
Antimalarial Properties of Dunnione Derivatives as NQO2 Substrates


 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
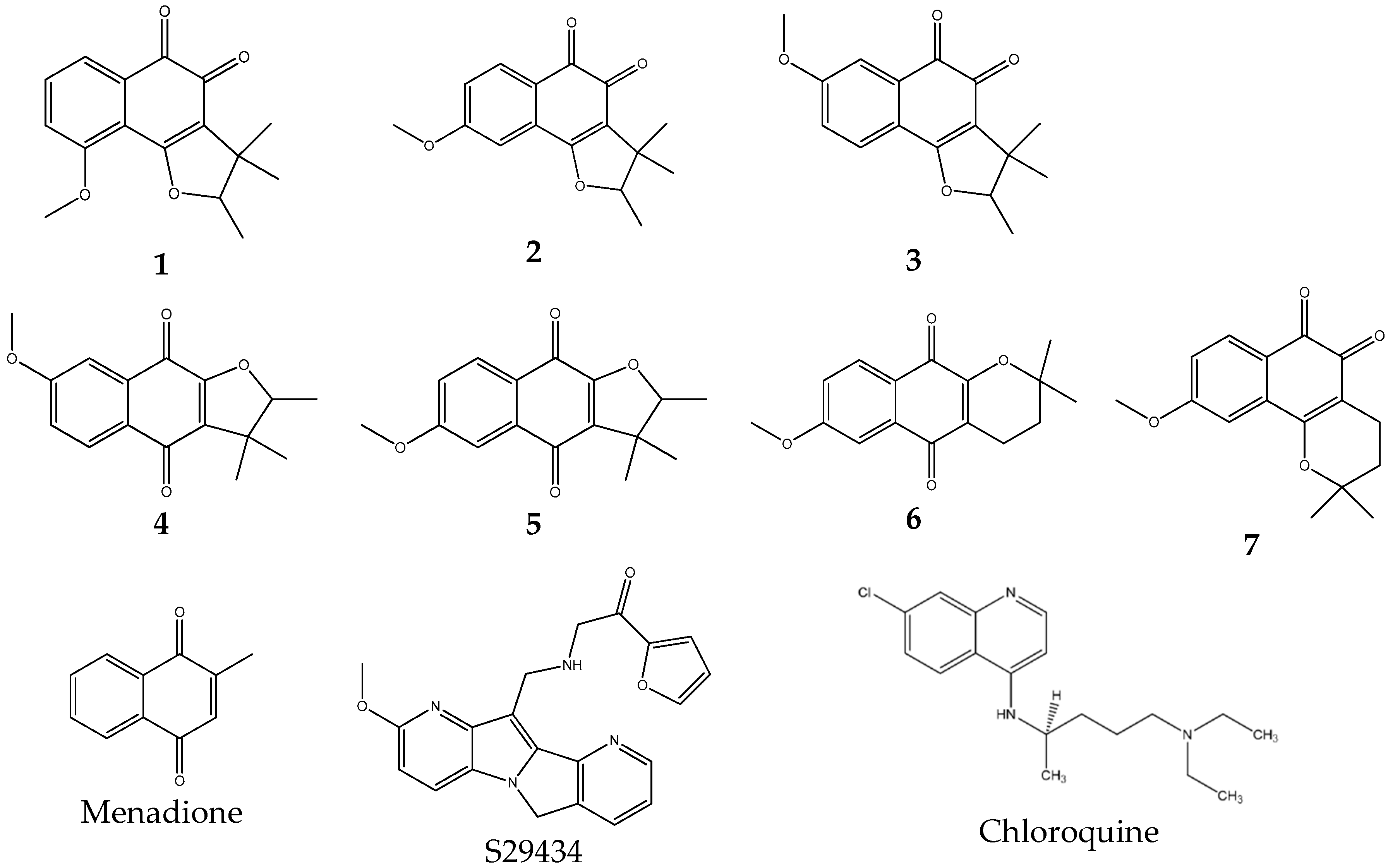
2.1. Rationale for Selecting NQO2 Substrates
2.2. Rationale for Selecting NQO2 Substrates for Testing In Vivo Antimalarial Properties
2.3. NQO2 Substrates As Antimalarian Compounds In Vivo
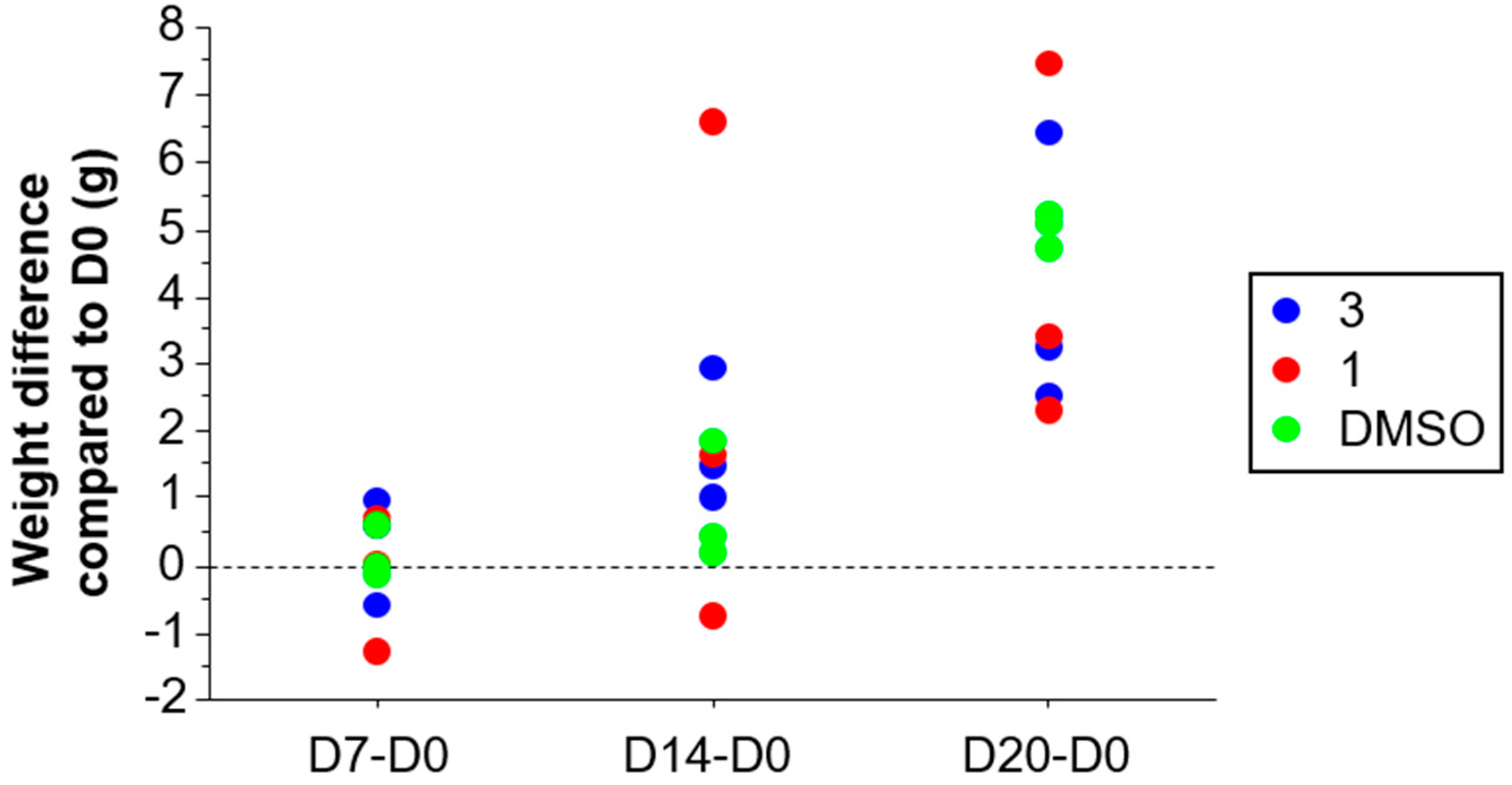
2.3.1. Acute Toxicity of Compound 1 and Compound 3
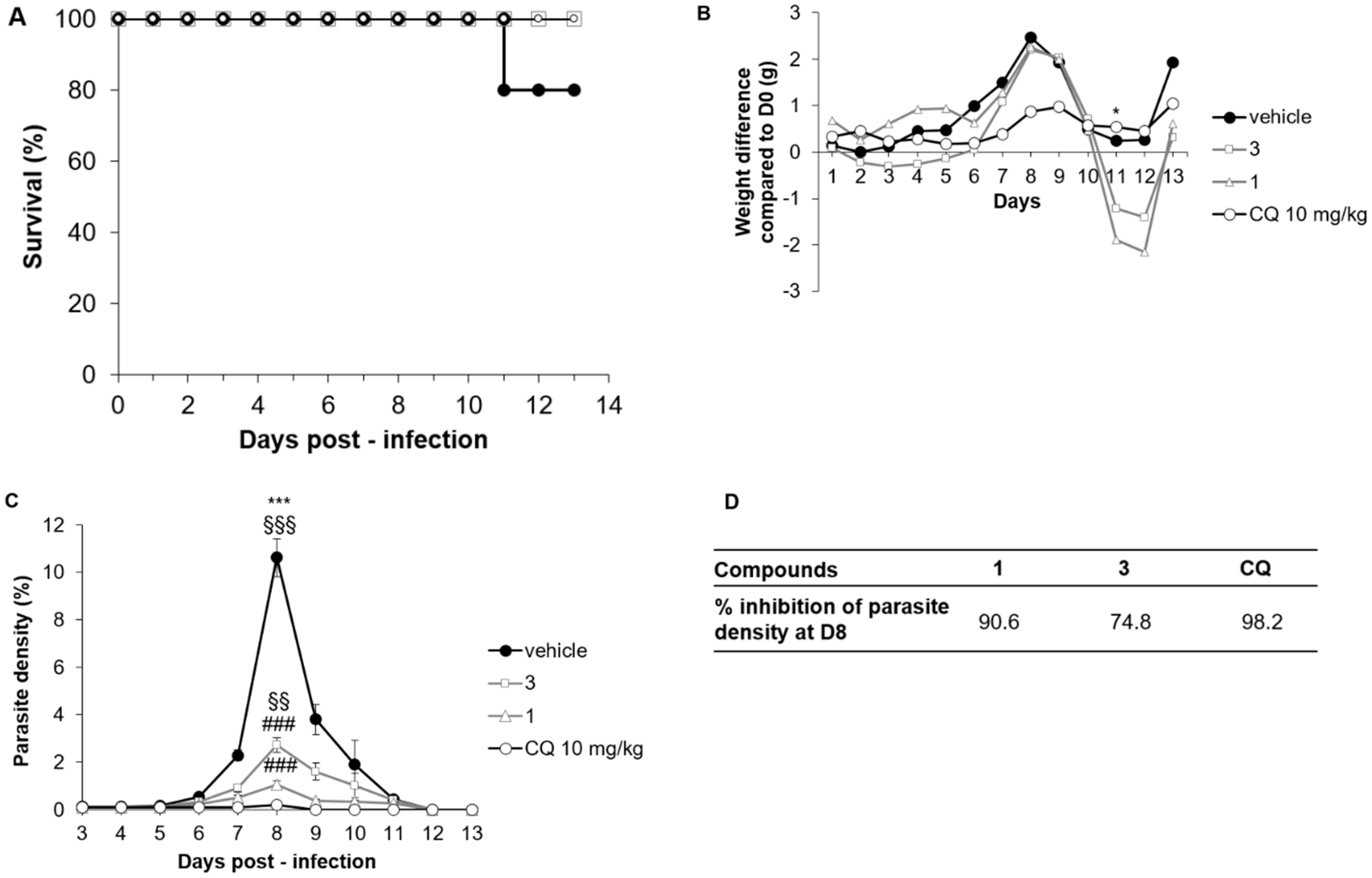
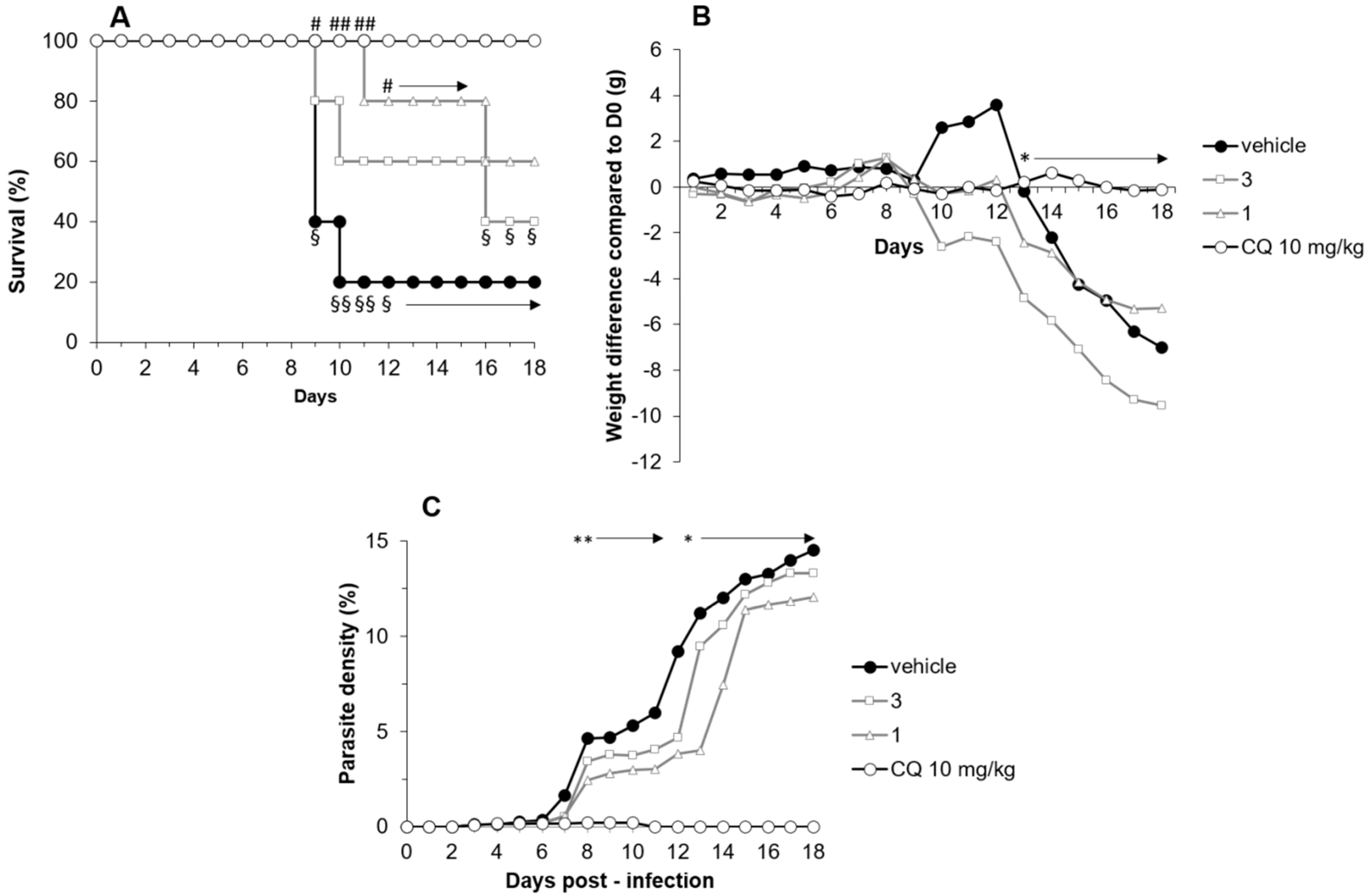
2.3.2. Compounds 1 and 3 Treatment Efficacy in the P. chabaudi chabaudi Mice Model
2.3.3. Compounds 1 and 3 Treatment Efficacy in a P. berghei ANKA Mice Model
3. Materials and Methods
3.1. Chemicals
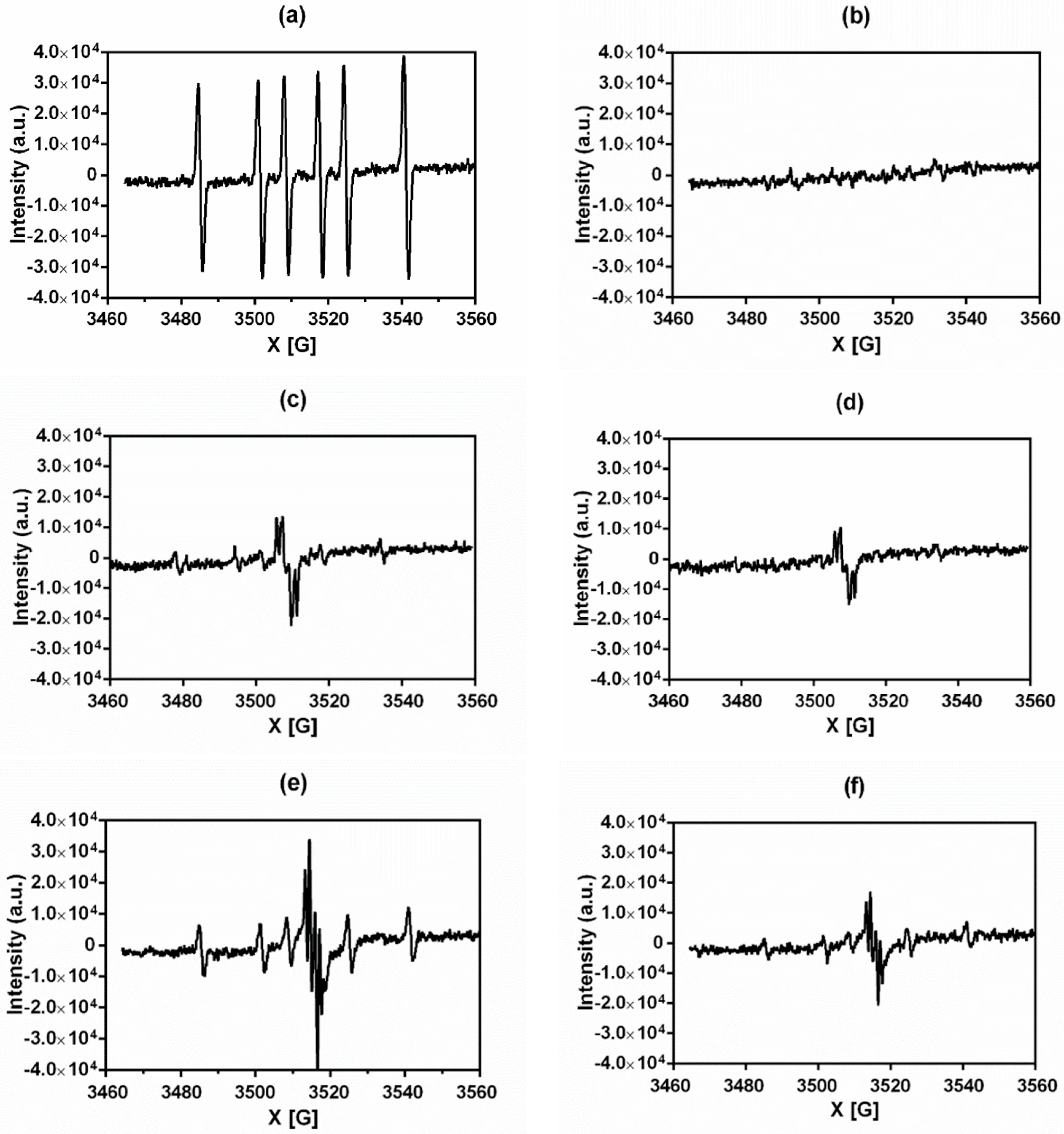
3.2. EPR Spectroscopy Experiments
3.3. In Vitro Antiplasmodial Activity
3.4. Cytotoxic Evaluation on the Hela Cell Line
3.5. Animals and Ethics Statement
3.6. Acute Oral Toxicity Measurement
3.7. Murine Malaria Models and In Vivo Antimalarial Tests
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization World Malaria Report 2018. Available online: http://www.who.int/malaria/publications/world-malaria-report-2018/report/en/ (accessed on 11 February 2019).
- Medicine for Malaria Venture MMV Annual Report 2017 | Medicines for Malaria Venture. Available online: https://www.mmv.org/newsroom/publications/mmv-annual-report-2017 (accessed on 15 February 2019).
- van der Pluijm, R.W.; Imwong, M.; Chau, N.H.; Hoa, N.T.; Thuy-Nhien, N.T.; Thanh, N.V.; Jittamala, P.; Hanboonkunupakarn, B.; Chutasmit, K.; Saelow, C.; et al. Determinants of dihydroartemisinin-piperaquine treatment failure in Plasmodium falciparum malaria in Cambodia, Thailand, and Vietnam: A prospective clinical, pharmacological, and genetic study. Lancet Infect. Dis. 2019, 19, 952–961. [Google Scholar] [CrossRef]
- Ibrahim, N.; Ibrahim, H.; Dormoi, J.; Briolant, S.; Pradines, B.; Moreno, A.; Mazier, D.; Legrand, P.; Nepveu, F. Albumin-bound nanoparticles of practically water-insoluble antimalarial lead greatly enhance its efficacy. Int. J. Pharm. 2014, 464, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Nepveu, F.; Najahi, E.; Valentin, A. Antimalarial activities of indolones and derivatives. Curr. Top Med. Chem. 2014, 14, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Nepveu, F.; Turrini, F. Targeting the redox metabolism of Plasmodium falciparum. Future Med. Chem. 2013, 5, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Nepveu, F.; Kim, S.; Boyer, J.; Chatriant, O.; Ibrahim, H.; Reybier, K.; Monje, M.-C.; Chevalley, S.; Perio, P.; Lajoie, B.H.; et al. Synthesis and antiplasmodial activity of new indolone N-oxide derivatives. J. Med. Chem. 2010, 53, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Cassagnes, L.-E.; Rakotoarivelo, N.; Sirigu, S.; Pério, P.; Najahi, E.; Chavas, L.M.G.; Thompson, A.; Gayon, R.; Ferry, G.; Boutin, J.A.; et al. Role of Quinone Reductase 2 in the antimalarial properties of indolone-type derivatives. Molecules 2017, 22, 210. [Google Scholar] [CrossRef] [PubMed]
- Reybier, K.; Nguyen, T.H.Y.; Ibrahim, H.; Perio, P.; Montrose, A.; Fabre, P.-L.; Nepveu, F. Electrochemical behavior of indolone-N-oxides: Relationship to structure and antiplasmodial activity. Bioelectrochemistry 2012, 88, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, H.; Pantaleo, A.; Turrini, F.; Arese, P.; Nallet, J.-P.; Nepveu, F. Pharmacological properties of indolone-N-oxides controlled by a bioreductive transformation in red blood cells? Med. Chem. Comm. 2011, 2, 860–869. [Google Scholar] [CrossRef]
- Cassagnes, L.-E.; Perio, P.; Ferry, G.; Moulharat, N.; Antoine, M.; Gayon, R.; Boutin, J.A.; Nepveu, F.; Reybier, K. In cellulo monitoring of quinone reductase activity and reactive oxygen species production during the redox cycling of 1,2 and 1,4 quinones. Free Radic. Biol. Med. 2015, 89, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Price, J.R.; Robinson, R. A new natural colouring matter of the naphthalene group. Nature 1938, 142, 147. [Google Scholar] [CrossRef]
- Inoue, K.; Ueda, S.; Nayeshiro, H.; Inouyet, H. Quinones of streptocarpus dunnii. Phytochemistry 1983, 22, 737–741. [Google Scholar] [CrossRef]
- Bian, J.; Xu, L.; Deng, B.; Qian, X.; Fan, J.; Yang, X.; Liu, F.; Xu, X.; Guo, X.; Li, X.; et al. Synthesis and evaluation of (±)-dunnione and its ortho-quinone analogues as substrates for NAD(P)H:quinone oxidoreductase 1 (NQO1). Bioorg. Med. Chem. Lett. 2015, 25, 1244–1248. [Google Scholar] [CrossRef] [PubMed]
- Graves, P.R.; Kwiek, J.J.; Fadden, P.; Ray, R.; Hardeman, K.; Coley, A.M.; Foley, M.; Haystead, T.A.J. Discovery of novel targets of quinoline drugs in the human purine binding proteome. Mol. Pharmacol. 2002, 62, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Dufour, M.; Yan, C.; Siegel, D.; Colucci, M.A.; Jenner, M.; Oldham, N.J.; Gomez, J.; Reigan, P.; Li, Y.; De Matteis, C.I.; et al. Mechanism-based inhibition of quinone reductase 2 (NQO2): Selectivity for NQO2 over NQO1 and structural basis for flavoprotein inhibition. Chem. Biochem. 2011, 12, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Boutin, J.A.; Bouillaud, F.; Janda, E.; Gacsalyi, I.; Guillaumet, G.; Hirsch, E.C.; Kane, D.A.; Nepveu, F.; Reybier, K.; Dupuis, P.; et al. S29434, a Quinone Reductase 2 inhibitor: Main biochemical and cellular characterization. Mol. Pharmacol. 2019, 95, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Reybier, K.; Perio, P.; Ferry, G.; Bouajila, J.; Delagrange, P.; Boutin, J.A.; Nepveu, F. Insights into the redox cycle of human quinone reductase 2. Free Radic. Res. 2011, 45, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Villamil, S.F.; Stoppani, A.O.M.; Dubin, M. Redox cycling of β-lapachone and structural analogues in microsomal and cytosol liver preparations. In Methods in Enzymology; Quinones and Quinone Enzymes, Part A; Academic Press: Cambridge, MA, USA, 2004; Volume 378, pp. 67–87. [Google Scholar]
- Haidara, M.; Haddad, M.; Denou, A.; Marti, G.; Bourgeade-Delmas, S.; Sanogo, R.; Bourdy, G.; Aubouy, A. In vivo validation of anti-malarial activity of crude extracts of Terminalia macroptera, a Malian medicinal plant. Malar. J. 2018, 17, 68. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.J.; Peters, W. The antimalarial activity of N-benzyloxydihydrotriazines. Ann. Trop. Med. Parasit. 1980, 74, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.E.; Dei-Cas, E.; Maurois, P.; Slomianny, C.; Prensier, G.; Houcke-Lecomte, M.; Vernes, A.; Camus, D. Histopathology of the liver and kidney during malaria: Relation to malaria-induced dyslipoproteinemia. Ann. Parasitol. Hum. Comp. 1988, 63, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.R.; Ye, X.Y.; Chu, Y.H. Comparison of effects of pyronaridine, amodiaquine, mefloquine and qinghaosu on rodent malaria. Southeast Asian J. Trop. Med. Public Health 1992, 23, 59–63. [Google Scholar] [PubMed]
Sample Availability: Compounds might be obtained in reasonable amount from les Laboratoires Servier. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ROS after NQO2 Reduction | ROS after Inhibition of NQO2 with S29434 |
|---|---|---|
| Menadione | + + + + + | - |
| 1 | + + | + |
| 2 | - | - |
| 3 | + + + | + |
| 4 | + + + | + |
| 5 | - | - |
| 6 | + + + | + + |
| 7 | - | - |
| Chloroquine | - | - |
| Compound | CC50 (µM) | IC50 (µM) | TI 1 |
|---|---|---|---|
| Doxorubicin | 0.3109 ± 0.478 | n.a. | n.a. |
| Chloroquine | >0.625 | 0.000164 ± 0.000055 | >4000 |
| 1 | 2.424 ± 0.905 | 0.633 ± 0.740 | 3.83 |
| 2 | 1.249 ± 0.160 | 0.580 ± 0.503 | 2.15 |
| 3 | 1.432 ± 0.314 | 0.845 ± 0.334 | 1.70 |
| 4 | 11.752 ± 2.196 | 1.377 ± 0.627 | 8.53 |
| 5 | 14.861 ± 3.226 | 1.515 ± 0.853 | 9.81 |
| 6 | >18.362 | 19.464 ± 7.090 | >0.94 |
| 7 | 1.665 ± 0.368 | 0.753 ± 0.484 | 2.21 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chhour, M.; Aubouy, A.; Bourgeade-Delmas, S.; Pério, P.; Ternet-Fontebasso, H.; Haidara, M.; Ferry, G.; Nepveu, F.; Boutin, J.A.; Reybier, K. Antimalarial Properties of Dunnione Derivatives as NQO2 Substrates. Molecules 2019, 24, 3697. https://doi.org/10.3390/molecules24203697
Chhour M, Aubouy A, Bourgeade-Delmas S, Pério P, Ternet-Fontebasso H, Haidara M, Ferry G, Nepveu F, Boutin JA, Reybier K. Antimalarial Properties of Dunnione Derivatives as NQO2 Substrates. Molecules. 2019; 24(20):3697. https://doi.org/10.3390/molecules24203697
Chicago/Turabian StyleChhour, Monivan, Agnès Aubouy, Sandra Bourgeade-Delmas, Pierre Pério, Hélène Ternet-Fontebasso, Mahamane Haidara, Gilles Ferry, Françoise Nepveu, Jean A. Boutin, and Karine Reybier. 2019. "Antimalarial Properties of Dunnione Derivatives as NQO2 Substrates" Molecules 24, no. 20: 3697. https://doi.org/10.3390/molecules24203697