Supramolecular Complexes of Graphene Oxide with Porphyrins: An Interplay between Electronic and Magnetic Properties

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
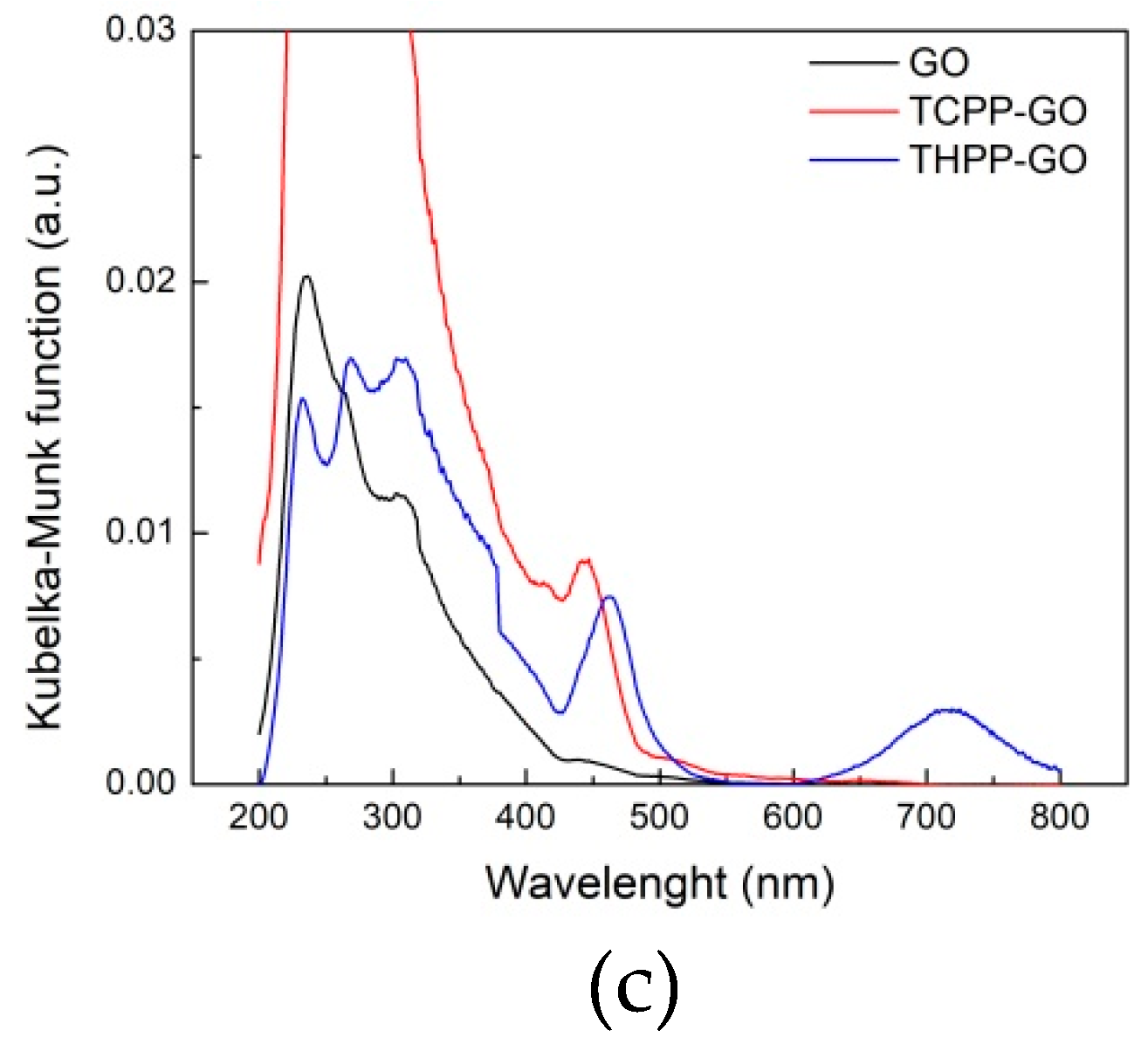
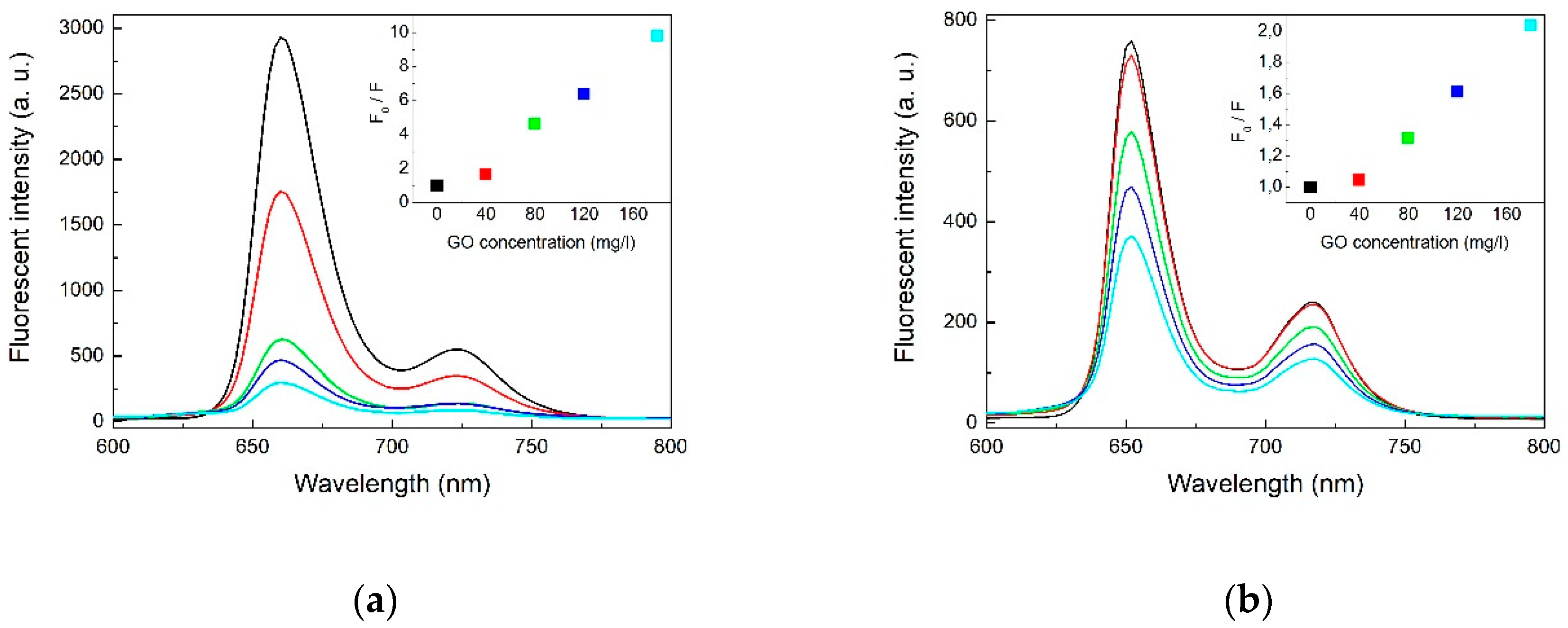
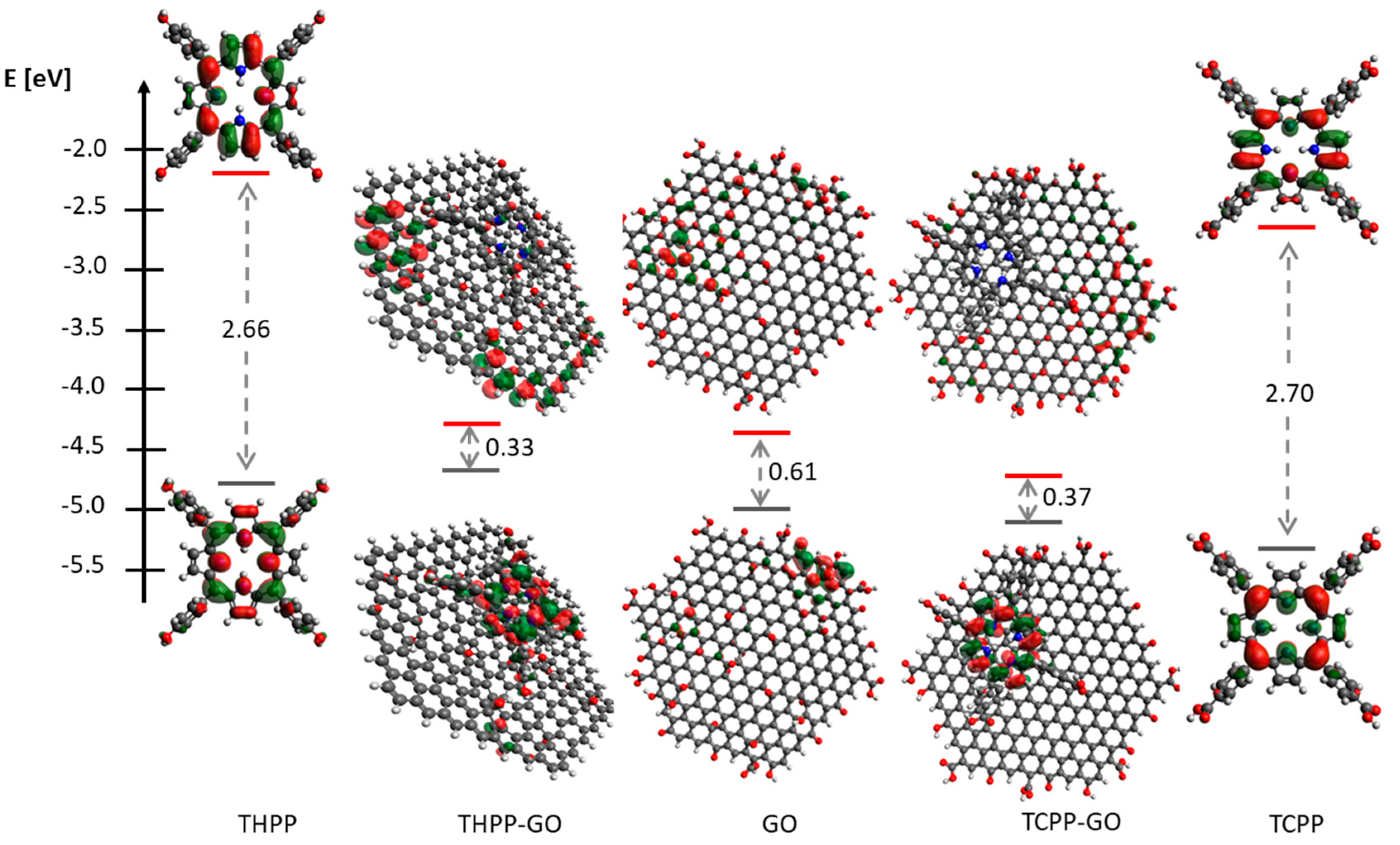
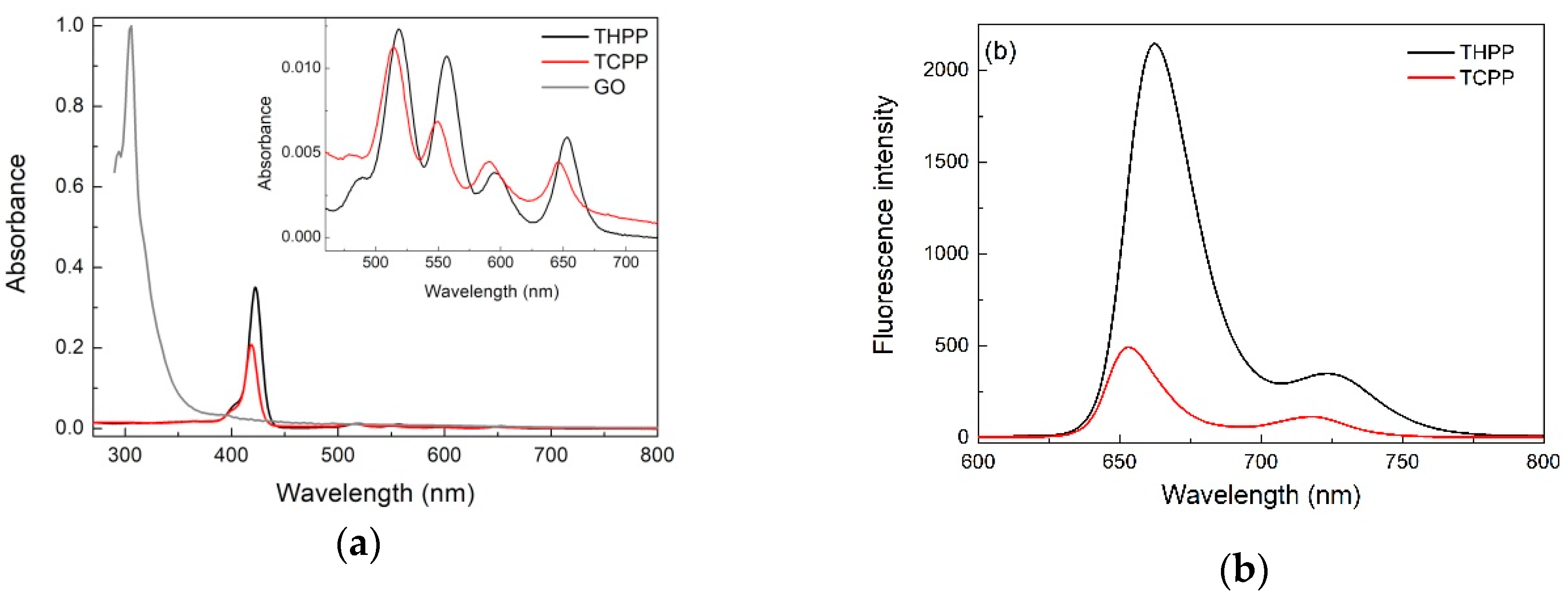
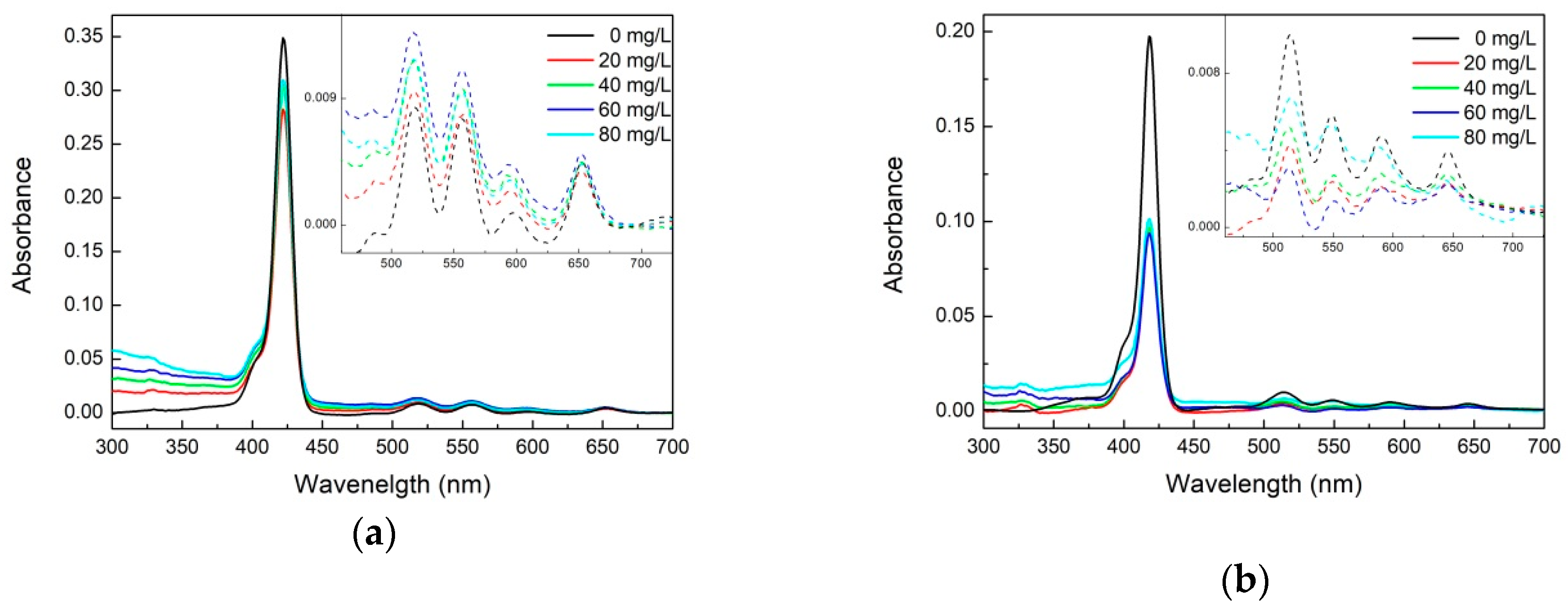
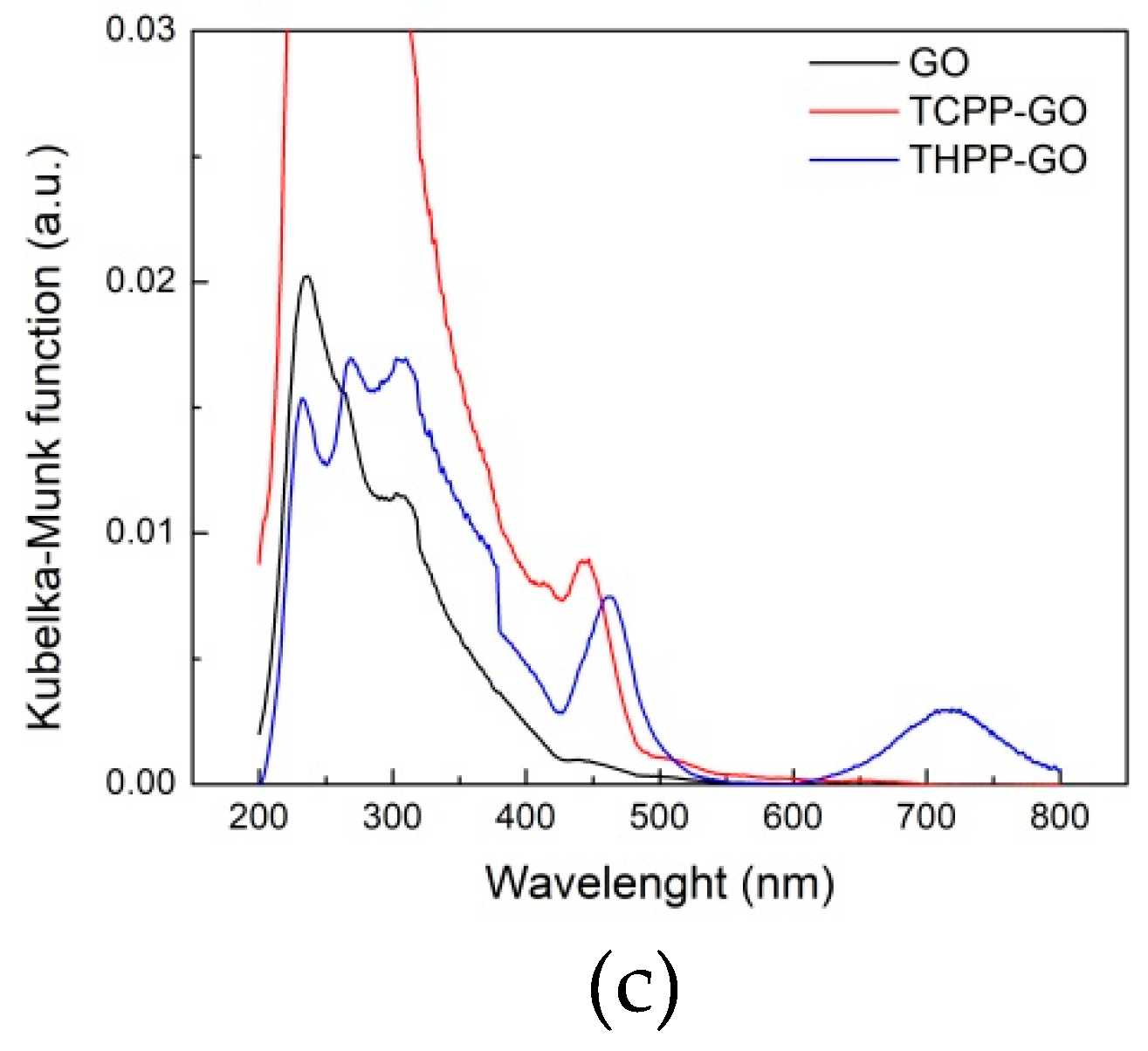
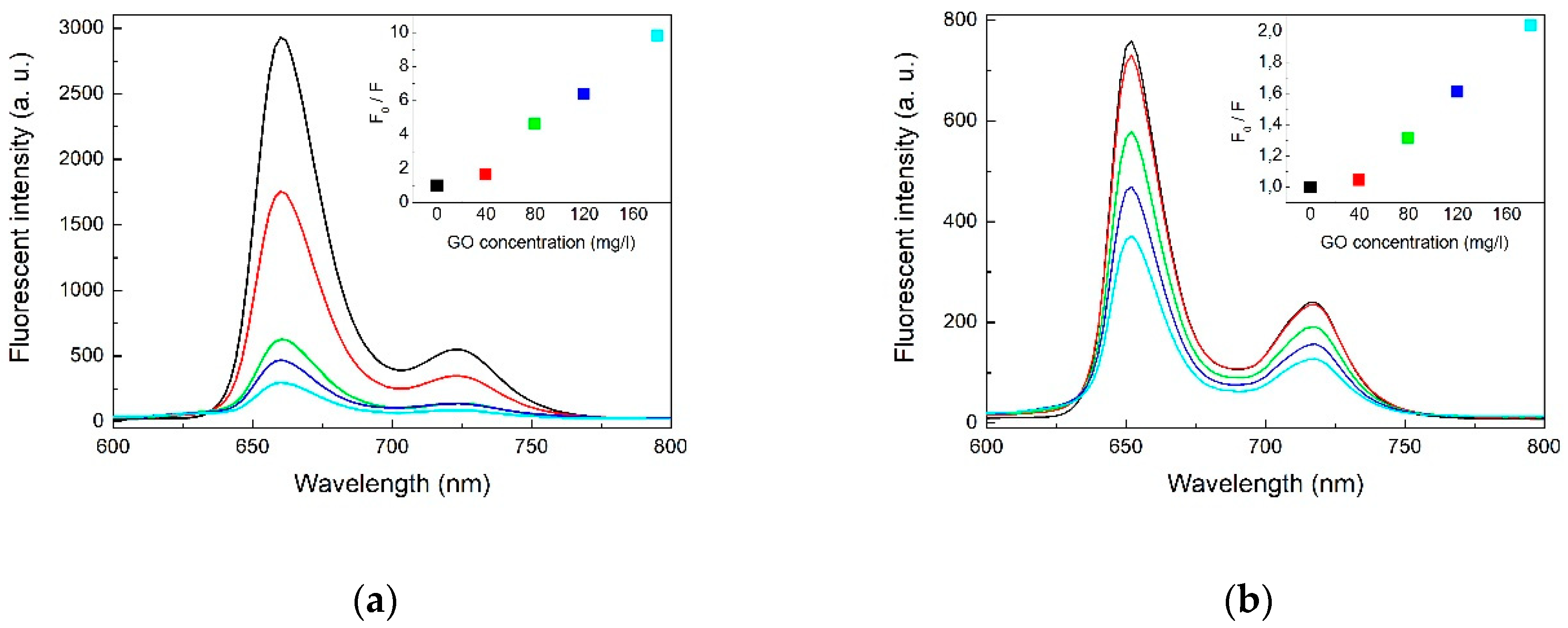
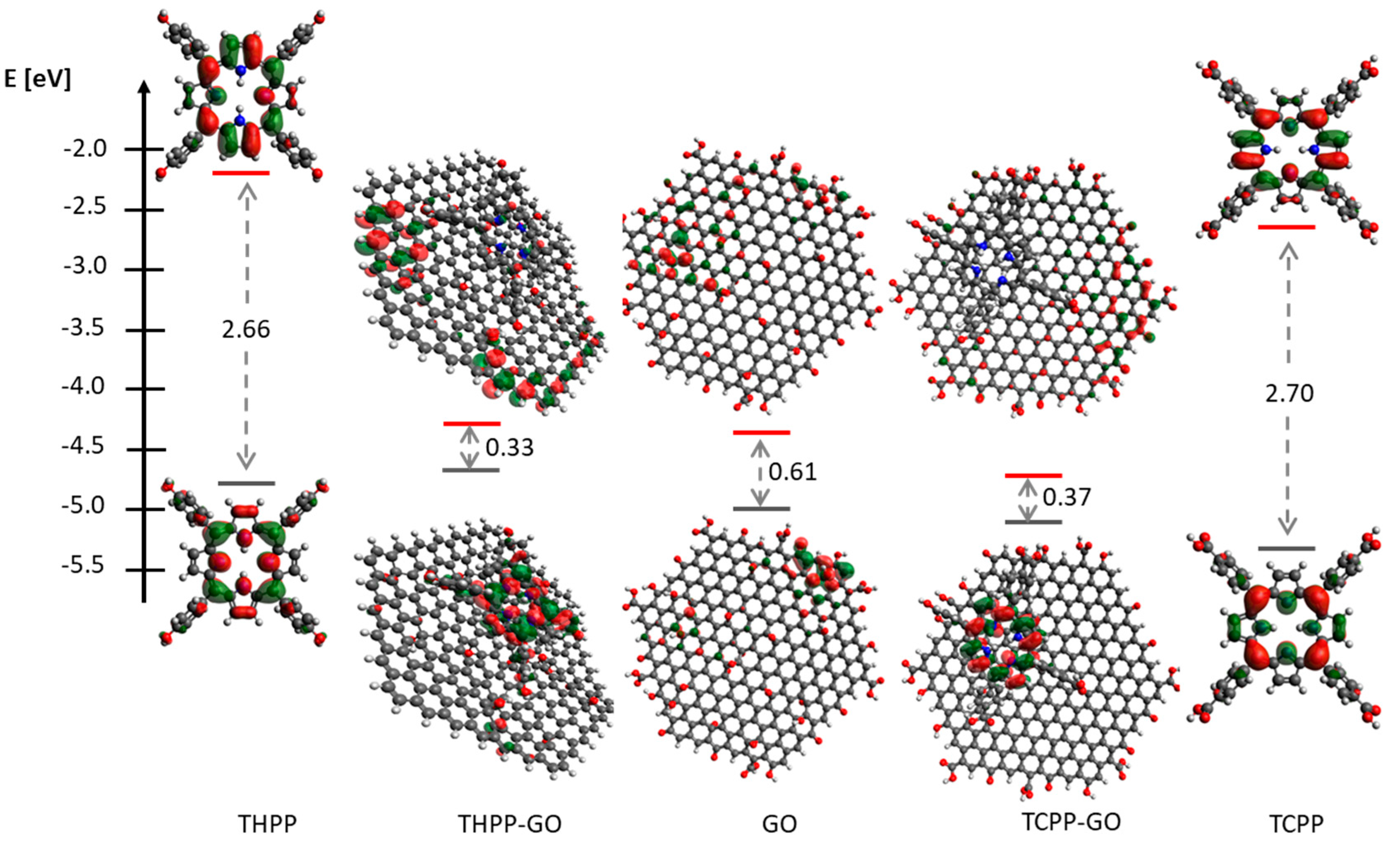
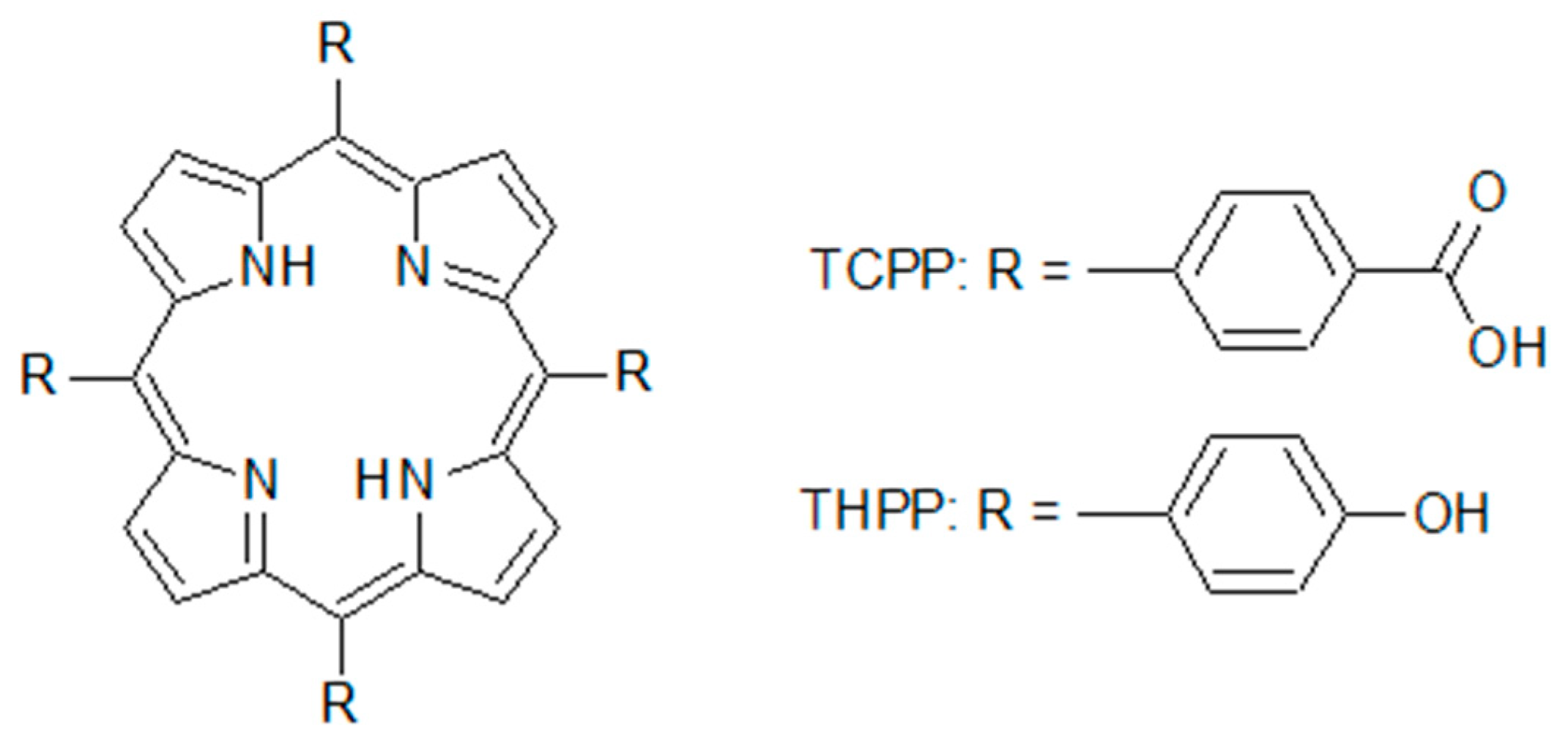
2.1. Optical Spectroscopy
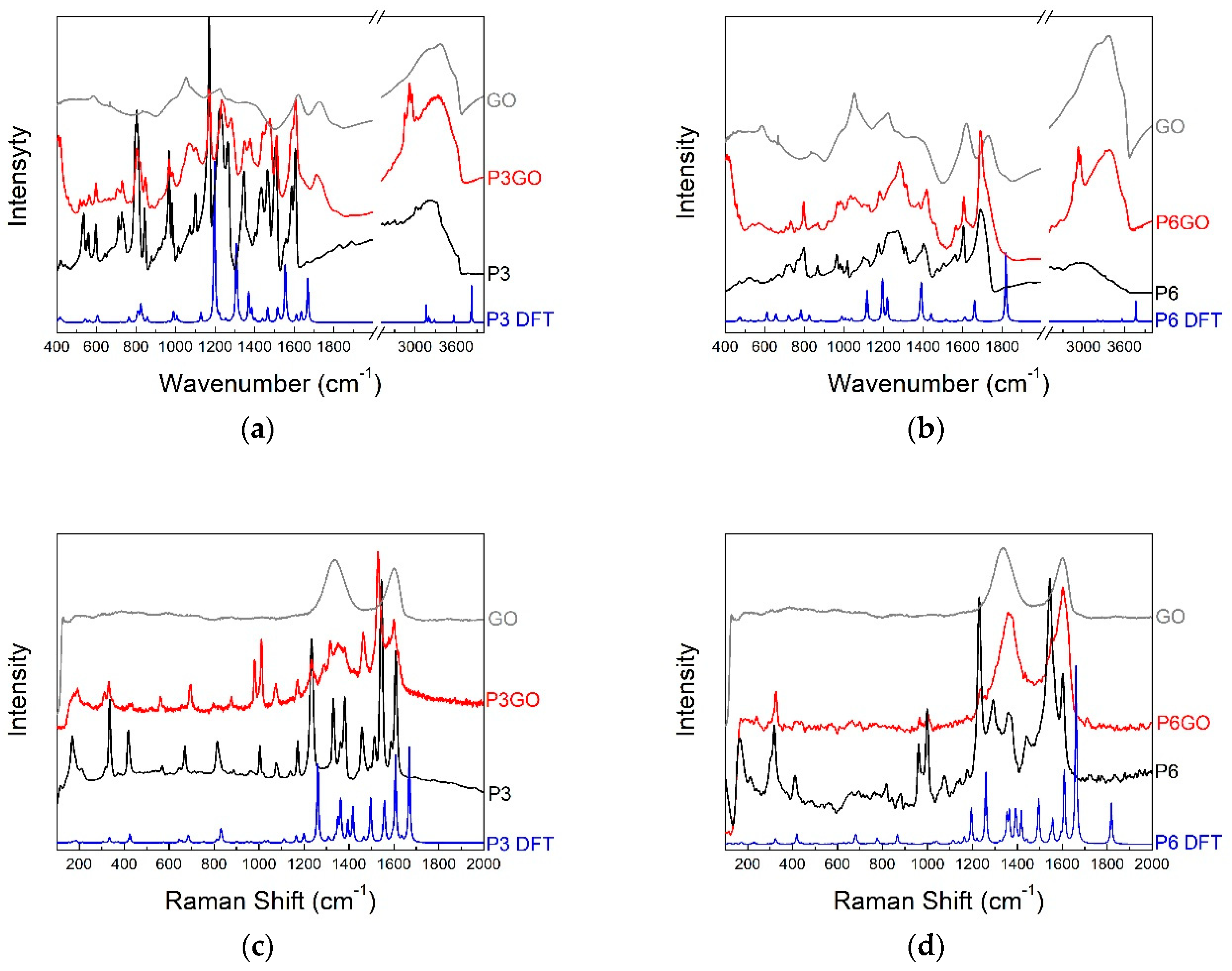
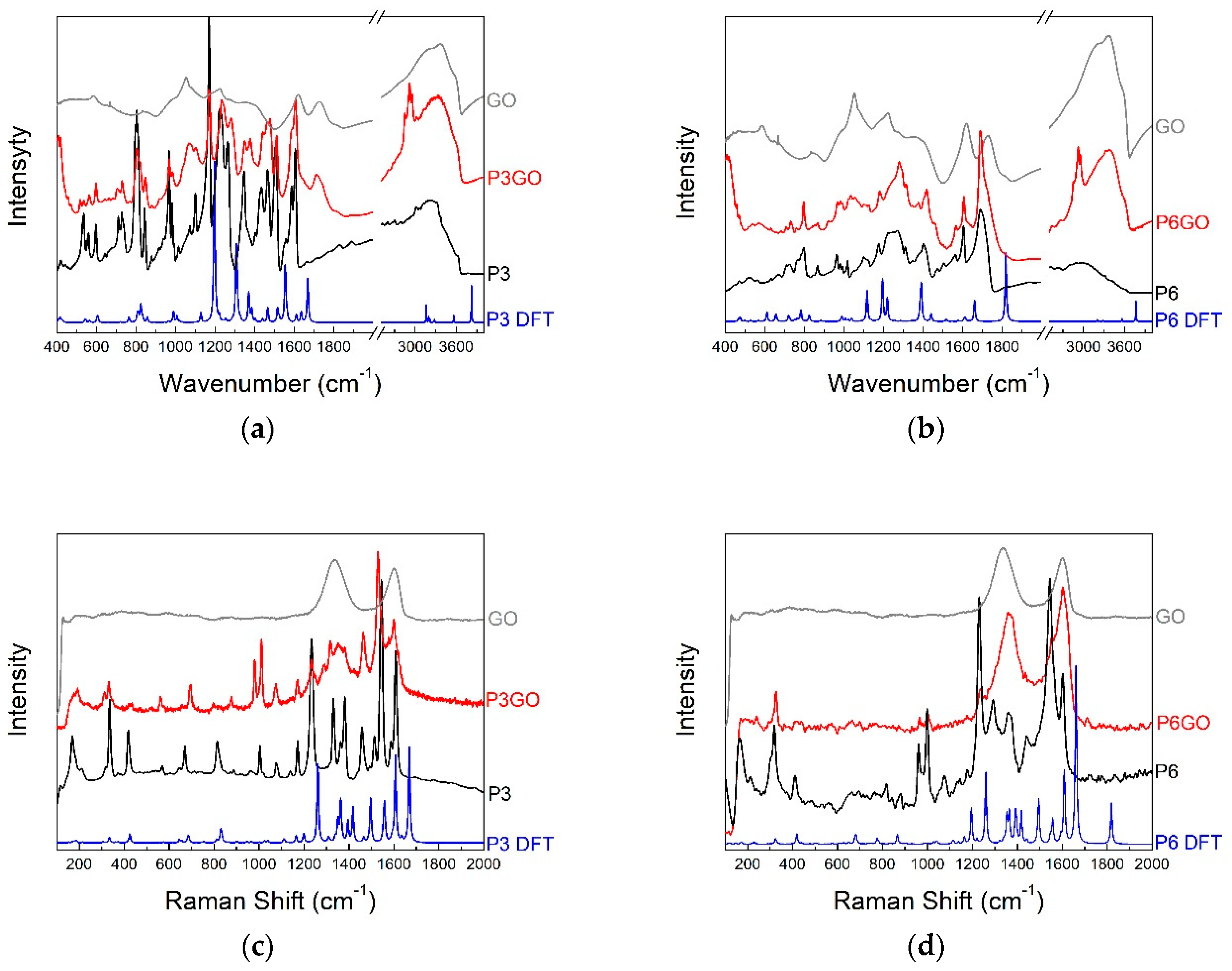
2.2. Vibrational Spectroscopy
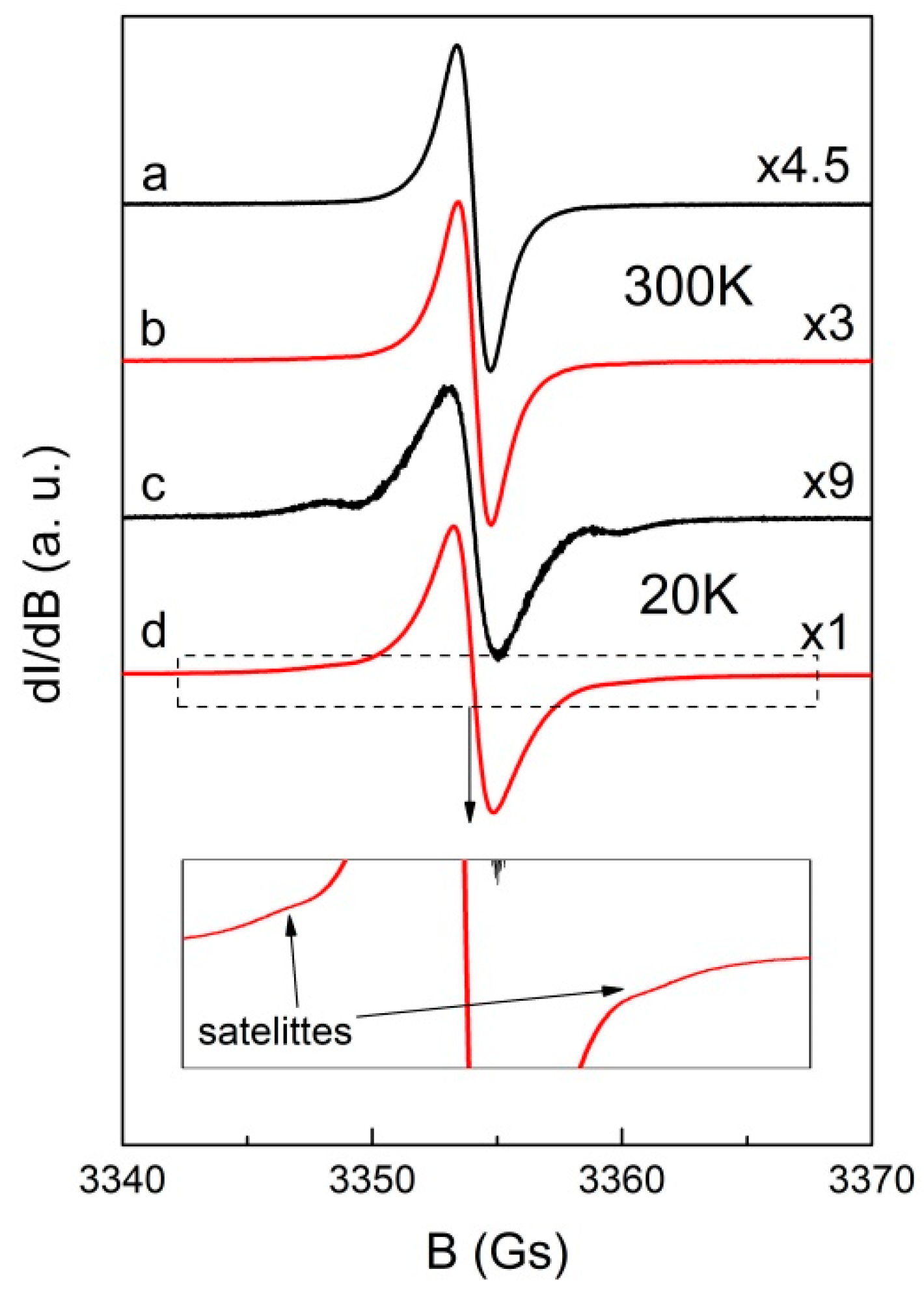
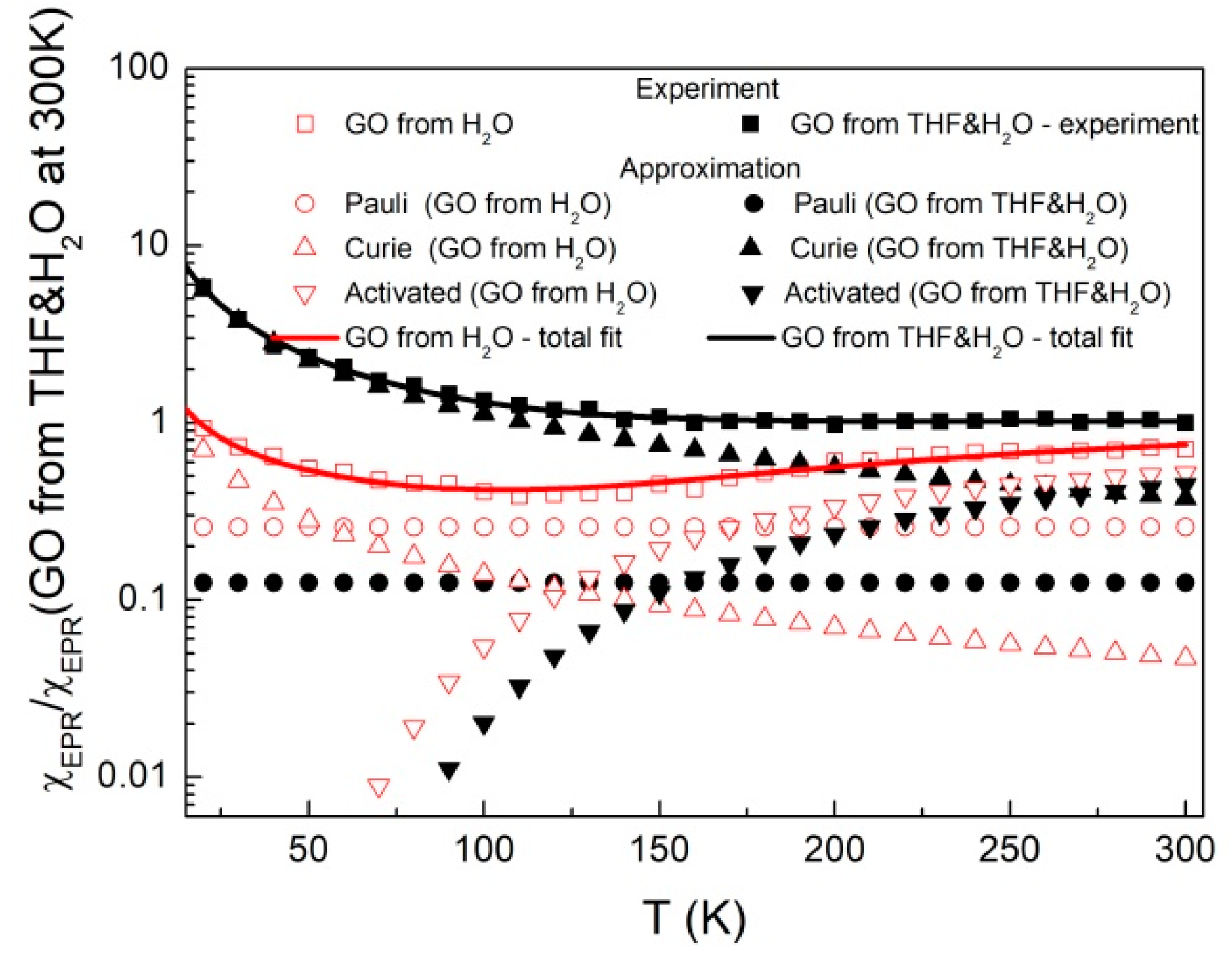
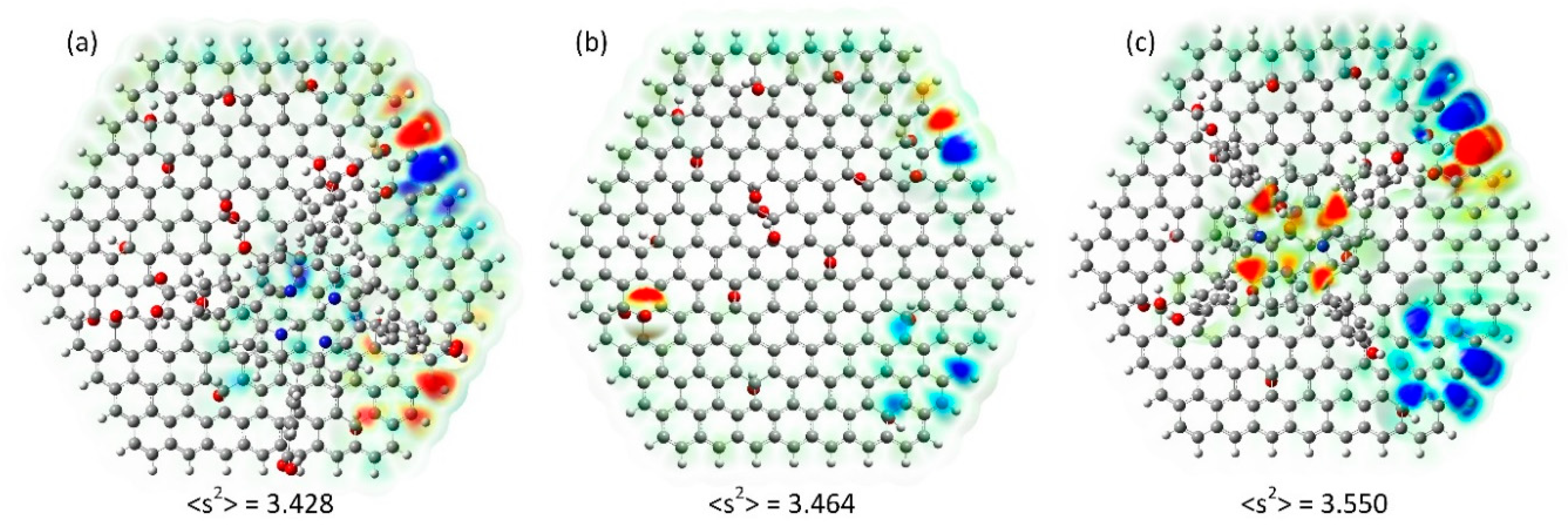
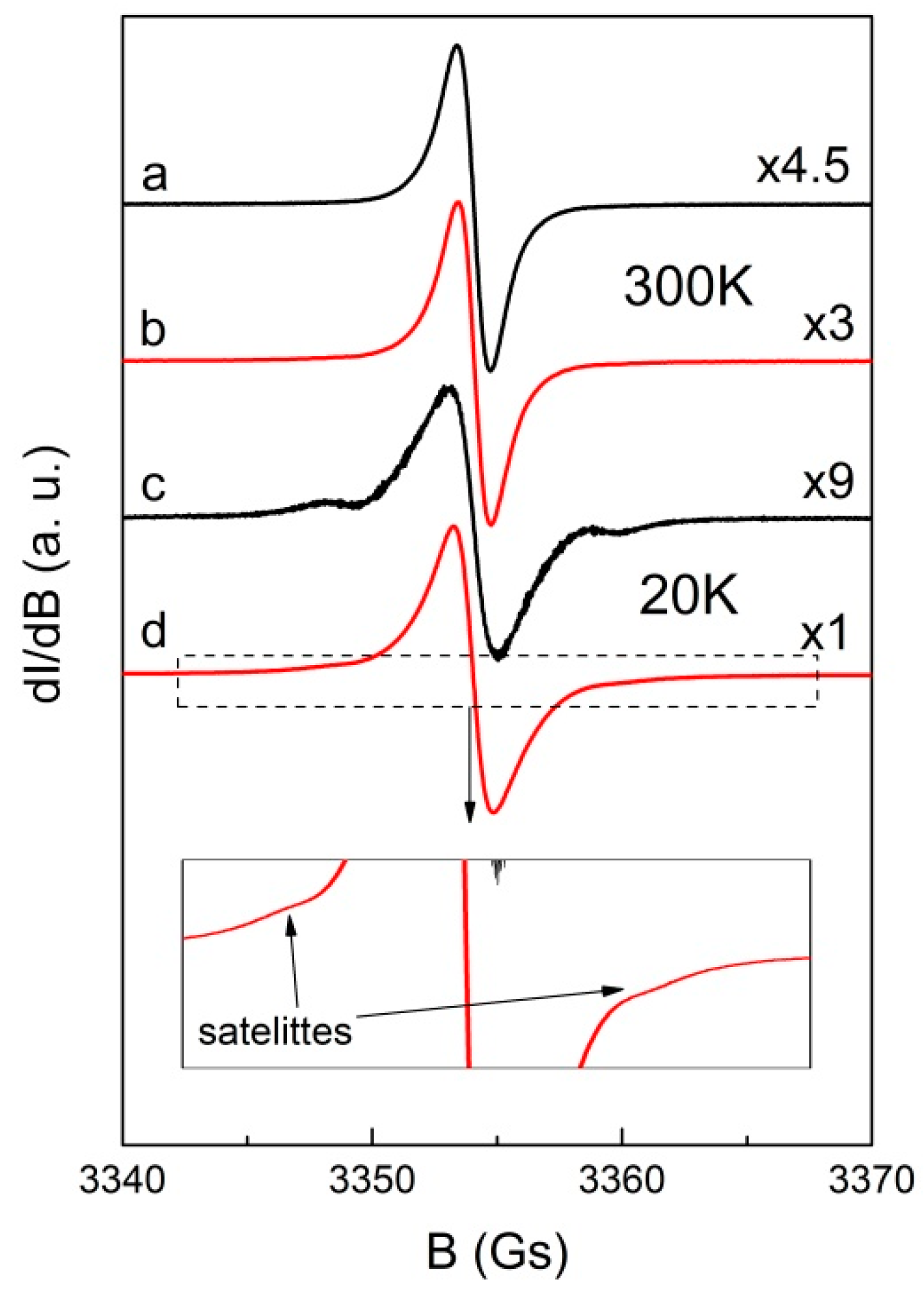
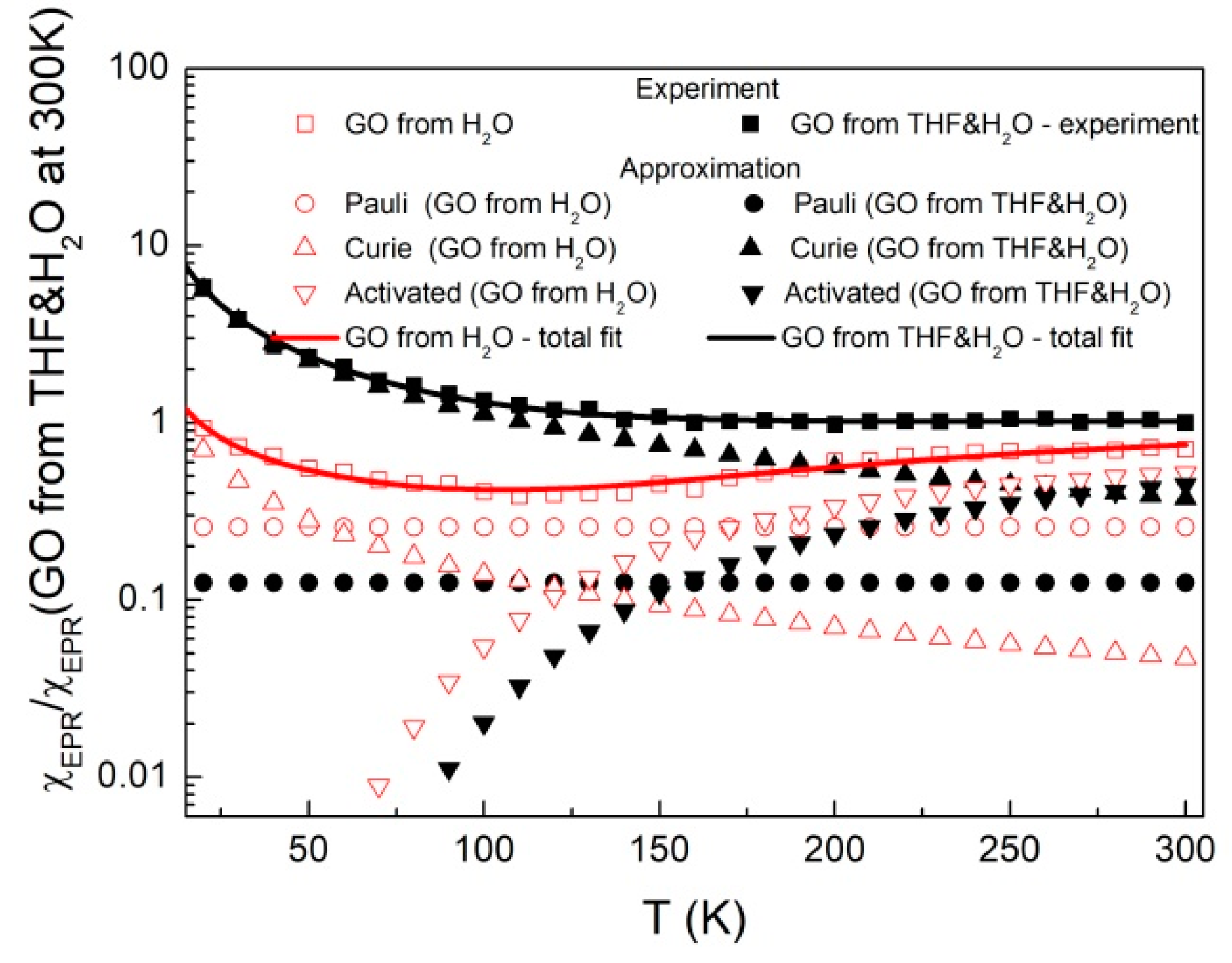
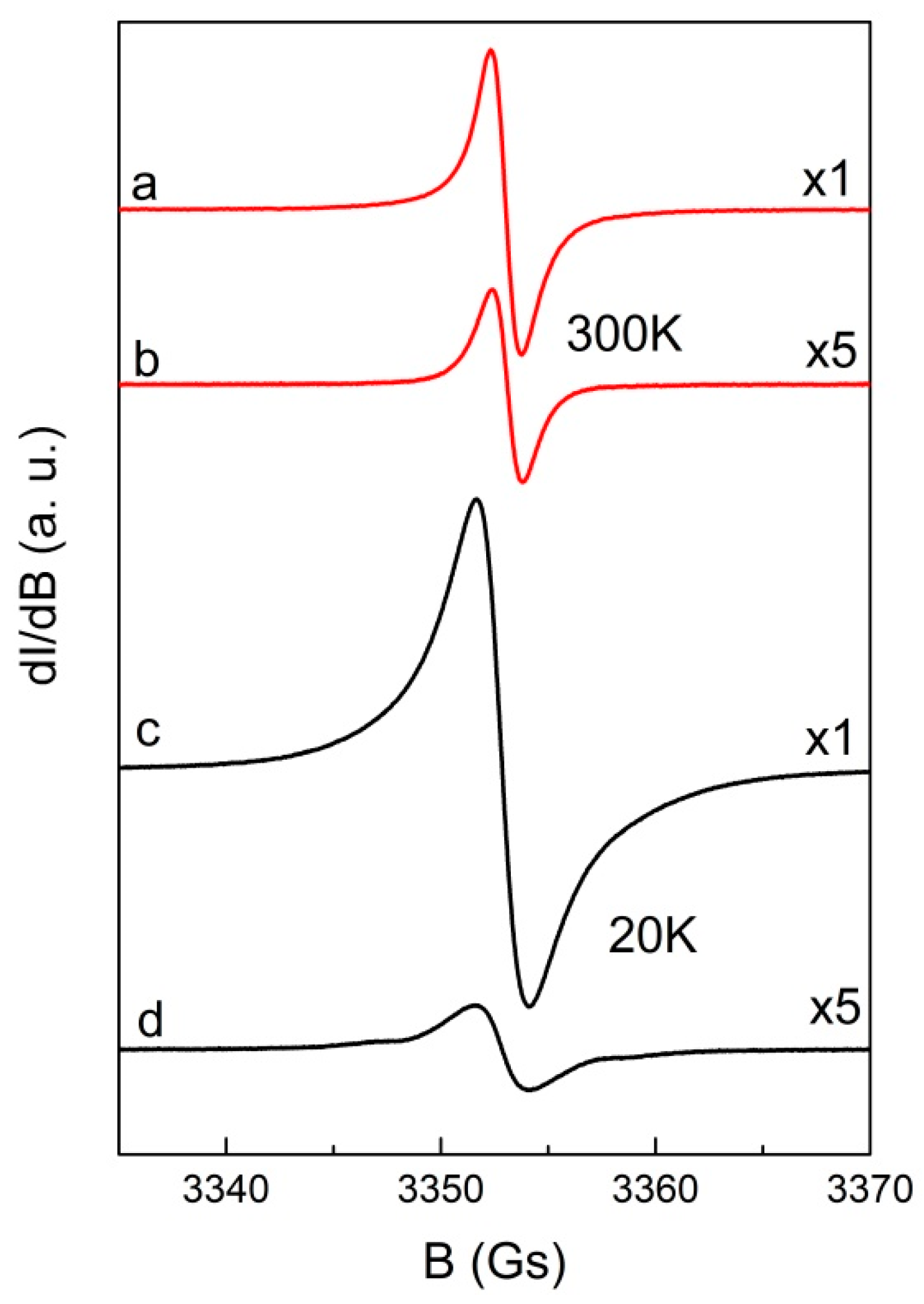
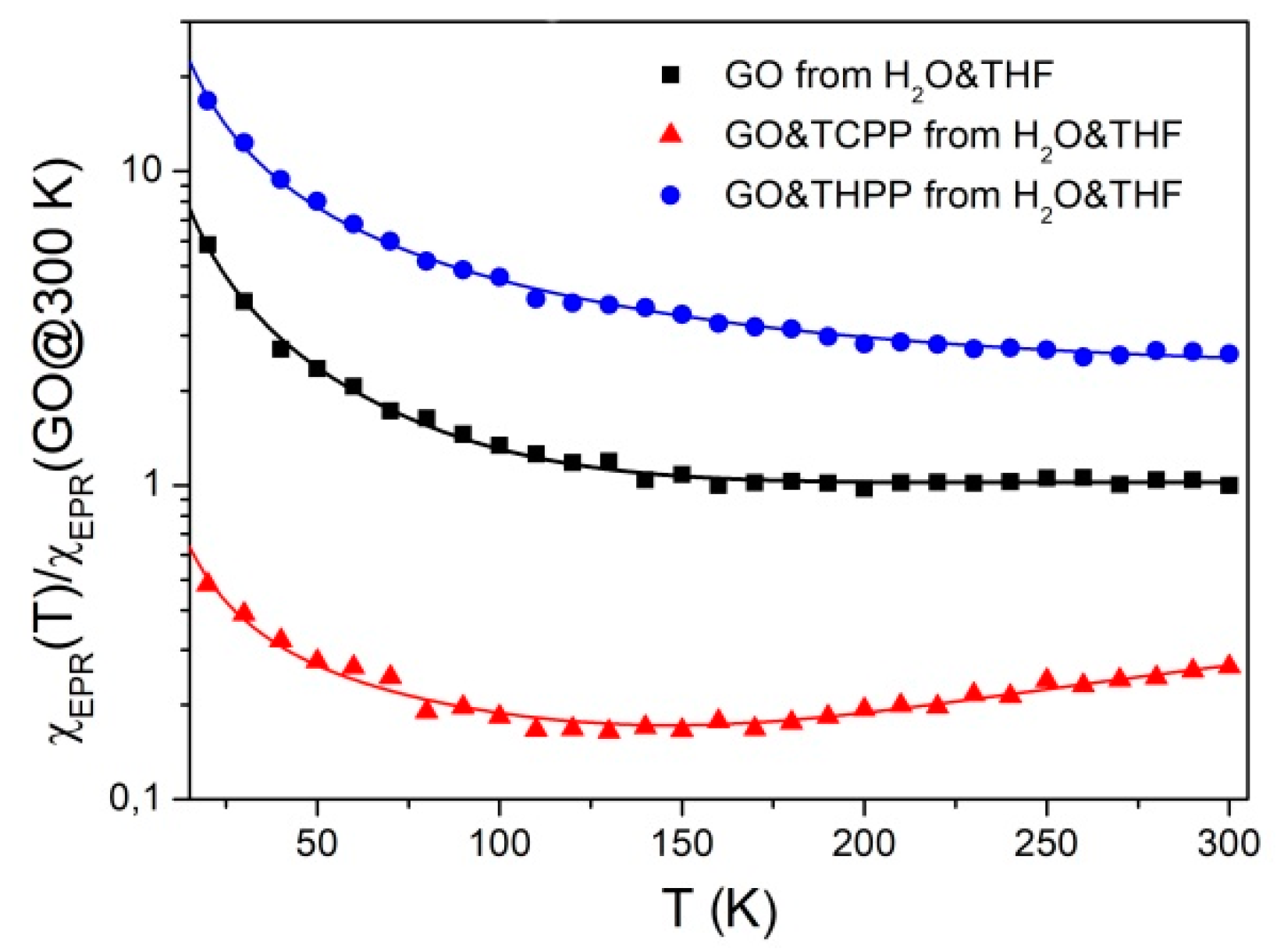
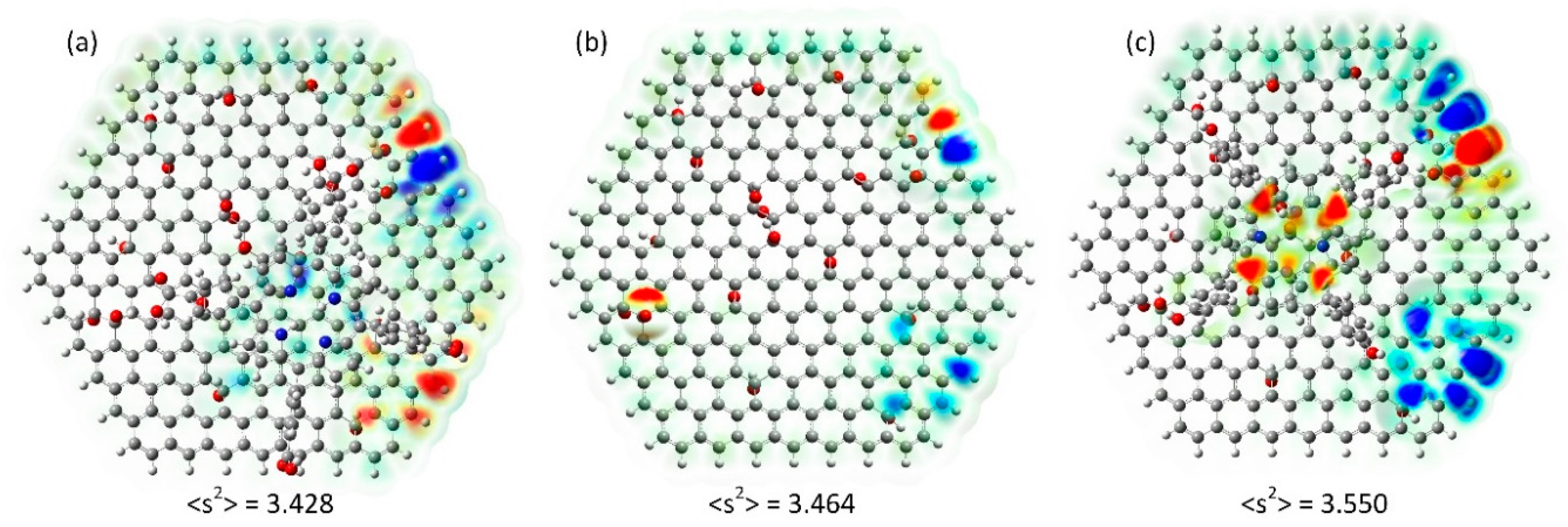
2.3. Magnetism
3. Materials and Methods
3.1. Materials
3.2. Instrumentation
3.3. Computation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferrari, A.C.; Bonaccorso, F.; Fal’ko, V.; Novoselov, K.S.; Roche, S.; Bøggild, P.; Borini, S.; Koppens, F.H.L.; Palermo, V.; Pugno, N.; et al. Kinare, Science and technology roadmap for graphene, related two-dimensional crystals, and hybrid systems. Nanoscale 2015, 7, 4598–4810. [Google Scholar] [CrossRef] [PubMed]
- Hummers, W.S., Jr.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Georgakilas, V.; Perman, J.A.; Tucek, J.; Zboril, R. Broad family of carbon nanoallotropes: Classification, chemistry, and applications of fullerenes, carbon dotys, nanotubes, graphene, nanodiamonds and combined structures. Chem. Rev. 2015, 115, 4744–4822. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.T.; Shah, S.; Chhowalla, M.; Lee, K.-B. Design, synthesis, and characterization of graphene–nanoparticle hybrid materials for bioapplications. Chem. Rev. 2015, 115, 2483–2531. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, V.; Tiwari, J.N.; Kemp, K.C.; Perman, J.A.; Bourlinos, A.B.; Kim, K.S.; Zboril, R. Noncovalent functionalization of graphene and graphene oxide for energy materials, biosensing, catalytic, and biochemical applications. Chem. Rev. 2016, 116, 5464–5519. [Google Scholar] [CrossRef] [PubMed]
- Stangel, C.; Schubert, C.; Kuhri, S.; Rotas, G.; Margraf, J.T.; Regulska, E.; Clark, T.; Torres, T.; Tagmatarchis, N.; Coutsolelos, A.G. Tuning the reorganization energy of electron transfer in supramolecular ensembles–metalloporphyrin, oligophenylenevinylenes, and fullerene–and the impact on electron transfer kinetics. Nanoscale 2015, 7, 2597–2608. [Google Scholar] [CrossRef]
- Hasobe, T.; Imahori, H.; Kamat, P.V.; Ahn, T.K.; Kim, S.K.; Kim, D.; Fujimoto, A.; Hirakawa, T.; Fukuzumi, S. Photovoltaic cells using composite nanoclusters of porphyrins and fullerenes with gold nanoparticles. J. Am. Chem. Soc. 2005, 127, 1216–1228. [Google Scholar] [CrossRef]
- Ge, R.; Wang, X.; Zhang, C.; Kang, S.-Z.; Qin, L.; Li, G.; Li, X. The influence of combination mode on the structure and properties of porphyrin–graphene oxide composites. Colloid Surf. A 2015, 483, 45–52. [Google Scholar] [CrossRef]
- Qin, Y.; Cheng, Y.; Jiang, L.; Jin, X.; Li, M.; Luo, X.; Liao, G.; Wei, T.; Li, Q. Top-down strategy toward versatile graphene quantum dots for organic/inorganic hybrid solar cells. ACS Sustain. Chem. Eng. 2015, 3, 637–644. [Google Scholar] [CrossRef]
- Ruan, C.; Zhang, L.; Qin, Y.; Xu, C.; Zhang, X.; Wan, J.; Peng, Z.; Shi, J.; Li, X.; Wang, L. Synthesis of porphyrin sensitized TiO2/graphene and its photocatalytic property under visible light. Mater. Lett. 2015, 141, 362–365. [Google Scholar] [CrossRef]
- Pang, S.; Hernandez, Y.; Feng, X.; Müllen, K. Graphene as transparent electrode material for organic electronics. Adv. Mater. 2011, 23, 2779–2795. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Pisula, W.; Müllen, K. Graphenes as potential material for electronics. Chem. Rev. 2007, 107, 718–747. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Li, Z.; Xiao, B.; Lu, Y.; Du, Y.; Yang, P.; Wang, X. Surfactant assistance in improvement of photocatalytic hydrogen production with the porphyrin noncovalently functionalized graphene nanocomposite. ACS Appl. Mater. Interface 2013, 5, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Feng, Y.; Tang, S.; Feng, W. Preparation of a graphene oxide–phthalocyanine hybrid through strong π–π interactions. Carbon 2010, 48, 211–216. [Google Scholar] [CrossRef]
- Xu, Y.; Bai, H.; Lu, G.; Li, C.; Shi, G. Flexible graphene films via the filtration of water-soluble noncovalent functionalized graphene sheets. J. Am. Chem. Soc. 2008, 130, 5856–5857. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Liu, Z.; Ma, Y.; Huang, Y.; Chen, Y. High-efficiency loading and controlled release of doxorubicin hydrochloride on graphene oxide. J. Phys. Chem. C 2008, 112, 17554–17558. [Google Scholar] [CrossRef]
- Hao, R.; Qian, W.; Zhang, L.; Hou, Y. Aqueous dispersions of TCNQ-anion-stabilized graphene sheets. Chem. Commun. 2008, 6576–6578. [Google Scholar] [CrossRef]
- Mani, V.; Chen, S.-M.; Lou, B.-S. Three dimensional graphene oxide-carbon nanotubes and graphene-carbon nanotubes hybrids. Int. J. Electrochem. Sci. 2013, 8, e60. [Google Scholar]
- Latos-Grazynski, L.; Kadish, K.; Smith, K.; Guilard, R. The porphyrin handbook. Acad. Press 2000, 361–416. [Google Scholar]
- El-Khouly, M.E.; Ito, O.; Smith, P.M.; D’Souza, F. Intermolecular and supramolecular photoinduced electron transfer processes of fullerene–porphyrin/phthalocyanine systems. J. Photochem. Photobiol. C Photochem. Rev. 2004, 5, 79–104. [Google Scholar] [CrossRef]
- Claessens, C.G.; Hahn, U.; Torres, T. Phthalocyanines: From outstanding electronic properties to emerging applications. Chem. Rec. 2008, 8, 75–97. [Google Scholar] [CrossRef] [PubMed]
- Craciun, M.F.; Rogge, S.; den Boer, M.J.; Margadonna, S.; Prassides, K.; Iwasa, Y.; Morpurgo, A.F. Electronic Transport through Electron-Doped Metal Phthalocyanine Materials. Adv. Mater. 2006, 18, 320–324. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.D.; Fechtenkötter, A.; Müllen, K. Big Is Beautiful-“Aromaticity” Revisited from the Viewpoint of Macromolecular and Supramolecular Benzene Chemistry. Chem. Rev. 2001, 101, 1267–1300. [Google Scholar] [CrossRef] [PubMed]
- Müllen, K.; Rabe, J.P. Nanographenes as active components of single-molecule electronics and how a scannig tunneling microscope puts them to work. Acc. Chem. Res. 2008, 41, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Milgrom, L.R.; Warren, M.J. The colours of life: An introduction to the chemistry of porphyrins and related compounds. J. Chem. Educ. 1997. [Google Scholar]
- Mandal, S.; Bhattacharyya, S.; Borovkov, V.; Patra, A. Photophysical properties, self-assembly behavior, and energy transfer of porphyrin-based functional nanoparticles. J. Phys. Chem. C 2012, 116, 11401–11407. [Google Scholar] [CrossRef]
- Luo, Z.; Lu, Y.; Somers, L.A.; Johnson, A.T.C. High yield preparation of macroscopic graphene oxide membranes. J. Am. Chem. Soc. 2009, 131, 898–899. [Google Scholar] [CrossRef]
- Cuong, T.V.; Pham, V.H.; Tran, Q.T.; Hahn, S.H.; Chung, J.S.; Shin, E.W.; Kim, E.J. Photoluminescence and Raman studies of graphene thin films prepared by reduction of graphene oxide. Mater. Lett. 2010, 64, 399–401. [Google Scholar] [CrossRef]
- Li, D.; Müller, M.B.; Gilje, S.; Kaner, R.B.; Wallace, G.G. Processable aqueous dispersions of graphene nanosheets. Nat. Nanotechnol. 2008, 3, 101–105. [Google Scholar] [CrossRef]
- Zheng, W.; Shan, N.; Yu, L.; Wang, X. UV–visible, fluorescence and EPR properties of porphyrins and metalloporphyrins. Dyes Pigments 2008, 77, 153–157. [Google Scholar] [CrossRef]
- Karousis, N.; Sandanayaka, A.S.; Hasobe, T.; Economopoulos, S.P.; Sarantopoulou, E.; Tagmatarchis, N. Graphene oxide with covalently linked porphyrin antennae: Synthesis, characterization and photophysical properties. J. Mater. Chem. 2011, 21, 109–117. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, L.; Bai, H.; Hong, W.; Li, C.; Shi, G. Chemically converted graphene induced molecular flattening of 5, 10, 15, 20-tetrakis (1-methyl-4-pyridinio) porphyrin and its application for optical detection of cadmium (II) ions. J. Am. Chem. Soc. 2009, 131, 13490–13497. [Google Scholar] [CrossRef]
- Alvaro, M.; Atienzar, P.; de la Cruz, P.; Delgado, J.L.; Troiani, V.; Garcia, H.; Langa, F.; Palkar, A.; Echegoyen, L. Synthesis, photochemistry, and electrochemistry of single-wall carbon nanotubes with pendent pyridyl groups and of their metal complexes with zinc porphyrin. Comparison with pyridyl-bearing fullerenes. J. Am. Chem. Soc. 2006, 128, 6626–6635. [Google Scholar] [CrossRef]
- Cheng, F.; Adronov, A. Suzuki coupling reactions for the surface functionalization of single-walled carbon nanotubes. Chem. Mater. 2006, 18, 5389–5391. [Google Scholar] [CrossRef]
- Campidelli, S.; Sooambar, C.; Lozano Diz, E.; Ehli, C.; Guldi, D.M.; Prato, M. Dendrimer-functionalized single-wall carbon nanotubes: Synthesis, characterization, and photoinduced electron transfer. J. Am. Chem. Soc. 2006, 128, 12544–12552. [Google Scholar] [CrossRef] [PubMed]
- Pagona, G.; Sandanayaka, A.S.D.; Araki, Y.; Fan, J.; Tagmatarchis, N.; Charalambidis, G.; Coutsolelos, A.G.; Boitrel, B.; Yudasaka, M.; Iijima, S. Covalent functionalization of carbon nanohorns with porphyrins: Nanohybrid formation and photoinduced electron and energy transfer. Adv. Funct. Mater. 2007, 17, 1705–1711. [Google Scholar] [CrossRef]
- Cioffi, C.; Campidelli, S.; Sooambar, C.; Marcaccio, M.; Marcolongo, G.; Meneghetti, M.; Paolucci, D.; Paolucci, F.; Ehli, C.; Rahman, G.M.A. Synthesis, characterization, and photoinduced electron transfer in functionalized single wall carbon nanohorns. J. Am. Chem. Soc. 2007, 129, 3938–3945. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.B.; Tian, J.G.; Guo, Z.; Ren, D.M.; Du, F.; Zheng, J.Y.; Chen, Y.S. Enhanced Optical Limiting Effects in Porphyrin-Covalently Functionalized Single-Walled Carbon Nanotubes. Adv. Mater. 2008, 20, 511–515. [Google Scholar] [CrossRef]
- Pagona, G.; Sandanayaka, A.S.D.; Hasobe, T.; Charalambidis, G.; Coutsolelos, A.G.; Yudasaka, M.; Iijima, S.; Tagmatarchis, N. Characterization and photoelectrochemical properties of nanostructured thin film composed of carbon nanohorns covalently functionalized with porphyrins. J. Phys. Chem. C 2008, 112, 15735–15741. [Google Scholar] [CrossRef]
- Vergeldt, F.J.; Koehorst, R.B.M.; van Hoek, A.; Schaafsma, T.J. Intramolecular interactions in the ground and excited states of tetrakis (N-methylpyridyl) porphyrins. J. Phys. Chem. 1995, 99, 4397–4405. [Google Scholar] [CrossRef]
- Kathiravan, A.; Kumar, P.S.; Renganathan, R.; Anandan, S. Photoinduced electron transfer reactions between meso-tetrakis (4-sulfonatophenyl) porphyrin and colloidal metal-semiconductor nanoparticles. Colloid Surface A 2009, 333, 175–181. [Google Scholar] [CrossRef]
- Shu, J.; Qiu, Z.; Wei, Q.; Zhuang, J.; Tang, D. Cobalt-porphyrin-platinum-functionalized reduced graphene oxide hybrid nanostructures: A novel peroxidase mimetic system for improved electrochemical immunoassay. Sci. Rep.-UK 2015, 5, 15113. [Google Scholar] [CrossRef]
- Casey, J.P.; Bachilo, S.M.; Weisman, R.B. Efficient photosensitized energy transfer and near-IR fluorescence from porphyrin–SWNT complexes. J. Mater. Chem. 2008, 18, 1510–1516. [Google Scholar] [CrossRef]
- Guldi, D.M.; Rahman, G.M.A.; Jux, N.; Tagmatarchis, N.; Prato, M. Integrating Single-Wall Carbon Nanotubes into Donor–Acceptor Nanohybrids. Angew. Chem.-Ger. Edit. 2004, 116, 5642–5646. [Google Scholar] [CrossRef]
- Lewandowska, K.; Pilarczyk, K.; Podborska, A.; Kim, T.-D.; Lee, K.-S.; Szaciłowski, K. Tuning of electronic properties of fullerene-oligothiophene layers. Appl. Phys. Lett. 2015, 106, 103719. [Google Scholar] [CrossRef]
- Lewandowska, K.; Barszcz, B.; Graja, A.; Nam, S.Y.; Han, Y.-S.; Kim, T.-D.; Lee, K.-S. Spectroscopic properties and orientation of molecules in Langmuir-Blodgett layers of selected functionalized fullerenes. Spectrochim. Acta A 2014, 118, 204–209. [Google Scholar] [CrossRef]
- Lewandowska, K.; Graja, A.; Barszcz, B.; Biadasz, A.; Wróbel, D. Raman and infrared studies of molecular orientation in fullerene-thiophene films. New J. Chem. 2011, 36, 1291–1295. [Google Scholar] [CrossRef]
- Wróbel, D.; Lewandowska, K. Covalent dyads of porphyrin–fullerene and perylene–fullerene for organic photovoltaics: Spectroscopic and photocurrent studies. Opt. Mater. 2011, 33, 1424–1428. [Google Scholar] [CrossRef]
- Lewandowska, K.; Barszcz, B.; Graja, A.; Bursa, B.; Biadasz, A.; Wróbel, D.; Bednarski, W.; Waplak, S.; Grzybowski, M.; Gryko, D.T. Absorption and emission properties of the corrole–fullerene dyad. Synth. Met. 2013, 166, 70–76. [Google Scholar] [CrossRef]
- Bredas, J.-L. Mind the gap! Mater. Horiz. 2014, 1, 17–19. [Google Scholar] [CrossRef]
- Endicott, J.F. Molecular electron transfer. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 7, pp. 657–730. [Google Scholar]
- Ebenso, E.E.; Kabanda, M.M.; Murulana, L.C.; Singh, A.K.; Shukla, S.K. Electrochemical and quantum chemical investigation of some azine and thiazine dyes as potential corrosion inhibitors for mild steel in hydrochloric acid solution. Ind. Eng. Chem. Res. 2012, 51, 12940–12958. [Google Scholar] [CrossRef]
- Kabanda, M.M.; Ebenso, E.E. Density functional theory and quantitative structure-activity relationship studies of some quinoxaline derivatives as potential corrosion inhibitors for copper in acidic medium. Int. J. Electrochem. Sci 2012, 7, 8713–8733. [Google Scholar]
- Kabanda, M.M.; Shukla, S.K.; Singh, A.K.; Murulana, L.C.; Ebenso, E.E. Electrochemical and quantum chemical studies on calmagite and fast sulphone black F dyes as corrosion inhibitors for mild steel in hydrochloric medium. Int. J. Electrochem. Sci. 2012, 7, 8813–8831. [Google Scholar]
- Lonfat, M.; Marsen, B.; Sattler, K. The energy gap of carbon clusters studied by scanning tunneling spectroscopy. Chem. Phys. Lett. 1999, 313, 539–543. [Google Scholar] [CrossRef]
- Stranks, S.D.; Sprafke, J.K.; Anderson, H.L.; Nicholas, R.J. Electronic and mechanical modification of single-walled carbon nanotubes by binding to porphyrin oligomers. ACS Nano 2011, 5, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.-Y.; Han, T.H.; Hong, J.; Kim, J.E.; Lee, S.H.; Kim, H.W.; Kim, S.O. Noncovalent functionalization of graphene with end-functional polymers. J. Mater. Chem. 2010, 20, 1907–1912. [Google Scholar] [CrossRef]
- Krishna, M.B.M.; Venkatramaiah, N.; Venkatesan, R.; Rao, D.N. Synthesis and structural, spectroscopic and nonlinear optical measurements of graphene oxide and its composites with metal and metal free porphyrins. J. Mater. Chem. 2012, 22, 3059–3068. [Google Scholar] [CrossRef]
- Dubis, A. Zastosowanie spektroskopii w podczerwieni w jakościowej i ilościowej analizie organicznej. Zakład Chemii Produktów Naturalnych Instytutu Chemii, Uniwersytet w Białymstoku. 2012. [Google Scholar]
- Zhou, Y.-G.; Chen, J.-J.; Wang, F.-b.; Sheng, Z.-H.; Xia, X.-H. A facile approach to the synthesis of highly electroactive Pt nanoparticles on graphene as an anode catalyst for direct methanol fuel cells. Chem. Commun. 2010, 46, 5951–5953. [Google Scholar] [CrossRef]
- Yuan, W.H.; Gu, Y.J.; Li, L. Green synthesis of graphene/Pt nanocomposites with vitamin C. Appl. Surf. Sci. 2014, 28, 258–263. [Google Scholar]
- Lomeda, J.R.; Doyle, C.D.; Kosynkin, D.V.; Hwang, W.-F.; Tour, J.M. Diazonium functionalization of surfactant-wrapped chemically converted graphene sheets. J. Am. Chem. Soc. 2008, 130, 16201–16206. [Google Scholar] [CrossRef]
- Ćirić, L.; Sienkiewicz, A.; Djokić, D.M.; Smajda, R.; Magrez, A.; Kaspar, T.; Nesper, R.; Forró, L. Size dependence of the magnetic response of graphite oxide and graphene flakes–an electron spin resonance study. Phys. Status Solidi B 2010, 247, 2958–2961. [Google Scholar] [CrossRef]
- Panich, A.M.; Shames, A.I.; Sergeev, N.A. Paramagnetic impurities in graphene oxide. App. Magn. Reson. 2013, 44, 107–116. [Google Scholar] [CrossRef]
- Majchrzycki, Ł.; Augustyniak-Jabłokow, M.A.; Strzelczyk, R.; Maćkowiak, M. Magnetic Centres in Functionalized Graphene. Acta Phys. Pol. A 2015, 127. [Google Scholar] [CrossRef]
- Oszajca, M.; Kwolek, P.; Mech, J.; Szaciłowski, K. Substituted polyacenes as prospective modifiers of TiO2 surface. Curr. Phys. Chem. 2011, 1, 242–260. [Google Scholar] [CrossRef]
- Kwolek, P.; Oszajca, M.; Szaciłowski, K. Catecholate and 2,3-acenediolate complexes of d0 ions as prospective materials for molecular electronics and spintronics. Coord. Chem. Rev. 2012, 56, 1706–1731. [Google Scholar] [CrossRef]
- Pilevarshahri, R.; Rungger, I.; Archer, T.; Sanvito, S.; Shahtahmassebi, N. Spin transport in higher n-acene molecules. Phys. Rev. B 2011, 84, 174437. [Google Scholar] [CrossRef]
- Yazyev, O.V. Emergence of magnetism in graphene materials and nanostructures. Rev. Mod. Phys. 2010, 73, 056501. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Li, C.M. Magnetism in graphene oxide. New. J. Phys. 2010, 12, 083049. [Google Scholar] [CrossRef]
- Tajima, K.; Isaka, T.; Yamashina, T.; Ohta, Y.; Matsuo, Y.; Takai, K. Functional group dependence of spin magnetism in graphene oxide. Polyhedron 2017, 136, 155–158. [Google Scholar] [CrossRef]
- Lee, D.; Seo, J.; Zhu, Z.; Cole, J.M.; Su, H. Magnetism in graphene oxide induced by epoxy groups. Appl. Phys. Lett. 2015, 106, 172402. [Google Scholar] [CrossRef]
- Diamantopolou, A.; Glenis, S.; Zolnierkiwicz, G.; Guskos, N.; Likodimos, V. Magnetism in prostine and chemically reduced graphene oxide. J. Appl. Phys. 2017, 121, 043906. [Google Scholar] [CrossRef]
- Qin, S.; Guo, X.; Cao, Y.; Ni, Z.; Xu, Q. Strong ferromagnetism of reduced graphene oxide. Carbon 2014, 78, 559–565. [Google Scholar] [CrossRef]
- Khurana, G.; Kumar, N.; Kotnala, R.K.; Nautiyal, T.; Katiyar, R.S. Temperature tuned defect induced magnetism in reeguced graphene oxide. Nanoscale 2013, 5, 3346–3351. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-W.; Kim, H.-K.; Lee, K.; Roh, K.C.; Han, J.T.; Kim, K.-B.; Lee, S.; Jung, M.-H. Studying reduction of graphene oxide with magnetic measurements. Carbon 2019, 142, 373–378. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C. Gaussian 03, Revision D. 01. 2004. Available online: http://www. gaussian. com (accessed on 14 February 2019).
- Dennington, R.T.; Keith, T.; Millam, J. GaussView 5; Semichem Inc.: Shawnee, KS, USA, 2009. [Google Scholar]
Sample Availability: Samples of THPP-GO and TCPP-GO are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IR (cm−1) | Raman (cm−1) | DFT (cm−1) | Bands Assignment |
|---|---|---|---|
| THPP/THPP-GO | THPP/THPP-GO | THPP | |
| 334/330 | 333 | breathing porphyrin ring | |
| 417 | 424 | def. benzene ring | |
| 535/536 | 542 | C-H w in benzene ring + def. porphyrin | |
| 560/566 | 568 | C-H w in benzene ring + def. porphyrin | |
| 597/599 | 607 | breathing benzene ring + def. porphyrin | |
| 647 | 645 | N-H w | |
| 669/695 | 687 | N-H w + C-H w | |
| 729/729 | 764 | N-H w | |
| 804/805 | 824 | C-H w + N-H w | |
| 814 | 835 | C-C-C b in benzene ring + N-H w + C-O s | |
| 843/846 | 857 | C-H w | |
| 966/968 | 989 | C-C s in porphyrin | |
| 983/985 | 1008 | C-C s in porphyrin | |
| 1004/1010 | 1027 | C-C s in porphyrin | |
| 1018 | 1039 | C-H r + N-H r | |
| 1075/1068 | 1075/1075 | 1109 | C-H r |
| 1100/1102 | 1128 | C-H r | |
| 1139 | 1165 | N-H r + C-H r | |
| 1169/1170 | 1197 | C-O-H b + C-H r | |
| 1171/1171 | 1199 | C-O-C b + C-H r | |
| 1223/1233 | 1221 | N-H r + C-H r + C-O-H b | |
| 1234/1234 | 1261 | C-C s between porphyrin and aryl group | |
| 1263/1281 | 1308 | C-O s + C-H r | |
| 1346/1350 | 1370 | C-H r + C-O-H b | |
| 1363 | 1395 | C-C s | |
| 1381/1383 | 1417 | C-N s + C-H r | |
| 1402/1442 | 1439 | C-C s + C-H r | |
| 1433/1463 | 1466 | C-H r + C-O-H b | |
| 1459/1452 | 1496 | C-C s + C=C s | |
| 1465/1477 | 1515 | C=C s | |
| 1508/1511 | 1555 | C-C s + C-H r + C-C s between porphyrin and aryl group | |
| 1516/1527 | 1559 | C-C s + C=C s + C-C s between porphyrin and aryl group | |
| 1544 | 1607 | C=C s + C-N-H b | |
| 1558 | 1610 | C=C s + C-N-H b | |
| 1586/1588 | 1635 | C-C s + C-O-H b | |
| 1605/1606 | 1608/1601 | 1668 | C-C s + C=C s |
| 1662/1648 | C=N s | ||
| -/1731 | C=O s in GO |
| IR (cm−1) | Raman (cm−1) | DFT (cm−1) | Bands Assignment |
|---|---|---|---|
| TCPP/TCPP-GO | TCPP/TCPP-GO | TCPP | |
| 319/324 | 323 | breathing porphyrin ring | |
| 410 | 418 | def. benzene ring | |
| 671/690 | 687 | N-H w | |
| 723/732 | 763 | N-H w | |
| 797/796 | 824 | N-H w+ C-H w | |
| 817 | 837 | C- C-C b + N-H w | |
| 866/866 | 884 | C-H w + def. pophyrin | |
| 964/968 | 989 | breathing porphyrin and benzene rings | |
| 980/982 | 1008 | breathing porphyrin ring | |
| 994/994 | 1022 | C-N s + C-H r + N-H r | |
| 999/1003 | 1027 | C-C s | |
| 1019/1023 | 1039 | C-H + N-H r | |
| 1073 | 1114 | C-H r | |
| 1101/1101 | 1118 | C-O s + C-H r | |
| 1145 | 1164 | N-H r | |
| 1176/1181 | 1176 | 1195 | C-H r + C-O-H b |
| 1221/1225 | 1220 | C-H r + C-O-H b + N-H r + C-N s | |
| 1226 | 1231/1236 | 1258 | C-C s between porphyrin and aryl group |
| 1270/1279 | 1287 | C-C s + C-N s + N-H b | |
| 1293 | 1363 | C-H r | |
| 1310/1314 | 1318 | 1391 | C-O-H b + C-C s + C-N s + C-H r |
| 1358/1363 | 1417 | C-C s + C-N s + C-H r | |
| 1403/1418 | 1441 | C-C s + C=C s + C-H r | |
| 1440 | 1495 | C-C s + C-H r i N-H r | |
| 1473/1470 | 1518 | C=C s w | |
| 1495 | 1558 | C=C s | |
| 1505 | 1579 | C=C s + C-C s + C-H r + N-H r | |
| 1545/1555 | 1609 | C=C s | |
| 1564/1567 | 1614 | C=C s | |
| 1605/1607 | 1605/1603 | 1660 | C=C s + C-C s |
| 1691/1690 | 1661 | C=O s | |
| 1727/1720 | 1818 | C=O s in GO |
| Sample | C1 | C2 | ΔE/kB [K] | A | Total Spin Concentration at RT [1017/g] | Concentration of Delocalized Electrons [1017/g] |
|---|---|---|---|---|---|---|
| GO from H2O | 14.1 ± 0.8 | 1088 ± 168 | 629 ± 47 | 0.26 ± 0.02 | 2.33 | 0.89 ± 0.07 |
| GO from H2O&THF | 112.6 ± 1.8 | 838 ± 156 | 504 ± 58 | 0.13 ± 0.05 | 3.44 | 0.45 ± 0.17 |
| THPP-GO from H2O&THF | 318 ± 4 | 1111 ± 822 | 946 ± 355 | 1.33 ± 0.07 | 9.02 | 4.58 ± 0.24 |
| TCPP-GO from H2O&THF | 7.96 ± 0.28 | 945 ± 326 | 951 ± 101 | 0.108 ± 0.006 | 0.91 | 0.37 ± 0.02 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewandowska, K.; Rosiak, N.; Bogucki, A.; Cielecka-Piontek, J.; Mizera, M.; Bednarski, W.; Suchecki, M.; Szaciłowski, K. Supramolecular Complexes of Graphene Oxide with Porphyrins: An Interplay between Electronic and Magnetic Properties. Molecules 2019, 24, 688. https://doi.org/10.3390/molecules24040688
Lewandowska K, Rosiak N, Bogucki A, Cielecka-Piontek J, Mizera M, Bednarski W, Suchecki M, Szaciłowski K. Supramolecular Complexes of Graphene Oxide with Porphyrins: An Interplay between Electronic and Magnetic Properties. Molecules. 2019; 24(4):688. https://doi.org/10.3390/molecules24040688
Chicago/Turabian StyleLewandowska, Kornelia, Natalia Rosiak, Andrzej Bogucki, Judyta Cielecka-Piontek, Mikołaj Mizera, Waldemar Bednarski, Maciej Suchecki, and Konrad Szaciłowski. 2019. "Supramolecular Complexes of Graphene Oxide with Porphyrins: An Interplay between Electronic and Magnetic Properties" Molecules 24, no. 4: 688. https://doi.org/10.3390/molecules24040688