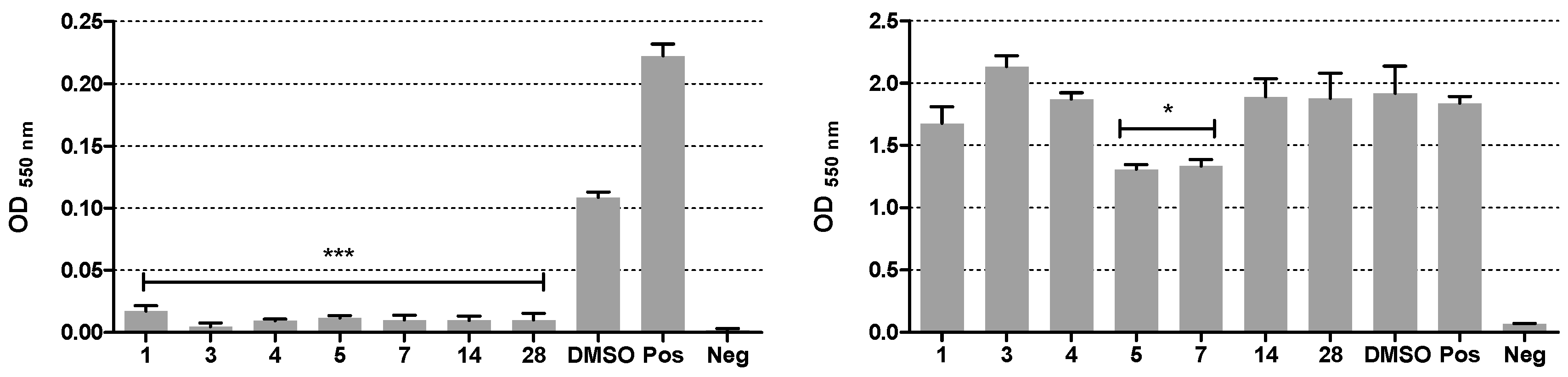
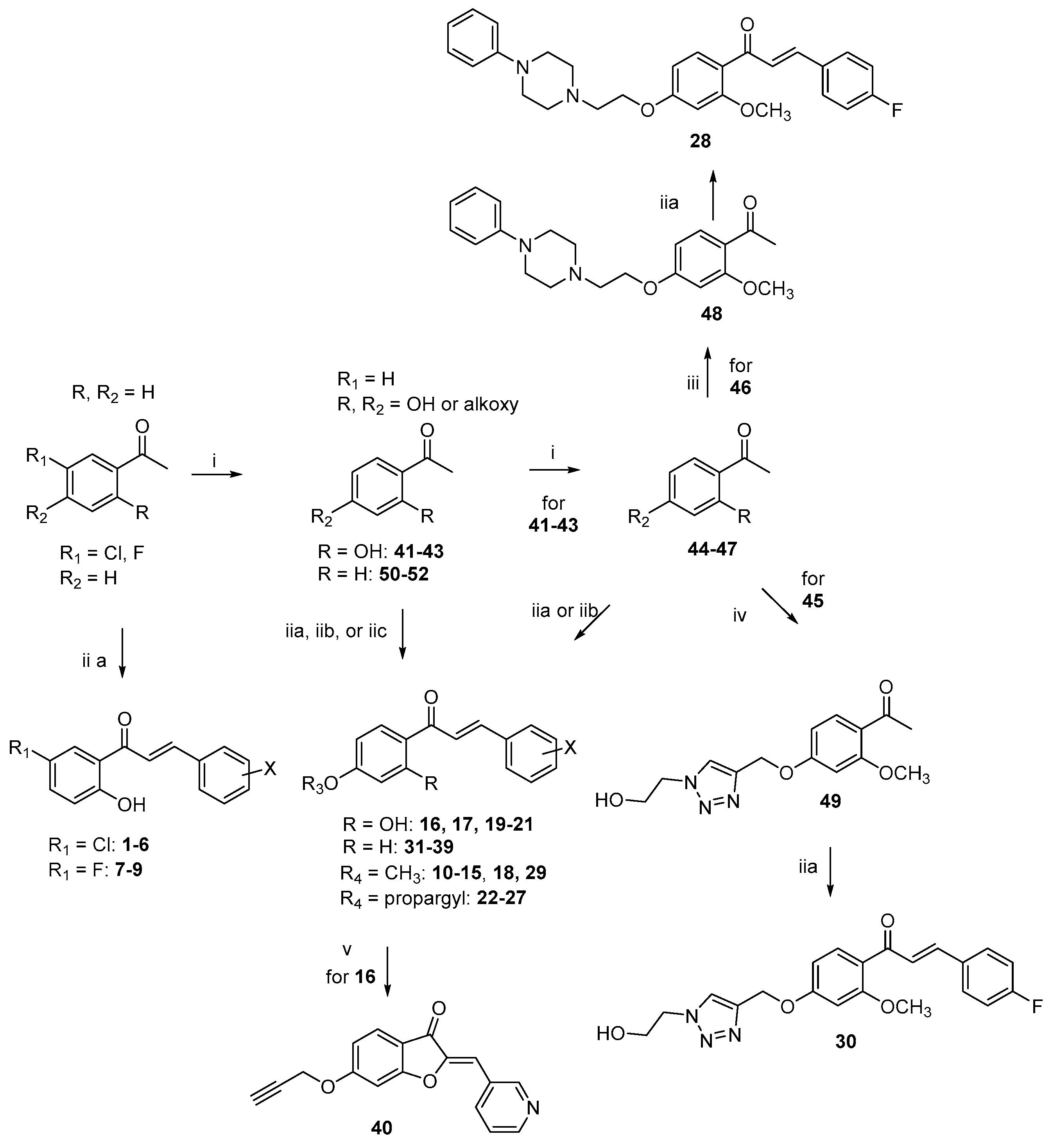
Starting materials, unless otherwise specified, were used as high-grade commercial products. Solvents were of analytical grade. Reaction progress was followed by thin-layer chromatography (TLC) on precoated silica gel plates (Merck Silica Gel 60 F254, Darmstadt, Germany) and then visualized with a UV 254 lamplight. Chromatographic separations were performed on Merck silica gel columns by the flash method (Kieselgel 40, 0.040–0.063 mm, Merck, Darmstadt, Germany). Melting points were determined in open glass capillaries, using a Büchi SMP-20 apparatus (Büchi Italia s.r.l., Cornaredo, Mi, Italy) and are uncorrected. 1H-NMR and 13C-NMR spectra were recorded on a Varian Gemini spectrometer (Scientific instruments, Palo Alto, CA, USA) 400 MHz and 101 MHz, respectively, and chemical shifts (δ) are reported as parts per million (ppm) values relative to tetramethylsilane (TMS) as internal standard; unless otherwise indicated, CDCl3 was used as the solvent. Standard abbreviations indicating spin multiplicities are given as follows: s (singlet), d (doublet), t (triplet), br (broad), q (quartet), or m (multiplet); coupling constants (J) are reported in Hertz (Hz). Mass spectra were recorded on a Waters ZQ 4000 apparatus Waters Alliance, San Diego, CA, USA) operating in electrospray mode (ES). Analyses indicated by the symbols of the elements were within ± 0.4% of the theoretical values. Compounds were named relying on the naming algorithm developed by CambridgeSoft Corporation and used in Chem-BioDraw Ultra 14.0 (PerkinElmer Inc., Waltham, MA, USA).
4.1.2. Claisen–Schmidt Reaction: General Procedure
Route A. To a solution of the selected acetophenone (1.0 eq) and aldehyde (1.1 eq) in EtOH (10 mL) a KOH aqueous solution (50% p/v, 1 mL) was added dropwise and reaction mixture was stirred overnight at rt, diluted with H2O, and neutralized with aqueous 6N HCl. The formed solid/semisolid product was collected by vacuum filtration or extracted with a suitable solvent. The crude of reaction was then purified by column chromatography or by crystallization.
Route B. To a solution the selected acetophenone (1.0 eq) and aldehyde (1.1 eq) in EtOH (10 mL), a solution of piperidine (0.5 mL) and acetic acid (0.3 mL) in EtOH (1 mL) was added. The reaction mixture was heated under reflux for 12–18 h and the reaction progress was monitored by TLC. The solvent was removed under reduced pressure and the crude of reaction was purified by column chromatography or crystallization.
Route C. A mixture the selected acetophenone (1.0 eq) and aldehyde (1.1 eq) in EtOH (10 mL), Ba(OH)2 (3.3 eq) was added. The reaction mixture was stirred at rt overnight; then diluted with H2O and neutralized with aqueous 6N HCl. The formed solid/semisolid product was collected by vacuum filtration or extracted with a suitable solvent. The crude of reaction was then purified by column chromatography or by crystallization.
(E)-1-(5-Chloro-2-hydroxyphenyl)-3-phenylprop-2-en-1-one (1). Following route A, and starting from 5′-chloro-2′-hydroxyacetophenone (0.17 g, 1.0 mmol) and benzaldehyde (0.12 g, 1.1 mmol) a solid product was obtained that was collected by vacuum filtration. The crude of reaction was purified by crystallization from EtOH producing 1 as a yellowish solid (0.22 g), 85% yield, mp 97–98 °C. 1H-NMR δ 6.98 (d, J = 8.4 Hz, 1H, H-3), 7.43 (dd, J = 1.2 and 8.4 Hz, 2H), 7.33–7.38 (m, 2H, H-3′-H5′), 7-50–7.54 (m, 2H, H-2′ and H-6′), 7.60 (d, J = 16.0 Hz, 1H, β-CH=), 7.86 (1H, d, J = 1.2 Hz, H-6), 8.04 (d, J = 15.6 Hz, 1H, α-CH=), 12.55 (br, 1H, OH). 13C-NMR δ 119.21, 124.45, 127.21, 128.75, 128.77, 130.76, 131.32, 137.14, 145.16, 161.08, 192.11. ESI-MS (m/z) 259 (M + 1). HRMS ESI + [M + 1]: calcd for C15H11ClO2, 259.0526. Found: 259.0528.
(E)-1-(5-Chloro-2-hydroxyphenyl)-3-(2-fluorophenyl)prop-2-en-1-one (2). Following route A, and starting from 5′-chloro-2′-hydroxyacetophenone (0.17 g, 1.0 mmol) and 2-fluorobenzaldehyde (0.14 g, 1.1 mmol), a solid product was obtained that was collected by vacuum filtration and crystallized from EtOH, producing 2 as a white solid (0.13 g), 47% yield, mp 86–87 °C. 1H-NMR δ 7.00 (d, J = 8.8 Hz, 1H, H-3), 7.15–7.20 (m, 1H, Ar), 7.46–7.48 (m, 2H, Ar), 7.67–7.72 (m, 2H, Ar), 7.70 (d, J = 16.0 Hz, 1H, β-CH=), 7.76–7.86 (m, 1H, Ar), 8.04 (d, J = 15.6 Hz, 1H, α-CH=), 12.67 (br, 1H, OH). 13C-NMR δ 115.16, 119.45, 121.21, 123.11, 124.25, 128.27, 128.75, 131.86, 137.32, 145.16, 161.58, 192.31. ESI-MS (m/z): 399 (M+23). HRMS ESI + [M + 1]: calcd for C15H10ClFO2, 277.0431. Found: 277.04533.
(E)-1-(5-Chloro-2-hydroxyphenyl)-3-(3-fluorophenyl)prop-2-en-1-one (3). Following route A, and starting from 5′-chloro-2′-hydroxyacetophenone (0.17 g, 1.0 mmol) and 3-fluorobenzaldehyde (0.14 g, 1.1 mmol), a solid product was obtained that was collected by vacuum filtration, purified by column chromatography (PE/EtOAc 9.5:0.5), and crystallized from EtOH, producing 3 as a yellowish solid (0.13 g), 46% yield, mp 75–77 °C. 1H-NMR δ 7.02 (d, J = 8.8 Hz, 1H, H-5), 7.14-7.17 (m, 1H, Ar), 7.37–7.44 (m, 3H, Ar), 7.49 (dd, J = 2.4 and 8.8 Hz, 1H, H-4), 7.57 (d, J = 15.2 Hz, 1H, β-CH=), 7.87 (d, J = 2.4 Hz, 1H, H-6), 7.91 (d, J = 15.2 Hz, 1H, α-CH=). 13C-NMR δ 113.26, 114.45, 119.91, 123.81, 124.28, 127.77, 130.20, 131.86, 136.22, 161.16, 162.58, 192.51. ESI-MS (m/z) 277 (M + 1). HRMS ESI + [M + 1]: calcd for C15H10ClFO2, 277.0431. Found: 277.0432.
(E)-1-(5-Chloro-2-hydroxyphenyl)-3-(4-fluorophenyl)prop-2-en-1-one (4). Following route A, and starting from 5′-chloro-2′-hydroxyacetophenone (0.17 g, 1.0 mmol) and 4-fluorobenzaldehyde (0.14 g, 1.1 mmol), a solid product was obtained that was collected by vacuum filtration and crystallized from EtOH, to obtain 4 as a white solid (0.21 g), 75% yield, mp 79–81 °C. 1H-NMR δ 6.99 (d, J = 8.8 Hz, 1H, H-5), 7.13 (d, J = 8.8 Hz, 1H, H-3′), 7.15 (d, J = 8.8 Hz, 1H, H-5′), 7.45 (dd, J = 2.4 and 8.8 Hz, 1H, H-4) 7.49 (d, J = 15.2 Hz, 1H, β-CH=), 7.68 (d, J = 8.4 Hz, 1H, H-2′), 7.69 (d, J = 8.8 Hz, 1H, H-6′), 7.80 (d, J = 2.4 Hz, 1H, H-6), 7.92 (d, J = 15.2 Hz, 1H, α-CH=). 13C-NMR δ 116.25, 116.47, 119.19, 120.29, 120.54, 123.57, 128.75, 130.85 136.14, 145.16, 162.08, 192.64. ESI-MS (m/z) 277 (M + 1). HRMS ESI + [M + 1]: calcd for C15H10ClFO2, 277.0431. Found: 277.0433.
(E)-1-(5-Chloro-2-hydroxyphenyl)-3-(3,5-difluorophenyl)prop-2-en-1-one (5). Following route A, and starting from 5′-chloro-2′-hydroxyacetophenone (0.17 g, 1.0 mmol) and 3,5-di-fluorobenzaldehyde (0.16 g, 1.1 mmol), a solid product was obtained that was collected by vacuum filtration and crystallized from EtOH, to obtain 5 as a white solid (0.20 g), 67% yield, mp 102–103 °C. 1H-NMR δ 6.92 (t, J = 8.4 Hz 1H, H-4′), 7.02 (d, J = 9.2 Hz, 1H, H-3), 7.20 (d, J = 6.0 Hz, 2H, H-2′ and H-6′), 7.48 (dd, J = 2.0 and 8.8 Hz, 1H, H-4), 7.55 (d, J = 15.6 Hz, 1H, β-CH=), 7.85 (s, 1H, H-6), 7.83 (d, J = 15.6 Hz, 1H, α-CH=), 12.98 (br, 1H, OH). 13C-NMR δ 103.25, 109.47, 119.33, 124.29, 127.53, 130.57, 131.75, 137.14, 138.99, 160.68, 192.33. ESI-MS (m/z) 295 (M + 1). HRMS ESI + [M + 1]: calcd for C15H9ClF2O2, 295.0337. Found: 295.0339.
(E)-1-(5-chloro-2-hydroxyphenyl)-3-(4-(dimethylamino)phenyl)prop-2-en-1-one (6). Following route B, and starting from 5′-chloro-2′-hydroxyacetophenone (0.17 g, 1.0 mmol) and 4-(dimethylamino)benzaldehyde (0.16 g, 1.1 mmol), a crude of reaction was obtained purified by column chromatography (PE/EtOAc 7:3), and crystallized from EtOH to obtain 6 as a red solid (0.20 g), 67% yield, mp 125–127 °C. 1H-NMR δ 3.10 (s, 6H, CH3), 6.79 (d, J = 8.4 Hz, 2H, H-2′ and H-6′), 6.97 (d, J = 9.2 Hz, 1H, H-3), 7.38 (d, J = 15.2 Hz, 1H, β-CH=), 7.41 (dd, J = 2.4 and 8.8 Hz, 1H, H-4), 7.62 (d, J = 8.4 Hz, 2H, H-3′ and H-5′), 7.88 (d, J = 2.8 Hz, 1H, H-6), 7.95 (d, J = 14.8 Hz, 1H, α-CH=), 13.10 (br, 1H, OH). 13C-NMR δ 40.08, 111.77, 113.41, 120.01, 121.03, 122.05, 123.15, 128.52, 131.14, 135.31, 147.58, 152.54, 161.94. ESI-MS (m/z) 302 (M + 1). HRMS ESI + [M + 1]: calcd for C17H16ClNO2, 302.0948. Found: 302.0950.
(E)-1-(5-Fluoro-2-hydroxyphenyl)-3-(3-fluorophenyl)prop-2-en-1-one (7). Following route A, and starting from 5′-fluoro-2′-hydroxyacetophenone (0.15 g, 1.0 mmol) and 3-fluorobenzaldehyde (0.14 g, 1.1 mmol), a solid product was obtained that was collected by vacuum filtration and crystallized from EtOH to obtain 7 as a white solid (0.18 g), 70% yield, mp 92–94 °C. 1H-NMR δ 7.00–7.04 (m, 1H, Ar), 7.15–7.19 (m, 1H, Ar), 7.26–7.25 (m, 1H, Ar), 7.37–7.39 (m, 1H, Ar), 7.41–7.45 (m, 2H, Ar), 7.54 (d, J = 15.6 Hz, 1H, β-CH=), 7.56–7.29 (m, 1H), 7.90 (d, J = 15.2 Hz, 1H, α-CH=). 13C-NMR δ 110.22, 114.44, 118.29, 120.29, 123.44, 123.56, 125.57, 130.45, 136.14, 145.76, 159.11, 162.08, 192.67. ESI-MS (m/z) 261 (M + 1). HRMS ESI + [M + 1]: calcd for C15H10F2O2, 261.0727. Found: 261.0725.
(E)-1-(5-Fluoro-2-hydroxyphenyl)-3-(4-fluorophenyl)prop-2-en-1-one (8). Following route A, and starting from 5′-fluoro-2′-hydroxyacetophenone (0.15 g, 1.0 mmol) and 4-fluorobenzaldehyde (0.14 g, 1.1 mmol), a semisolid product was obtained that was extracted with DCM (3 × 30 mL). The unified organic layers were dried over anhydrous Na2SO4 and evaporated to dryness to give a crude that was purified by column chromatography (PE/EtOAc 95:0.5) and then crystallized from EtOAc/n-hexane, to produce 8 as a white solid (0.21 g), 80% yield, mp 94–96 °C. 1H-NMR δ 7.02 (dd, J = 8.8 and 9.6 Hz, 1H, H-3), 7.17 (d, J = 8.4 Hz, 1H, H-3′), 7.18 (d, J = 8.4 Hz, 1H, H-5′), 7.23-7.26 (m, 1H, H-4), 7.48 (d, J = 15.2 Hz, 1H, β-CH=), 7.58 (dd, J = 2.4 and 8.8 Hz, 1H, H-6), 7.68 (d, J = 8.8 Hz, 1H, H-2′), 7.69 (d, J = 8.4 Hz, 1H, H-6′), 7.92 (d, J = 15.2 Hz, 1H, α-CH=), 12.55 (s, 1H, OH). 13C-NMR δ 116.26, 116.67, 119.21, 120.27, 120.76, 123.69, 128.69, 130.23, 136.44, 159.90, 162.69, 192.44. ESI-MS (m/z) 261 (M + 1). HRMS ESI + [M + 1]: calcd for C15H10F2O2, 261.0727. Found: 261.0728.
(E)-3-(4-(Dimethylamino)phenyl)-1-(5-fluoro-2-hydroxyphenyl)prop-2-en-1-one (9). Following route B, and starting from 5′-fluoro-2′-hydroxyacetophenone (0.15 g, 1.0 mmol) and 4-(dimethylamino)benzaldehyde (0.16 g, 1.1 mmol) a crude of reaction was obtained, purified by column chromatography (PE/EtOAc 7:3), and crystallized from EtOH to obtain 9 as a red solid (0.14 g), 50% yield, mp 131–135 °C. 1H-NMR δ 3.09 (s, 6H, CH3), 6.75 (d, J = 9.2 Hz, 2H, H-3′ and H-5′), 6.97 (dd, J = 8.8 and 8.8 Hz, 1H, H-3), 7.24 (dd, J = 7.6 and 8.8 Hz, 1H, H-4), 7.34 (d, J = 15.2 Hz, 1H, β-CH=), 7.57–7.60 (m, 3H, H-6, H-3′, and H-5′), 7.94 (d, J = 15.2 Hz, 1H, α-CH=), 12.89 (s, 1H, OH). 13C-NMR δ 41.35, 110.41, 117.55, 117.23, 124.78, 125.88, 145.21, 150.61, 159.41, 159.95, 193.57. ESI-MS (m/z) 286 (M + 1). HRMS ESI + [M + 1]: calcd for C17H16FNO2, 286.1243. Found: 286.1244.
(E)-3-(4-fluorophenyl)-1-(2-methoxy-4-(prop-2-yn-1-yloxy)phenyl)prop-2-en-1-one (10). Following route A, and starting from 45 (0.20 g, 1.0 mmol) and 4-fluorobenzaldehyde (0.14 g, 1.1 mmol), a semisolid product was obtained that was extracted with EtOAc (3 × 30 mL). The unified organic layers were dried over anhydrous Na2SO4 and evaporated to dryness to give a crude that was purified by column chromatography (PE/EtOAc 8:2), to produce 10 as a solid (0.29 g), 94% yield, mp 196–168 °C. 1H-NMR δ 2.58 (t, J = 2.4 Hz, 1H, CH), 3.92 (s, 3H, OCH3), 4.77 (d, J = 2.0 Hz, 2H, OCH2), 6.61 (d, J = 2.0 Hz, 1H, H-3), 6.65 (dd, J = 2.4 and 8.8 Hz, 1H, H-5), 7.09 (d, J = 8.8 Hz, 1H, H-3′), 7.10 (d, J = 8.8 Hz, 1H, H-6′), 7.43 (d, J = 16.0 Hz, 1H, β-CH=), 7.50-7.59 (m, 2H, H-2′ and H-6′), 7.65 (d, J = 16 Hz, 1H, α-CH=), 7.76 (d, J = 8.8 Hz, 1H, H-6). 13C-NMR δ 56.21, 76.54, 101.11, 107.45, 115.88, 118.66, 123.67, 130.11, 130.54, 131.87, 145.64, 162.46, 163.78, 188.32. ESI-MS (m/z) 311 (M + 1). HRMS ESI + [M + 1]: calcd for C19H15FO3, 311.1083. Found: 311.1085.
(E)-3-(2,4-dichlorophenyl)-1-(2-methoxy-4-(prop-2-yn-1-yloxy)phenyl)prop-2-en-1-one (11). Following route A, and starting from 45 (0.20 g, 1.0 mmol) and 2,4-dichlorobenzaldehyde (0.19 g, 1.1 mmol) a solid product was obtained that was collected by vacuum filtration and crystallized from EtOH to give 11 as a pale yellow solid (0.29 g), 84% yield, mp 174–75 °C. 1H-NMR δ 2.59 (t, J = 2.4 Hz, 1H, CH), 3.92 (s, 3H, OCH3), 4.78 (s, 2H, OCH2), 6.61 (d, J = 2.0 Hz, 1H, H-3), 6.66 (dd, J = 2.4 and 8.8 Hz, 1H, H-5), 7.28 (dd, J = 2.2 and 8.4 Hz 1H, H-5′), 7.45 (d, J = 2.2 Hz, 1H, H-3′), 7.47 (d, J = 16.0 Hz, 1H, β-CH=), 7.64 (d, J = 8.8 Hz, 1H, H-6′), 7.79 (d, J = 8.8 Hz, 1H, H-6), 7.98 (d, J = 16.0 Hz, 1H, α-CH=). 13C-NMR δ 55.81, 56.71, 76.42, 77.66, 101.10, 107.54, 121.56, 122.47, 124.94, 125.98, 127.15, 129.74, 130.45, 1.3.31, 136.60, 161.74, 167.21, 194.11. ESI-MS (m/z) 361 (M+1). HRMS ESI + [M + 1]: calcd for C19H14Cl2O3, 361.0398. Found: 361.0399.
(E)-3-(4-(dimethylamino)phenyl)-1-(2-methoxy-4-(prop-2-yn-1-yloxy) phenyl)prop-2-en-1-one (15). Following route B, and starting from 45 (0.19 g, 1.0 mmol) 4-(dimethylamino)benzaldehyde (0.16 g, 1.1 mmol) a crude of reaction was obtained that was purified by column chromatography (PE/EtOAc 7:3), and crystallized from EtOH to obtain 15 as a red solid (0.20 g), 62% yield, mp 137–149 °C. 1H-NMR δ 2.57 (t, J = 2.4 Hz, 1H, CH), 3.04 (s, 6H, CH3), 3.89 (s, 3H, OCH3), 4.76 (d, J = 2.4 Hz, 2H, OCH2), 6.60 (s, 1H, H-3), 6.63 (dd, J= 2.4 and 10.4 Hz, 1H, H-5), 6.69 (d, J = 8.8 Hz, 2H, H-6′ and H-2′), 7.26 (d, J = 15.6 Hz, 1H, β-CH=), 7.63 (d, J = 15.6 Hz, 1H, α-CH=), 7.69 (d, J = 8.8 Hz, 1H, H-6). 13C-NMR δ 41.34, 56.18, 55.21, 76.44, 78.76, 101.38, 106,96, 111.15, 113.24, 118.91, 123.96, 124.99, 145.12, 152.45, 163.07, 166.21, 198.10. ESI-MS (m/z) 336 (M + 1). HRMS ESI + [M + 1]: calcd for C21H21NO3, 336.1599. Found: 336.1597.
(E)-3-(4-(dimethylamino)phenyl)-1-(2-hydroxy-4-(prop-2-yn-1-yloxy)phenyl)prop-2-en-1-one (17). Following route B, and starting from 42 (0.20 g, 1.0 mmol) 4-(dimethylamino)benzaldehyde (0.16 g, 1.1 mmol), a crude of reaction was obtained that was purified by column chromatography (PE/EtOAc 7:3), and crystallized from EtOH to obtain 17 as a red solid (0.20 g), 62% yield, mp 137–149 °C. 1H-NMR δ 2.59 (t, J = 2.4 Hz, 1H, CH), 3.92 (s, 3H, OCH3), 4.78 (s, 2H, OCH2), 6.61 (d, J = 2.0 Hz, 1H, H-3), 6.66 (dd, J = 2.4 and 8.8 Hz, 1H, H-5), 7.28 (dd, J = 2.2 and 8.4 Hz 1H, H-5′), 7.45 (d, J = 2.2 Hz, 1H, H-3′), 7.47 (d, J = 16.0 Hz, 1H, β-CH=), 7.64 (d, J = 8.8 Hz, 1H, H-6′), 7.79 (d, J = 8.8 Hz, 1H, H-6), 7.98 (d, J = 16.0 Hz, 1H, α-CH=). 13C-NMR δ 55.81, 56.71, 76.42, 77.66, 101.10, 107.54, 121.56, 122.47, 124.94, 125.98, 127.15, 129.74, 130.45, 1.3.31, 136.60, 161.74, 167.21, 194.11. ESI-MS (m/z) 322 (M + 1). HRMS ESI + [M + 1]: calcd for C20H19NO3, 322.1443. Found: 322.1445.
(E)-3-(2,4-dichlorophenyl)-1-(2-hydroxy-4-((3-methylbut-2-en-1-yl)oxy)phenyl)prop-2-en-1-one (19). Following route A, and starting from 41 (0.22 g, 1.0 mmol) and 2,4-dichlorobenzaldehyde (0.19 g, 1.1 mmol) a solid product was obtained that was collected by vacuum filtration and crystallized from EtOH to give 19 as a pale yellow solid (0.33 g), 88% yield, mp 191–93 °C. 1H-NMR δ 1.77 (s, 3H, CH3), 1.83 (s, 3H, CH3), 4.59 (d, J = 6.8 Hz, 2H, OCH2), 5.50 (t, J = 6.8 Hz, 1H, CH), 6.51 (d, J = 7.2 Hz, 1H, H-5), 6.52 (s, 1H, H-3), 7.32 (dd, J = 2.2 and 6.8 Hz, 1H, H-5′), 7.50 (d, J = 2.0 Hz, 1H, H-3′), 7.69 (d, J = 8.8 Hz, 1H, H-6′), 7.55 (d, J = 15.6 Hz, 1H, β-CH=), 7.80 (d, J = 9.2 Hz, 1H, H-6), 8.19 (d, J = 15.6 Hz, 1H, α-CH=), 13.3 (s, 1H, OH). 13C-NMR δ 18.23, 24.67, 64.64, 103.12, 107.75, 113.93, 119.45, 121.43, 124.11, 128.31, 130.00, 131.05, 131. 77, 138.54, 145.46, 165.92, 169.03, 194.34. ESI-MS (m/z) 377 (M + 1). HRMS ESI + [M + 1]: calcd for C20H18Cl2O3, 377.0711. Found: 377.0713.
(E)-1-(2,4-bis(prop-2-yn-1-yloxy)phenyl)-3-(2,4-dichlorophenyl)prop-2-en-1-one (23). Following route A, and starting from 47 (0.23 g, 1.0 mmol) and 2,4-dichlorobenzaldehyde (0.19 g, 1.1 mmol) a solid product was obtained that was collected by vacuum filtration and crystallized from EtOH to give 23 as a pale yellow solid (0.30 g), 78% yield, mp 166–68 °C. 1H-NMR δ 2.58-2.62 (m, 2H, CH), 4.77 (d, J = 2.4 Hz, 2H, OCH2), 4.79 (d, J = 2.4 Hz, 2H, OCH2), 6.70 (d, J = 2.0 Hz, 1H, H-3), 6.73 (dd, J = 2.0 and 8.0 Hz, 1H, H-5), 7.26 (dd, J = 2.0 and 8.0 Hz, 1H, H-5′), 7.45 (d, J = 2.0 Hz, 1H, H-3′), 7.54 (d, J = 16.0 Hz, 1H, β-CH=), 7.70 (d, J = 8.4 Hz, 1H, H-6′), 7.82 (d, J = 8.4 Hz, 1H, H-6), 7.99 (d, J = 15.6 Hz, 1H, α-CH=). 13C-NMR δ 56.44, 75.16, 78.92, 101.33, 107.21, 130.55, 121.98, 124.27, 126.19, 127.54, 128.23, 131.29, 137.37, 148.32, 162.62, 166.88, 193.71. ESI-MS (m/z) 385 (M + 1). HRMS ESI + [M + 1]: calcd for C21H14Cl2O3, 385.0398. Found: 385.0398.
(E)-1-(2,4-bis(prop-2-yn-1-yloxy)phenyl)-3-(4-(dimethylamino)phenyl)prop-2-en-1-one (27). Following route B, and starting from 47 (0.23 g, 1.0 mmol) and 4-(dimethylamino)benzaldehyde (0.16 g, 1.1 mmol), a crude of reaction was obtained that was purified by column chromatography (PE/EtOAc 7:3), and crystallized from EtOH to obtain to obtain 27 as a red solid (0.19 g), 55% yield, mp 127–129 °C. 1H-NMR δ 2.56–2.61 (m, 2H, CH), 3.02 (s, 3H, CH3), 3.02 (s, 3H, CH3), 4.72–4.78 (m, 4H, OCH2), 6.53 (dd, J = 2.0 and 8.4 Hz, 1H, H-5), 6.56 (d, J = 2.4 Hz, 1H, H-3), 6.7(d, J = 8.4 Hz, 2H, H-3′ and H-5′), 7.36 (d, J = 15.2 Hz, 1H, β-CH=), 7.58 (d, J = 8.4 Hz, 2H, H-2′ and H-6′), 7.87 (d, J = 8.8 Hz, 1H, H-6), 7.89 (d, J = 15.2 Hz, 1H, α-CH=). 13C-NMR δ 41.41, 41.49, 56.43, 56.63, 76.11, 76.32, 78.78, 78.92, 101.33, 107.64, 111.32, 118.32, 118.54, 123.564, 124.32, 129.95, 131.12, 145.63, 151.01, 162.90, 165.17, 196.15. ESI-MS (m/z) 360 (M + 1). HRMS ESI + [M + 1]: calcd for C23H21NO3, 360.1599. Found: 360.1597.
(E)-3-(4-Fluorophenyl)-1-(2-methoxy-4-(2-(4-phenylpiperazin-1-yl)ethoxy)phenyl)prop-2-en-1-one (28). Following route A, and starting from 48 (0.35 g, 1.0 mmol) and 4-fluorobenzaldehyde (0.14 g, 1.1 mmol), a semisolid product was obtained that was extracted with DCM (3 × 30 mL). The unified organic layers were dried over anhydrous Na2SO4 and evaporated to dryness to give a crude that was purified by column chromatography (DCM/MeOH 95:5), to produce 28 as a semisolid product (0.15 g), 32% yield, that was converted into the hydrochloride salt, mp 174–176 °C. 1H-NMR δ 3.41–3.45 (m, 2H, piperazine), 3.61–3.45 (m, 2H, piperazine), 3.91–3.95 (m, 4H, piperazine), 3.96 (s, 3H, OCH3), 4.19 (t, J = 5.6 Hz, 2H, OCH2), 4.89(t, J = 5.6 Hz, 2H, NCH2), 6.55 (dd, J = 8.4 and 2.2 Hz, 1H, H-5), 6.62 (d, J = 2.2 Hz, 1H, H-3), 6.94–7.06 (m, 1H, Ph), 7.06–7.11 (m, 2H, Ph), 7.34–7.38 (m, 2H, Ph), 7.36 (d, J = 15.6 Hz, 1H, β-CH=), 7.52–7.56 (m, 2H, H-2′ and H-6′), 7.61 (d, J = 15.6 Hz, 1H, α-CH=), 7.69 (d, J = 8.8 Hz, 1H, H-6). 13C-NMR δ 49.27, 53.81, 57.03, 58.47, 66.49, 101.67, 108.12, 114.19, 116.25, 116.47, 119.94, 128.75, 129.26, 130.80, 136.24, 132.45, 145.16, 151.40, 165.36. ESI-MS (m/z) 461 (M + 1). HRMS ESI + [M + 1]: calcd for C28H29FN2O3, 461.2240. Found: 461.2142.
(E)-3-(4-Fluorophenyl)-1-(4-hydroxy-2-methoxyphenyl)prop-2-en-1-one (29). Following route A, and starting from 1-(4-hydroxy-2-methoxyphenyl)ethan-1-one (0.17 g, 1.0 mmol) and 4-fluorobenzaldehyde (0.14 g, 1.1 mmol), a semisolid product was obtained that was extracted with EtOAc (3 × 30 mL). The unified organic layers were dried over anhydrous Na2SO4 and evaporated to dryness to give a crude that was purified by column chromatography (PE/EtOAc 9:1), and crystallized from EtOH to produce 29 as a solid (0.035 g), 13% yield, mp 166–168 °C. 1H-NMR δ 3.89 (s, 3H, OCH3). 6.50 (dd, J = 2.2 and 8.4 Hz, 1H, H-5), 6.51 (d, J =2.2 Hz, 1H, H-3), 7.06-7.10 (m, 2H, H-3′and H-5′), 7.44 (d, J = 16.0 Hz, 1H, β-CH=), 7.55-7.58 (m, 2H, H-2′ and H-6′), 7.61 (d, J = 16.0 Hz, 1H, β-CH=), 764 (d, J = 15.6 Hz, 1H, α-CH=), 7.69 (d, J = 8.8 Hz, 1H, H-6). 13C-NMR δ 102.64, 113.45, 115.24, 115.28, 118.67, 130.25, 130.56, 145.21, 162.49, 162.77, 165.45, 168.88. ESI-MS (m/z) 273 (M + 1). HRMS ESI + [M + 1]: calcd for C16H13FO3, 273.0927. Found: 273.0925.
(E)-3-(4-fluorophenyl)-1-(4-((1-(2-hydroxyethyl)-1H-1,2,3-triazol-4-yl)methoxy)-2-methoxyphenyl)prop-2-en-1-one (30). Following route A, and starting from 49 (0.29 g, 1.0 mmol) and 4-fluorobenzaldehyde (0.14 g, 1.1 mmol), a solid product was obtained that was collected by vacuum filtration and purified by column chromatography (PE/EtOAc 1:1), to produce 30 as a white solid (0.079 g), 20% yield, mp 100–102 °C. 1H-NMR δ 2.93 (br, 1H, OH), 3.88 (s, 3H, OCH3), 4.10 (t, J = 5.2 Hz, 2H, NCH2), 4.55 (t, J = 5.2 Hz, 2H, CH2OH), 5.21 (s, 2H, OCH2), 6.52 (d, J = 2.4 Hz, 1H, H-3), 6.62 (dd, J = 2.4 and 7.6 Hz, 1H, H-5), 7.42–7.49 (m, 2H, H-3′and H-5′), 7.60 (d, J = 15.6 Hz, 1H, β-CH=), 7.63–7.69 (m, 2H, H-2′ and H-6′), 7.80 (s, 1H, triazole CH), 7.74 (d, J = 15.6 Hz, 1H, α-CH=), (7.83 (d, J = 8.0 Hz, 1H, H-6). 13C-NMR δ 54.11, 55.64, 60.57, 72.12, 100.54, 105.21, 115.64, 115.74, 118.52, 123.77, 127.32, 130.47, 131.25, 131.55, 142.84, 145.37, 145.22, 162.34, 167.88, 195.94. ESI-MS (m/z) 398 (M + 1). HRMS ESI + [M + 1]: calcd for C21H20FN3O4, 398.1516. Found: 398.1515.
(E)-3-(4-fluorophenyl)-1-(4-(2-hydroxyethoxy)phenyl)prop-2-en-1-one (31). Following route A, and starting from 50 (0.18 g, 1.0 mmol) and 4-fluorobenzaldehyde (0.14 g, 1.1 mmol), a solid product was obtained that was collected by vacuum filtration and purified by column chromatography (PE/EtOAc 4:1), to produce 31 as a white solid (0.19 g), 67% yield. 1H-NMR (acetone d6) δ 3.92–3.96 (m, 2H, CH2OH), 4.02–4.04 (m, 1H, OH), 4.18 (t, J = 6.4 Hz, 2H, CH2O), 7.05 (d, J = 6.8 Hz, 2H, H-3 and H-5), 7.20 (t, J = 8.8 Hz, 2H, H-3′ and H-5′), 7.72 (d, J = 15.6 Hz, 1H, β-CH=), 7.81 (d, J = 15.6 Hz, 1H, α-CH=), 7.83-7.88 (m, 2H, H-2′ and H-6′), 8.12 (d, J = 6.8 Hz, 2H, H-2 and H-6). 13C-NMR δ (acetone d6) 60.9, 69.9, 114.12, 116.80, 120.54, 129.65, 130.22, 130.97, 131.15, 131.45, 145.16, 160.21, 168. 01. ESI-MS (m/z) 287 (M + 1). HRMS ESI + [M + 1]: calcd for C17H15FO3, 287.1083. Found: 287.1085.
(E)-1-(4-((3-methylbut-2-en-1-yl)oxy)phenyl)-3-(pyridin-3-yl)prop-2-en-1-one (32). Following Route C, and starting from 51 (0.20 g, 1.0 mmol) and pyridine-3-carboxyaldehyde (0.12 g, 1.1 mmol) a solid product was obtained that was collected by vacuum filtration and crystallized from EtOH to give 32 (0.17 g), 60% yield, mp 87–88 °C. 1H-NMR δ 1.78 (s, 3H, CH3), 1.83 (s, 3H, CH3), 4.62 (d, J = 6.8 Hz, 2H, OCH2), 5.11 (t, J = 1.2 Hz, 1H, CH), 7.01 (d, J = 8.8 Hz, 2H, H-3 and H-5), 7.35-7.39 (m, 1H, H-5′), 7.62 (d, J = 15.6 Hz, 1H, β-CH=), 7.79 (d, J = 15.6 Hz, 1H, α-CH=), 7.95 (dd, J = 1.6 and 7.6 Hz, 1H, H-4′), 8.05 (d, J = 8.8 Hz, 2H, H-2 and H-6), 8.63 (dd, J = 1.6 and 7.6 Hz, 1H, H-4′), 8.87 (d, J = 2.0 Hz, 1H, H-2′). 13C-NMR δ 54.21, 55.87, 60.02, 72.15, 101.33, 105.74, 113.11, 118.21, 123.44, 128.64, 130.21, 130.31, 130.56, 131.64, 142.85, 162.18, 162.47. 163.54, 167.04, 195.32. ESI-MS (m/z) 294 (M + 1). HRMS ESI + [M + 1]: calcd for C19H19NO2, 294.1494. Found: 294.1495.
(E)-1-(4-((3-methylbut-2-en-1-yl)oxy)phenyl)-3-(pyridin-4-yl)prop-2-en-1-one (33). Following Route C, and starting from 51 (0.20 g, 1.0 mmol) and pyridine-4-carboxyaldehyde (0.12 g, 1.1 mmol) a solid was obtained that was filtered under vacuum and crystallized from EtOH to give 33 (0.21 g), 77% yield, mp 91–93 °C. 1H-NMR δ 1.78 (s, 3H, CH3), 1.83 (s, 3H, CH3), 4.62 (d, J = 7.2 Hz, 2H, OCH2), 5.51 (t, J = 6.8 Hz, 1H, CH), 7.01 (d, J = 8.8 Hz, 2H, H-3 and H-5), 7.48 (d, J = 7.2 Hz, 2H, H-2′ and H-6′), 7.63 (d, J = 15.6 Hz, 1H, β-CH=), 7.71 (d, J = 15.6 Hz, 1H, α-CH=), 8.04 (d, J = 8.4 Hz, 2H, H-2 and H-6), 8.69 (d, J = 7.2 Hz, 2H, H-3′ and H-5′). 13C-NMR δ 18.32, 18.57, 21.41, 64.21, 114.22, 119.65, 123.74, 127.88, 130.51, 138.08, 144.00, 149.16, 164.77, 191.66. ESI-MS (m/z) 294 (M + 1). HRMS ESI + [M + 1]: calcd for C19H19NO2, 294.1494. Found: 294.1492.
(E)-1-(4-((3-methylbut-2-en-1-yl)oxy)phenyl)-3-(4-nitrophenyl)prop-2-en-1-one (34). Following Route C and starting from 51 (0.20 g, 1.0 mmol) and of 4-nitrobenzaldheyde (0.17 g; 1.1 mmol) a solid was obtained that was filtered under vacuum and purified by crystallization from EtOH to give 34 as a brown powder, (0.26 g) 70% yield, mp 100–102 °C. 1H-NMR δ 1.79 (s, 3H, CH3), 1.85 (s, 3H, CH3), 4.62 (d, J = 7.2 Hz, 2H, OCH2), 5.51 (t, J = 6.8 Hz, 1H, CH), 7.00 (d, J = 8.4 Hz, 2H, H-3 and H-5), 7.63 (d, J = 15.6 Hz, 1H, β-CH=), 7.79 (d, J = 8.0 Hz, 2H, H-2′ and H-6′), (d, J = 15.6 Hz, 1H, β-CH=), 7.90 (d, J = 15.6 Hz, 1H, α-CH=), 8.07 (d, J = 9.2 Hz, 2H, H-2 and H-6), 8.29 (d, J = 8.8 Hz, 2H, H-3′ and H-5′). 13C-NMR δ 18.52, 24.11, 64.08, 114.63, 119.08, 121.08, 123.87, 129.45, 129.95, 138.11, 145.36, 163.21, 189.32. ESI-MS (m/z) 338 (M + 1). HRMS ESI + [M + 1]: calcd for C20H19NO4, 338.1392. Found: 338.1394.
(E)-3-(4-(dimethylamino)phenyl)-1-(4-((3-methylbut-2-en-1-yl)oxy)phenyl)prop-2-en-1-one (35). Following Route B, and starting from 51 (0.20 g, 1.0 mmol) and 4-(dimethylamino)benzaldehyde (0.16 g, 1.1 mmol), a red solid was obtained that was filtered under vacuum and crystallized from EtOH to give 35 as a red powder (0.03 g), 15% yield, 121–123 °C. 1H-NMR δ 1.77 (s, 3H, CH3), 1.82 (s, 3H, CH3), 3.05 (s, 6H, NCH3), 4.60 (d, J = 6.4 Hz, 2H, OCH2), 5.51 (t, J = 6.8 Hz, 1H, CH), 6.70 (d, J = 8.4 Hz, 2H, H-2′ and H-6′), 6.98 (d, J = 8.8 Hz, 2H, H-3 and H-5), 7.36 (d, J = 15.6 Hz, 1H, β-CH=), 7.55 (d, J = 9.2 Hz, 2H, H-3′ and H-5′), 7.79 (d, J = 15.6 Hz, 1H, α-CH=), 8.02 (d, J = 8.8 Hz, 2H, H-2 and H-6). 13C-NMR δ 18.8, 24.36, 41.35, 64.02, 11.33, 111.45, 111.66, 114.25, 119.57, 121.18, 124.61, 129.72, 130.12, 130.97, 131.25, 145.74, 150.01, 163.75, 189.22. ESI-MS (m/z) 336 (M + 1). HRMS ESI + [M + 1]: calcd for C22H25NO2, 336.1963. Found: 336.1964.
(E)-1-(4-(prop-2-yn-1-yloxy)phenyl)-3-(pyridin-3-yl)prop-2-en-1-one (36). Following Route C, and starting from 52 (0.17 g, 1.0 mmol) and pyridine-3-carboxyaldehyde (0.12 g, 1.1 mmol) a solid was obtained that was filtered under vacuum and crystallized from EtOH to give 36 (0.17 g), 67% yield, mp 96–98 °C. 1H-NMR δ 2.58 (d, J = 2.0 Hz, 1H, CH), 4.82 (d, J = 2.4 Hz, 2H, OCH2), 7.09 (d, J = 9.2 Hz, 2H, H-3 and H-5), 7.33–7.39 (m, 1H, H-5′), 7.61 (d, J = 16.0 Hz, 1H, β-CH=), 7.80 (d, J = 15.6 Hz, 1H, α-CH=), 7.93 (dd, J = 1.6 and 8.8 Hz, 1H, H-4′), 8.07 (d, J = 8.8 Hz, 2H, H-2 and H-6), 8.64 (dd, J = 1.6 and 8.8 Hz, 1H, H-3′), 8.87 (d, J = 1.6 Hz, 1H, H-2′). 13C-NMR δ 56.98, 76.11, 87.91, 114.22, 114.65, 123.45. 123.87, 129.64, 130.45, 130.76, 132.16, 141.74, 142.03, 149.84, 163.25, 189.99. ESI-MS (m/z) 264 (M + 1). HRMS ESI + [M + 1]: calcd for C17H13NO2, 264.1024. Found: 264.1022.
(E)-1-(4-(prop-2-yn-1-yloxy)phenyl)-3-(pyridin-4-yl)prop-2-en-1-one (37). Following Route C, and starting from 52 (0.17 g, 1.0 mmol) and pyridine-4-carboxyaldehyde (0.12 g, 1.1 mmol) a solid was obtained that was filtered under vacuum and crystallized from EtOH to give 37 (0.20 g), 80% yield, mp 99–101 °C. 1H-NMR δ 2.57 (t, J = 2.0 Hz, 1H, CH), 4.79 (d, J = 2.4 Hz, 2H, OCH2), 7.09 (d, J = 9.2 Hz, 2H, H-3 and H-5), 7.47 (d, J = 8.0 Hz, 2H, H-2′ and H-6′), 7.66 (d, J = 16.4 Hz, 1H, β-CH=), 7.70 (d, J = 16.4 Hz, 1H, α-CH=), 8.06 (d, J = 8.8 Hz, 2H, H-2 and H-6), 8.69 (d, J = 8.6 Hz, 2H, H-3′ and H-5′). 13C-NMR δ 56.41, 76.11, 78.78, 114.21, 114.68, 123.54, 123.98, 124.54, 127.88, 128.45, 130.02. 130.64, 131.11, 144.56, 149.00, 149.857, 163.24, 189.69. ESI-MS (m/z) 264 (M + 1). HRMS ESI + [M + 1]: calcd for C17H13NO2, 264.1024. Found: 264.1025.
(E)-3-(4-nitrophenyl)-1-(4-(prop-2-yn-1-yloxy)phenyl)prop-2-en-1-one (38). Following Route C, and starting from 52 (0.17 g, 1.0 mmol) and 4-nitrobenzaldheyde (0.17 g; 1.1 mmol) a solid was obtained that was filtered under vacuum and crystallized from EtOH to give 38 as a brown powder, (0.16 g) 54% yield, mp 103–105 °C. 1H-NMR δ 2.58 (d, J = 2.0 Hz, 1H, CH), 4.82 (d, J = 2.4 Hz, 2H, OCH2), 7.09 (d, J = 8.4 Hz, 2H, H-3 and H-5), 7.64 (d, J = 15.6 Hz, 1H, β-CH=), 7.78 (d, J = 8.8 Hz, 2H, H-2′ and H-6′), 7.80 (d, J = 15.6 Hz, 1H, α-CH=), 8.07 (d, 2H, H-3′ and H-5′), 8.87 (s, 1H, H-2), 8.07 (d, J = 9.2 Hz, 2H, H-2 and H-6), 8.28 (d, J = 8.8 Hz, 2H, H-3′ and H-5′). 13C-NMR (CDCl3) δ 56.21, 76.54, 78.11, 114.77, 121.18, 123.87, 129.11, 130.54, 141.07, 145.98, 147.64, 163.74, 189.60. ESI-MS (m/z) 308 (M + 1). HRMS ESI + [M + 1]: calcd for C18H13NO4, 308.0923. Found: 308.0922.
(E)-3-(4-(dimethylamino)phenyl)-1-(4-(prop-2-yn-1-yloxy)phenyl)prop-2-en-1-one (39). Following Route B, and starting from 52 (0.17 g, 1.0 mmol) and 4-(dimethylamino)benzaldehyde (0.16 g, 1.1 mmol), a red solid was obtained that was filtered under vacuum and crystallized from EtOH to give 39 as a red powder (0.03 g), 15% yield, 133–135 °C. 1H-NMR δ 2.56 (t, J = 2.4 Hz, 1H, CH), 3.05 (d, 6H, NCH3), 4.82 (d, J = 2.8 Hz, 2H, OCH2), 6.70 (d, J = 8.4 Hz, 2H, H-3 and H-5), 7.05 (d, J = 9.2 Hz, 2H, H-3′ and H-5′), 7.35 (d, J = 15.6 Hz, 1H, β-CH=), 7.55 (d, J = 8.8 Hz, 2H, H-2′ and H-6′), 7.80 (d, J = 15.6 Hz, 1H, α-CH=), 8.04 (d, J = 8.8 Hz, 2H, H-2 and H-6). 13C-NMR δ 41.12,56.21, 76.44, 78.64, 111.10, 114.26, 121.87, 124.68, 129.39, 129.85, 130.22, 130.51, 145.74, 150.63, 164.08, 189.89. ESI-MS (m/z) 306 (M + 1). HRMS ESI + [M + 1]: calcd for C20H19NO2, 306.1494. Found: 306.1492.
(Z)-6-(prop-2-yn-1-yloxy)-2-(pyridin-3-ylmethylene)benzofuran-3(2H)-one (40). To a solution of 16 (0.17 g, 0.60 mmol) in EtOH (17 mL) at 0 °C, NaOH aq solution (0.16 g, 3.33 mmol, dissolved in 1 mL of H2O) and H2O2 (0.07 mL, 3.33 mmol) were added dropwise. The reaction was stirred at rt for 12 h, then water was added and the solution was acidified with 2N HCl. The solid formed was separated by vacuum filtration, dried, and purified by column chromatography (EtOAc/MeOH 9:1) to obtain 40 as a red solid (0.05 g), 29% yield, mp 121.123 °C. 1H-NMR δ 3.42 (t, J = 2.4, 1H, CH), 4.69 (d, J = 2.4 Hz, 2H, OCH2), 6.26 (d, J = 8.8 Hz, 1H, H-5), 6.28 (s, 1H, H-7), 7.31-7.34 (m, 1H, H-5′), 7.59 (d, J = 8.4 Hz, 1H, H-4), 8.12 (d, J = 8.0 Hz, 1H, H-6′), 8.38 (s, 1H, CH), 8.46 (s, 1H, H-4′), 8.91 (S, 1H, H-2′). 13C-NMR δ 56.21, 76.11, 78.84, 100.00, 109.54, 112.57, 114.08, 123.22, 125.08, 132.11, 132.55, 132.98, 148.64, 149.74, 152.32, 165.09, 167.46, 18.02. ESI-MS (m/z) 278 (M + 1)). HRMS ESI + [M + 1]: calcd for C17H11NO3, 278.0817. Found: 278.0815.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







































