1. Introduction
In main group chemistry, trends within a group play an important role for the understanding of how particular properties of a molecule can be tuned. By selecting a congener, in which an atom is replaced by another element from the same group, the geometric shape of the molecules can be changed. For instance, in organic compounds, carbon atoms can be replaced by heavier tetrels (Si, Ge, Sn and Pb) to give semi-inorganic hydrocarbons [
1]. However, size is not the only change; optophysical properties can also be influenced substantially. For example, it has been shown that for cyclopentadiene analogs, in which the sp
3 substituted C atom is replaced by a heavier group 14 element, the HOMO-LUMO gap (HOMO = highest occupied molecular orbital; LUMO = lowest unoccupied molecular orbital) is reduced due to σ*-π* conjugation [
2,
3,
4,
5,
6]. Due to our interest in both main group chemistry and photoswitchable molecules, we systematically investigated the influence that could be exerted by tetrels in the
ortho-position of azobenzenes, as well as the structural influences this substitution might trigger: Azobenzenes are thermally and photochemically stable molecular switches that can undergo a reversible
trans–
cis isomerization under illumination. The properties of azobenzenes, such as absorption maxima and switching behavior, can be tuned by their substitution pattern [
7,
8,
9,
10,
11].
Ortho-substituted azobenzenes distinguish themselves from most molecular switches by their significantly longer thermal relaxation times of the
cis-isomer back to their
trans-isomers [
12,
13]. This property makes
ortho-substituted azobenzenes suitable for several applications where long half-life times of the thermal relaxation are required, giving rise to new materials and dyes, e.g., external stimuli responsive polymers [
14,
15].
In general,
ortho-azobenzenes can be accessed either by using C–H activation methods with late transition metals like nickel, palladium, platinum or ruthenium [
16,
17], by deprotonation with metal bases [
18,
19], or by direct halogen-lithium exchange [
19,
20]. The latter has been shown to proceed with high efficiency in the synthesis of several
ortho-substituted organometallic azobenzenes (
Scheme 1) [
21].
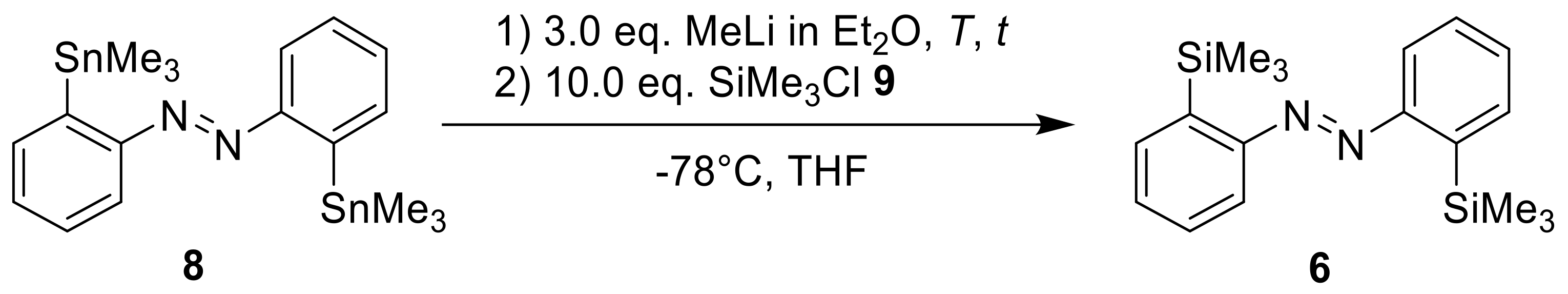
This methodology is superior to the classical azocoupling approach and could be used with a high variety of electrophiles. It should also be mentioned that in this study, the
ortho-bis(silyl) azobenzene (compound
6 in our study) was synthesized by oxidation of the respective amine. This product was used for mono-desilylation to give the monosilylated azobenzene displayed above [
21]. Other metalation methods were used to introduce pentacoordinated silyl atoms in azobenzene motifs [
20,
22]. It was also found that substitution with heavier group 14 elements, which convey a positive inductive effect on the aromatic ring, resulted in a slight bathochromic shift of the absorption maxima compared to unsubstituted azobenzene.
Since we developed a method for the lithiation of azobenzenes by halogen-lithium exchange reactions for
para- and
meta-substituted azobenzenes [
23,
24], we were interested to transfer this methodology towards disubstituted
ortho-azobenzenes. This process involved the synthesis of tin-substituted azobenzenes through a Kelly-Stille cross-coupling reaction, followed by a low temperature tin-lithium exchange. Functionalization could be achieved by quenching the lithiated species with a variety of electrophiles at low temperatures. Initial results showed that the structure-reactivity relationship of the
meta-,
para- and
ortho-azobenzenes in the Kelly-Stille cross-coupling reaction led to significant differences in yields (
para: 93%,
meta: 88% and
ortho: 43%). An advantage of a functionalization methodology that involves stannylated groups in the reagents is the good shelf-life and the potential use of these reagents as nucleophilic components in Stille type cross-coupling reactions. This was already reported for
para-azobenzenes and gives access to substitutions with aromatic or vinylic groups [
25].
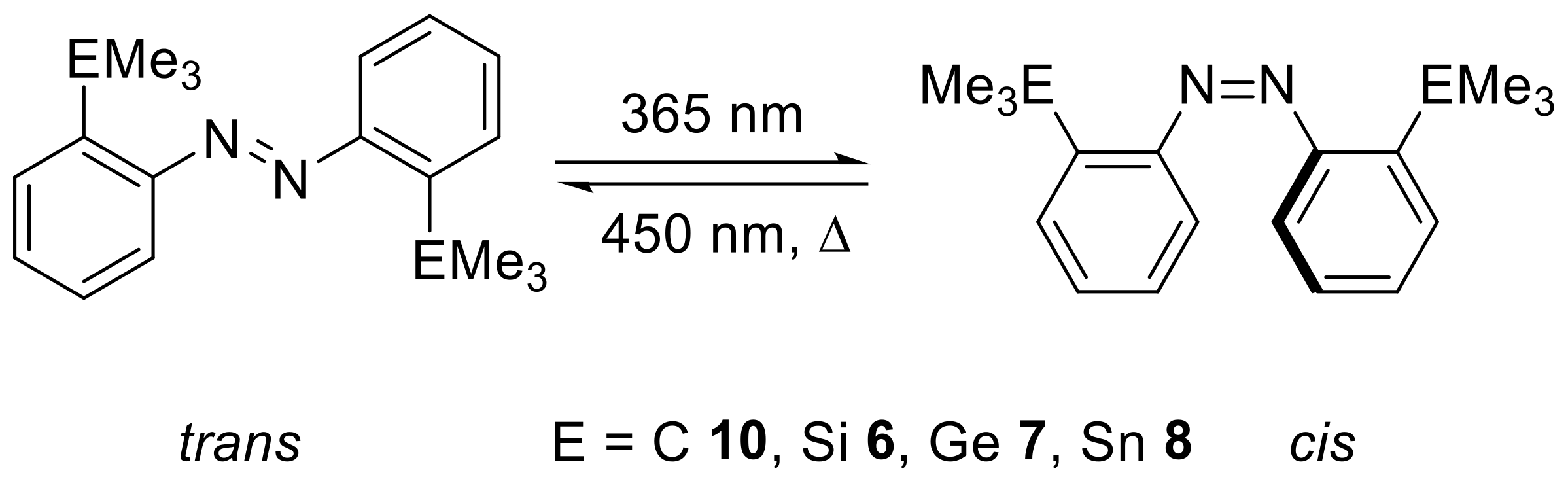
In this work, we were interested in the detail of the structure/property relationship of different tetrel-substituted azobenzenes. In contrast to previous studies, the carbon congener is included and both ortho-positions of the azobenzene are substituted.
2. Results
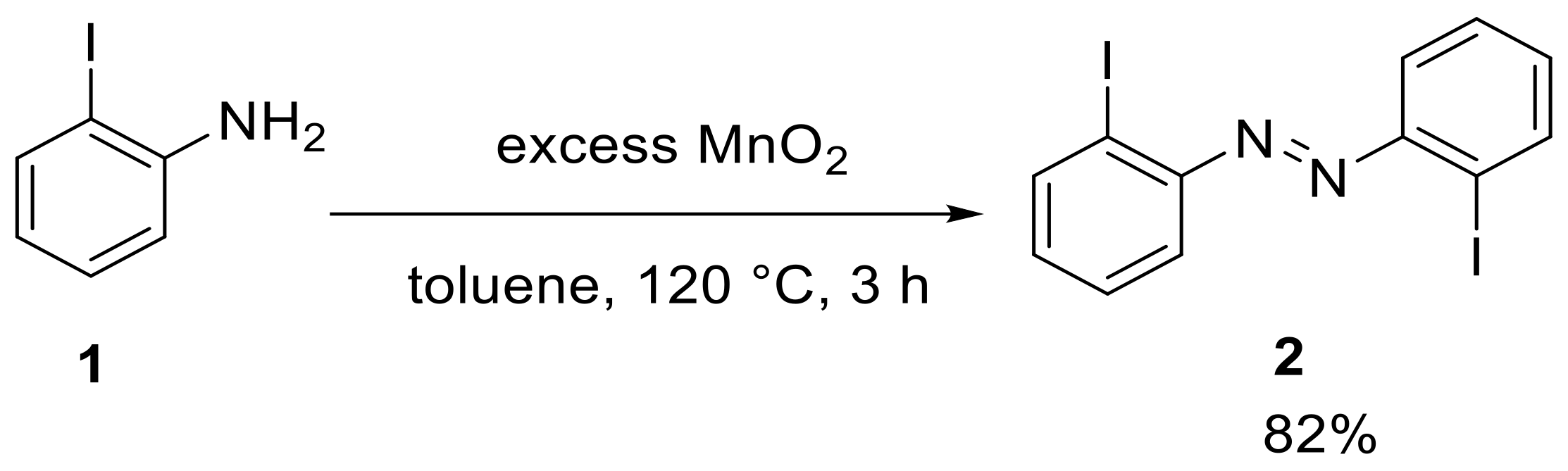
The investigation started with the synthesis of
ortho-substituted azobenzenes, in which the substituent was always EMe
3 with E = C, Si, Ge and Sn (E = Pb proved inaccessible). Therefore, we prepared a 2,2′-substituted difunctional azobenzene through the oxidation of 2-iodoaniline (
1) with manganese dioxide to give 2,2′-diiodoazobenzene (
2) in a yield of 82% (
Scheme 2) [
26].
It needs to be pointed out that azobenzenes with di-
ortho-substituents are more difficult to prepare than those with the same substituents in
meta- and
para-position. Oxidizing reagents, like copper(I) bromide/air [
25], copper(II)/silver(I)/tetra-
n-butylammonium bromide [
27] or diacetoxyiodobenzene [
28], were explored in the synthesis, but in all cases no conversion was observed.
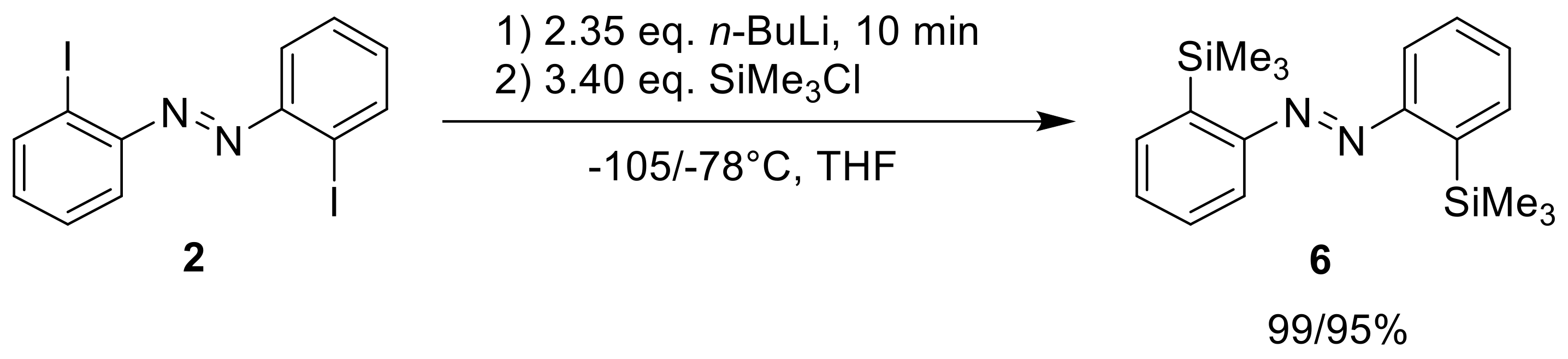
Following the iodo-lithium exchange, which was already reported [
20], 2,2′-diiodo-azobenzene (
2) was treated with
n-butyllithium at a low temperature (−105 or −78°C) and quenched with a trimethylsilyl chloride electrophile (
Scheme 3). The reactions proceeded very cleanly, which in our experience is different to
meta- and
para-iodinated azobenzenes, where the electrophilic N=N group can be attacked by the nucleophilic
n-butyllithium. This suggests a significant stabilization of the
ortho-lithiated species by an N→Li coordination.
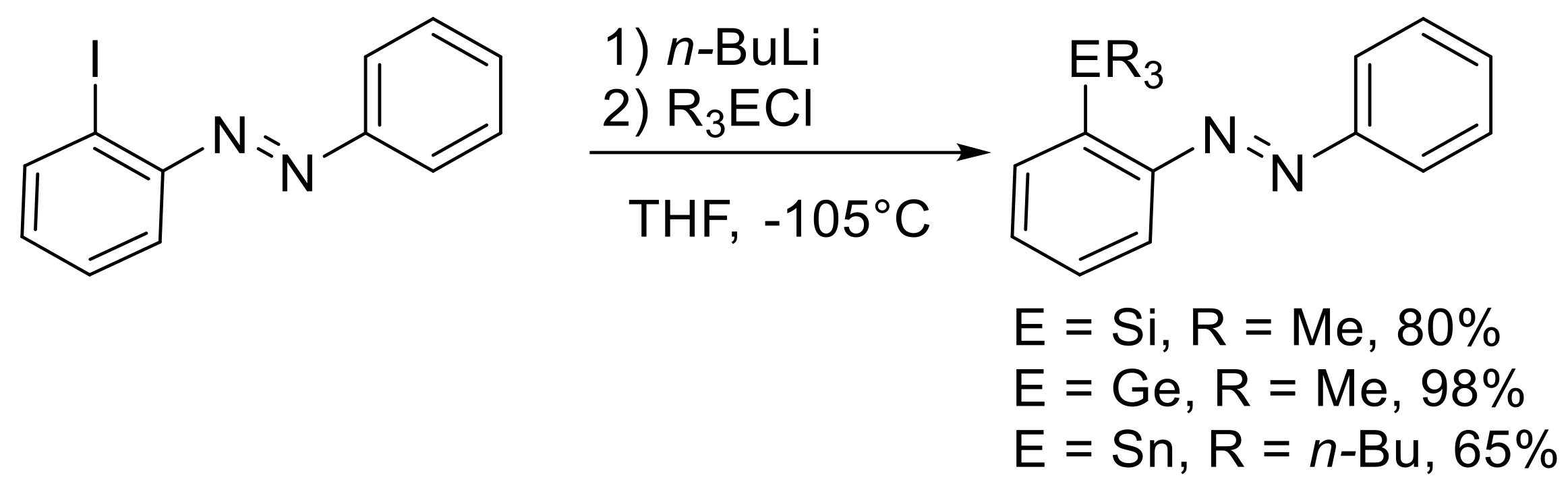
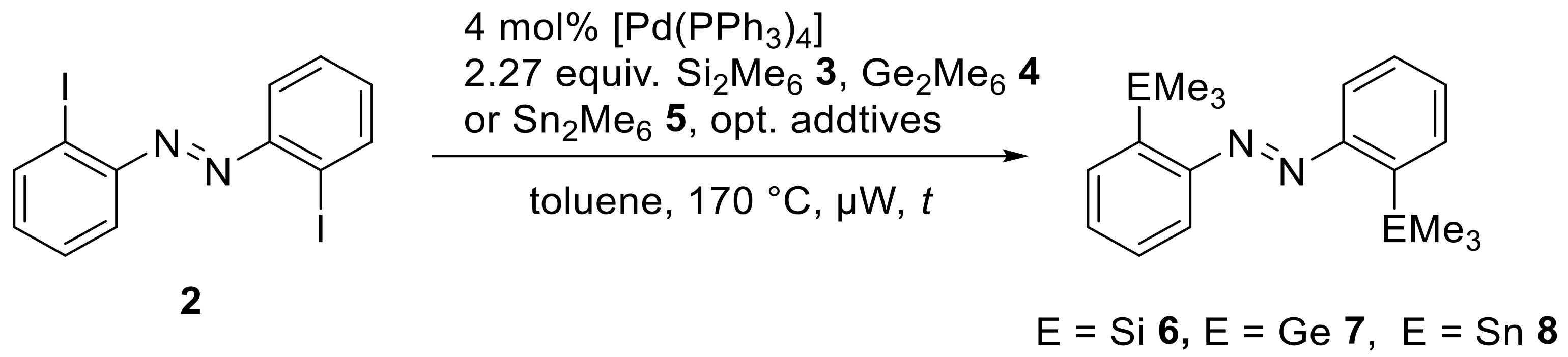
Although this is a very effective route to synthesize these
ortho-substituted azobenzenes, we were interested in obtaining the tetrel-substituted compounds under cross-coupling conditions. Therefore, the next step to generate the EMe
3 ortho-substituted azobenzenes was a palladium catalyzed cross-coupling procedure with Me
3E-EMe
3 reagents. Since this reaction has been already reported for
meta- and
para-substituted azobenzenes for E = Sn, we adapted to the reaction conditions also for other tetrels, for which this type of reagent was available (Si, Ge and Sn) (
Scheme 4,
Table 1).
We initially assumed that the metal-metal bond dissociation energy (BDE) might have a substantial impact on the reactivity towards the oxidative addition of the catalyst. The bond dissociation energies for hexamethyl dielement species have been reported: BDE (Me
3C-CMe
3) = 76.0 kcal/mol [
33], BDE (Me
3Si-SiMe
3) = 69.1 kcal/mol [
34] and 79.7 kcal/mol [
35], BDE (Me
3Ge-GeMe
3) = 69.1 kcal/mol [
34], BDE (Me
3Sn-SnMe
3) = 61.6 kcal/mol and BDE (Me
3Pb-PbMe
3) = 54.6 kcal/mol [
34]. Since the BDE of the tin dimer is relatively lower than the other hexamethyl dielement compounds, we assumed that a more labile tin-tin bond would react more easily, but it was surprising that neither the Me
3Si-SiMe
3 (
3) reagent, nor the Me
3Ge-GeMe
3 (
4) reagent provided any conversion. Steric reasons may be a potential issue: The C–Sn bond is longer and thus may lead to less congestion in the
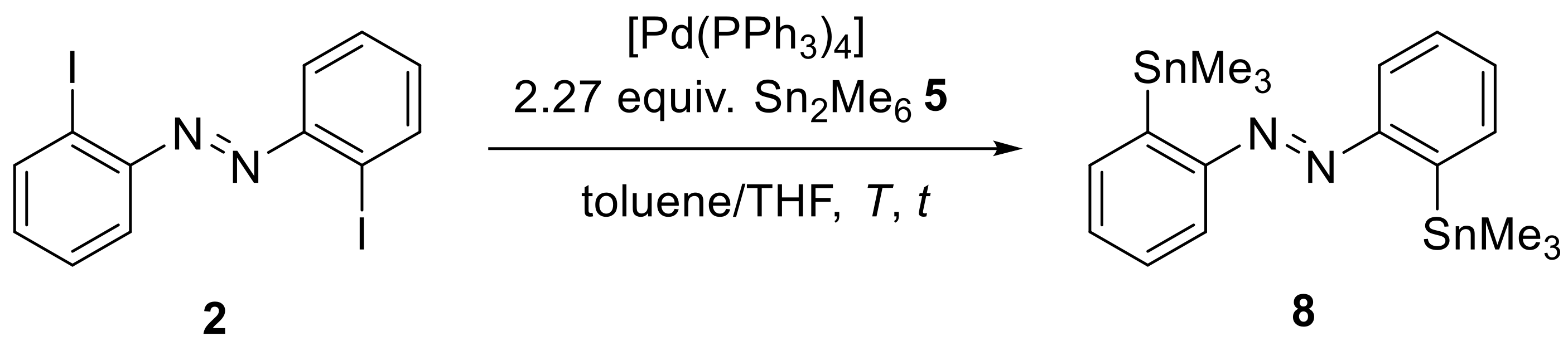
ortho-positions. Thus, the Stille-Kelly type cross-coupling reaction with the hexamethylditin (
5) was optimized towards the reaction temperature, reaction time and catalyst load (
Scheme 5,
Table 2): We envisaged that with this reagent in hand, alternative routes to the other
ortho-substituted azobenzenes may be also accessible via tin-lithium exchange and electrophilic quenching processes (see below).
It was found that increasing the reaction temperature from 130 °C to 170 °C, along with changing from conventional to microwave assisted heating (entries 2 and 4), led to an increase in yield and a decrease in reaction time. Furthermore, the catalyst to hexamethyldistannane (
5) ratio had a crucial impact on the success of the reaction. Increasing only the catalyst load led to a decrease in the yield (60% to 14%, entries 6 and 8). Additives like copper (I) halides [
36] and lithium chloride [
37] also lowered the yield of the product (entry 7). Interestingly, a high catalyst load in combination with a high amount of hexymethyldistannane (
5) resulted in nearly quantitative yield. Combined with the fact that the reaction vessels always contained an insoluble black precipitate after the reaction, we assumed that the autocatalytic agglomeration of the palladium catalyst that can occur more easily with higher catalyst loadings may be the cause of this observation [
38]. With an excess of hexamethyldistannane (
5), agglomeration was not observed (see the high difference in yield for entries 6, 8 and 9), presumably because the desired reaction was accelerated compared to the agglomeration process. Since hexamethyldistannane (
5) is an expensive and toxic compound, a high excess and high amount of organotin waste is not desirable. Therefore, we performed further reactions with only 2.27 equivalents of this organotin compound, accepting the somewhat lower yield. We also tested whether the stannylated product
8 might react with the iodinated starting material
2 in a standard Stille reaction, but this was not the case, which excludes this reaction as a cause for the lower yields from entries 7 to 8 (see
SI).
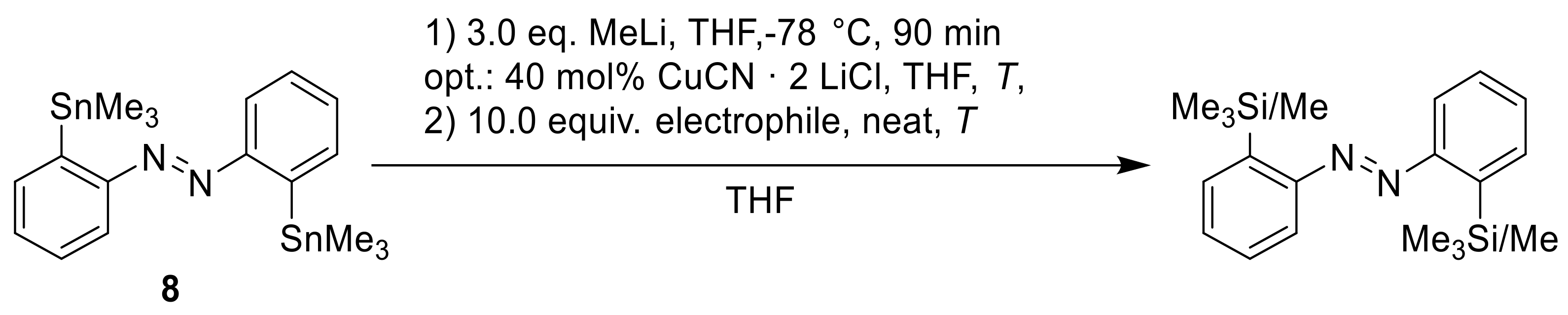
To access the other
ortho-substituted azobenzenes for EMe
3 (E = C, Si, Ge), we followed a transmetalation procedure which was already reported for di-stannylated
meta- and
para-azobenzenes [
23]. Initially, the metalation temperature and time were optimized, with trimethylsilyl chloride (
9) being used as an electrophile (
Scheme 6,
Table 3).
It was found that increasing the temperature for the lithiation reaction led to a decrease in product and an emergence of unassignable signals in the
1H NMR spectra. However, the metalation time played only a minor role in the formation of the product (Entries 6–10). The best conditions were found to when the lithiation temperature was −78 °C and a metalation time of 90 min was used. To expand the variety of electrophiles to less hard organometals, we made use of a transmetalation from the hard, reactive lithium towards the softer and less reactive cuprate, which was initially reported by Knochel [
39]. This methodology has so far not been applied to azobenzene chemistry (
Scheme 7).
We tested this reaction sequence with several electrophiles and different metalation temperatures (
Table 4).
The yield decreased if the temperature for the lithiated species was changed from −78 °C to −40 °C (Entries 1 and 2), which may be an indication of a reductive attack by the organolithium reagent on the azo group [
18]. The transmetalation to copper at −78 °C provided largely the same yields (Entry 3), although at −40 °C, using the cuprate gave a very low yield of 7% (Entry 4). For Me
3SiI, a decrease in yield after the transmetalation to copper was observed (Entries 5–7), which was unexpected as this electrophile is softer. With methyl iodide as an electrophile, we could observe an increase in the yield by increasing the transmetalation temperature from −78°C to −40 °C (Entries 8 and 9). In that case, using the cuprate was essential for the reaction, because the lithiated species did not react with the electrophiles (Entry 10).
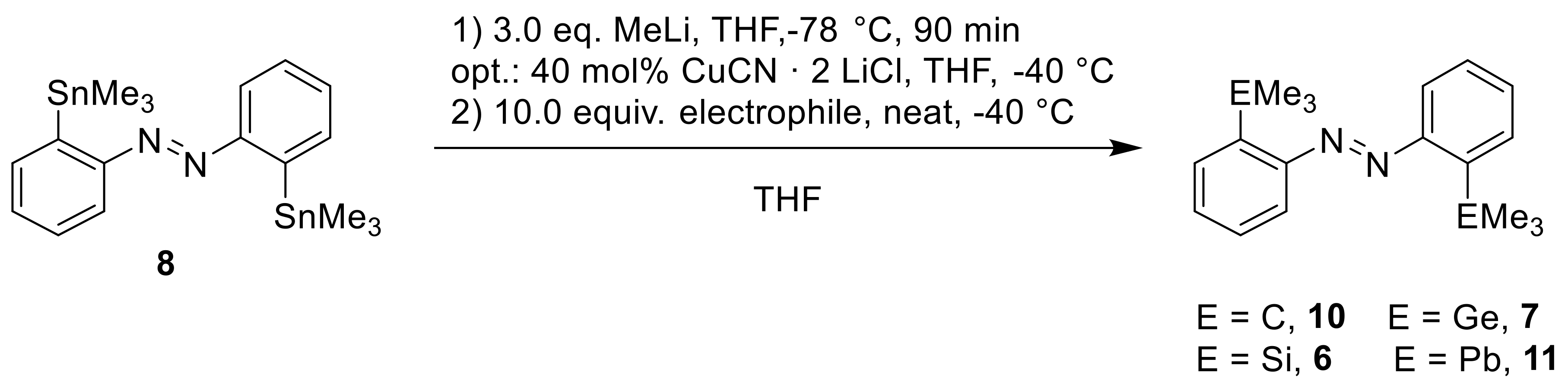
With both methods in hand, we incorporated new trimethyltetrel halides. In dependence of the nature of the respective electrophile, we were interested in the reactivity towards the hard organolithium or the soft organocuprate (
Scheme 8,
Table 5).
Besides the above mentioned trimethylsilyl chloride (
9), the reaction of trimethylgermanyl chloride (
12) to give the digermanylated azobenzene (
7) could be performed in a good yield of 60% (Entry 3). With
tert-butyl chloride (
13) and trimethyllead bromide (
14), neither reaction was successful, irrespective of whether the lithium or copper species (Entries 1 and 4) were used, but for different reasons: The nucleophilic substitution of
tert-butyl chloride (
13) would be highly disfavored for an S
N2 reaction due to its steric hindrance, and also because a S
N1 reaction is unlikely under these nonpolar reaction conditions. The reaction with trimethyllead bromide (
14) was unsuccessful with both the lithium or copper nucleophile. It is possible that the trimethyllead bromide did react, but that the lead(IV)-substituted azobenzene was then too reactive to be isolated; recent examples of this are given in Reference [
41]. This hypothesis is supported by the fact that after the reactions, a shiny insoluble black precipitate could be observed, which are probably decomposition products from lead.
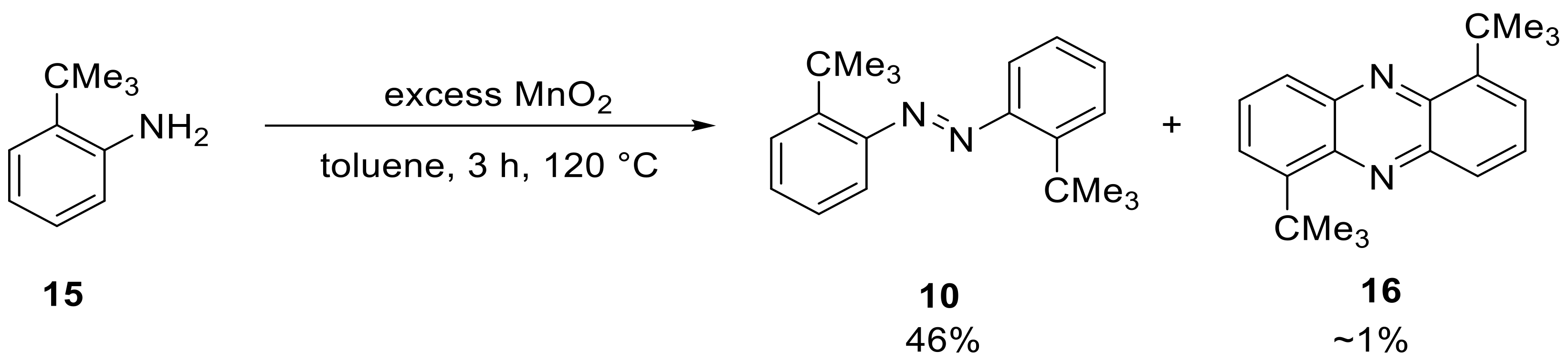
Since we were interested in comparing the influence of different trimethyltetrel substituents in
ortho-position of the azobenzenes, we obtained the carbon-derivative via oxidation of the respective aniline (
Scheme 9).
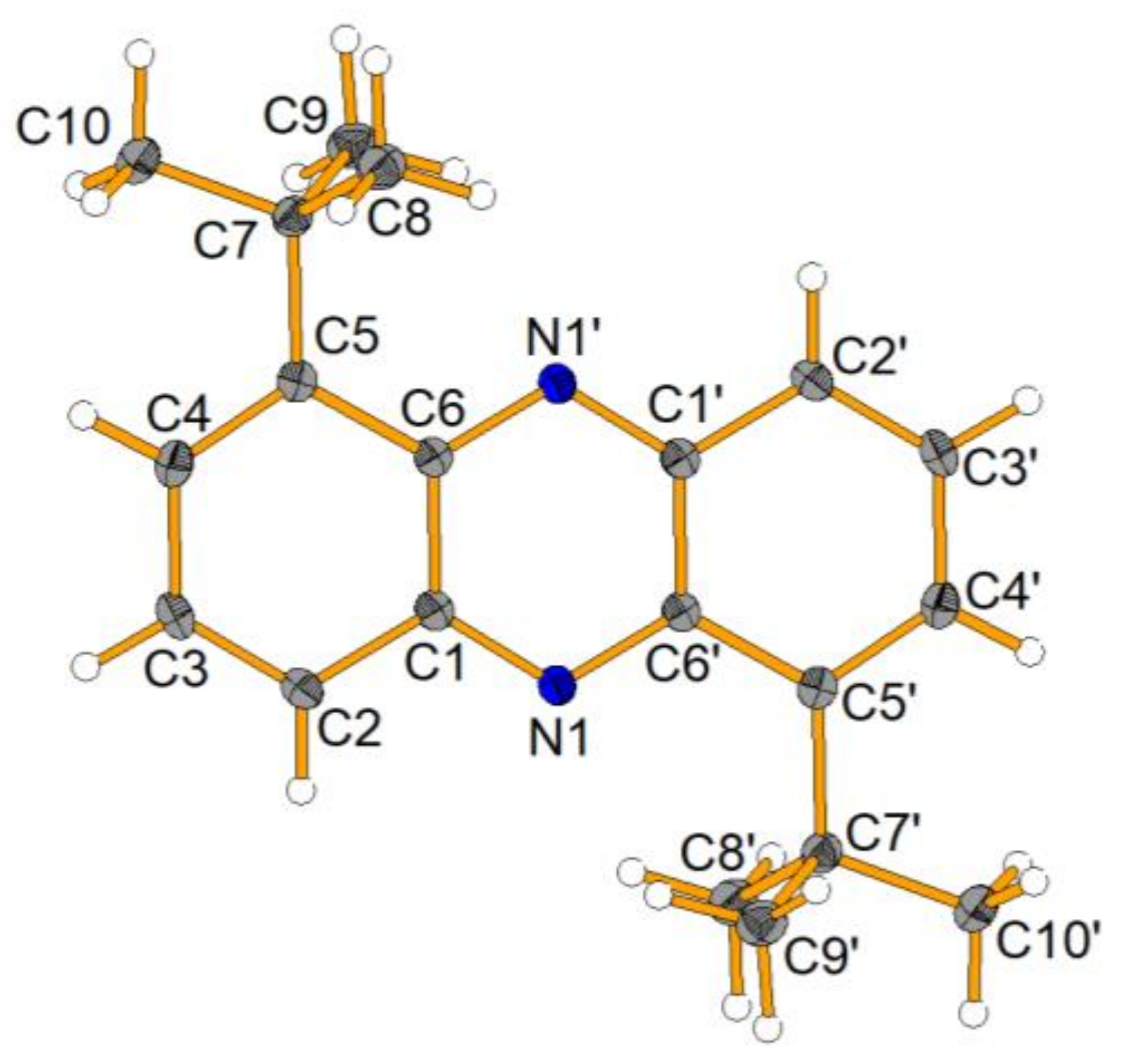
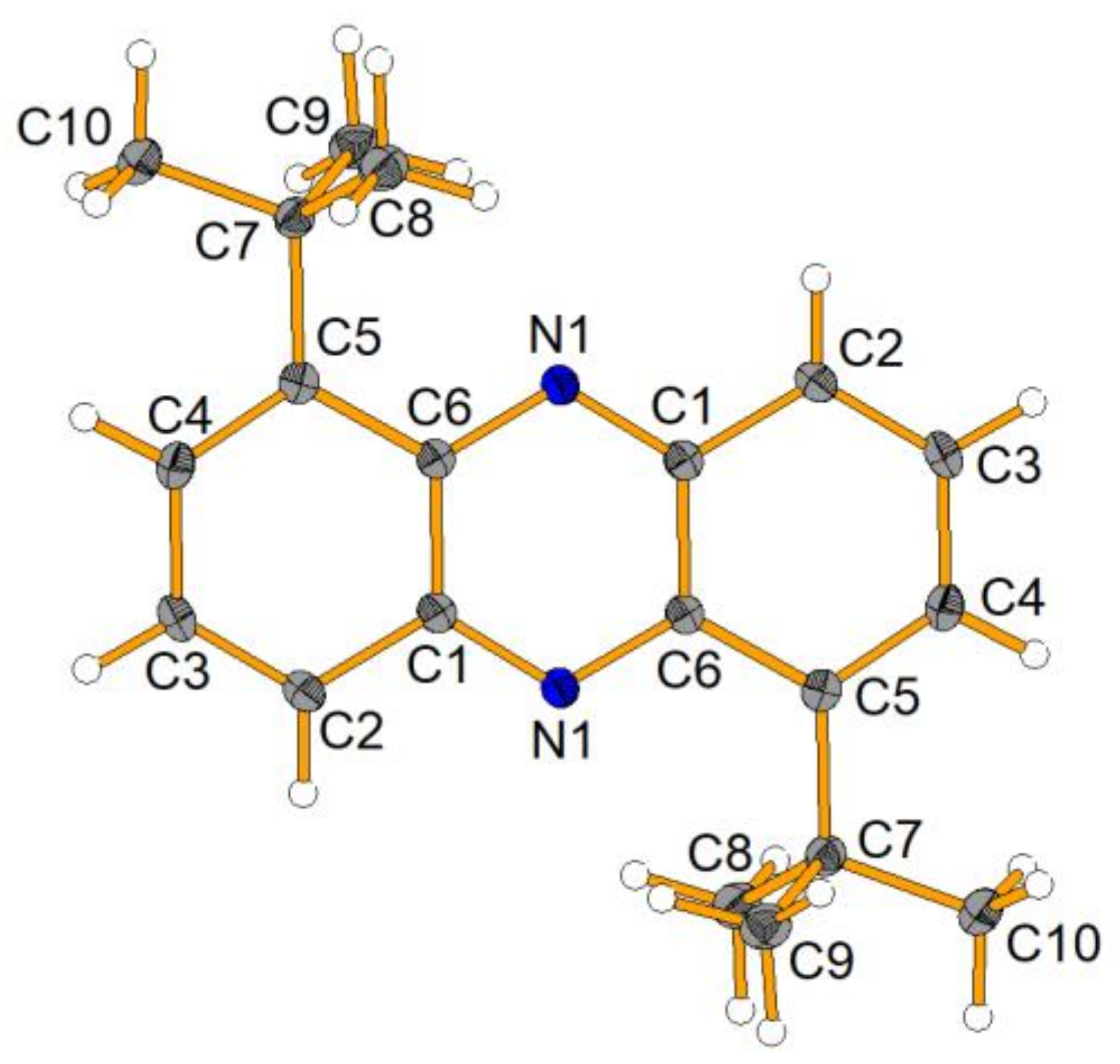
The product
10 was isolated in a moderate yield of 46%, but surprisingly, we could also isolate a side product, which was characterized by NMR, HRMS, FTIR and XRD and was found to be a phenazine derivative (
Scheme 9 and
Figure 1).
It is not clear at present how this side product formed, but the literature suggests that elevated temperature and oxidative conditions favor the formation of the very thermodynamically stable phenazines [
27].
With the series of ortho-tetrel substituted azobenzenes in hand, we were interested in the comparison of their structures and the effect of the group 14 elements on the physical properties of the azobenzenes.
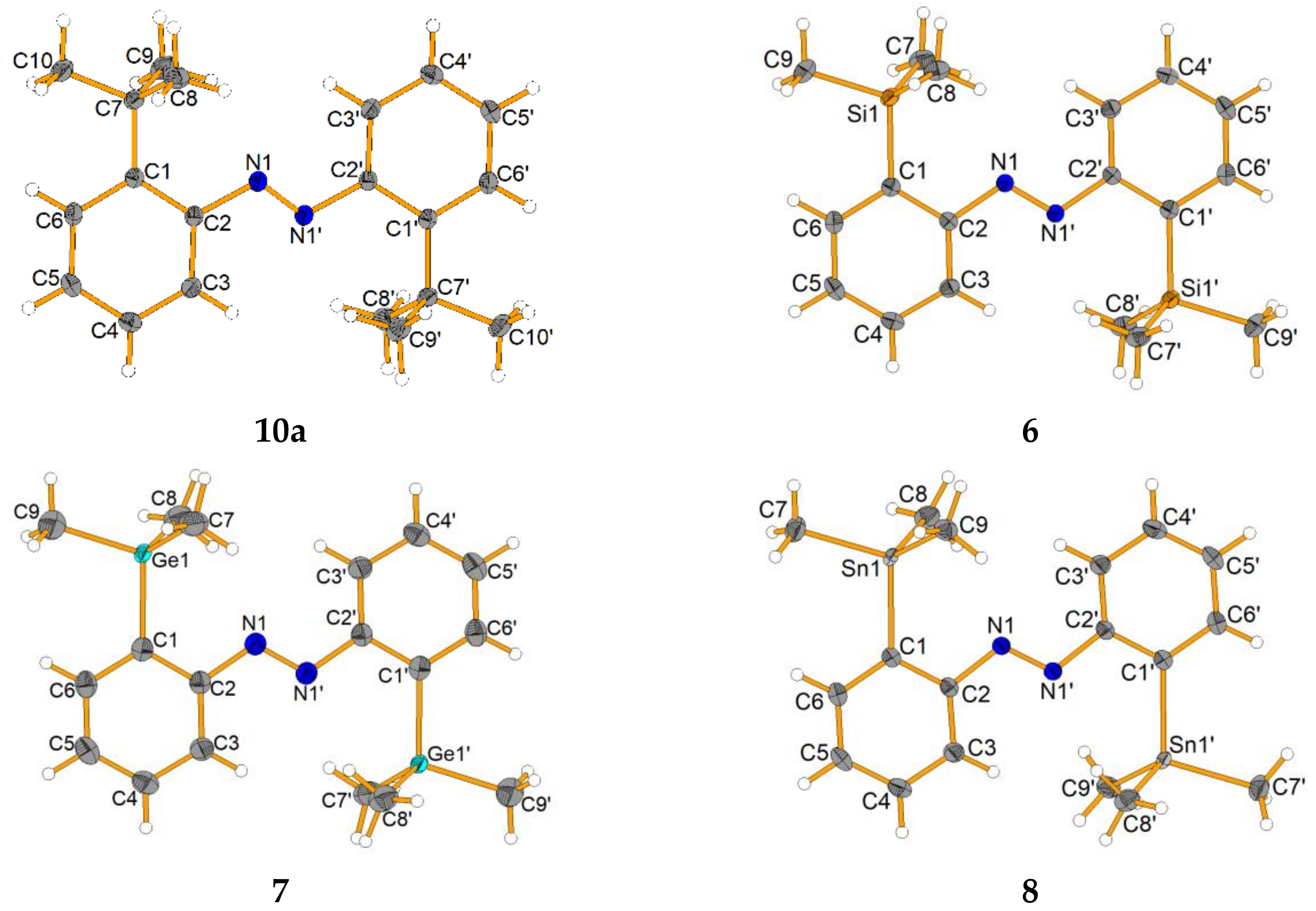
The crystal structures of all molecules were determined (
Figure 2). Since the interaction of tetrels in the
ortho-position of azobenzenes has been reported for electron deficient fluorinated silyl groups [
42,
43], we were interested in how the structural parameters of our crystals would compare with the mono-substituted compounds described by Kano. A special interest was the N=N and carbon-tetrel bond, as well as the possible interaction of the group 14 element with the azo group, which is here indicated by the distance and the torsion from the phenyl ring and the azo group (
Table 6).
The
ortho-substitution had only a marginal effect on the length of the azo group (from 1.2509(18) Å to 1.258(2) Å). However, as expected, the distance of the carbon-tetrel bond increased from the lighter to the heavier elements (1.5348(14) Å (For E = C) to 2.144(1) Å (for E = Sn)). The nitrogen-tetrel distance did not indicate any interaction of the azo group with the respective tetrels (2.9091(18) Å for Si to 3.0471 Å for Sn). This can be compared with azobenzenes with SiMe
2F (2.585(3) Å) and SiF
3 (2.371(4) Å) [
41]: These much-shortened distances indicated that the lone pair of the nitrogen atom was able to coordinate to the Lewis acidic silicon center, which was not observed in our case. Furthermore, the
29Si NMR shift of this compound was to be found at
29Si{
1H} NMR: δ = −4.04 ppm, which is a typical shift for tetracoordinated silicon species. This is well aligned with Kano’s work [
41], who reported a
29Si{
1H} NMR-shift of −3.8 ppm for a trimethylsilyl substituted azobenzene, as compared to −16.8 ppm if one of the Me groups was replaced by F, or even −57.8 ppm for a SiF
3 group in this position. Furthermore, the crystal structure differs in the fact that in the case of a nitrogen-tetrel interaction, a four-membered ring would result, whereas the structures from Kano showed interaction with the second nitrogen atom of the azo group, resulting in a five membered ring; for this to happen, the aromatic rings would need to rotate by approximately 180°, which was not observed. Upon substitution with higher congeners than carbon or silicon, the germanium and tin substituted azobenzene exhibited a carbon-tetrel bond length of 2.0234(18) Å and 2.144(1) Å, which can be considered as an ordinary organotetrel bond length. However, the torsion angles for the CMe
3 substituted azobenzene were surprisingly far from planar (by ca. 15–17°), which was in contrast to Kano’s work on Si and also our results on the SiMe
3 substituted compounds, which show almost no distortion. We assumed that the sterical hinderance of the
tert-butyl group, in comparison with the short C(Ph)–C(CMe
3) bond, forced the system to undergo distortion. Due to more flexible and longer C–Si, C–Ge and C–Sn bonds, we observed less planar distortion (3–6°).
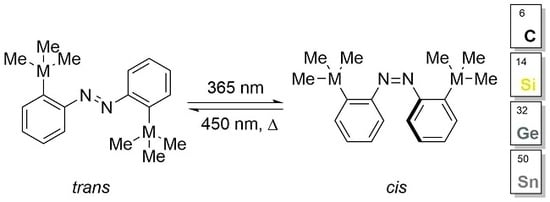
Since azobenzenes can undergo
trans/
cis isomerization, we studied the effect of the different group 14 elements on the general absorption properties, the
cis/
trans equilibrium in the dark and the photostationary state (PSS) at 365 nm and 450 nm through UV and NMR spectroscopy (
Scheme 10).
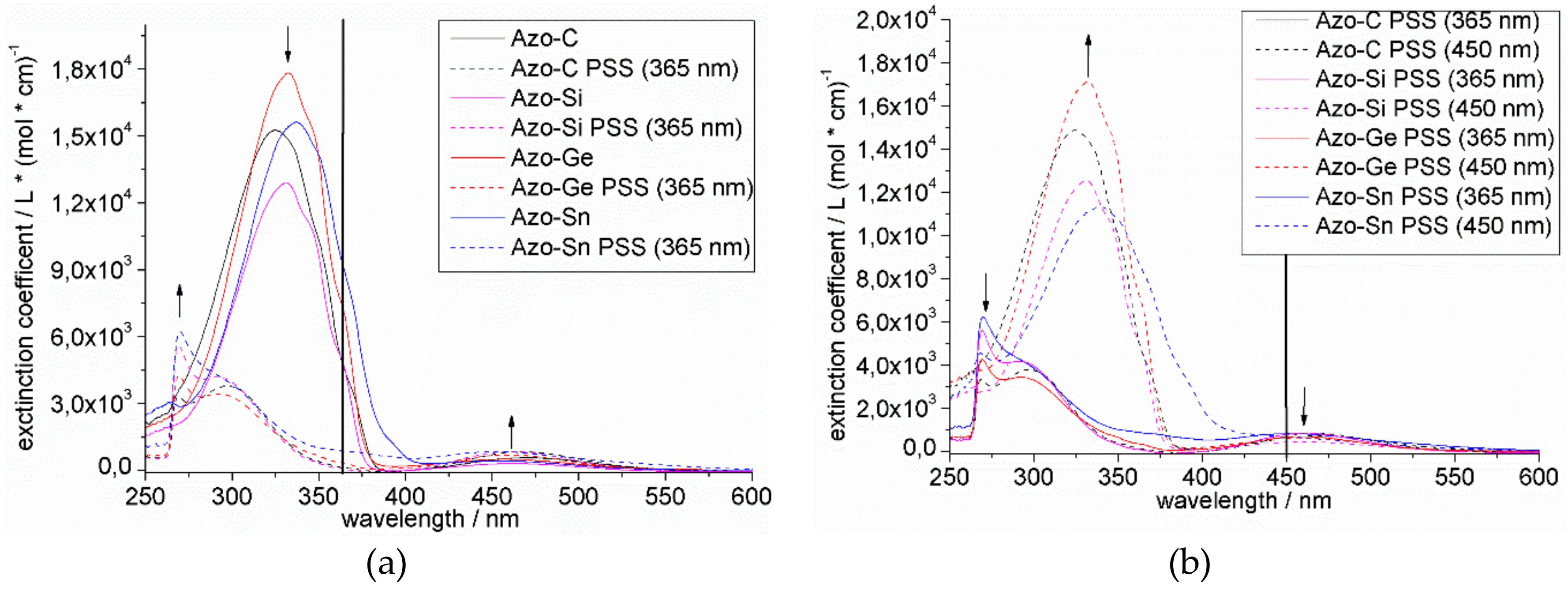
In general, the UV spectra of the azobenzenes exhibit a strong ππ* absorption around 330 nm and a weak absorption for the nπ* band of the
cis-isomer around 475 nm (
Figure 3a,b). Upon irradiation of the azobenzenes with UV light (365 nm), we observed a significant decrease in the absorbance of these molecules (
Figure 3a), which can be assumed to switch to the respective
cis-isomer. Blue light (450 nm) shifts the photostationary state (PSS) to the
trans-isomers (
Figure 3b).
In all cases, the switching was totally reversible but the tin-substituted azobenzene
8 showed a decrease and broadening of the ππ* band (
Figure 3b), which was not expected, and may indicate a photochemical decomposition process.
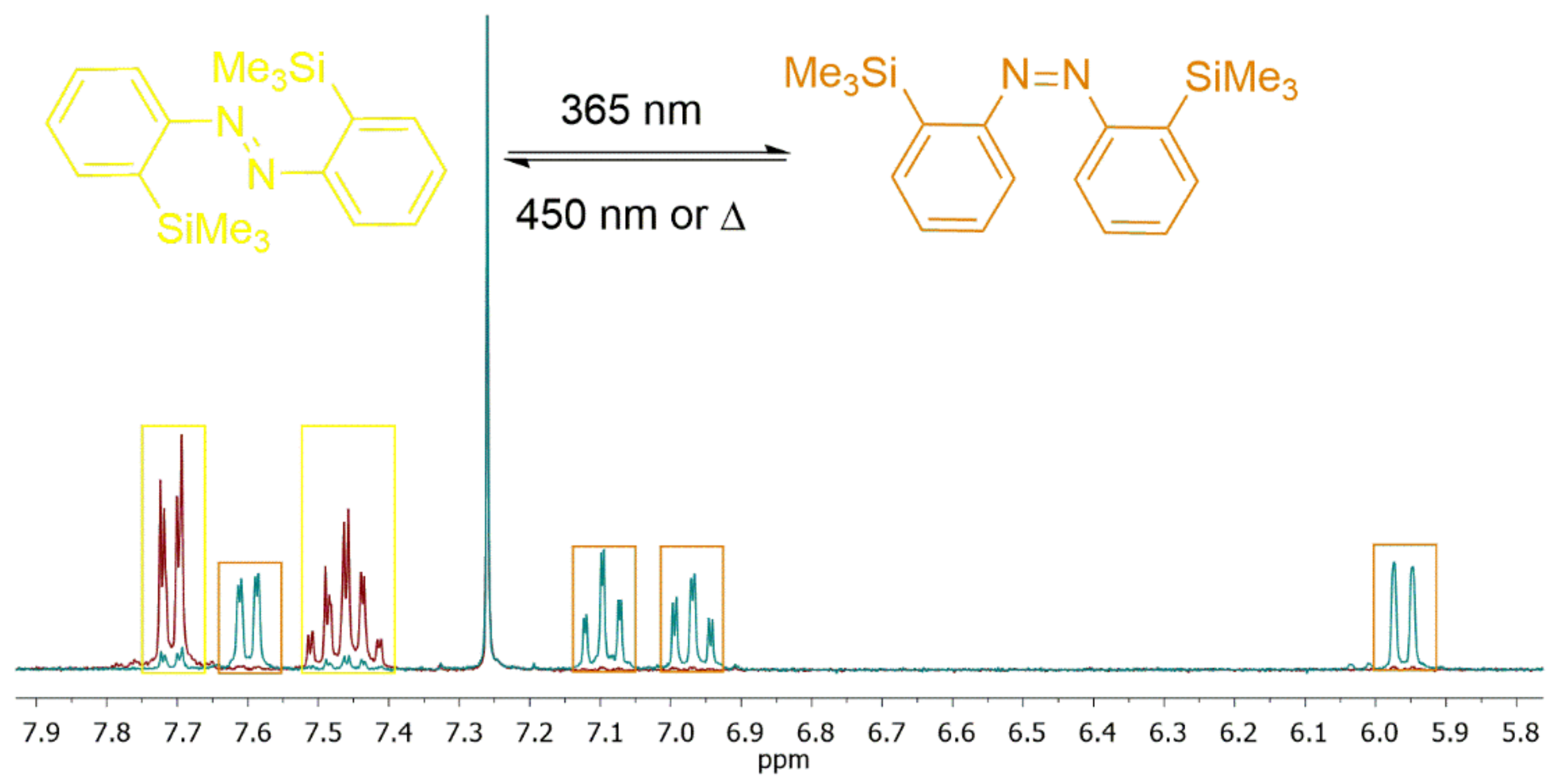
To obtain the ratio of the
cis/
trans-isomer in the PSS, we combined irradiation experiments with
1H NMR analysis. The result of this experiment for molecule
6 is shown below (
Figure 4).
To compare the general trends upon substitution with different tetrels on the azobenzenes, we compared the collected isomerization information (
Table 7).
Upon substitution of these compounds with heavier group 14 elements, we observed a slight bathochromic shift of the ππ* band (325 nm to 338 nm). This could be explained by the effect of the decreasing torsion angle due to the bulky trimethyltetrel groups, allowing extended π-conjugation (
Table 8). However, the effect of the heavier tetrels on the extinction coefficient of the azobenzene system is rather small.
All systems contained only a small amount of the
cis-isomer ‘as synthesized’. Upon irradiation with 365 nm light, the maximum amount of the
cis-isomer was found to range from 80% to 91% from E = C to E = Ge, respectively. Therefore, the PSS is substantially influenced by the tetrel. For E = Sn, however, the ratio appeared much lower, but this could also be attributed to photodecomposition. Irradiation with 450 nm light did not result in a complete switching to the
trans-isomer, which can be observed by the existence of the nπ*-band in the UV-VIS spectra. Unfortunately, irradiation with 365 nm and 450 nm light caused the tin-substituted azobenzene
8 to experience some decomposition, which was detected by UV and
1H NMR spectroscopy. This may be caused by a small C–Sn bond-dissociation energy. We further observed the thermal relaxation of the
cis-azobenzenes that had been obtained after irradiation with 365 nm light with UV-VIS and NMR spectroscopy. The half-life times (τ) in the NMR experiments were determined to be between 55.61 h to 65.84 h, in which an increasing half-life time with heavier tetrels trend could be observed. The UV experiments for compounds
6,
7 and
10 revealed half-life times of 17.07 h to 20.45 h. In this case, no correlation between the tetrel substitution and the half-life time could be found (for all data see the
SI). There is a substantial discrepancy between the half-life times measured by the two techniques. It needs to be pointed out in this context that the concentrations in the NMR were higher by approximately 2 orders of magnitude. Although half-life times should not be concentration dependent in general, they can be different if agglomerations and the stacking of molecules occur, which is likely to be the case for the compounds under investigation.
These systems were also analyzed by dynamic scanning calorimetry (DSC). To ensure thermal stability in the temperature range of investigation of these compounds, thermogravimetric analysis (TGA) was conducted (see
SI). The compounds showed mass loss at 208 °C (C,
10), 188 °C (Si,
6), 196 °C (Ge,
7) and 248 °C (Sn,
8). However, when a TGA experiment on molecule
10 was interrupted at mass loss and the residue was analyzed by NMR spectroscopy, no decomposition was observed. Therefore, we assumed that the azobenzene evaporated at the higher temperature, accounting for the observed loss of mass.
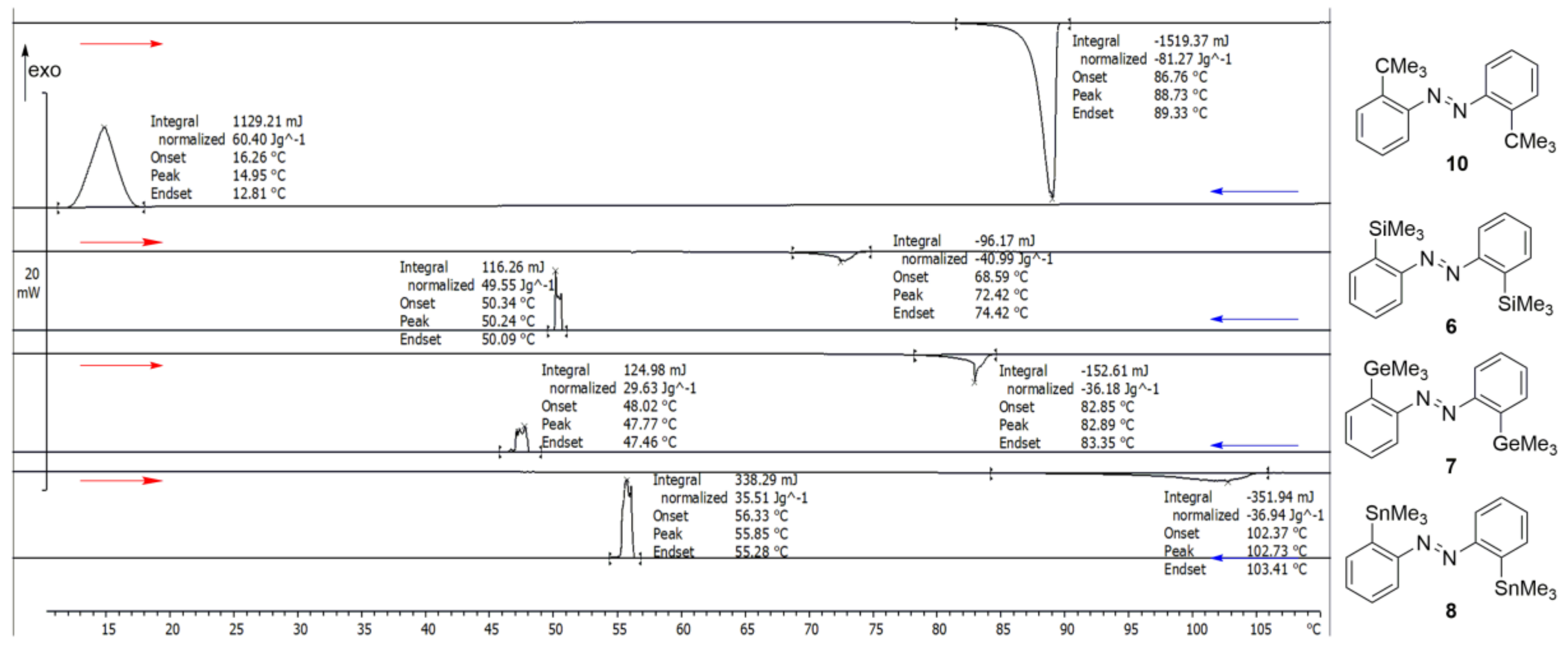
In general, these compounds showed endothermic melting signals at onset temperatures of 87 °C (E = C), 69 °C (E = Si
6), 83 °C (E = Ge) and 102 °C (E = Sn
8), and exothermic crystallization peaks of 16 °C (E = C
10), 50 (E = Si), (48 °C (E = Ge) and 56 °C (E = Sn
8) (
Figure 5,
Table 8)). In general, the melting temperature increased within the main group of compounds
6–
8, but compound
10 proved to be an exception. Although the temperature ramps were slow (0.5 K/min), the melting peaks were not completely sharp, additionally, the crystallization peaks showed multiple crystallization events due to spontaneous seeding (for all data see the
SI).
We observed that with faster DSC measuring modes (10 K/min) there was a high degree of amorphousness in the structures (see
SI). With slower DSC methods (0.5 K/min), the difference between the fusion and solidification measured with the slower DSC method enthalpies are still existent and may be related to a non-crystallizing part of the molecules. The fusion enthalpy of compound
10 was 23.93 KJ/mol, which is rather high in comparison of the other azobenzenes (
6: 13.37 KJ/mol,
7: 15.13 KJ/mol,
8: 15.05 KJ/mol).
4. Materials and Methods
4.1. General Information
For inert reactions, a nitrogen filled glovebox from Pure LabHE and standard Schlenk techniques were used. Except for the preparation of the 2,2′-diiodoazobenzene (2) and 2,2′-di(tert-butyl)azobenzene (10), all reactions were carried out under inert conditions.
All glassware for the inert reactions was dried in an oven at 200 °C for several hours prior to use. Additionally, before starting the reaction, the glassware was heated under vacuum (about 5 × 10−2 mbar) and flushed at least three times with argon. The NMR tubes were dried at 110 °C for several hours.
Microwave irradiated reactions were carried out using an Emrys
TM Optimizer instrument (Biotage, Uppsala, Sweden), in which the temperature was measured with an external IR detector. MeLi was titrated prior to usage with menthol, using 2,2′-bipyridine as an indicator [
50].
For chromatographic purification, silica gel 60 (MERCK, Darmstadt, Germany, 0.015–0.40 mm) was used. If stated, Celite® 503 (Carl Roth GmbH & Co. KG, Karlsruhe, Germany) was used as a filtration aid. Thin layer chromatography was performed by using thin layer chromatography (TLC) Silicagel 60 F254 from MERCK. A UV lamp (λ = 254 nm) was used for detection.
NMR spectra were recorded at 300 K on a Bruker AvanceNeo 500 (Bruker, Rheinstetten, Germany) (500 MHz (1H), 125 MHz (13C{1H}), 187 MHz (119Sn{1H}), 100 MHz (29Si {1H})). Where possible, NMR signals were assigned using 1H COSY, 1H/13C HSQC and 1H/13C HMBC experiments. 1H and 13C {1H} NMR spectra (125 MHz) were referenced against the residual solvent signal, CDCl3 (1H: δ = 7.26 ppm, 13C: δ = 77.16 ppm). 119Sn{1H} and 29Si{1H} NMR spectra (187 and 100 MHz) were measured based on the external reference of the 1H-NMR signal of tetramethylsilane.
Reaction controls by 1H-NMR and some 13C{1H} spectra were performed on a Bruker Avance WB 360 instrument (Bruker, Rheinstetten, Germany) (1H: 360 MHz, 13C{1H}: 91 MHz). The switching experiments were performed on a Bruker ARX300 (Bruker, Rheinstetten, Germany) (300 MHz (1H)) or Bruker AvanceNeo 500 (Burker, Rheinstetten, Germany) (500 MHz (1H).
Thermal analyses were performed on a standalone Mettler Toledo DSC 3+ STAR (Mettler-Toledo, Columbus, OH, USA) or a Mettler Toledo TGA/DSC 3+ System (Mettler-Toledo, Columbus, OH, USA), where 40 µL and 100 µL aluminum crucibles were used. For TGA experiments, no lids were used, whereas in DSC experiments pierced lids were used. Thermo analytical data was analyzed with the STARe software (Version 14.01, Mettler-Toledo, Columbus, OH, USA) by Mettler Toledo.
Infrared spectra were recorded on a NICOLET i510 FT-IR spectrometer from Thermo Fisher SCIENTIFIC (Thermo Fisher SCIENTIFIC, Waltham, MA, USA) with a diamond window in an area ranging from 500–4000 cm−1 with a resolution of 4 cm−1. All samples were measured 16 times against a background scan. Melting points were measured by DSC or using a BÜCHI Melting Point M-560 instrument (BÜCHI, Essen, Germany).
Electron impact (EI) mass experiments were measured using the direct inlet or indirect inlet methods, with a source temperature of 200 °C on a MAT95 XL double-focusing mass spectrometer from Finnigan MAT (Thermo Fisher SCIENTIFIC, Waltham, MA, USA). The ionization energy of the electron impact ionization was 70 eV. Atmospheric pressure chemical ionization (APCI) experiments were performed on a Bruker Impact II from Bruker Daltonics (Bruker Daltonics, Bremen, Germany).
For the UV switching experiments, a 365 nm (Ocean Optics USB 4000, Sahlmann Photochemical Solutions, Bad Segeberg, Germany, full width at half maximum (FWHM) 10 nm, 1.0 W) and a 443 nm LED lamp (Ocean Optics USB 4000, Sahlmann Photochemical Solutions, Bad Segeberg, Germany, FWHM 19 nm, 0.9 W) were used, while ensuring a constant distance towards the cuvettes of 1 cm. The NMR switching experiments were performed with a circular aligned lamp consisting of four high power UV-LEDs (365 nm) 300 mW in power each, produced by “Sahlmann Photochemichal Solutions”. For switching experiments, quartz cuvettes from Hellma Analytics (Hellma Analytics, Muehlheim an der Ruhr, Germany) (10 mm) or Quartz NMR tubes from Deutero (Deutero GmbH, Kastellaun, Germany) were used.
UV-VIS spectra were recorded at a resolution of 0.1 nm on a UV-2700 spectrometer from Shimadzu (Shimadzu, Kyoto, Japan) with a double monochromator. In all cases, cyclohexane was used as a solvent.
X-ray measurements were carried out at 100 K on a Bruker Venture D8 diffractometer (Bruker, Karlsruhe, Germany) with Mo-Kα (0.7107 Å) radiation. All structures were solved by intrinsic phasing and refined based on F2 by use of the SHELX program package, as implemented in OLex 1.2 [
51]. All non-hydrogen atoms were refined using anisotropic displacement parameters. Hydrogen atoms attached to carbon atoms were included in geometrically calculated positions using a riding model. All crystals were obtained by slow evaporation of an acetonitrile/dichloromethane mixture at 25 °C.
4.2. Syntheses
4.2.1. 2,2′-Diiodoazobenzene (2)
Adapted with changes from Takahashi et al. [
26]. A solution of 2-iodoaniline (
1) (2.00 g, 9.13 mmol) and MnO
2 (20.0 g, 230 mmol) in toluene (200 mL) was stirred for 3 h at 120 °C. After the reaction, the mixture was cooled to 25 °C, filtered through Celite
®, washed with toluene (200 mL) and the solvent was removed under reduced pressure. The red solid that was obtained was dissolved in DCM (5 mL) and purified by filtration through a short plug of silica (eluent:
n-pentane). After evaporation of the solvent, the dark orange solid was dried (4.6 × 10
−2 mbar, 25 °C, 4 h) to give a red compound (1.63 g, 3.76 mmol, 82%).
1H NMR (500 MHz, CDCl3) δ = 8.04 (dd, 3J = 7.9 Hz, 4J = 1.3 Hz, 2H, H-3), 7.77 (dd, 3J = 7.9 Hz, 4J = 1.6 Hz, 2H, H-6), 7.46 (ddd, 3J = 7.9, 7.3 Hz, 4J = 1.3 Hz, 2H, H-5), 7.20 (ddd, 3J = 7.9, 7.3 Hz, 4J = 1.6 Hz, 2H, H-4) ppm. 13C{1H} NMR (126 MHz, CDCl3) δ = 150.87 (C-1), 139.94 (C-3), 132.80 (C-4), 129.06 (C-5), 118.26 (C-6), 103.22 (C-2) ppm. IR (ATR): ν = 3055 (w), 2921 (w), 2851 (w), 1924 (w), 1838 (w), 1806 (w), 1699 (w), 1561 (m), 1455 (m), 1013 (s), 953 (m), 760 (s), 714 (s) cm−1. HRMS (EI, MAT 95 XL): m/z calcd. C12H8N2I2+ 433.87715 found 433.87701. Rf (n-pentane): 0.51. Mp (Büchi): 146 °C
4.2.2. 2,2′-Bis(trimethylstannyl)azobenzene (8)
Adapted with changes from Strüben et al. [
25] In a glovebox, a microwave vial was charged with 2,2′-diiodoazobenzene (
2) (200 mg, 0.46 mmol), hexamethylditin (
5) (422 mg, 1.03 mmol), tetrakis(triphenyl)phosphinopalladium(0) (15.7 mg, 13.0 µmol), THF (0.5 mL) and toluene (4.0 mL). The reaction mixture was heated for 1 h at 170 °C using microwave irradiation. The solution was cooled to 25 °C, filtered, rinsed with toluene (10.0 mL) and all volatiles were removed under reduced pressure. Then, the compound was dissolved in DCM (2 mL) and filtered through a short plug of silica (eluent:
n-pentane). After the solvent was removed, an orange solid (142 mg, 0.28 mmol, 61%) was received.
1H NMR (500 MHz, CDCl
3) δ = 7.78 (dd,
3J = 7.8 Hz,
4J = 1.4 Hz, 2H, H-6), 7.74 (dd,
3J = 7.1 Hz,
4J = 1.4 Hz, 2H, H-3), 7.47 (td,
3J = 7.1 Hz,
4J = 1.4 Hz, 2H, H-5), 7.43 (td,
3J = 7.1 Hz,
4J = 1.4 Hz, 2H, H-4), 0.32 (s, 18H, CH
3) ppm.
13C{
1H} NMR (126 MHz, CDCl
3) δ = 157.12 (C-1), 146.53 (C-2), 136.65 (C-3), 130.33 (C-4), 129.52 (C-2), 117.73 (C-6), −7.33 (C-7) ppm.
119Sn{
1H} NMR (187 MHz, CDCl
3) −34.36 ppm. IR (ATR): ν = 3050 (w), 2974 (w), 2909 (w), 2609 (w), 2354 (w), 1965 (w), 1932 (w), 1902 (w), 1853 (w), 1820 (w), 1432 (w), 1294 (w), 1188 (m), 1110 (m), 754 (s), 706 (s) cm
−1. HRMS (APCI):
m/z calcd. [C
18H
26N
2Sn
2 + H]
+ 509.02140 found 509.02126. R
f: (
n-pentane): 0.84. Mp (DSC; Onset): 102.37 °C
4.2.3. 2,2′-Bis(trimethylsilyl)azobenzene (6)
In an inert tube 2,2′-bis(trimethylstannyl)azobenzene (4) (80.0 mg, 0.16 mmol) was dissolved under Schlenk conditions in THF (5.00 mL) and cooled to −78 °C. MeLi (1.88 M in THF, 0.25 mL, 0.47 mmol) was added within 5 min and after 1 h at this temperature, trimethylsilyl chloride (9) (200 µL, 171 mg, 1.57 mmol) was added to the black reaction mixture in one portion. The reaction mixture was warmed to 25 °C over 14 h and the solvent was removed under reduced pressure. The brown solid, dissolved in DCM (3.00 mL), was purified by a short plug of silica (eluent: n-pentane). The first orange fraction was filtered through a PTFE filter (0.45 µm). From the filtrate, the solvent was removed to obtain an orange solid (43 mg, 0.132 mmol, 82%). 1H NMR (500 MHz, CDCl3): δ = 7.72 (dd, 3J = 7.7 Hz, 4J = 1.3 Hz, 4H, H-3 and H-6), 7.48 (ddd, 3J = 7.7, 7.2 Hz, 4J = 1.3 Hz, 2H, H-5), 7.44 (ddd, 3J = 7.7, 7.2 Hz, 4J = 1.3 Hz, 2H, H-4), 0.40 (s, 18H, CH3) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ = 157.27 (C-1), 142.95 (C-2), 134.97 (C-3), 130.14/130.11 (C-4 and C-5), 114.68 (C-6), 0.70 (C-7) ppm. 29Si{1H} NMR (100 MHz, CDCl3): δ = 4.04 ppm. IR (ATR): ν = 3059 (w), 2946 (w), 2987 (w), 2853 (w), 1968 (w), 1937 (w), 1859 (w), 1737 (w), 1581 (w), 1561 (w), 1465 (w), 1424 (w); 1296 (w), 1241 (m), 1119 (m), 1075 (w), 831 (s), 778 (s), 747 (m), 720 (s), 676 (m) cm−1. HRMS(EI): m/z calcd. C18H26N2Si2+ 326.16290 found 326.16245. Rf (n-pentane): 0.63. Mp (DSC; Onset): 68.59 °C
4.2.4. 2,2′-Bis(trimethylgermanyl)azobenzene (7)
A schlenk tube was filled with 2,2′-bis(trimethylstannyl)azobenzene (4) (80.0 mg, 0.16 mmol) in THF (5.00 mL) and cooled to −78 °C. Then, MeLi (1.88 M in THF, 0.25 mL, 0.47 mmol) was added within 5 min and after 1 h at this temperature, trimethylgermanium chloride (14) (200 µL, 171 mg, 1.57 mmol) was added to the dark reaction mixture in one portion. The reaction mixture was warmed to 25 °C over 14 h and the solvent was removed under reduced pressure. The brown solid, dissolved in DCM (3.00 mL), was purified by column chromatography (silica, n-pentane). From the filtrate, the solvent was removed to obtain an orange solid (31 mg, 0.09 mmol, 60%). 1H NMR (500 MHz, CDCl3): δ = 7.78–7.64 (m, 4H, H-3 and H-6), 7.49–7.39 (m, 4H, H4 and H5), 0.49 (s, 18H, CH3) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ = 156.59 (C-1), 146.12 (C-2), 134.21 (C-3), 130.11 (C-4), 129.52 (C-2), 114.98 (C-6), 0.32 (C-7) ppm. IR (ATR): ν = 3057 (w), 2962 (w), 2905 (w), 1563 (w), 1463 (w), 1432 (w), 1407 (w), 1295 (w), 1234 (m), 1114 (m), 1064 (w), 953 (w), 818 (m), 777 (s), 751 (m), 719 (m), 658 (m) cm−1. HRMS (APCI): m/z calcd. [C18H26N2Ge2 + H]+ 417.06085 found 417.06072. Rf (n-pentane): 0.78. Mp (DSC; Onset): 82.85 °C
4.2.5. 2,2′-Di(tert-butyl)azobenzene (10)
Adapted with changes from Takahashi et al. [
26] 2-
tert-butylaniline (
15) (5.00 g, 33.5 mmol) was dissolved in toluene (800 mL) and MnO
2 (50.0 g, 575 mmol) was added portionwise. The reaction mixture was heated to 120 °C. After 3 h at 120 °C, the orange reaction mixture was filtered with the help of Celite
®. The solvent was evaporated, the organic phase was washed with aq. HCl (2 M, 300 mL), and then the organic phase was dried over MgSO
4. After removal of the solvent, the product was dried in a vacuum (4 mbar, 110 °C, 5h) and could be isolated without further purification (2.26 g, 7.68 mmol, 46%, purity > 95%). For even higher purity, the compound was subjected to column chromatography (silica,
n-pentane). The first isolated band was the mentioned yellow byproduct (62 mg, 0.22 mmol, 1.31%). The second band was the orange product (620 mg, 2.11 mmol, 13%).
1H NMR (500 MHz, CDCl
3) δ = 7.54–7.50 (m, 4H, H-3 and H-6), 7.40 (td,
3J = 7.6 Hz,
4J = 1.4 Hz 2H, H-5), 7.32 (mc, 2H, H-4), 1.56 (s, 18 H, CH
3).
13C{
1H} NMR (126 MHz, CDCl
3): δ = 151.93 (
C-1), 148.60 (
C-2), 130.37 (
C-5), 126.80 (
C-4), 126.76 (
C-3), 117.05 (
C-6), 36.33 (
C(CH
3)
3, 32.16 (CH
3) ppm. IR (ATR): ν = 2948 (w), 2918 (w), 2904 (w), 2859 (w), 1478 (m), 1465 (w), 1435 (w), 1388 (w), 1354 (w), 1289 (w), 1277 (w), 1250 (w), 1196 (w), 1160 (w), 1086 (w), 1050 (w), 947 (w), 926 (w), 769 (s), 750 (s) cm
−1. HRMS (EI):
m/z calcd. C
20H
26N
2+ 294.20905 found 294.20851. R
f (
n-pentane): 0.76. Mp (DSC; Onset): 86.76 °C
4.2.6. Isolated By-Product: 1,6-Di-(tert-butyl)phenanzine (16)
1H NMR (500 MHz, CDCl3) δ = 8.11 (mc, 2H, H-4,9), 7.70 (mc, 4H, H-3,8 and H-2,7), 1.76 (s, 18H, CH3) ppm. 13C{1H} NMR (126 MHz, CDCl3) δ = 148.46 (C-1,6), 142.19 (C-4a,10a or C-5a,10), 141.89 (C-4a, 10a or C-5a,10a), 129.48 (C-3,8), 129.01 (C-4,9), 126.05 (C-2,7), 36.97 (C(CH3)3, 31.22 (CH3) ppm. IR (ATR): ν = 2997 (w), 2949 (w), 2902 (w), 2864 (w), 1616 (w), 1532 (w), 1481 (w), 1471 (w), 1383 (w), 1353 (w), 1343 (w), 1271 (w), 1126 (w), 997 (m), 932 (w), 857 (w), 813 (m), 748 (s) cm−1. HRMS (EI): m/z calcd. C20H24N2+ 292.19340 found 292.19284, calcd. [M-CH3]+ 277.16993 found 277.16993, calcd. [M-C3H6]+ 250.14645 found 250.14621, calcd. [M-C4H9]+ 235.12297 found 235.12263. Rf (n-pentane): 0.81. Mp (DSC; Onset): 193.89 °C





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}















