soc. 2002, 85, 1499–1504. [Google Scholar] [CrossRef]
- Zhuravlev, L.T. Concentration of hydroxyl groups on the surface of amorphous silicas. Langmuir 1987, 3, 316–318. [Google Scholar] [CrossRef]
- De Farias, R.F.; Airoldi, C. Thermogravimetry as a reliable tool to estimate the density of silanols on a silica gel surface. J. Therm. Anal. Calorim. 1998, 53, 751–756. [Google Scholar] [CrossRef]
- Morrow, B.A.; McFarlan, A.J. Surface vibrational modes of silanol groups on silica. J. Phys. Chem. 1992, 96, 1395–1400. [Google Scholar] [CrossRef]
- Mahadevan, T.S.; Garofalini, S.H. Dissociative chemisorption of water onto silica surfaces and formation of hydronium ions. J. Phys. Chem. C 2008, 112, 1507–1515. [Google Scholar] [CrossRef]
- Hensen, E.J.M.; Poduval, D.G.; Ligthart, D.A.J.M.; van Veen, J.A.R.; Rigutto, M.S. Quantification of strong Bronsted acid sites in aluminosilicates. J. Phys. Chem. C 2010, 114, 8363–8374. [Google Scholar] [CrossRef]
- Corma, A.; Fornes, V.; Navarro, M.T.; Perezpariente, J. Acidity and stability of MCM-41 crystalline aluminosilicates. J. Catal. 1994, 148, 569–574. [Google Scholar] [CrossRef]
- Perez-Beltran, S.; Balbuena, P.B.; Ramírez-Caballero, G.E. Surface structure and acidity properties of mesoporous silica SBA-15 modified with aluminum and titanium: First-principles calculations. J. Phys. Chem. C 2016, 120, 18105–18114. [Google Scholar] [CrossRef]
- Meziani, M.J.; Zajac, J.; Douillard, J.-M.; Jones, D.J.; Partyka, S.; Rozière, J. Evaluation of surface enthalpy of porous aluminosilicates of the MCM-41 type using immersional calorimetry: Effect of the pore size and framework Si:Al ratio. J. Colloid Interface Sci. 2001, 233, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W. The distribution of aluminum in the tetrahedra of silicates and aluminates. Am. Mineral. 1954, 39, 92–96. [Google Scholar] [CrossRef]
- Yang, R.T. Adsorbents: Fundamentals and Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; ISBN 0471297410. [Google Scholar] [CrossRef]
- Chen, L.Y.; Jaenicke, S.; Chuah, G.K. Thermal and hydrothermal stability of framework-substituted MCM-41 mesoporous materials. Microporous Mater. 1997, 12, 323–330. [Google Scholar] [CrossRef]
- Shen, S.; Kawi, S. Understanding of the effect of al substitution on the hydrothermal stability of MCM-41. J. Phys. Chem. B 1999, 103, 8870–8876. [Google Scholar] [CrossRef]
- Mokaya, R. Alumination pathways to mesoporous aluminosilicates with high-temperature hydrothermal stability. ChemPhysChem 2002, 3, 360–363. [Google Scholar] [CrossRef]
- Mokaya, R. Influence of pore wall thickness on the steam stability of Al-grafted MCM-41. Chem. Commun. 2001, 633–634. [Google Scholar] [CrossRef]
- Chua, H.T.; Ng, K.C.; Chakraborty, A.; Oo, N.M.; Othman, M.A. Adsorption Characteristics of Silica Gel + Water Systems. J. Chem. Eng. Data 2002, 1177–1181. [Google Scholar] [CrossRef]
- Ng, K.C.; Chua, H.T.; Chung, C.Y.; Loke, C.H.; Kashiwagi, T.; Akisawa, A.; Saha, B.B. Experimental investigation of the silica gel-water adsorption isotherm characteristics. Appl. Therm. Eng. 2001, 21, 1631–1642. [Google Scholar] [CrossRef]
- Ito, M.; Watanabe, F.; Hasatani, M. Improvement of both adsorption performance of silica gel and heat transfer characteristics by means of heat exchange modulation for a heat pump. Heat Transf. Jpn. Res. 1996, 25, 420–433. [Google Scholar] [CrossRef]
- Jaehnig, D.; Hausner, R.; Wagner, W.; Isaksson, C. Thermo-chemical storage for solar space heating in a single-family house. In Proceedings of the ECOSTOCK Conference, Pomona, NJ, USA, 31 May–2 June 2006. [Google Scholar]
- Hadorn, J. IEA Solar Heating and Cooling Programme Task 32: Advanced Storage Concepts for Solar and Low Energy Buildings; International Energy Agency: Paris, France, 2014. [Google Scholar]
- Aristov, Y.I. Novel materials for adsorptive heat pumping and storage: Screening and nanotailoring of sorption properties. J. Chem. Eng. Jpn. 2007, 40, 1242–1251. [Google Scholar] [CrossRef]
- Aristov, Y.I.; Glaznev, I.S.; Freni, A.; Restuccia, G. Kinetics of water sorption on SWS-1L (calcium chloride confined to mesoporous silica gel): Influence of grain size and temperature. Chem. Eng. Sci. 2006, 61, 1453–1458. [Google Scholar] [CrossRef]
- Gordeeva, L.G.; Aristov, Y.I. Composites’ salt inside porous matrix’ for adsorption heat transformation: A current state-of-the-art and new trends. Int. J. Low-Carbon Technol. 2012, 288–302. [Google Scholar] [CrossRef]
- Llewellyn, P.L.; Schueth, F.; Grillet, Y.; Rouquerol, F.; Rouquerol, J.; Unger, K.K. Water sorption on mesoporous aluminosilicate MCM-41. Langmuir 1995, 11, 574–577. [Google Scholar] [CrossRef]
- Kocherbitov, V.; Alfredsson, V. Hydration of MCM-41 studied by sorption calorimetry. J. Phys. Chem. C 2007, 111, 12906–12913. [Google Scholar] [CrossRef]
- Kittaka, S.; Ishimaru, S.; Kuranishi, M. Enthalpy and interfacial free energy changes of water capillary condensed in mesoporous silica, MCM-41 and SBA-15. Phys. Chem. Chem. Phys. 2006, 3223–3231. [Google Scholar] [CrossRef] [PubMed]
- Rozwadowski, M.; Lezanska, M.; Wloch, J.; Erdmann, K.; Golembiewski, R.; Kornatowski, J. Mechanism of adsorption of water, benzene, and nitrogen on Al-MCM-41 and effect of coking on the adsorption. Langmuir 2001, 17, 2112–2119. [Google Scholar] [CrossRef]
- Thach, U.D.; Hesemann, P.; Yang, G.; Geneste, A.; Le Caër, S.; Prelot, B. Ionosilicas as efficient sorbents for anionic contaminants: Radiolytic stability and ion capacity. J. Colloid Interface Sci. 2016, 482, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Auerbac, S.M.; Carrado, K.A.; Dutt, P.K. (Eds.) Handbook of Zeolite Science and Technology; Marcel Dekker: New York, NY, USA, 2003; ISBN 0824740203. [Google Scholar]
- Fletcher, R.E.; Ling, S.; Slater, B. Violations of Löwenstein’s rule in zeolites. Chem. Sci. 2017, 8, 7483–7491. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Pang, W.; Yu, J.; Huo, Q.; Chen, J. Chemistry of Zeolites and Related Porous Materials Synthesis and Structure; John Wiley & Sons (Asia) Pte Ltd.: Singapore, 2007; ISBN 9780470822333. [Google Scholar]
- Frising, T.; Leflaive, P. Extraframework cation distributions in X and Y faujasite zeolites: A review. Microporous Mesoporous Mater. 2008, 114, 27–63. [Google Scholar] [CrossRef]
- Culfaz, A.; Sand, L.B. Mechanism of nucleation and crystallization of zeolites from gels. Mol. Sieves 1973, 140–151. [Google Scholar] [CrossRef]
- Thompson, R.W. Recent advances in the understanding of zeolite synthesis. Mol. Sieves 1998, 1, 1–33. [Google Scholar] [CrossRef]
- Liu, Z.; Okabe, K.; Anand, C.; Yonezawa, Y.; Zhu, J.; Yamada, H.; Endo, A.; Yanaba, Y.; Yoshikawa, T.; Ohara, K.; et al. Continuous flow synthesis of ZSM-5 zeolite on the order of seconds. Proc. Natl. Acad. Sci. USA 2016, 113, 14267–14271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrioux, C. Modélisation moléculaire de l’adsorption et de la diffusion de molécules polaires dans un solide nanoporeux de type zéolithique. Ph.D. Thesis, Université Montpellier II, Montpellier, France, 2010. [Google Scholar]
- Santos, V. Caractérisation et modification de l’acidité résiduelle de zéolithes cationiques. Ph.D. Thesis, Université de Poitiers, Poitiers, France, 2008. [Google Scholar]
- Kramer, G.J.; Van Santen, R.A.; Emeis, C.A.; Nowak, A.K. Understanding the acid behaviour of zeolites from theory and experiment. Nature 1993, 363, 529–531. [Google Scholar] [CrossRef]
- Kim, K.M.; Oh, H.T.; Lim, S.J.; Ho, K.; Park, Y.; Lee, C.H. Adsorption equilibria of water vapor on zeolite 3A, zeolite 13X, and dealuminated Y zeolite. J. Chem. Eng. Data 2016, 61, 1547–1554. [Google Scholar] [CrossRef]
- Simonot-Grange, M.H.; Elm’Chaouri, A.; Weber, G.; Dufresne, P.; Raatz, F.; Joly, J.F. Characterization of the dealumination effect into H faujasites by adsorption: Part 1. The water molecule as a structural aluminum ion selective probe. Zeolites 1992, 12, 155–159. [Google Scholar] [CrossRef]
- Moıse, J.C.; Bellat, J.P.; Méthivier, A. Adsorption of water vapor on X and Y zeolites exchanged with barium. Microporous Mesoporous Mater. 2001, 43, 91–101. [Google Scholar] [CrossRef]
- Cindrella, L.; Dyer, A. Ion-exchanged and salt hydrates-encapsulated zeolites for solar refrigeration. Sol. Energy Mater. Sol. Cells 2009, 93, 161–166. [Google Scholar] [CrossRef]
- Zygmunt, S.A.; Curtiss, L.A.; Iton, L.E.; Erhardt, M.K. Computational studies of water adsorption in the zeolite H-ZSM-5. J. Phys. Chem. 1996 1996, 6663–6671. [Google Scholar] [CrossRef]
- Jungsuttiwong, S.; Limtrakul, J.; Truong, T.N. Theoretical study of modes of adsorption of water dimer on H-ZSM-5 and H-faujasite zeolites. J. Phys. Chem. B 2005, 109, 13342–13351. [Google Scholar] [CrossRef] [PubMed]
- Baerlocher, C.; McCusker, L.B. Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases/ (accessed on 11 September 2018).
- Di Lella, A.; Desbiens, N.; Boutin, A.; Demachy, I.; Ungerer, P.; Bellat, J.-P.; Fuchs, A.H. Molecular simulation studies of water physisorption in zeolites. Phys. Chem. Chem. Phys. 2006, 8, 5396–5406. [Google Scholar] [CrossRef] [PubMed]
- Abrioux, C.; Coasne, B.; Maurin, G.; Henn, F.; Jeffroy, M.; Boutin, A. Cation behavior in faujasite zeolites upon water adsorption: A combination of monte carlo and molecular dynamics simulations. J. Phys. Chem. C 2009, 113, 10696–10705. [Google Scholar] [CrossRef]
- Auroux, A. Microcalorimetry methods to study the acidity and reactivity of zeolites, pillared clays and mesoporous materials. Top. Catal. 2002, 19, 205–213. [Google Scholar] [CrossRef]
- Shirono, K.; Endo, A.; Daiguji, H. Molecular dynamics study of hydrated Faujasite-type zeolites. J. Phys. Chem. B 2005, 3446–3453. [Google Scholar] [CrossRef] [PubMed]
- Semmer, V.; Batamack, P.; Dorémieux-Morin, C.; Fraissard, J. NMR studies of the Brønsted acidity of solids. Application to superacidic solids. Top. Catal. 1998, 6, 119–125. [Google Scholar] [CrossRef]
- Batamack, P.; Vincent, R.; Fraissard, J. Acidity of partially and non-dealuminated HY zeolites measured by 4-K broad-line and 300-K high-resolution magic-angle spinning 1H nuclear magnetic resonance spectroscopy: Synergy between Briinsted and Lewis acid sites. Microporous Mater. 1994, 2, 515–524. [Google Scholar] [CrossRef]
- Rao, G.N.; Kotasthane, A.N. Thermal and hydrothermal stabilities of zeolite EU-1. Appl. Catal. Ageneral 1994, 119, 33–43. [Google Scholar] [CrossRef]
- Lutz, W. Zeolite Y: Synthesis, modification, and properties—A Case Revisited. Adv. Mater. Sci. Eng. 2014, 2014. [Google Scholar] [CrossRef]
- Pu, X.; Liu, N.; Shi, L. Microporous and mesoporous materials acid properties and catalysis of USY zeolite with different extra-framework aluminum concentration. Microporous Mesoporous Mater. 2015, 201, 17–23. [Google Scholar] [CrossRef]
- Dimitrijevic, R.; Lutz, W.; Ritzmann, A. Hydrothermal stability of zeolites: Determination of extra-framework species of H-Y faujasite-type steamed zeolite. J. Phys. Chem. Solids 2006, 67, 1741–1748. [Google Scholar] [CrossRef]
- Salman, N.; Rüscher, C.H.; Buhl, J.-C.; Lutz, W.; Toufar, H.; Stöcker, M. Effect of temperature and time in the hydrothermal treatment of HY zeolite. Microporous Mesoporous Mater. 2006, 90, 339–346. [Google Scholar] [CrossRef]
- Ristic, A.; Fischer, F.; Hauer, A.; Logar, N.Z. Improved performance of binder-free zeolite Y for low-temperature sorption heat storage. J. Mater. Chem. A 2018, 6, 11521–11530. [Google Scholar] [CrossRef]
- Buhl, J.; Gerstmann, M.; Lutz, W.; Ritzmann, A. Hydrothermal stability of the novel zeolite type LSX in comparison to the traditional 13X modification. Z. Anorg. Allg. Chem. 2004, 604–608. [Google Scholar] [CrossRef]
- Fischer, F.; Lutz, W.; Buhl, J.C.; Laevemann, E. Insights into the hydrothermal stability of zeolite 13X. Microporous Mesoporous Mater. 2018, 262, 258–268. [Google Scholar] [CrossRef]
- Gopal, R.; Hollebone, B.R.; Langford, C.H.; Shigeishi, R.A. The rates of solar energy storage and retrieval in a zeolite-water system. Sol. Energy 1982, 28, 421–424. [Google Scholar] [CrossRef]
- Gaeini, M.; Zondag, H.A.; Rindt, C.C.M. Effect of kinetics on the thermal performance of a sorption heat storage reactor. Appl. Therm. Eng. 2016, 102, 520–531. [Google Scholar] [CrossRef]
- Jänchen, J.; Schumann, K.; Thrun, E.; Brandt, A.; Unger, B.; Hellwig, U. Preparation, hydrothermal stability and thermal adsorption storage properties of binderless zeolite beads. Int. J. Low-Carbon Technol. Adv. Access 2012, 1–5. [Google Scholar] [CrossRef]
- Herzog, T.H.; Jänchen, J.; Kontogeorgopoulos, E.M.; Lutz, W. Steamed zeolites for heat pump applications and solar driven thermal adsorption storage. Energy Procedia 2014, 48, 380–383. [Google Scholar] [CrossRef]
- Gómez-Álvarez, P.; Perez-Carbajo, J.; Balestra, S.R.G.; Calero, S. Impact of the nature of exchangeable cations on LTA-type zeolite hydration. J. Phys. Chem. C 2016, 120, 23254–23261. [Google Scholar] [CrossRef]
- Li, X.; Narayanan, S.; Michaelis, V.K.; Ong, T.C.; Keeler, E.G.; Kim, H.; McKay, I.S.; Griffin, R.G.; Wang, E.N. Zeolite y adsorbents with high vapor uptake capacity and robust cycling stability for potential applications in advanced adsorption heat pumps. Microporous Mesoporous Mater. 2015, 201, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Stach, H.; Mugele, J.; Jänchen, J.; Weiler, E. Influence of cycle temperatures on the thermochemical heat storage densities in the systems water/microporous and water/mesoporous adsorbents. Adsorption 2005, 11, 393–404. [Google Scholar] [CrossRef]
- Lass-Seyoum, A.; Borozdenko, D.; Friedrich, T.; Langhof, T.; Mack, S. Practical test on a closed sorption thermochemical storage system with solar thermal energy. Energy Procedia 2016, 91, 182–189. [Google Scholar] [CrossRef]
- Jänchen, J.; Herzog, T.H.; Gleichmann, K.; Unger, B.; Brandt, A.; Fischer, G.; Richter, H. Performance of an open thermal adsorption storage system with Linde type A zeolites: Beads versus honeycombs. Microporous Mesoporous Mater. 2015, 207, 179–184. [Google Scholar] [CrossRef]
- Tatsidjodoung, P.; Le Pierrès, N.; Heintz, J.; Lagre, D.; Luo, L.; Durier, F. Experimental and numerical investigations of a zeolite 13X/water reactor for solar heat storage in buildings. Energy Convers. Manag. 2016, 108, 488–500. [Google Scholar] [CrossRef]
- Johannes, K.; Kuznik, F.; Hubert, J.L.; Durier, F.; Obrecht, C. Design and characterisation of a high powered energy dense zeolite thermal energy storage system for buildings. Appl. Energy 2015, 159, 80–86. [Google Scholar] [CrossRef]
- De Boer, R.; Smeding, S.F.; Zondag, H.A.; Krol, G. Development of a prototype system for seasonal solar heat storage using an open sorption process. Eurotherm Semin. 2014, 99, 28–30. [Google Scholar]
- Bales, C.; Gantenbein, P.; Jaenig, D.; Kerskes, H.; Summer, K.; Van Essen, M.; Weber, R. Laboratory Tests of Chemical Reactions and Prototype Sorption Storage Units; International Energy Agency: Paris, France, 2008. [Google Scholar]
- Wilson, S.T.; Lok, B.M.; Messina, C.A.; Cannan, T.R.; Flanigen, E.M. Aluminophosphate molecular sieves: A new class of microporous crystalline inorganic solids. J. Am. Chem. Soc. 1982, 104, 1146–1147. [Google Scholar] [CrossRef]
- Newalkar, B.L.; Jasra, R.V.; Kamath, V.; Bhat, S.G.T. Sorption of water in aluminophosphate molecular sieve AlPO4-5. Microporous Mesoporous Mater. 1998, 20, 129–137. [Google Scholar] [CrossRef]
- Kohler, T.; Hinze, M.; Müller, K.; Schwieger, W. Temperature independent description of water adsorption on zeotypes showing a type V adsorption isotherm. Energy 2017, 135, 227–236. [Google Scholar] [CrossRef]
- Ristić, A.; Logar, N.Z.; Henninger, S.K.; Kaučič, V. The performance of small-pore microporous aluminophosphates in low-temperature solar energy storage: The structure-property relationship. Adv. Funct. Mater. 2012, 22, 1952–1957. [Google Scholar] [CrossRef]
- Lok, B.M.; Messina, C.A.; Patton, R.L.; Gajek, R.T.; Cannan, T.R.; Flanigen, E.M. Silicoaluminophosphate molecular sieves: Another new class of microporous crystalline inorganic solids. J. Am. Chem. Soc. 1984, 106, 6092–6093. [Google Scholar] [CrossRef]
- Oliver, S.; Kuperman, A.; Coombs, N.; Lough, A.; Ozin, G.A. Lamellar aluminophosphates with surface patterns that mimic diatom and radiolarian microskeletons. Nature 1995, 378, 47–50. [Google Scholar] [CrossRef]
- Sayari, A.; Moudrakovski, I.; Reddy, J.S.; Gk, C.; Ratcliffe, C.I.; Ripmeester, J.A.; Preston, K.F. Synthesis of mesostructured lamellar aluminophosphates using supramolecular templates. Chem. Mater. 1996, 4756, 2080–2088. [Google Scholar] [CrossRef]
- Li, D.; Yao, J.; Wang, H. Hydrothermal synthesis of AlPO4-5: Effect of precursor gel preparation on the morphology of crystals. Prog. Nat. Sci. Mater. Int. 2012, 22, 684–692. [Google Scholar] [CrossRef] [Green Version]
- Afeworki, M.; Kennedy, G.J.; Dorset, D.L.; Strohmaier, K.G. Synthesis and characterization of a new microporous material. 2. AlPO and SAPO forms of EMM-3. Chem. Mater. 2006, 18, 1705–1710. [Google Scholar] [CrossRef]
- Lü, J.M.; Ranjit, K.T.; Rungrojchaipan, P.; Kevan, L. Synthesis of mesoporous aluminophosphate (AlPO) and investigation of zirconium incorporation into mesoporous AlPOs. J. Phys. Chem. B 2005, 109, 9284–9293. [Google Scholar] [CrossRef] [PubMed]
- Henninger, S.K.; Munz, G.; Ratzsch, K.-F.; Schossig, P. Cycle stability of sorption materials and composites for the use in heat pumps and cooling machines. Renew. Energy 2011, 36, 3043–3049. [Google Scholar] [CrossRef]
- Krese, G.; Koželj, R.; Butala, V.; Stritih, U. Thermochemical seasonal solar energy storage for heating and cooling of buildings. Energy Build. 2018, 164, 239–253. [Google Scholar] [CrossRef]
- Tatsidjodoung, P.; Le Pierrès, N.; Luo, L. A review of potential materials for thermal energy storage in building applications. Renew. Sustain. Energy Rev. 2013, 18, 327–349. [Google Scholar] [CrossRef]
- Shimooka, S.; Oshima, K.; Hidaka, H.; Takewaki, T.; Kakiuchi, H.; Kodama, A.; Kubota, M.; Matsuda, H. The evaluation of direct cooling and heating desiccant device coated with FAM. J. Chem. Eng. Jpn. 2007, 40, 1330–1334. [Google Scholar] [CrossRef]
- Mitsubishi Plastics Zeolitic Water Vapor Adsorbent AQSOATM. Available online: http://www.mpi.co.jp/infopdf/AQSOA.pdf (accessed on 22 September 2018).
- Bauer, J.; Herrmann, R.; Mittelbach, W.; Schwieger, W. Zeolite/aluminum composite adsorbents for application in adsorption refrigeration. Int. J. Energy Res. 2009, 33, 1233–1249. [Google Scholar] [CrossRef]
- Jänchen, J.; Stach, H. Shaping adsorption properties of nano-porous molecular sieves for solar thermal energy storage and heat pump applications. Sol. Energy 2013, 104, 16–18. [Google Scholar] [CrossRef]
- Floquet, N.; Coulomb, J.P.; Dufau, N.; Andre, G. Structure and dynamics of confined water in AlPO4-5 zeolite. J. Phys. Chem. B 2004, 108, 13107–13115. [Google Scholar] [CrossRef]
- Freni, A.; Bonaccorsi, L.; Calabrese, L.; Caprì, A.; Frazzica, A.; Sapienza, A. SAPO-34 coated adsorbent heat exchanger for adsorption chillers. Appl. Therm. Eng. 2015, 82, 1–7. [Google Scholar] [CrossRef]
- Goldsworthy, M.J. Measurements of water vapour sorption isotherms for RD silica gel, AQSOA-Z01, AQSOA-Z02, AQSOA-Z05 and CECA zeolite 3A. Microporous Mesoporous Mater. 2014, 196, 59–67. [Google Scholar] [CrossRef]
- Baerloher, C.; Meier, W.M.; Olson, D.H. Atlas of Zeolite Frameworks Type, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2001; ISBN 978-0-444-53064-6. [Google Scholar] [CrossRef]
- Li, J.; Yu, J.; Xu, R. Progress in heteroatom-containing aluminophosphate molecular sieves. Proc. R. Soc. A Math. Phys. Eng. Sci. 2012, 468, 1955–1967. [Google Scholar] [CrossRef] [Green Version]
- Goldfarb, D.; Li, H.; Davis, M.E. Dynamics of water molecules in VPI-5 and AlPO4-5 studied by NMR spectroscopy. J. Am. Chem. Soc. 1992, 3690–3697. [Google Scholar] [CrossRef]
- Askari, S.; Halladj, R.; Sohrabi, M. An overview of the effects of crystallization time, template and silicon sources on hydrothermal synthesis of SAPO-34 molecular sieve with small crystals. Rev. Adv. Mater. Sci. 2012, 32, 83–93. [Google Scholar]
- Van Heyden, H.; Munz, G.; Schnabel, L.; Schmidt, F.; Mintova, S.; Bein, T. Kinetics of water adsorption in microporous aluminophosphate layers for regenerative heat exchangers. Appl. Therm. Eng. 2009, 29, 1514–1522. [Google Scholar] [CrossRef] [Green Version]
- Rouhani, M.; Huttema, W.; Bahrami, M. Thermal conductivity of AQSOA FAM-Z02 packed bed adsorbers in open and closed adsorption thermal energy storage systems. Int. J. Refrig. 2018, 1–11. [Google Scholar] [CrossRef]
- Yaghi, O.M.; O’Keeffe, M.; Ockwig, N.W.; Chae, H.K.; Eddaoudi, M.; Kim, J. Reticular synthesis and the design of new materials. Nature 2003, 423, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y.; Jensen, S.; Tan, K.; Wojtas, L.; Roveto, M.; Cure, J.; Thonhauser, T.; Chabal, Y.J.; Zaworotko, M.J. Modulation of water vapor sorption by a 4th generation metal-organic material with a rigid framework and self-switching pores. J. Am. Chem. Soc. 2018, 140, jacs.8b07290. [Google Scholar] [CrossRef]
- Liu, J.; Chen, L.; Cui, H.; Zhang, J.; Zhang, L.; Su, C.Y. Applications of metal-organic frameworks in heterogeneous supramolecular catalysis. Chem. Soc. Rev. 2014, 43, 6011–6061. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Kondo, M. Functional micropore chemistry of crystalline metal complex-assembled compounds. Chem. Soc. Jpn. 1998, 71, 1739–1753. [Google Scholar] [CrossRef]
- Bousquet, D.; Coudert, F.X.; Fossati, A.G.J.; Neimark, A.V.; Fuchs, A.H.; Boutin, A. Adsorption induced transitions in soft porous crystals: An osmotic potential approach to multistability and intermediate structures. J. Chem. Phys. 2013, 138. [Google Scholar] [CrossRef] [PubMed]
- Boulé, R.; Roland, C.; Le Pollés, L.; Audebrand, N.; Ghoufi, A. Thermal and guest-assisted structural transition in the NH2-MIL-53(Al) Metal Organic Framework: A molecular dynamics simulation investigation. Nanomaterials 2018, 8, 531. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M. The postsynthetic renaissance in porous solids. J. Am. Chem. Soc. 2017, 139, 2855–2863. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M. Postsynthetic methods for the functionalization of metal-organic frameworks. Chem. Rev. 2012, 112, 970–1000. [Google Scholar] [CrossRef] [PubMed]
- Stock, N.; Biswas, S. Synthesis of metal-organic frameworks (MOFs): Routes to various MOF topologies, morphologies, and composites. Chem. Rev. 2012, 112, 933–969. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cohen, S.M. Postsynthetic modification of metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1315–1329. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Xu, D.; Chung, J.D.; Kaviany, M.; Huang, B. H2O adsorption/desorption in MOF-74: Ab Initio Molecular Dynamics and experiments. J. Phys. Chem. C 2015, 119, 13021–13031. [Google Scholar] [CrossRef]
- Eddaoudi, M.; Li, H.; Yaghi, O.M. Highly porous and stable metal-organic frameworks: Structure design and sorption properties. J. Am. Chem. Soc. 2000, 122, 1391–1397. [Google Scholar] [CrossRef]
- Planchais, A.; Devautour-Vinot, S.; Salles, F.; Ragon, F.; Devic, T.; Serre, C.; Maurin, G. A joint experimental/computational exploration of the dynamics of confined water/Zr-based MOFs systems. J. Phys. Chem. C 2014, 118, 14441–14448. [Google Scholar] [CrossRef]
- Cadiau, A.; Lee, J.S.; Damasceno Borges, D.; Fabry, P.; Devic, T.; Wharmby, M.T.; Martineau, C.; Foucher, D.; Taulelle, F.; Jun, C.H.; et al. Design of hydrophilic Metal Organic Framework water adsorbents for heat reallocation. Adv. Mater. 2015, 27, 4775–4780. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.; Bezverkhyy, I.; Bellat, J.P.; Ballandras, A.; Ortiz, G.; Chaplais, G.; Patarin, J.; Coudert, F.X.; Fuchs, A.H.; Boutin, A. Mechanism of water adsorption in the large pore form of the gallium-based MIL-53 metal-organic framework. Microporous Mesoporous Mater. 2016, 222, 145–152. [Google Scholar] [CrossRef]
- Salles, F.; Bourrelly, S.; Jobic, H.; Devic, T.; Guillerm, V.; Llewellyn, P.; Serre, C.; Ferey, G.; Maurin, G. Molecular insight into the adsorption and diffusion of water in the versatile hydrophilic/hydrophobic flexible MIL-53(Cr) MOF. J. Phys. Chem. C 2011, 115, 10764–10776. [Google Scholar] [CrossRef]
- De Lange, M.F.; Gutierrez-Sevillano, J.-J.; Hamad, S.; Vlugt, T.J.H.; Calero, S.; Gascon, J.; Kapteijn, F. Understanding adsorption of highly polar vapors on mesoporous MIL-100(Cr) and MIL-101(Cr): Experiments and molecular simulations. J. Phys. Chem. C 2013, 117, 7613–7622. [Google Scholar] [CrossRef]
- Li, S.; Chen, Y.; Pei, X.; Zhang, S.; Feng, X.; Zhou, J.; Wang, B. Water purification: Adsorption over Metal-Organic Frameworks. Chin. J. Chem. 2016, 34, 175–185. [Google Scholar] [CrossRef]
- Canivet, J.; Bonnefoy, J.; Daniel, C.; Legrand, A.; Coasne, B.; Farrusseng, D. Structure-property relationships of water adsorption in metal-organic frameworks. New J. Chem. 2014, 38, 3102–3111. [Google Scholar] [CrossRef]
- Canivet, J.; Fateeva, A.; Guo, Y.; Coasne, B.; Farrusseng, D. Water adsorption in MOFs: Fundamentals and applications. Chem. Soc. Rev. 2014, 5594–5617. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Gándara, F.; Zhang, Y.-B.; Jiang, J.; Queen, W.L.; Hudson, M.R.; Yaghi, O.M. Water adsorption in porous metal-organic frameworks and related materials. J. Am. Chem. Soc. 2014, 136, 4369–4381. [Google Scholar] [CrossRef] [PubMed]
- Burtch, N.C.; Jasuja, H.; Walton, K.S. Water stability and adsorption in metal-organic frameworks. Chem. Rev. 2014, 114, 10575–10612. [Google Scholar] [CrossRef] [PubMed]
- Chui, S.S.-Y.; Lo, S.M.-F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. A chemically functionalizable nanoporous material [Cu3(TMA)2(H2O)3]n. Science 1999, 3, 1148–1150. [Google Scholar] [CrossRef]
- Schoenecker, P.M.; Carson, C.G.; Jasuja, H.; Flemming, C.J.J.; Walton, K.S. Effect of water adsorption on retention of structure and surface area of Metal−Organic Frameworks. Ind. Eng. Chem. Res. 2012, 6513–6519. [Google Scholar] [CrossRef]
- Küsgens, P.; Rose, M.; Senkovska, I.; Fröde, H.; Henschel, A.; Siegle, S.; Kaskel, S. Characterization of metal-organic frameworks by water adsorption. Microporous Mesoporous Mater. 2009, 120, 325–330. [Google Scholar] [CrossRef]
- Al-Janabi, N.; Martis, V.; Servi, N.; Siperstein, F.R.; Fan, X. Cyclic adsorption of water vapour on CuBTC MOF: Sustaining the hydrothermal stability under non-equilibrium conditions. Chem. Eng. J. 2018, 333, 594–602. [Google Scholar] [CrossRef]
- Soubeyrand-lenoir, E.; Vagner, C.; Yoon, J.W.; Bazin, P.; Ragon, F.; Hwang, Y.K.; Serre, C.; Chang, J.; Llewellyn, P.L. How water fosters a remarkable 5-Fold increase in low-pressure. J. Am. Chem. Soc. 2012, 10174–10181. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Marshall, M.; Chaffee, A.L. CO2 adsorption-based separation by metal organic framework (Cu-BTC) versus zeolite (13X). Energy Fuels 2009, 2785–2789. [Google Scholar] [CrossRef]
- Gul-E-Noor, F.; Jee, B.; Pöppl, A.; Hartmann, M.; Himsl, D.; Bertmer, M. Effects of varying water adsorption on a Cu3(BTC)2 metal–organic framework ( MOF ) as studied by 1H and 13C solid-state NMR spectroscopy. Phys. Chem. Chem. Phys 2011, 7783–7788. [Google Scholar] [CrossRef] [PubMed]
- Al-Janabi, N.; Alfutimie, A.; Siperstein, F.R.; Fan, X. Underlying mechanism of the hydrothermal instability of Cu3(BTC)2 metal-organic framework. Front. Chem. Sci. Eng. 2016, 10, 103–107. [Google Scholar] [CrossRef]
- Vishnyakov, A.; Ravikovitch, P.I.; Neimark, A.V.; Bu, M.; Wang, Q.M. Nanopore structure and sorption properties of Cu−BTC Metal−Organic Framework. Nano Lett. 2003, 3, 713–718. [Google Scholar] [CrossRef]
- DeCoste, J.B.; Peterson, G.W.; Schindler, B.J.; Killops, K.L.; Broweb, M.A.; Mahleb, J.J. The effect of water adsorption on the structure of the carboxylate containing metal–organic frameworks Cu-BTC, Mg-MOF-74, and UiO-66. J. Mater. Chem. A 2013, 1, 11922–11932. [Google Scholar] [CrossRef]
- Elsayed, E.; AL-Dadah, R.; Mahmoud, S.; Anderson, P.A.; Elsayed, A.; Youssef, P.G. CPO-27(Ni), aluminium fumarate and MIL-101(Cr) MOF materials for adsorption water desalination. Desalination 2017, 406, 25–36. [Google Scholar] [CrossRef]
- Shi, B.; Al-Dadah, R.; Mahmoud, S.; Elsayed, A.; Elsayed, E. CPO-27(Ni) metal-organic framework based adsorption system for automotive air conditioning. Appl. Therm. Eng. 2016, 106, 325–333. [Google Scholar] [CrossRef]
- Dietzel, P.D.C.; Blom, R.; Fjellvåg, H. Base-induced formation of two magnesium metal-organic framework compounds with a bifunctional tetratopic ligand. Eur. J. Inorg. Chem. 2008, 3624–3632. [Google Scholar] [CrossRef]
- Dietzel, P.D.C.; Panella, B.; Hirscher, M.; Blom, R.; Fjellvåg, H. Hydrogen adsorption in a nickel based coordination polymer with open metal sites in the cylindrical cavities of the desolvated framework. Chem. Commun. 2006, 1, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Dietzel, P.D.C.; Morita, Y.; Blom, R.; Fjellvåg, H. An in situ high-temperature single-crystal investigation of a dehydrated metal-organic framework compound and field-induced magnetization of one-dimensional metal-oxygen chains. Angew. Chem. Int. Ed. 2005, 44, 6354–6358. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Y.; Benin, A.I.; Jakubczak, P.; Willis, R.R.; LeVan, M.D. CO2/H2O adsorption equilibrium and rates on metal-organic frameworks: HKUST-1 and Ni/DOBDC. Langmuir 2010, 26, 14301–14307. [Google Scholar] [CrossRef] [PubMed]
- Zuluaga, S.; Fuentes-Fernandez, E.M.A.; Tan, K.; Arter, C.A.; Li, J.; Chabal, Y.J.; Thonhauser, T. Chemistry in confined spaces: Reactivity of the Zn-MOF-74 channels. J. Mater. Chem. A 2016, 4, 13176–13182. [Google Scholar] [CrossRef]
- Tan, K.; Zuluaga, S.; Gong, Q.; Canepa, P.; Wang, H.; Li, J.; Chabal, Y.J.; Thonhauser, T. Water reaction mechanism in metal organic frameworks with coordinatively unsaturated metal ions: MOF-74. Chem. Mater. 2014, 26, 6886–6895. [Google Scholar] [CrossRef]
- Zuluaga, S.; Fuentes-Fernandez, E.M.A.; Tan, K.; Xu, F.; Li, J.; Chabal, Y.J.; Thonhauser, T. Understanding and controlling water stability of MOF-74. J. Mater. Chem. A 2016, 4, 5176–5183. [Google Scholar] [CrossRef] [Green Version]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef] [PubMed]
- Cmarik, G.E.; Kim, M.; Cohen, S.M.; Walton, K.S. Tuning the adsorption properties of UiO-66 via ligand functionalization. Langmuir 2012, 28, 15606–15613. [Google Scholar] [CrossRef] [PubMed]
- Jeremias, F.; Lozan, V.; Henninger, S.K.; Janiak, C. Programming MOFs for water sorption: Amino-functionalized MIL-125 and UiO-66 for heat transformation and heat storage applications. Dalton Trans. 2013, 42, 15967–15973. [Google Scholar] [CrossRef] [PubMed]
- Decoste, J.B.; Peterson, G.W.; Jasuja, H.; Glover, T.G.; Huang, Y.G.; Walton, K.S. Stability and degradation mechanisms of metal-organic frameworks containing the Zr6O4(OH)4 secondary building unit. J. Mater. Chem. A 2013, 1, 5642–5650. [Google Scholar] [CrossRef]
- Férey, G.; Mellot-Draznieks, C.; Serre, C.; Millange, F.; Dutour, J.; Surblé, S.; Margiolaki, I. A Chromium Terephthalate–based solid with unusually large pore volumes and surface area. Science 2005, 309, 2040. [Google Scholar] [CrossRef] [PubMed]
- Soubeyrand-Lenoir, E. Adsorption et Separation de gaz en Mode Dynamique sur des Materiaux Hybrides. Ph.D. Thesis, Université d’Aix-Marseille, Marseille, France, 2012. [Google Scholar]
- Akiyama, G.; Matsuda, R.; Sato, H.; Hori, A.; Takata, M.; Kitagawa, S. Effect of functional groups in MIL-101 on water sorption behavior. Microporous Mesoporous Mater. 2012, 157, 89–93. [Google Scholar] [CrossRef]
- Cui, S.; Qin, M.; Marandi, A.; Steggles, V.; Wang, S.; Feng, X.; Nouar, F.; Serre, C. Metal-Organic Frameworks as advanced moisture sorbents for energy-efficient high temperature cooling. Sci. Rep. 2018, 8, 15284. [Google Scholar] [CrossRef] [PubMed]
- Khutia, A.; Rammelberg, H.U.; Schmidt, T.; Henninger, S.; Janiak, C. Water sorption cycle measurements on functionalized MIL-101Cr for heat transformation application. Chem. Mater. 2013, 25, 790–798. [Google Scholar] [CrossRef]
- Férey, G. Structural flexibility in crystallized matter: From history to applications. Dalton Trans. 2016, 45, 4073–4089. [Google Scholar] [CrossRef] [PubMed]
- Bourrelly, S.; Moulin, B.; Rivera, A.; Maurin, G.; Devautour-Vinot, S.; Serre, C.; Devic, T.; Horcajada, P.; Vimont, A.; Clet, G.; et al. Explanation of the adsorption of polar vapors in the highly flexible metal organic framework MIL-53(Cr). J. Am. Chem. Soc. 2010, 132, 9488–9498. [Google Scholar] [CrossRef] [PubMed]
- Boudjema, L.; Long, J.; Salles, F.; Larionova, J.; Guari, Y.; Trens, P. Switch in the hydrophobic-hydrophilic character of the Prussian Blue Analogues: An affinity control for smart gas sorption. Chem. A Eur. J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lee, J.S.; Wahiduzzaman, M.; Park, J.; Muschi, M.; Martineau-corcos, C.; Tissot, A.; Cho, K.H.; Marrot, J.; Shepard, W.; et al. A robust large-pore zirconium carboxylate metal-organic framework for energy-efficient water-sorption-driven refrigeration. Nat. Energy 2018, 1–10. [Google Scholar] [CrossRef]
- Pryor, T.L.; Close, D.J. The behaviour of adsorbent energy storage beds. Sol. Energy 1976, 18, 287–292. [Google Scholar] [CrossRef]
- Solomon, I.; Ribeiro, A.M.; Santos, J.C.; Loureiro, J.M.; Rodrigues, A.E.; Sandu, I.; Mamaliga, I. Adsorption equilibrium of water vapor on activated carbon and alumina and carbon and alumina impregnated with hygroscopic salt. Turk. J. Chem. 2013, 37, 358–365. [Google Scholar] [CrossRef]
- Shi, Y.F.; Liu, X.J.; Nie, H.J.; Liu, Y.S. The water adsorption isotherm models on activated alumina. Appl. Mech. Mater. 2012, 229–231, 100–104. [Google Scholar] [CrossRef]
- Li, G.; Xiao, P.; Webley, P. Binary adsorption equilibrium of carbon dioxide and water vapor on activated alumina. Langmuir 2009, 25, 10666–10675. [Google Scholar] [CrossRef] [PubMed]
- Marcussen, L. The kinetics of water adsorption on porous alumina. Chem. Eng. Sci. 1970, 25, 1487–1499. [Google Scholar] [CrossRef]
- Rouquerol, J.; Sing, K.S.W.; Llewellyn, P. Adsorption by metal oxides. In Adsorption by Powders and Porous Solids: Principles, Methodology and Applications; Academic Press: London, UK, 1999; pp. 287–354. ISBN 9780080970356. [Google Scholar] [CrossRef]
- Carruthers, J.D.; Payne, D.A.; Sing, K.S.W.; Stryker, L.J. Specific and nonspecific interactions in the adsorption of argon, nitrogen, and water vapor on oxides. J. Colloid Interface Sci. 1971, 36, 205–216. [Google Scholar] [CrossRef]
- Pryor, T.L.; Close, D.J. Measurements of the behaviour of adsorbent energy storage beds. Sol. Energy 1978, 20, 151–155. [Google Scholar] [CrossRef]
- Desai, R.; Hussain, M.; Ruthven, D.M. Adsorption of water vapour on activated alumina. I-equilibrium behaviour. Can. J. Chem. Eng. 1992, 70, 699–706. [Google Scholar] [CrossRef]
- Moore, J.D.; Serbezov, A. Correlation of adsorption equilibrium data for water vapor on F-200 activated alumina. Adsorption 2005, 11, 65–75. [Google Scholar] [CrossRef]
- Ferreira, D.; Magalhães, R.; Taveira, P.; Mendes, A. Effective adsorption equilibrium isotherms and breakthroughs of water vapor and carbon dioxide on different adsorbents. Ind. Eng. Chem. Res. 2011, 50, 10201–10210. [Google Scholar] [CrossRef]
- Sadek, O.M.; Mekhemer, W.K. Ca-montmorillonite clay as thermal energy storage material. Thermochim. Acta 2000, 363, 47–54. [Google Scholar] [CrossRef]
- Konta, J. Clay and man: Clay raw materials in the service of man. Appl. Clay Sci. 1995, 10, 275–335. [Google Scholar] [CrossRef]
- Salles, F.; Douillard, J.M.; Bildstein, O.; Gaudin, C.; Prelot, B.; Zajac, J.; Van Damme, H. Driving force for the hydration of the swelling clays: Case of montmorillonites saturated with alkaline-earth cations. J. Colloid Interface Sci. 2013, 395, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Musyoka, N.M.; Langmi, H.W. Clay and clay-supported materials for energy storage applications. In Bentonite; Mishra, A.K., Ed.; Nova Science Publishers, Inc.: Hauppaug, NY, USA, 2015; ISBN 9781634821421. [Google Scholar]
- Hensen, E.J.M.; Smit, B. Why clays swell. J. Phys. Chem. B 2002, 106, 12664–12667. [Google Scholar] [CrossRef]
- Hendricks, S.B.; Nelson, R.A.; Alexander, L.T. Hydration mechanism of the clay mineral Montmorillonite saturated with various cations. J. Am. Chem. Soc. 1940, 62, 1457–1464. [Google Scholar] [CrossRef]
- Sadek, O.M.; Mekhemer, W.K. Na-montmorillonite clay as thermal energy storage material. Thermochim. Acta 2001, 370, 57–63. [Google Scholar] [CrossRef]
- Caballero, E.; Jiménez de Cisneros, C. Hydration properties of bentonites from Cabo de Gata (SE, Spain). Isotopic study (18O/16O;2H/H) of the hydration water. Chem. Erde 2011, 71, 389–395. [Google Scholar] [CrossRef]
- Desai, R.R.; Erwin Desa, J.A.; Aswal, V.K. Hydration studies of Bentonite clay. AIP Conf. Proc. 2012, 1447, 197–198. [Google Scholar] [CrossRef]
- Chiou, C.T.; Rutherford, D.W. Effects of exchanged cation and layer charge on the sorption of water and EGME vapors on montmorillonite clays. Clays Clay Miner. 1997, 45, 867–880. [Google Scholar] [CrossRef]
- Tajeddine, L.; Gailhanou, H.; Blanc, P.; Lassin, A.; Gaboreau, S.; Vieillard, P. Hydration-dehydration behavior and thermodynamics of MX-80 montmorillonite studied using thermal analysis. Thermochim. Acta 2015, 604, 83–93. [Google Scholar] [CrossRef]
- Salles, F.; Douillard, J.-M.; Denoyel, R.; Bildstein, O.; Jullien, M.; Beurroies, I.; Van Damme, H. Hydration sequence of swelling clays: Evolutions of specific surface area and hydration energy. J. Colloid Interface Sci. 2009, 333, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, N.; Mercado, A.; Bolzone, C. Reversibility studies of clay hydration degree in its natural and composite condition. Procedia Mater. Sci. 2015, 9, 135–141. [Google Scholar] [CrossRef]
- Rouquerol, J.; Sing, K.S.W.; Llewellyn, P. Adsorption by active carbons. In Adsorption by Powders and Porous Solids: Principles, Methodology and Applications; Academic Press: London, UK, 1999; pp. 237–280. [Google Scholar]
- Salame, I.I.; Bandosz, T.J. Experimental study of water adsorption on activated carbons. Langmuir 1999, 15, 587–593. [Google Scholar] [CrossRef]
- Huber, L.; Ruch, P.; Hauert, R.; Saucke, G.; Matam, S.K.; Michel, B.; Koebel, M.M. Monolithic nitrogen-doped carbon as a water sorbent for high-performance adsorption cooling. RSC Adv. 2016, 6, 25267–25278. [Google Scholar] [CrossRef]
- Critoph, R.E. Activated carbon adsorption cycles for refrigeration and heat pumping. Carbon 1989, 27, 63–70. [Google Scholar] [CrossRef]
- De Lange, M.F.; Van Velzen, B.L.; Ottevanger, C.P.; Verouden, K.J.F.M.; Lin, L.C.; Vlugt, T.J.H.; Gascon, J.; Kapteijn, F. Metal-organic frameworks in adsorption-driven heat pumps: The potential of alcohols as working fluids. Langmuir 2015, 31, 12783–12796. [Google Scholar] [CrossRef] [PubMed]
- Reinsch, H.; Van Der Veen, M.A.; Gil, B.; Marszalek, B.; Verbiest, T.; De Vos, D.; Stock, N. Structures, sorption characteristics, and nonlinear optical properties of a new series of highly stable aluminum MOFs. Chem. Mater. 2013, 25, 17–26. [Google Scholar] [CrossRef]
- Hongois, S. Stockage de Chaleur Inter-Saisonnier par voie Thermochimique pour le Chauffage Solaire de la Maison Individuelle. Ph.D. Thesis, Institut National des Sciences Appliquées de Lyon, Villeurbanne, France, 2011. [Google Scholar]
- Agence de l’Environnement et de la Maîtrise de l’Energie (ADEME). L’effacement de consommation électrique en France: Rapport Final; E-CUBE Strategy Consultants, CEREN: Anger, France, 2017. [Google Scholar]

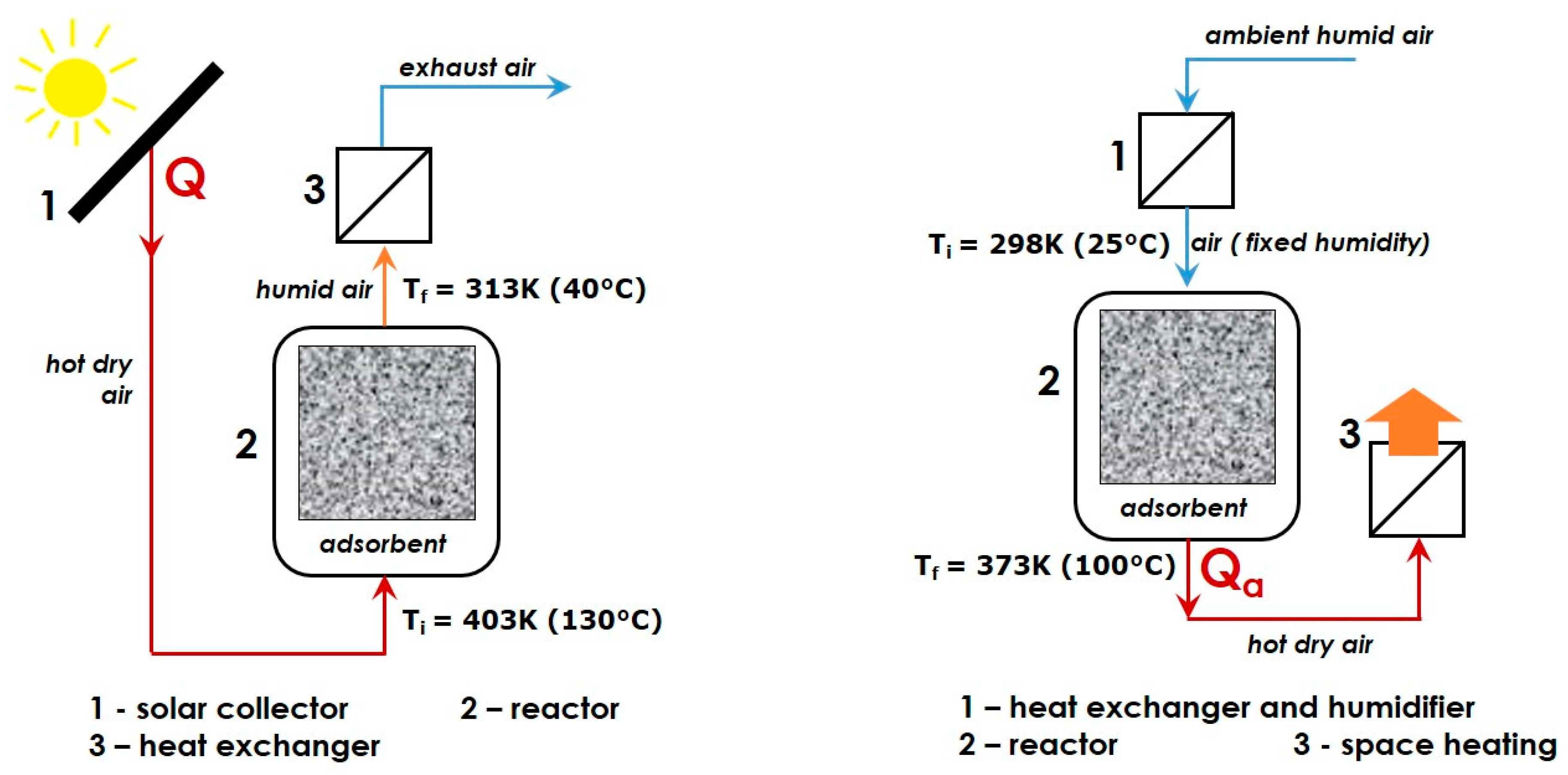



{kind=link}
{kind=link}
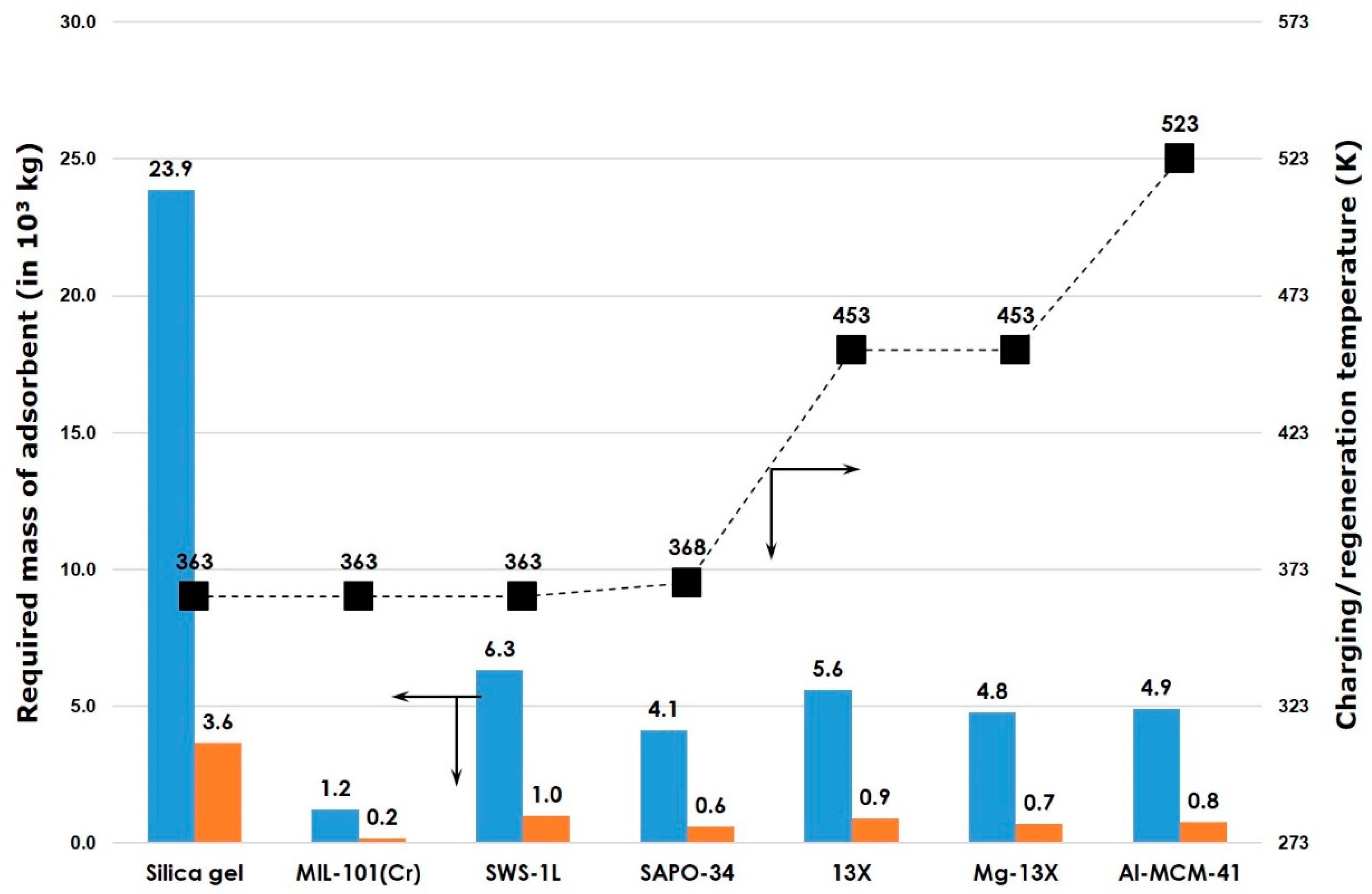
{kind=link}
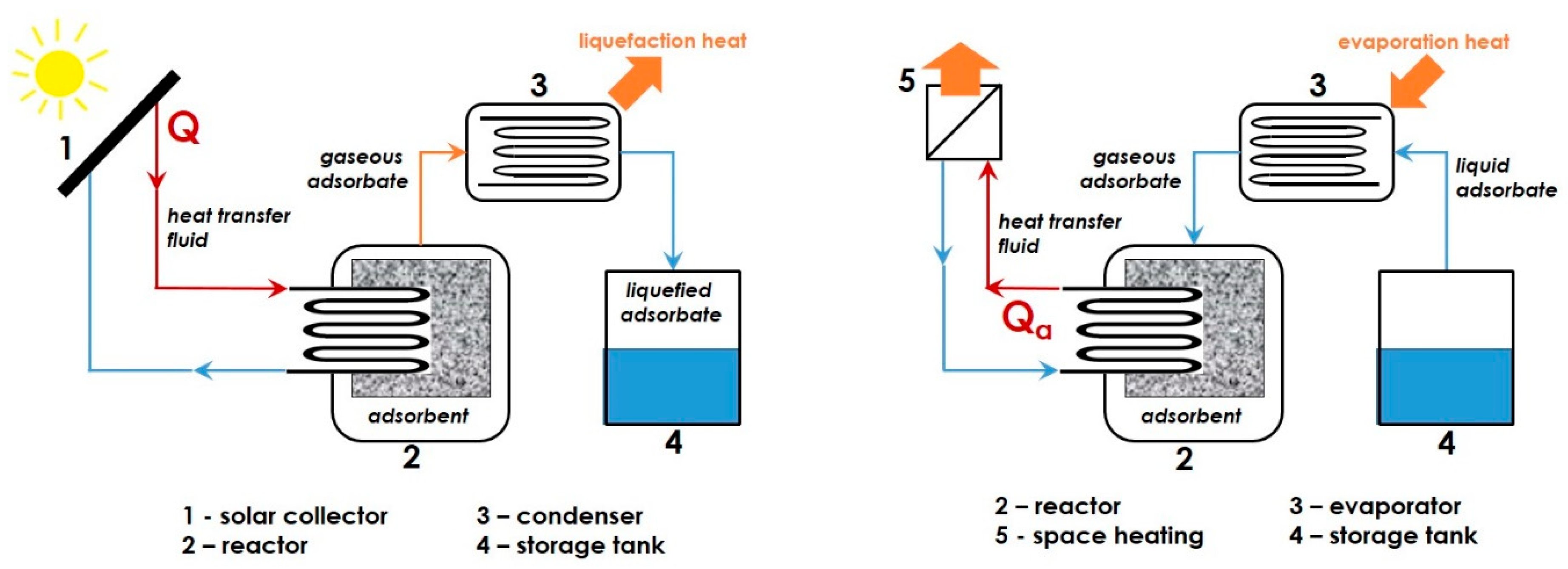
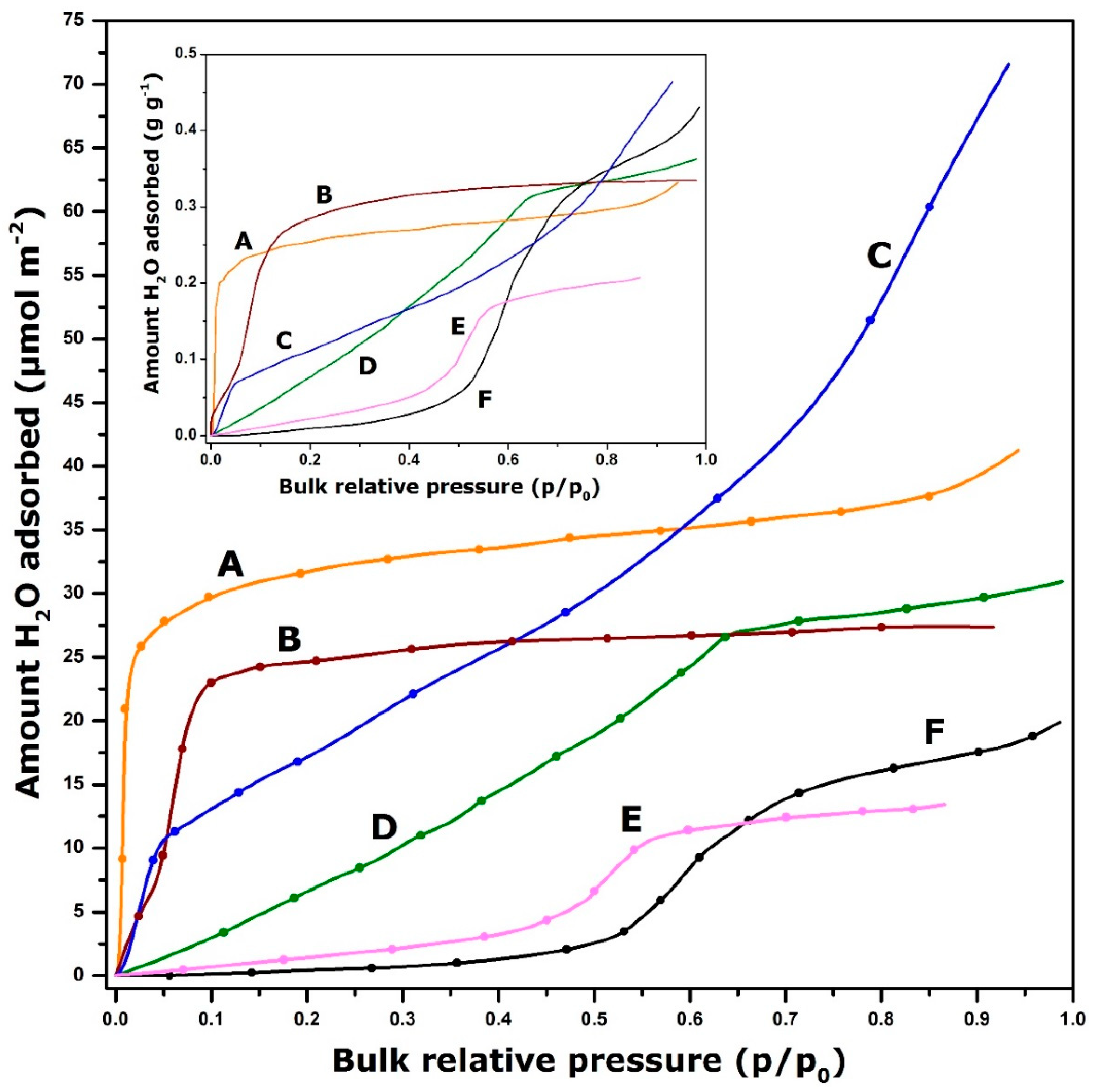{kind=link}
{kind=link}
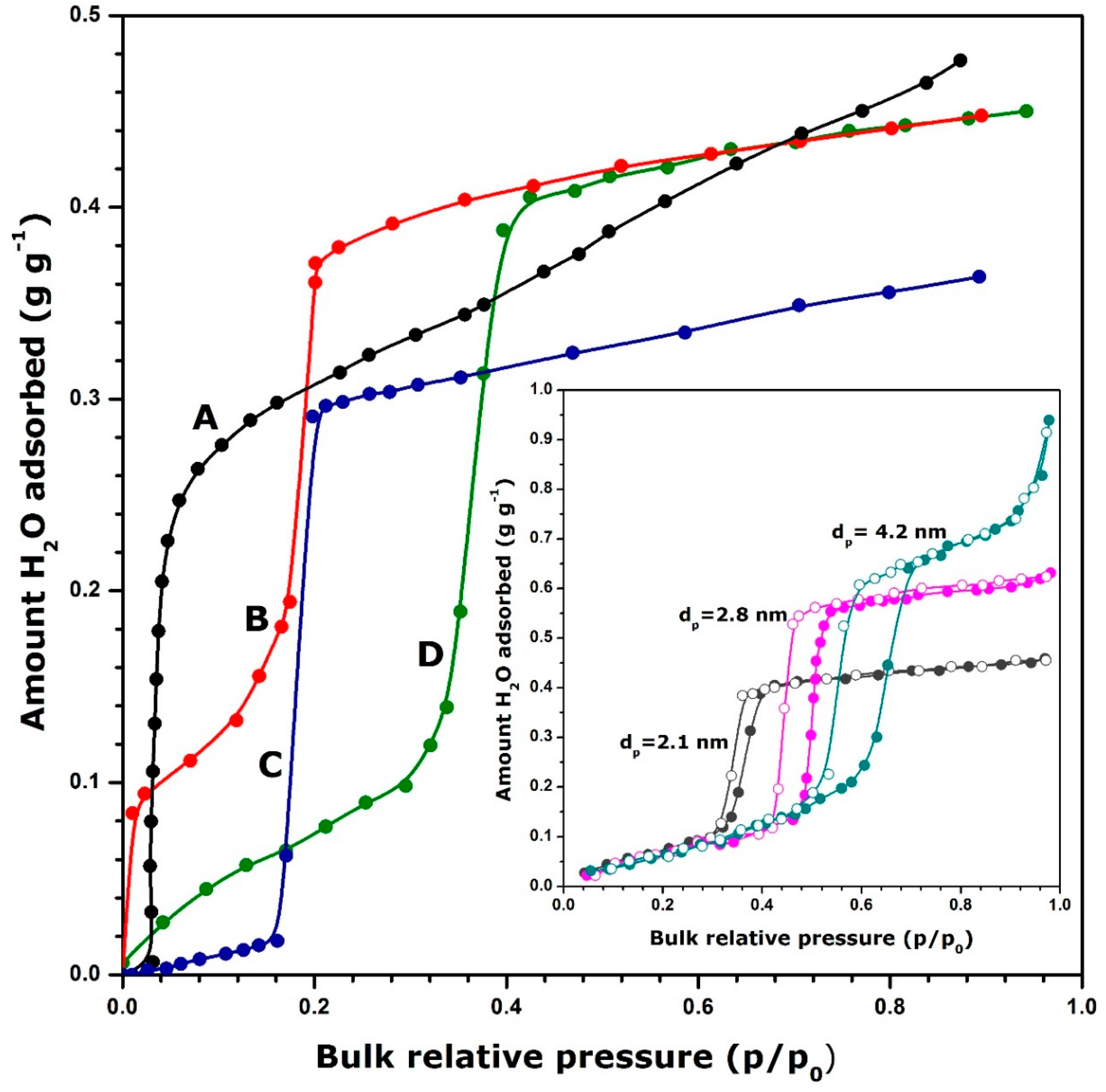{kind=link}
{kind=link}
| Physical Property or Characteristics | Heat Storage Principle and Working Materials | |||||
|---|---|---|---|---|---|---|
| Sensible Heat Storage | Latent Heat Storage | Adsorption Heat Storage | ||||
| Water | Rock | Paraffin Wax | CaCl2·6H2O | Silica Gel/Water | Zeolite/Water | |
| Latent heat of fusion (kJ kg−1) | - | - | 174.4 | 160 | - | - |
| Specific heat capacity (kJ kg−1 K−1) | 4.18 | 0.9 | - | - | 1.13 | 1.07 |
| Heat of adsorption (kJ kg−1 solid) | - | - | - | - | 1380 | 1107 |
| Density (kg m−3) | 1000 | 2240 | 1802 | 1830 | 600 | 650 |
| Volume of material for storing 1GJ (m3) | 4.8 | 9.9 | 3.2 | 3.4 | 1.2 | 1.4 |
| Heat storage density (MJ m−3 solid) | 209 | 100 | 310 | 292 | 767 | 713 |
| Advantages | (1) simplicity of design and use (2) low implementation costs | (1) lower heat loss | (1) higher storage density (2) smaller volume of working materials (3) very low heat loss (4) lower charging and discharging temperatures | |||
| Weaknesses | (1) low energy density (2) high heat loss (self-discharging effects possible) (3) space limitations (large volume of working materials) | (1) low thermal stability (risk of chemical decomposition at high temperatures) (2) costly investment in thermal insulation | (1) complexity of design and use (2) higher investment costs (3) heat and mass transfer limitations | |||
| Substance | H2O | NH3 | CO2 | C2H5OH | CH3OH | N2 | O2 | He | Air (Dry) |
|---|---|---|---|---|---|---|---|---|---|
| Critical temperature (K) | 647 | 405.3 | 304 | 513.9 | 512.4 | 126 | 154.4 | 2 | 132.5 |
| Boiling point at 105 Pa (K) | 373 | 239.7 | 194.5 | 351.3 | 337.6 | 77 | 90 | 5 | - |
| Enthalpy of vaporization (kJ mol−1) | 40.65 | 23.4 | 15.3 | 38.56 | 35.21 | 5.57 | 6.82 | 0.08 | - |
| Saturated vapour pressure at 293 K (kPa) | 2.34 | 857.1 | 5.7∙103 | 5.8 | 12.8 | - | - | - | - |
| Isobaric heat capacity in the gas phase at 300 K and 105 Pa (J mol−1 K−1) | 33.6 | 37.0 | 36.94 | 74 | 44.1 | 29.2 | 29.4 | 20.8 | 9.15 |
| Thermal conductivity in the gas phase at 300 K and 105 Pa (mW m−1 K−1) | 18.7 | 24.4 | 16.8 | 14.4 | 26.2400 K | 25.8 | 26.3 | 156.7 | 26.2 |
| Kinetic diameter of molecules (nm) | 0.265 | 0.260 | 0.330 | 0.450 | 0.360 | 0.364 | 0.346 | 0.260 | - |
| Dipole moment for molecules in the gas phase (in debyes) | 1.85 | 1.47 | 0 | 1.69 | 1.7 | 0 | 0 | - | - |
| Proton affinity (kJ mol−1) | 697 | 853.5 | 548 | 788 | 761 | 495 | 422 | 178 | - |
| Absolute hardness parameter (eV) | 9.5 | 8.2 | 8.8 | 8.0 | 5.8 | 8.9 | 5.9 | - | - |
| Adsorbent Group | Working Couple | Textural Properties of Adsorbent | Adsorption Performance | Storage Performance Tests | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| Porosity Type | Specific Surface Area (m2 g−1) | Capacity (g g−1) | Heat (kJ mol−1) | Target Storage Density (kJ kg−1) | Operating Conditions | Information on Stability | |||
| Amorphous SiO2 | silica aerogel & H2O | micro-/meso- | 783 | 1.35 | - | - | charging: vacuum, 343 K, 24 h discharging: 293 K | Adsorption greatly decreasing after the first cycle (raw sample: 42%; calcined one: 26%), then remained stable over next 25 cycles | [111] |
| silica gel LE32 & H2O | macro- | - | 0.6 | 25 | 86 | charging: 363 K discharging: 313 K | - | [23] | |
| Amorphous Al2O3 | alumina aerogel & H2O | micro-/meso- | 453 | 1.25 | - | - | charging: 343 K discharging: 293 K | adsorption slowly decreasing within 10 cycles, then remained stable | [111] |
| 30% alumina + 70% silica aerogel & H2O | micro-/meso- | 577 | 1.15 | - | - | stable over 25 cycles | |||
| Ordered SiO2 | MCM41 & H2O | meso- | 1137 | 0.04 at p/p0 = 0.3 | 47–54 | - | charging: 523 K, 3 h discharging: 293 K | - | [92] |
| SBA15 & H2O | meso- | 554 | 0.02 at p/p0 = 0.3 | - | - | - | |||
| Salt-oxide hybrids | CaCl2-slica gel KSK (SWS-1L) & H2O | meso- | 230 | 0.7 | 43.9–63.1 | 475 | charging: 353–423 K | - | [112,113,114] |
| Ca(NO3)2-silica gel KSK (SWS-8L) & H2O | meso- | 60 | 0.23 | 47–52 | - | charging: 348–353 K | - | [115] | |
| Al-MCM41 & H2O | meso- | 941–1021 | 0.17 at p/p0 = 0.3 | 65 | 450–612 | charging: 253 K discharging: 293 K | - | [92] | |
| Al-SBA15 & H2O | meso- | 541–550 | 0.09 at p/p0 = 0.3 | 67 | 227–335 | - | |||
| Zeolites | 4A & H2O | micro- | - | 0.22 | 72 | 670 | charging: 463 K discharging: 298 K | - | [116] |
| 13X & H2O | micro- | - | 0.34 | 51.3 | 536 | charging: 723 K discharging: 300 K | - | [69] | |
| MgNa-X & H2O | micro- | - | 0.212 | 53 | 630 | - | |||
| MgSO4-13X & H2O | micro- | - | 0.15 | 81.6 | 648 | charging: 423 K | adsorption unchanged over 3 cycles | [117] | |
| Zeotype materials | AlPO-18 & H2O | meso- | - | 0.34 | 55 | 875 | charging: 268 K discharging: 313 K | - | [87] |
| SAPO-34 & H2O | meso- | - | 0.3 | 56 | 730 | - | |||
| MOF materials | MIL-101 (Cr) & H2O | meso- | 4150 | 1.6 | 44.5 | - | charging: 423 K discharging: 303 K | stable over 10 cycles | [118] |
| MIL-100 (Fe) & H2O | meso- | 2300 | 0.87 | 49.5 | - | stable over 10 cycles | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.; Salles, F.; Zajac, J. A Critical Review of Solid Materials for Low-Temperature Thermochemical Storage of Solar Energy Based on Solid-Vapour Adsorption in View of Space Heating Uses. Molecules 2019, 24, 945. https://doi.org/10.3390/molecules24050945
Wu H, Salles F, Zajac J. A Critical Review of Solid Materials for Low-Temperature Thermochemical Storage of Solar Energy Based on Solid-Vapour Adsorption in View of Space Heating Uses. Molecules. 2019; 24(5):945. https://doi.org/10.3390/molecules24050945
Chicago/Turabian StyleWu, Hao, Fabrice Salles, and Jerzy Zajac. 2019. "A Critical Review of Solid Materials for Low-Temperature Thermochemical Storage of Solar Energy Based on Solid-Vapour Adsorption in View of Space Heating Uses" Molecules 24, no. 5: 945. https://doi.org/10.3390/molecules24050945