
Cyclic Peptides Acting as Allosteric Inhibitors of Human Thymidylate Synthase and Cancer Cell Growth

 ,
,  , , , ,
, , , ,
Abstract
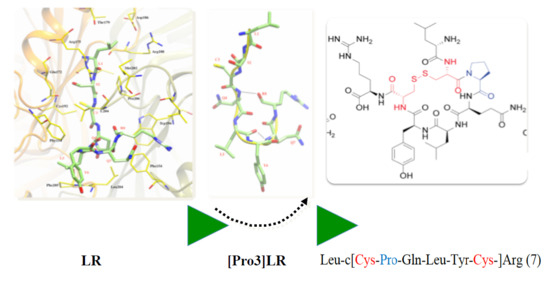
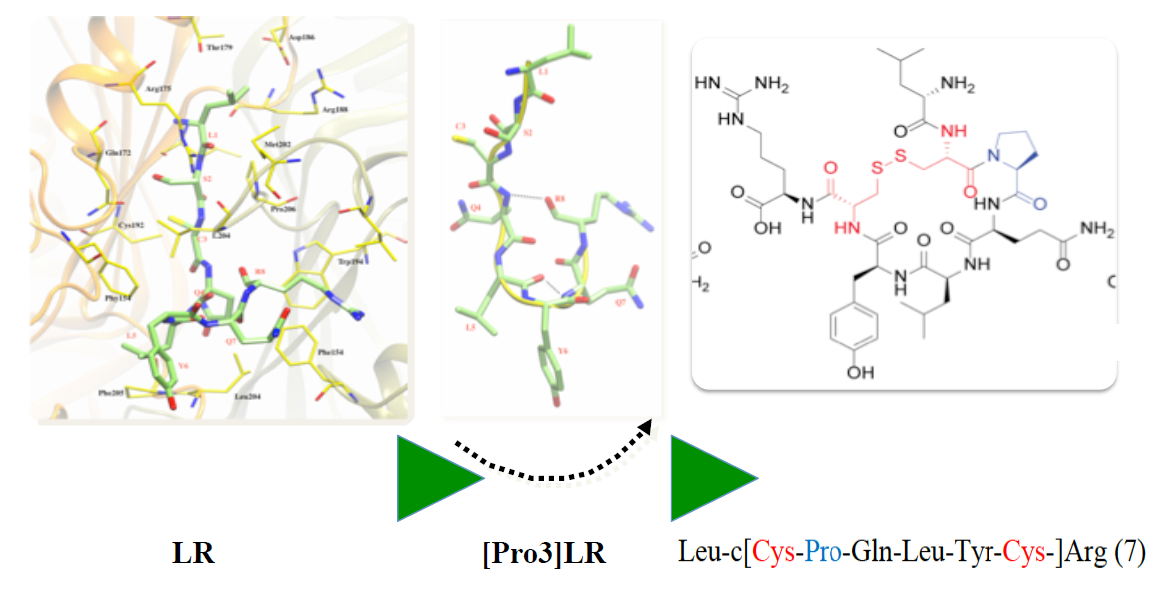
1. Introduction
2. Results and Discussion
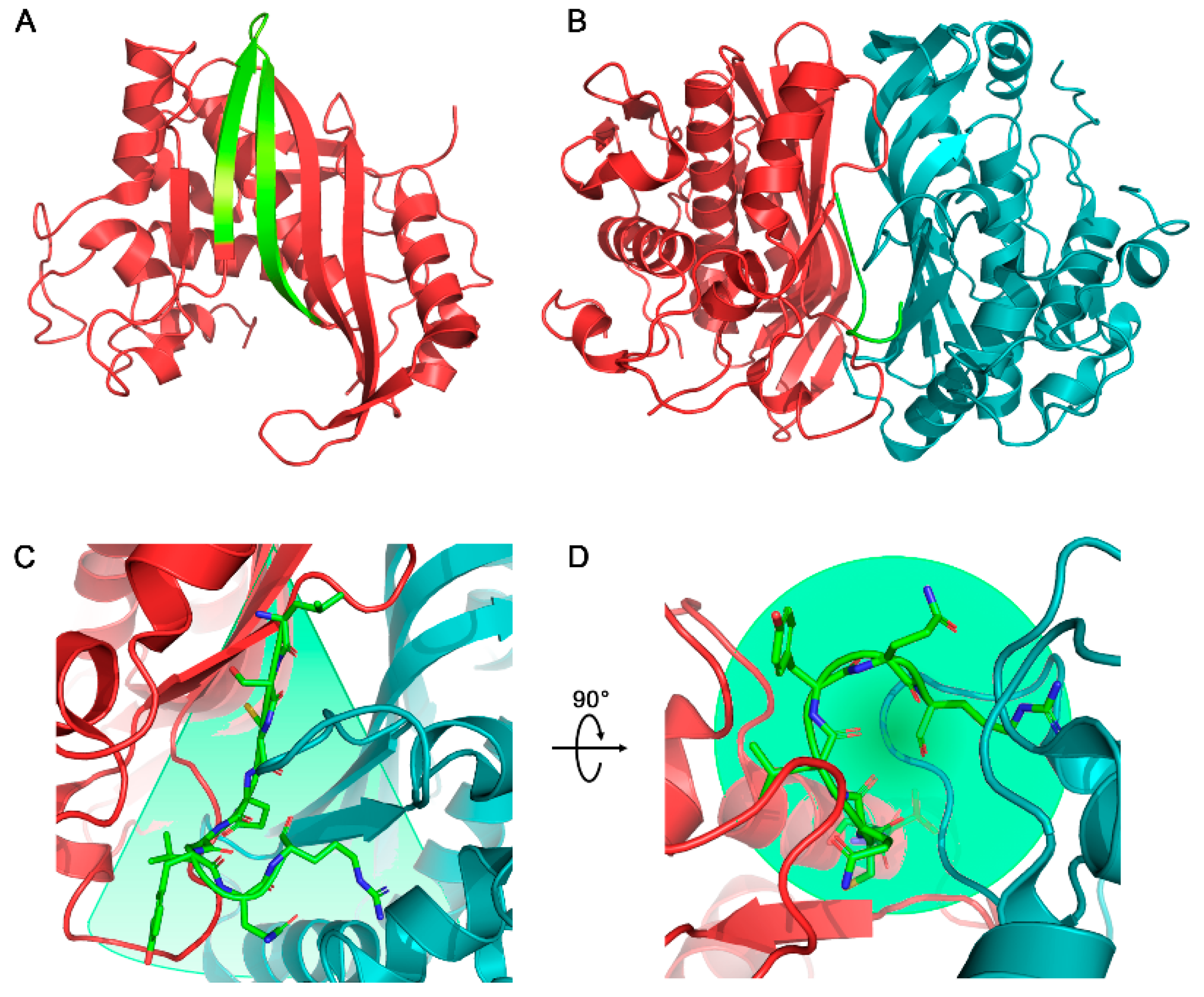
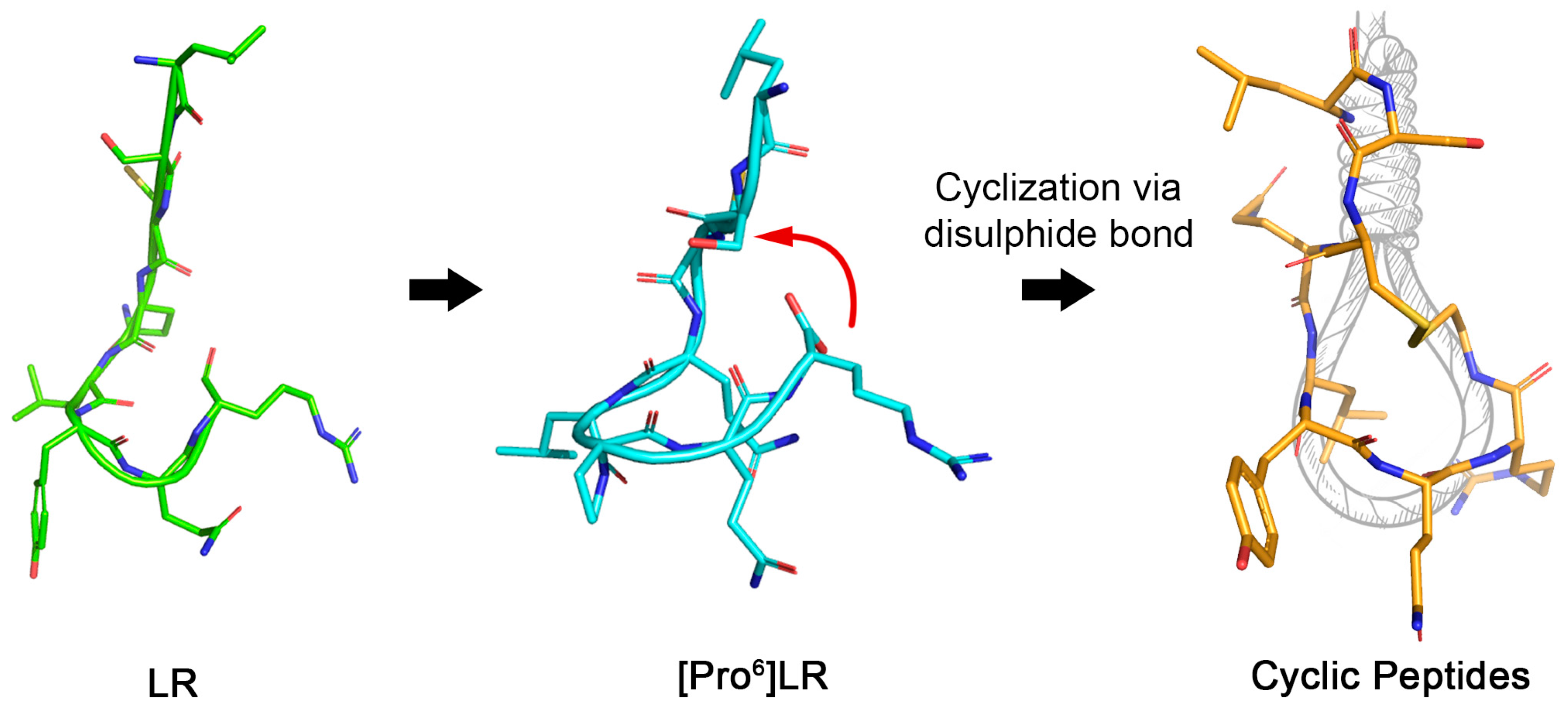
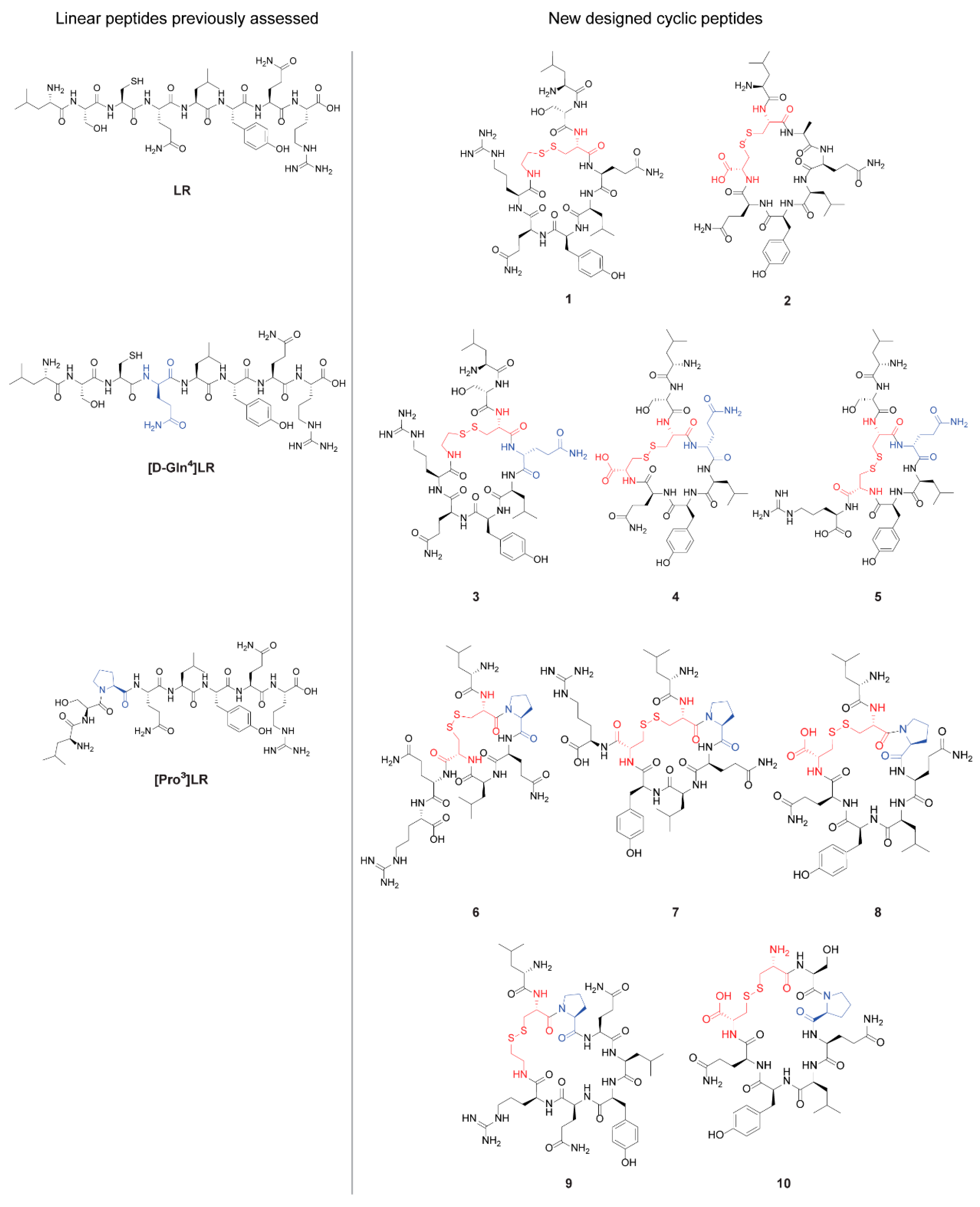
2.1. Design of Cyclic Peptides
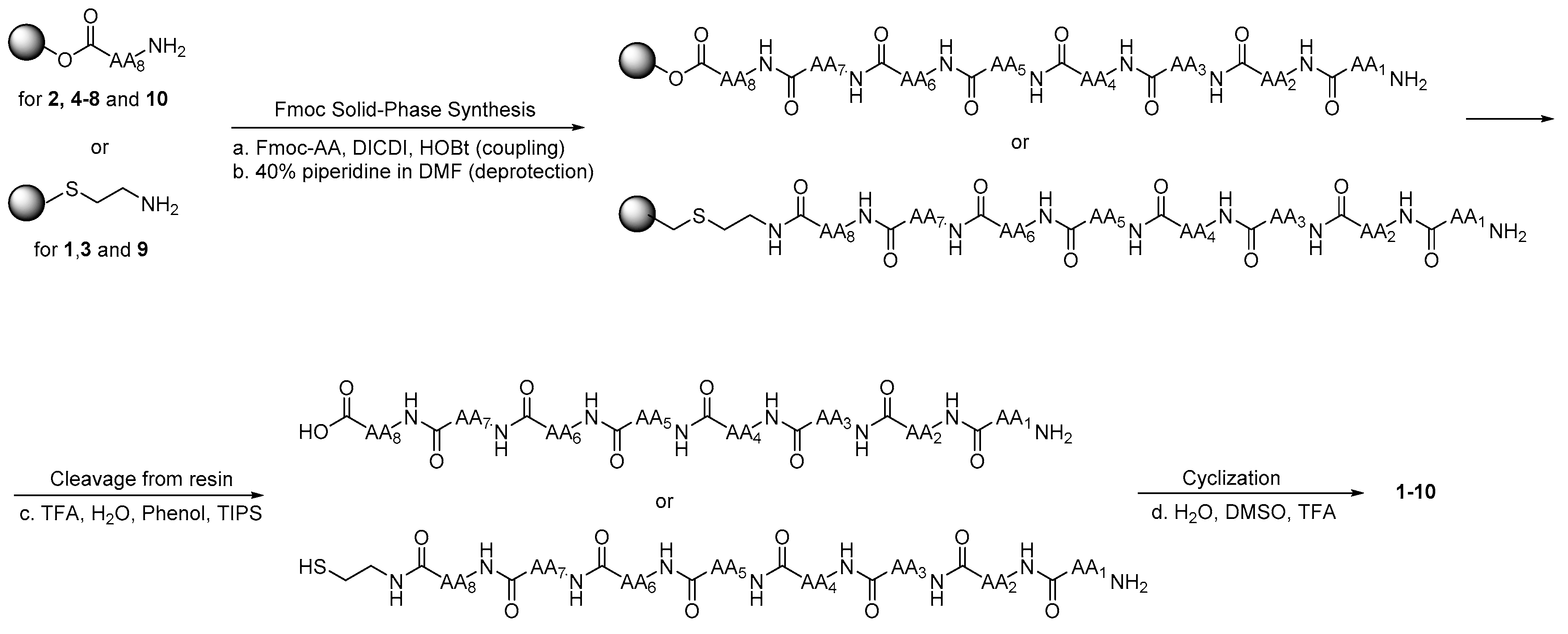
2.2. Chemistry
2.3. Human Thymidylate Synthase Inhibitory Activity
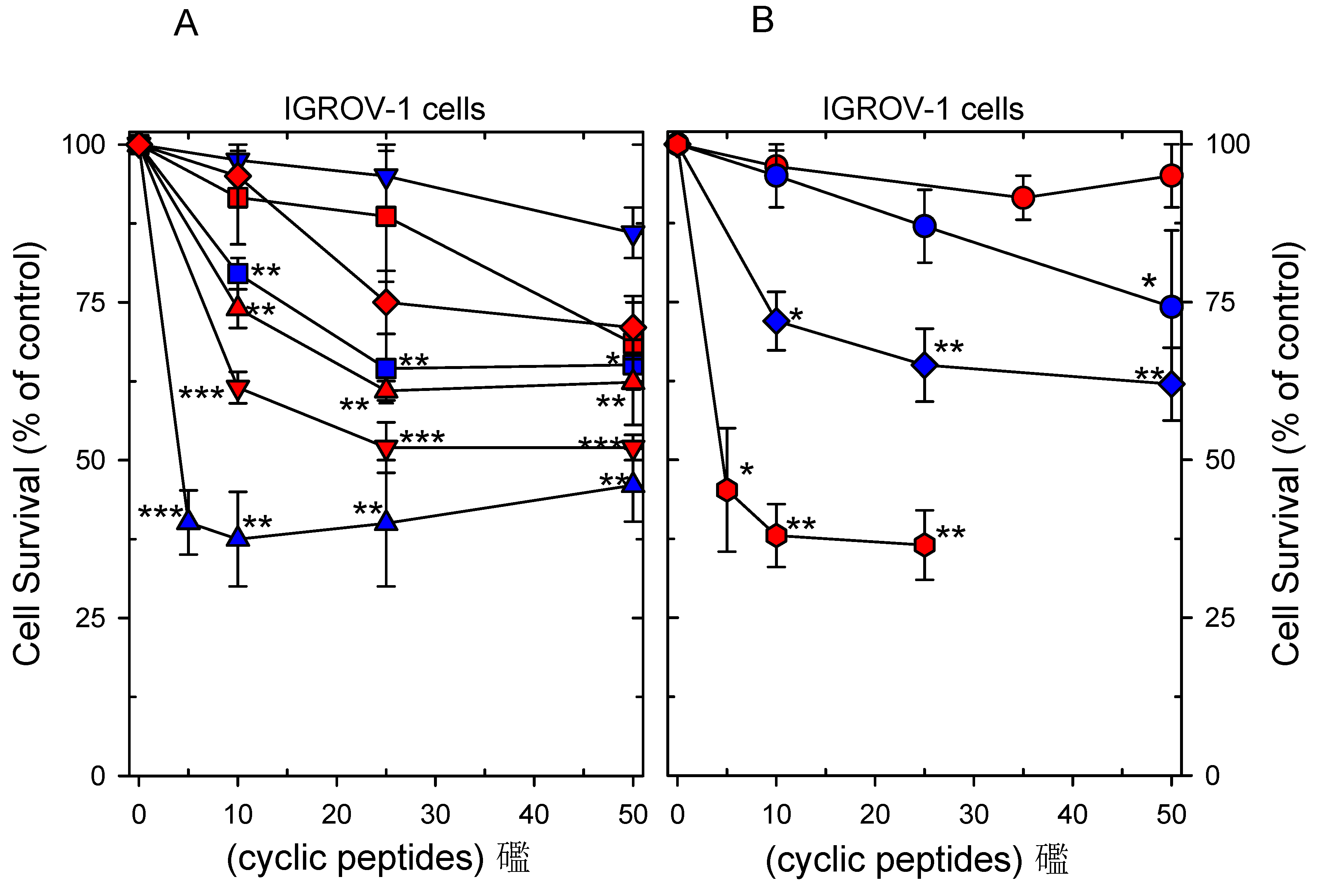
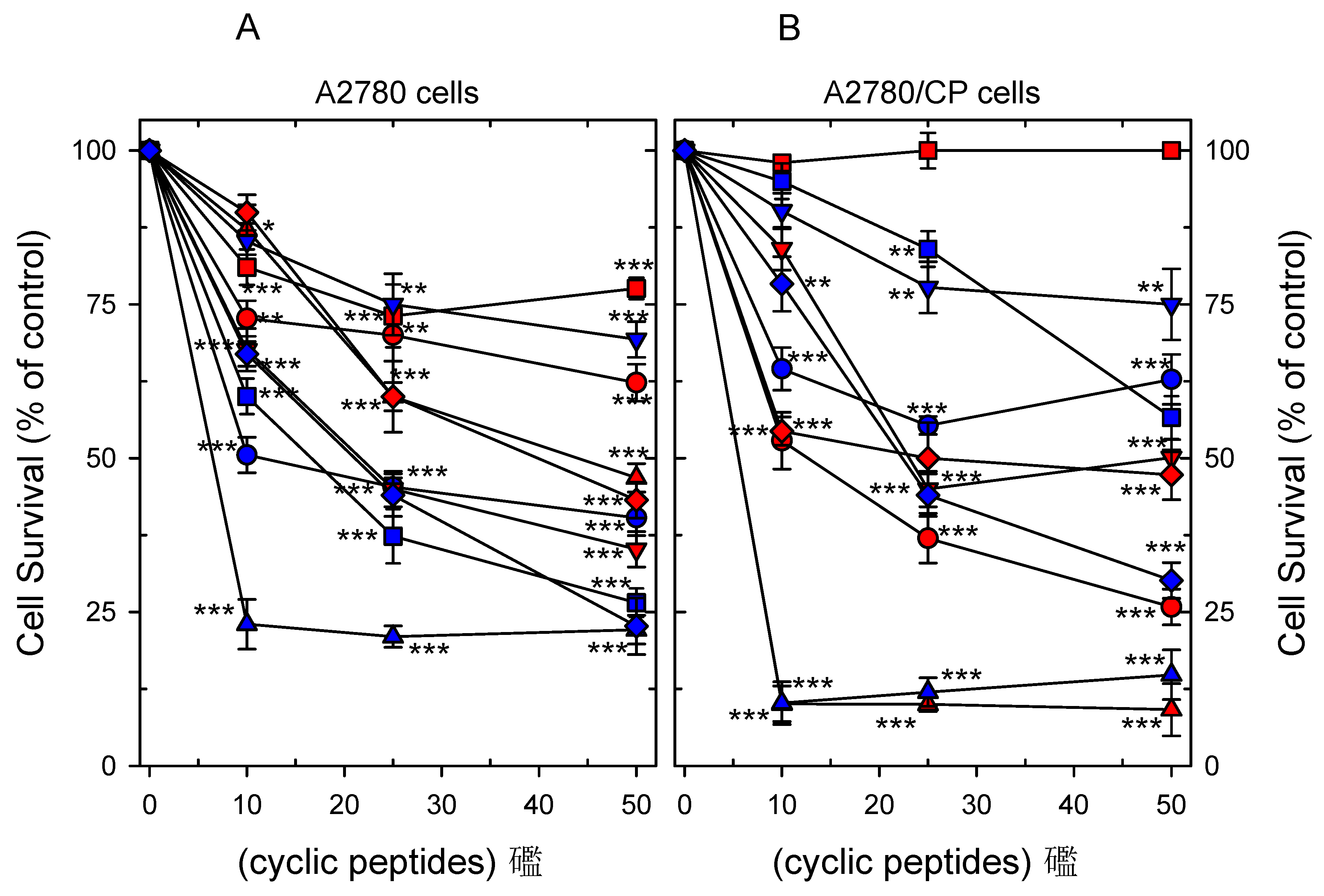
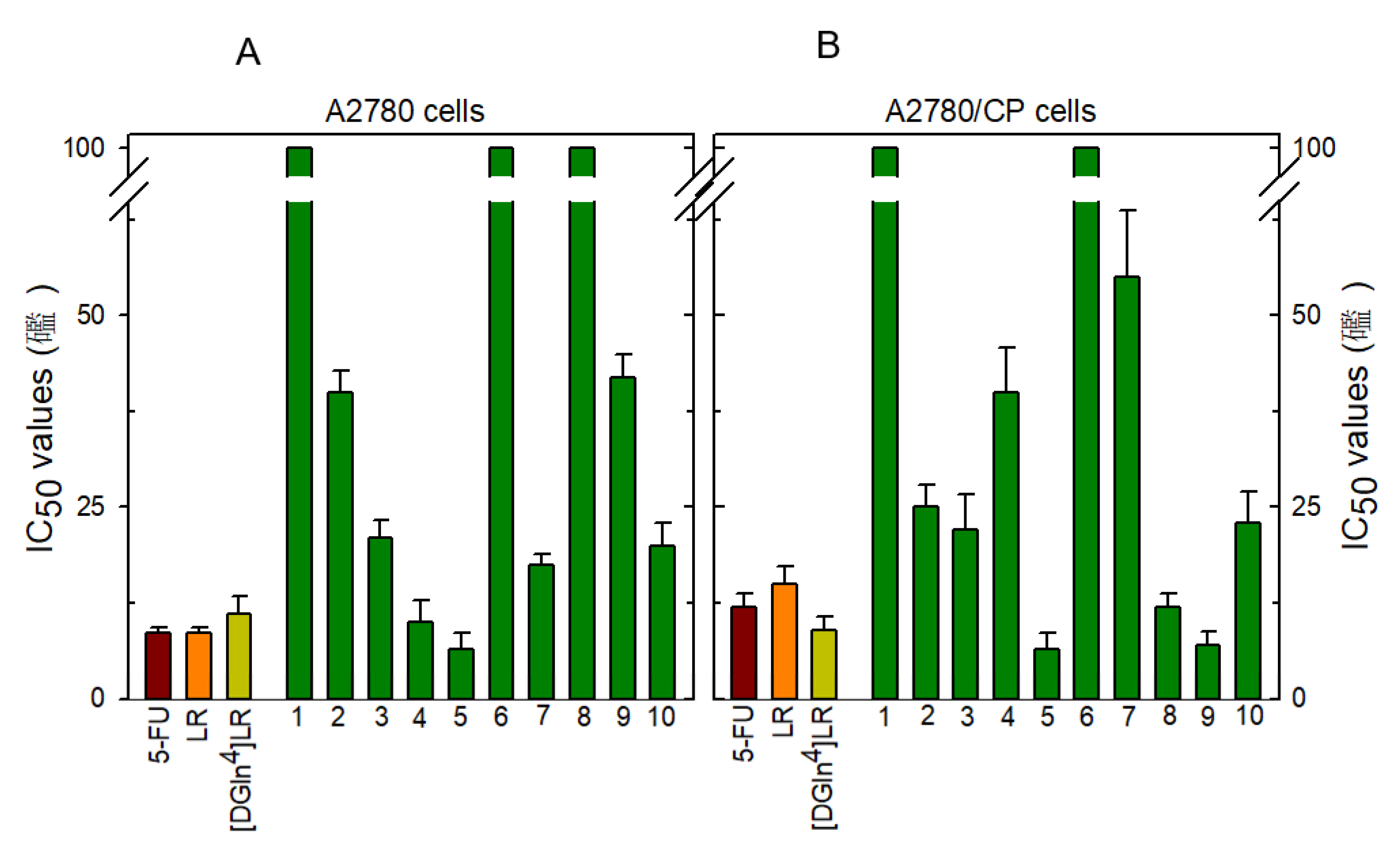
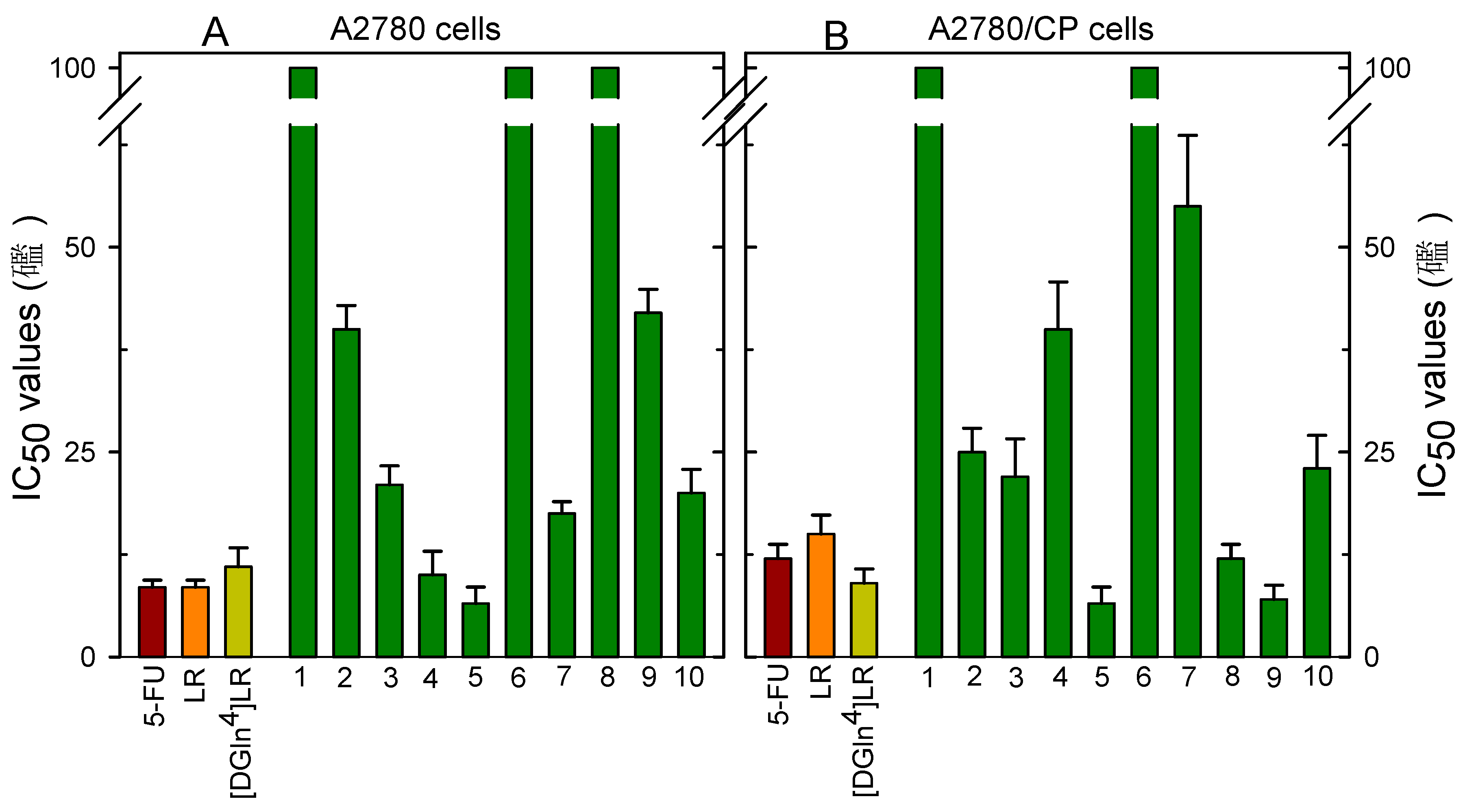
2.4. Cytotoxicity
3. Conclusions
4. Materials and Methods
4.1. Synthesis and Purification
4.1.1. Linear Peptides
4.1.2. Cyclic Peptides (1–10)
- Leu-Ser-c[Cys-Gln-Leu-Tyr-Gln-Arg-CAM-] (1)
- 15% yield. Retention time (Rt): 8.63 min. ESI-MS [M + H]+ calcd: 1067.513; found: 1068.006.
- Leu-c[Cys-Ala-Gln-Leu-Tyr-Gln-Cys] (2)
- 30% yield. Rt: 11.08 min. ESI-MS [M + H]+ calcd: 939.407; found: 939.852.
- Leu-Ser-c[Cys-D-Gln-Leu-Tyr-Gln-Arg-CAM-] (3)
- 13% yield. Rt: 8.66 min. ESI-MS [M + H]+ calcd: 1067.513; found: 1067.974.
- Leu-Ser-c[Cys-D-Gln-Leu-Tyr-Gln-Cys-] (4)
- 18% yield. Rt: 12.47 min. ESI-MS [M + H]+ calcd: 955.402; found: 955.402.
- Leu-Ser-c[Cys-D-Gln-Leu-Tyr-Cys-]Arg (5)
- 29% yield. Rt: 10.74 min. ESI-MS [M + H]+ calcd: 983.444; found: 983.904.
- Leu-c[Cys-Pro-Gln-Leu-Cys-]Gln-Arg (6)
- 25% yield. Rt: 8.42 min. ESI-MS [M + H]+ calcd: 958.460; found: 958.756.
- Leu-c[Cys-Pro-Gln-Leu-Tyr-Cys-]Arg (7)
- 18% yield. Rt: 9.33 min. ESI-MS [M + H]+ calcd: 993.465; found: 993.561.
- Leu-c[Cys-Pro-Gln-Leu-Tyr-Gln-Cys-] (8)
- 22% yield. Rt: 12.47 min. ESI-MS [M + H]+ calcd: 965.422; found: 966.074.
- Leu-c[Cys-Pro-Gln-Leu-Tyr-Gln-Arg-CAM] (9)
- 10% yield. Rt: 9.11 min. ESI-MS [M + H]+ calcd: 1078.331; found: 1078.251.
- c[Cys-Ser-Pro-Gln-Leu-Tyr-Gln-Cys] (10)
- 25% yield. Rt: 9.23 min. ESI-MS [M + H]+ calcd: 939.370; found: 939.730.
4.2. Thymidylate Synthase Purification, Characterization, and Inhibition
4.3. Cell Lines
4.4. Treatment with Peptides in Presence of SAINT-PhD Delivery System
4.5. Cytotoxicity Assay of Peptides
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carreras, C.W.; Santi, D.V. The Catalytic Mechanism and Structure of Thymidylate Synthase. Annu. Rev. Biochem. 1995, 64, 721–762. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.; Cogliati, T.; Copur, S.M.; Borre, A.; Voeller, D.M.; Allegra, C.J.; Segal, S. Identification of in vivo target RNA sequences bound by thymidylate synthase. Nucleic Acids Res. 1996, 24, 3222–3228. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chu, E.; Koeller, D.M.; Casey, J.L.; Drake, J.C.; Chabner, B.A.; Elwood, P.C.; Zinn, S.; Allegra, C.J. Autoregulation of human thymidylate synthase messenger RNA translation by thymidylate synthase. Proc. Natl. Acad. Sci. USA 1991, 88, 8977–8981. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.; Voeller, D.; Koeller, D.M.; Drake, J.C.; Takimoto, C.H.; Maley, G.F.; Maley, F.; Allegra, C.J. Identification of an RNA binding site for human thymidylate synthase. Proc. Natl. Acad. Sci. USA 1993, 90, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Schmitz, J.C.; Lin, X.; Tai, N.; Yan, W.; Farrell, M.; Bailly, M.; Chen, T.; Chu, E. Thymidylate synthase as a translational regulator of cellular gene expression. Biochim. Biophys. Acta Mol. Basis Dis. 2002, 1587, 174–182. [Google Scholar] [CrossRef]
- Taddia, L.; D’Arca, D.; Ferrari, S.; Marraccini, C.; Severi, L.; Ponterini, G.; Assaraf, Y.G.; Marverti, G.; Costi, M.P. Inside the biochemical pathways of thymidylate synthase perturbed by anticancer drugs: Novel strategies to overcome cancer chemoresistance. Drug Resist. Updates 2015, 23, 20–54. [Google Scholar] [CrossRef]
- Rose, M.G.; Farrell, M.P.; Schmitz, J.C. Thymidylate Synthase: A Critical Target for Cancer Chemotherapy. Clin. Colorectal Cancer 2002, 1, 220–229. [Google Scholar] [CrossRef]
- Wilson, P.M.; Danenberg, P.V.; Johnston, P.G.; Lenz, H.-J.; Ladner, R.D. Standing the test of time: Targeting thymidylate biosynthesis in cancer therapy. Nat. Rev. Clin. Oncol. 2014, 11, 282–298. [Google Scholar] [CrossRef]
- Li, Q.; Boyer, C.; Lee, J.Y.; Shepard, H.M. A novel approach to thymidylate synthase as a target for cancer chemotherapy. Mol. Pharmacol. 2001, 59, 446–452. [Google Scholar] [CrossRef]
- Jackman, A.L.; Taylor, G.A.; Gibson, W.; Kimbell, R.; Brown, M.; Calvert, A.H.; Judson, I.R.; Hughes, L.R. ICI D1694, a quinazoline antifolate thymidylate synthase inhibitor that is a potent inhibitor of L1210 tumor cell growth in vitro and in vivo: A new agent for clinical study. Cancer Res. 1991, 51, 5579–5586. [Google Scholar]
- Shih, C.; Chen, V.J.; Gossett, L.S.; Gates, S.B.; MacKellar, W.C.; Habeck, L.L.; Shackelford, K.A.; Mendelsohn, L.G.; Soose, D.J.; Patel, V.F.; et al. LY231514, a pyrrolo[2,3-d]pyrimidine-based antifolate that inhibits multiple folate-requiring enzymes. Cancer Res. 1997, 57, 1116–1123. [Google Scholar] [PubMed]
- Sayre, P.H.; Finer-Moore, J.S.; Fritz, T.A.; Biermann, D.; Gates, S.B.; MacKellar, W.C.; Patel, V.F.; Stroud, R.M. Multi-targeted antifolates aimed at avoiding drug resistance form covalent closed inhibitory complexes with human and Escherichia coli thymidylate synthases. J. Mol. Biol. 2001, 313, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Mullen, M.M.; Kuroki, L.M.; Thaker, P.H. Novel treatment options in platinum-sensitive recurrent ovarian cancer: A review. Gynecol. Oncol. 2019, 152, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, K.J.; Kashani-Sabet, M. Elevated expression of thymidylate synthase cycle genes in cisplatin-resistant human ovarian carcinoma A2780 cells. Proc. Natl. Acad. Sci. USA 1988, 85, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L.R.; Kimbell, R.; Hardcastle, A.; Aherne, G.W.; Jackman, A.L. Relationship between resistance to cisplatin and antifolates in sensitive and resistant tumour cell lines. Eur. J. Cancer. 1995, 31A, 981–986. [Google Scholar] [CrossRef]
- Marverti, G.; Ligabue, A.; Paglietti, G.; Corona, P.; Piras, S.; Vitale, G.; Guerrieri, D.; Luciani, R.; Costi, MP.; Frassineti, C.; et al. Collateral sensitivity to novel thymidylate synthase inhibitors correlates with folate cycle enzymes impairment in cisplatin-resistant human ovarian cancer cells. Eur. J. Pharmacol. 2009, 615, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Pela, M.; Saxena, P.; Luciani, R.; Santucci, M.; Ferrari, S.; Marverti, G.; Marraccini, C.; Martello, A.; Pirondi, S.; Genovese, F.; et al. Optimization of peptides that target human thymidylate synthase to inhibit ovarian cancer cell growth. J. Med. Chem. 2014, 57, 1355–1367. [Google Scholar] [CrossRef]
- Tochowicz, A.; Santucci, M.; Saxena, P.; Guaitoli, G.; Trande, M.; Finer-Moore, J.; Stroud, R.M.; Costi, M.P. Alanine mutants of the interface residues of human thymidylate synthase decode key features of the binding mode of allosteric anticancer peptides. J. Med. Chem. 2015, 58, 1012–1018. [Google Scholar] [CrossRef]
- Ponterini, G.; Martello, A.; Pavesi, G.; Lauriola, A.; Luciani, R.; Santucci, M.; Pela, M.; Gozzi, G.; Pacifico, S.; Guerrini, R.; et al. Intracellular quantitative detection of human thymidylate synthase engagement with an unconventional inhibitor using tetracysteine-diarsenical-probe technology. Sci. Rep. 2016, 6, 27198. [Google Scholar] [CrossRef]
- Saxena, P.; Severi, L.; Santucci, M.; Taddia, L.; Ferrari, S.; Luciani, R.; Marverti, G.; Marraccini, C.; Tondi, D.; Mor, M.; et al. Conformational Propensity and Biological Studies of Proline Mutated LR Peptides Inhibiting Human Thymidylate Synthase and Ovarian Cancer Cell Growth. J. Med. Chem. 2018, 61, 7374–7380. [Google Scholar] [CrossRef]
- Cardinale, D.; Guaitoli, G.; Tondi, D.; Luciani, R.; Henrich, S.; Salo-Ahen, O.M.H.; Ferrari, S.; Marverti, G.; Guerrieri, D.; Ligabue, A.; et al. Protein-protein interface-binding peptides inhibit the cancer therapy target human thymidylate synthase. Proc. Natl. Acad. Sci. USA 2011, 108, E542–E549. [Google Scholar] [CrossRef]
- Genovese, F.; Gualandi, A.; Taddia, L.; Marverti, G.; Pirondi, S.; Marraccini, C.; Perco, P.; Pela, M.; Guerrini, R.; Amoroso, M.R.; et al. Mass spectrometric/bioinformatic identification of a protein subset that characterizes the cellular activity of anticancer peptides. J. Proteome Res. 2014, 13, 5250–5261. [Google Scholar] [CrossRef]
- Cannazza, G.; Cazzato, A.S.; Marraccini, C.; Pavesi, G.; Pirondi, S.; Guerrini, R.; Pelà, M.; Frassineti, C.; Ferrari, S.; Marverti, G.; et al. Internalization and stability of a thymidylate synthase peptide inhibitor in ovarian cancer cells. J. Med. Chem. 2014, 57, 10551–10556. [Google Scholar] [CrossRef]
- Martí-Centelles, V.; Pandey, M.D.; Burguete, M.I.; Luis, S. V Macrocyclization Reactions: The Importance of Conformational, Configurational, and Template-Induced Preorganization. Chem. Rev. 2015, 115, 8736–8834. [Google Scholar] [CrossRef]
- Driggers, E.M.; Hale, S.P.; Lee, J.; Terrett, N.K. The exploration of macrocycles for drug discovery—An underexploited structural class. Nat. Rev. Drug Discov. 2008, 7, 608–624. [Google Scholar] [CrossRef]
- Pauletti, G.M.; Gangwar, S.; Siahaan, T.J.; Aubé, J.; Borchardt, R.T. Improvement of oral peptide bioavailability: Peptidomimetics and prodrug strategies. Adv. Drug Deliv. Rev. 1997, 27, 235–256. [Google Scholar] [CrossRef]
- Chan, L.Y.; Gunasekera, S.; Henriques, S.T.; Worth, N.F.; Le, S.J.; Clark, R.J.; Campbell, J.H.; Craik, D.J.; Daly, N.L. Engineering pro-angiogenic peptides using stable, disulfide-rich cyclic scaffolds. Blood 2011, 18, 6709–6717. [Google Scholar] [CrossRef]
- Haÿ, E.; Buczkowski, T.; Marty, C.; Da Nascimento, S.; Sonnet, P.; Marie, P.J. Peptide-based mediated disruption of N-cadherin-LRP5/6 interaction promotes Wnt signaling and bone formation. J. Bone Miner. Res. 2012, 27, 1852–1863. [Google Scholar]
- Benoiton, N.L. Chemistry of Peptide Synthesis, 1st ed.; CRC Press: Boca Raton, FL, USA, 2005; ISBN 9781574444544. [Google Scholar]
- Sole, N.A.; Barany, G. Optimization of solid-phase synthesis of [Ala8]-dynorphin A. J. Org. Chem. 1992, 57, 5399–5403. [Google Scholar] [CrossRef]
- Marzola, E.; Camarda, V.; Batuwangala, M.; Lambert, D.G.; Calo’, G.; Guerrini, R.; Trapella, C.; Regoli, D.; Tomatis, R.; Salvadori, S. Structure-activity relationship study of position 4 in the urotensin-II receptor ligand U-II(4-11). Peptides 2008, 29, 674–679. [Google Scholar] [CrossRef]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Beaufort, B.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.J.; Heine, A.A.J.; Smid, M.; et al. Ovarian Cancer Cell Line Panel (OCCP): Clinical Importance of In Vitro Morphological Subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef]
- Synvolux Products. Available online: https://www.synvoluxproducts.com/saint-protein/ (accessed on 24 September 2019).
- Marverti, G.; Ligabue, A.; Lombardi, P.; Ferrari, S.; Monti, M.G.; Frassineti, C.; Costi, M.P. Modulation of the expression of folate cycle enzymes and polyamine metabolism by berberine in cisplatin-sensitive and-resistant human ovarian cancer cells. Int. J. Oncol. 2013, 43, 1269–1280. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Primary Sequence | % in hTS a (IC50 in µM) a |
|---|---|---|
| LR | Leu-Ser-Cys-Gln-Leu-Tyr-Gln-Arg | (75) b |
| [d-Gln4]LR | Leu-Ser-Cys-d-Gln-Leu-Tyr-Gln-Arg | (68) b |
| [Pro3]LR | Leu-Ser-Pro-Gln-Leu-Tyr-Gln-Arg | 25% at 100 µM c |
| 1 | Leu-Ser-c[Cys-Gln-Leu-Tyr-Gln-Arg-CAM] | 25% at 400 µM and r.t. under stirring (2125) |
| 2 | Leu-c[Cys-Ala-Gln-Leu-Tyr-Gln-Cys] | 36% at 400 µM and r.t. under stirring (900) |
| 3 | Leu-Ser-c[Cys-d-Gln-Leu-Tyr-Gln-Arg-CAM] | N.I. at 500 µM and r.t. N.I. at 500 µM and 37 °C under stirring |
| 4 | Leu-Ser-c[Cys-d-Gln-Leu-Tyr-Gln-Cys] | N.I. at 500 µM and r.t. N.I. at 500 µM and 37 °C under stirring |
| 5 | Leu-Ser-c[Cys-d-Gln-Leu-Tyr-Cys]-Arg | N.I. at 500 µM and r.t. N.I. at 500 µM and 37 °C under stirring |
| 6 | Leu-c[Cys-Pro-Gln-Leu-Cys]-Gln-Arg | N.I. at 500 µM and r.t. N.I. at 500 µM and 37 °C under stirring |
| 7 | Leu-c[Cys-Pro-Gln-Leu-Tyr-Cys]-Arg | 76% at 500 µM, 59% at 100 µM and r.t. under stirring (148) Allosteric inhibition profile (see Figure S1) |
| 8 | Leu-c[Cys-Pro-Gln-Leu-Tyr-Gln-Cys] | 38% at 500 µM and r.t. under stirring (856) |
| 9 | Leu-c[Cys-Pro-Gln-Leu-Tyr-Gln-Arg-CAM] | N.I. at 500 µM and r.t. N.I. at 500 µM and 37 °C under stirring |
| 10 | c[Cys-Ser-Pro-Gln-Leu-Tyr-Gln-Cys] | 50% at 500 µM and r.t. under stirring (474) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacifico, S.; Santucci, M.; Luciani, R.; Saxena, P.; Linciano, P.; Ponterini, G.; Lauriola, A.; D’Arca, D.; Marverti, G.; Guerrini, R.; et al. Cyclic Peptides Acting as Allosteric Inhibitors of Human Thymidylate Synthase and Cancer Cell Growth. Molecules 2019, 24, 3493. https://doi.org/10.3390/molecules24193493
Pacifico S, Santucci M, Luciani R, Saxena P, Linciano P, Ponterini G, Lauriola A, D’Arca D, Marverti G, Guerrini R, et al. Cyclic Peptides Acting as Allosteric Inhibitors of Human Thymidylate Synthase and Cancer Cell Growth. Molecules. 2019; 24(19):3493. https://doi.org/10.3390/molecules24193493
Chicago/Turabian StylePacifico, Salvatore, Matteo Santucci, Rosaria Luciani, Puneet Saxena, Pasquale Linciano, Glauco Ponterini, Angela Lauriola, Domenico D’Arca, Gaetano Marverti, Remo Guerrini, and et al. 2019. "Cyclic Peptides Acting as Allosteric Inhibitors of Human Thymidylate Synthase and Cancer Cell Growth" Molecules 24, no. 19: 3493. https://doi.org/10.3390/molecules24193493
APA StylePacifico, S., Santucci, M., Luciani, R., Saxena, P., Linciano, P., Ponterini, G., Lauriola, A., D’Arca, D., Marverti, G., Guerrini, R., & Costi, M. P. (2019). Cyclic Peptides Acting as Allosteric Inhibitors of Human Thymidylate Synthase and Cancer Cell Growth. Molecules, 24(19), 3493. https://doi.org/10.3390/molecules24193493