
Advances in the Asymmetric Total Synthesis of Natural Products Using Chiral Secondary Amine Catalyzed Reactions of α,β-Unsaturated Aldehydes


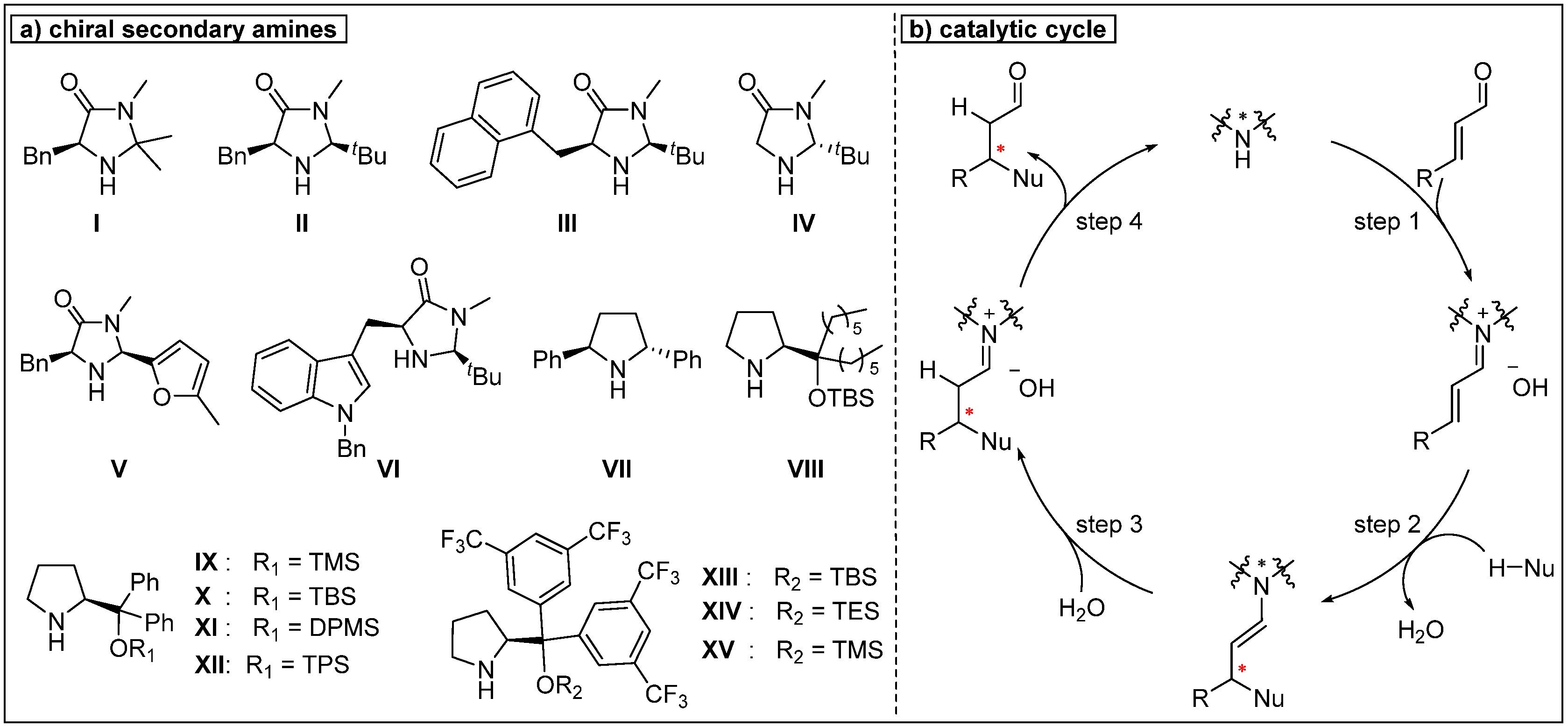{kind=link}
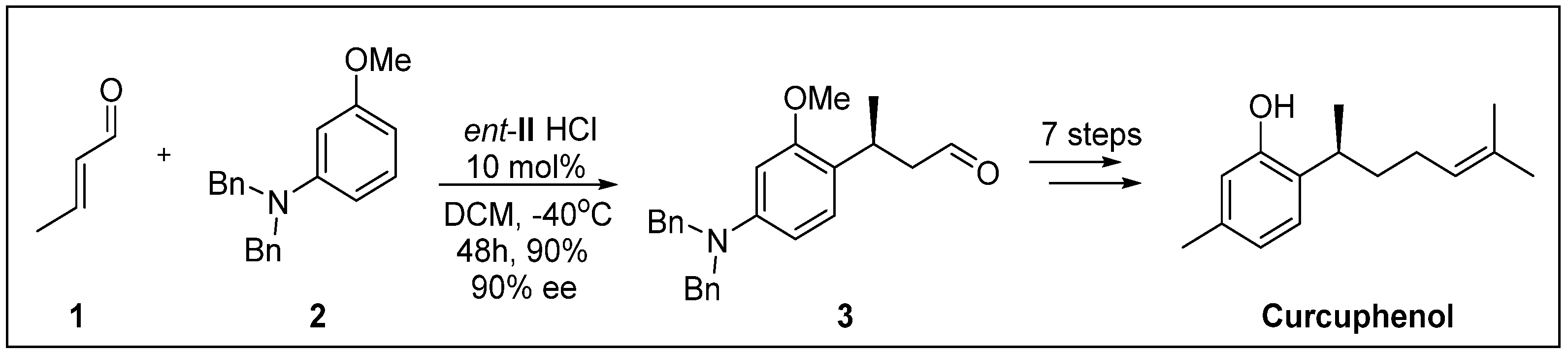{kind=link}
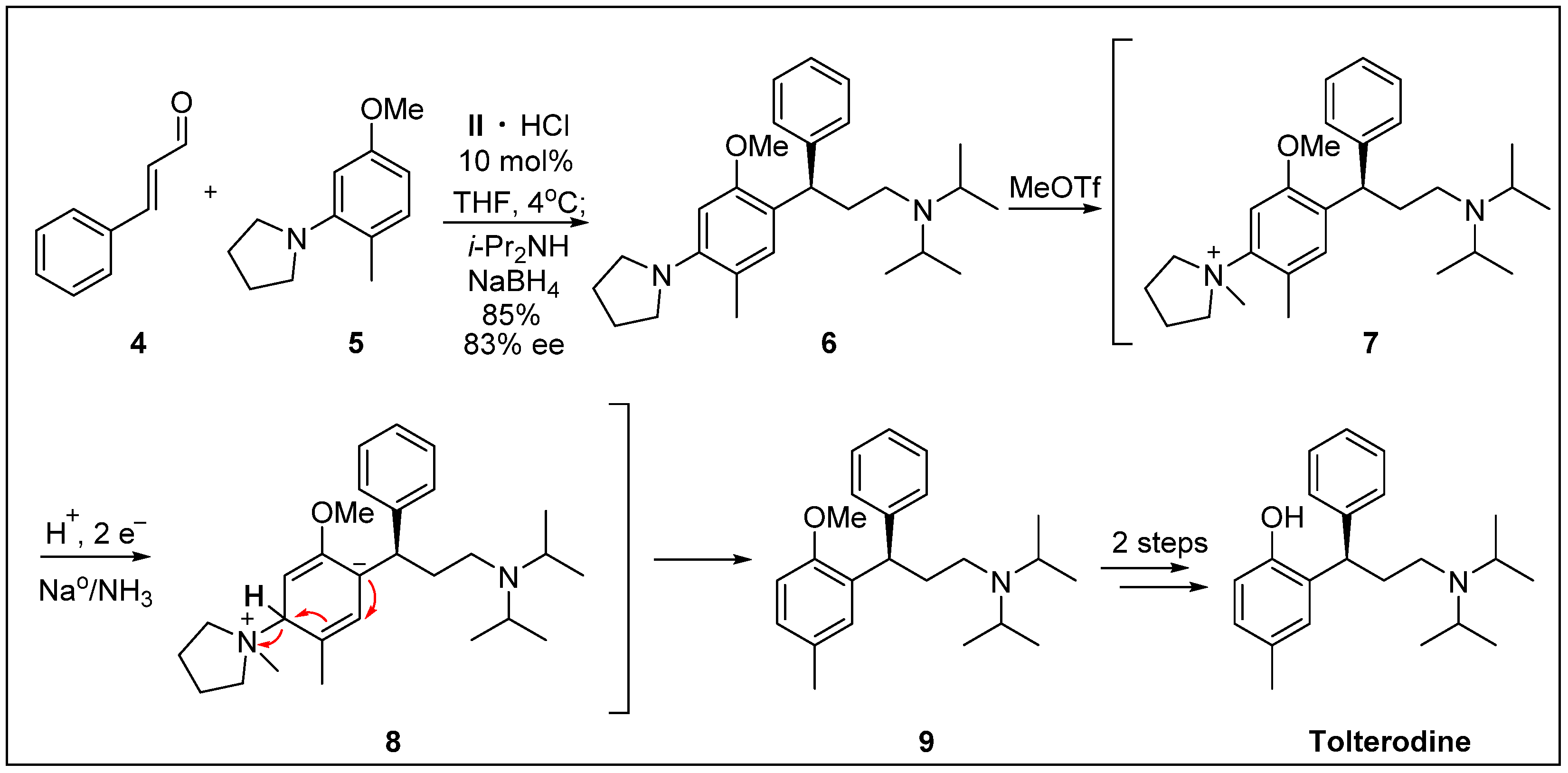{kind=link}
{kind=link}
{kind=link}
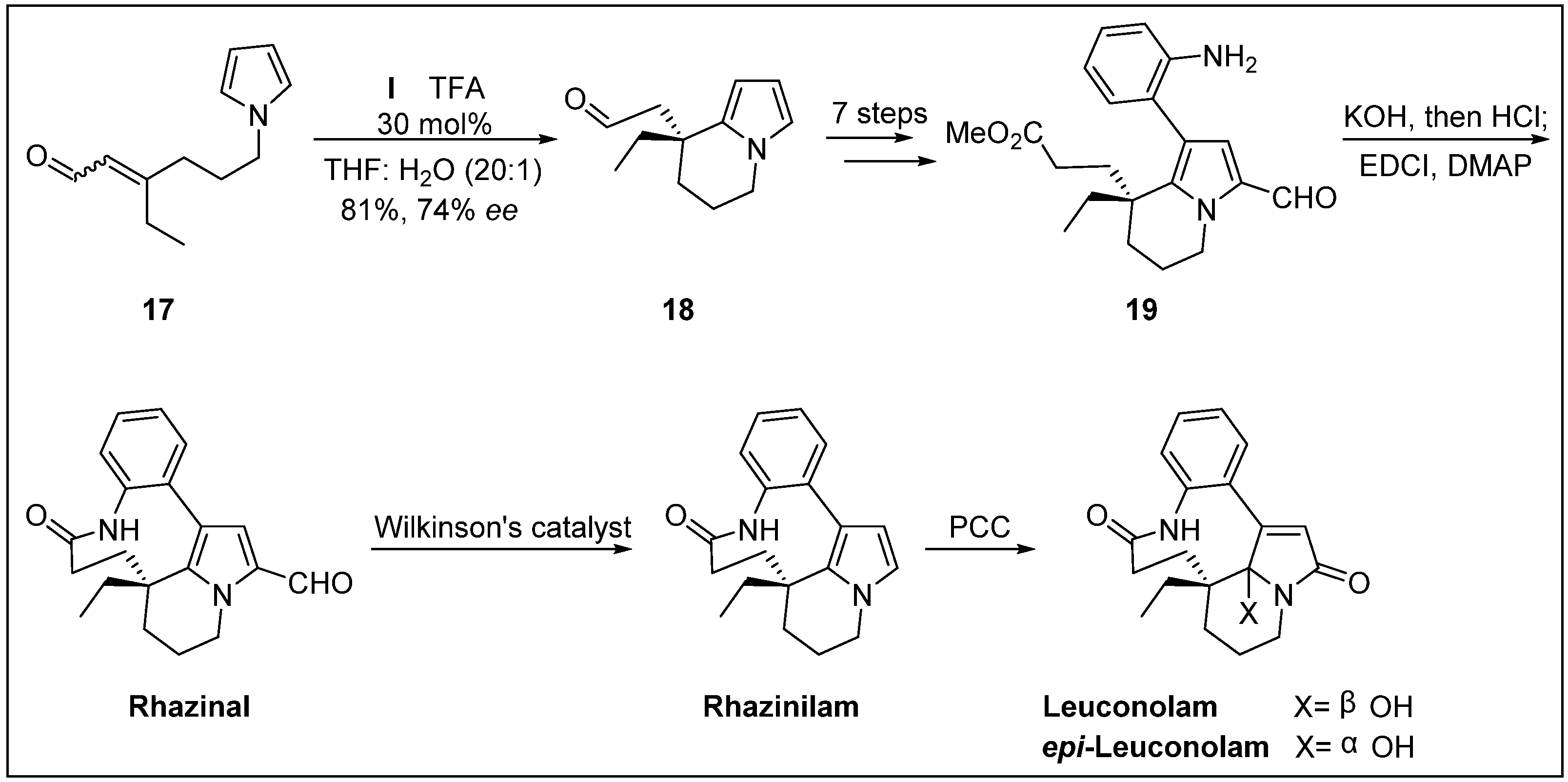{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Asymmetric Total Synthesis of Bioactive Natural Products and Pharmaceuticals
2.1. Friedel-Crafts Alkylation Reaction
2.2. Michael Addition Reaction
2.2.1. Oxa-Michael Addition
2.2.2. Aza-Michael Addition
2.2.3. Michael Addition
2.3. Hydrogenation
2.4. Cycloadditions
2.4.1. Cyclopropanations
2.4.2. Epoxidations
2.4.3. Diels-Alder Reactions
2.4.4. [3+2]-Cycloadditions
2.4.5. [3+3]-Cycloadditions
2.5. Organocascade Reactions
3. Conclusions
Funding
Conflicts of Interest
References
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W. New strategies for organic catalysis: The first highly enantioselective organocatalytic diels—alder reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Diphenylprolinol silyl ethers as efficient organocatalysts for the asymmetric Michael reaction of aldehydes and nitroalkenes. Angew. Chem. Int. Ed. 2005, 44, 4212–4215. [Google Scholar] [CrossRef]
- Franzén, J.; Marigo, M.; Fielenbach, D.; Wabnitz, T.C.; Kjærsgaard, A.; Jørgensen, K.A. A general organocatalyst for direct α-functionalization of aldehydes: Stereoselective C−C, C−N, C−F, C−Br, and C−S bond-forming reactions. scope and mechanistic insights. J. Am. Chem. Soc. 2005, 127, 18296–18304. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, D.W. The advent and development of organocatalysis. Nature 2008, 455, 304. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Cabrera, S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem. Soc. Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef] [PubMed]
- Lelais, G.; MacMillan, D.W. Modern strategies in organic catalysis: The advent and development of iminium activation. Aldrichim. Acta 2006, 39, 79–87. [Google Scholar]
- Erkkilä, A.; Majander, I.; Pihko, P.M. Iminium catalysis. Chem. Rev. 2007, 107, 5416–5470. [Google Scholar] [CrossRef] [PubMed]
- Klier, L.; Tur, F.; Poulsen, P.H.; Jørgensen, K.A. Asymmetric cycloaddition reactions catalysed by diarylprolinol silyl ethers. Chem. Soc. Rev. 2017, 46, 1080–1102. [Google Scholar] [CrossRef] [PubMed]
- Moyano, A.; Rios, R. Asymmetric organocatalytic cyclization and cycloaddition reactions. Chem. Rev. 2011, 111, 4703–4832. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Wang, C.; Liebich, J.X. Organocatalytic asymmetric aza—Michael additions. Chem. Eur. J. 2009, 15, 11058–11076. [Google Scholar] [CrossRef] [PubMed]
- Grondal, C.; Jeanty, M.; Enders, D. Organocatalytic cascade reactions as a new tool in total synthesis. Nat. Chem. 2010, 2, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.-F. Total synthesis of natural and pharmaceutical products powered by organocatalytic reactions. Tetrahedron Lett. 2015, 56, 2133–2140. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, O.V.; Beletskaya, I.P.; Zlotin, S.G. Organocatalytic Michael and Friedel-Crafts reactions in enantioselective synthesis of biologically active compounds. Russ. Chem. Rev. 2011, 80, 1067–1113. [Google Scholar] [CrossRef]
- Reyes-Rodríguez, G.J.; Rezayee, N.M.; Vidal-Albalat, A.; Jørgensen, K.A. Prevalence of diarylprolinol silyl ethers as catalysts in total synthesis and patents. Chem. Rev. 2019, 119, 4221–4260. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-H.; Tan, F.; Xiao, W.-J. Enantioselective organocatalytic Friedel-Crafts alkylations. Curr. Org. Chem. 2011, 15, 4022–4045. [Google Scholar] [CrossRef]
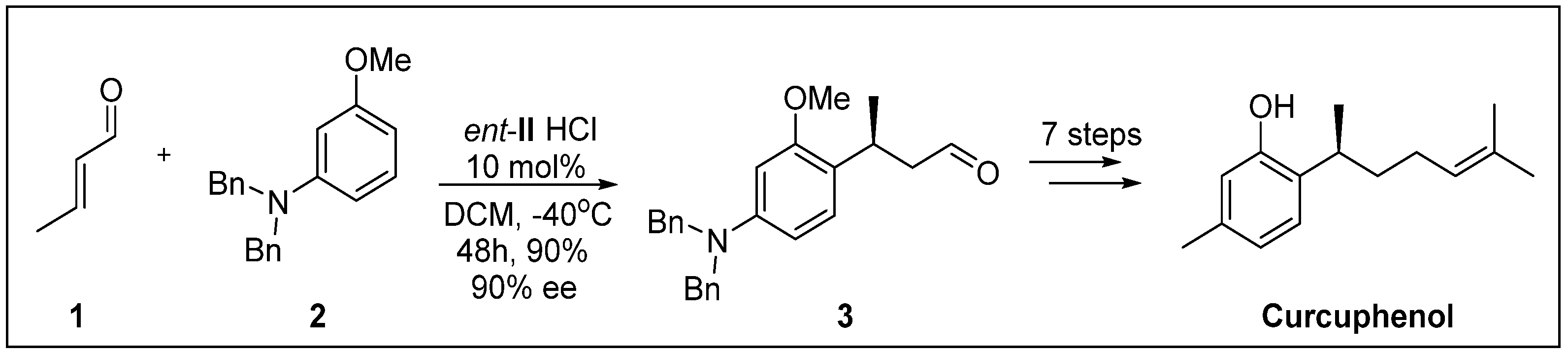
- Kim, S.G.; Kim, J.; Jung, H. Efficient total synthesis of (+)-curcuphenol via asymmetric organocatalysis. Tetrahedron Lett. 2005, 46, 2437–2439. [Google Scholar] [CrossRef]
- Wright, A.E.; Pomponi, S.A.; McConnell, O.J.; Kohmoto, S.; McCarthy, P.J. (+)-Curcuphenol and (+)-Curcudiol, Sesquiterpene phenols from shallow and deep water collections of the marine sponge Didiscus flavus. J. Nat. Prod. 1987, 50, 976–978. [Google Scholar] [CrossRef]
- Gaspar, H.; Feio, S.; Rodrigues, A.; Van Soest, R. Antifungal activity of (+)-curcuphenol, a metabolite from the marine sponge Didiscus oxeata. Mar. Drugs 2004, 2, 8–13. [Google Scholar] [CrossRef]
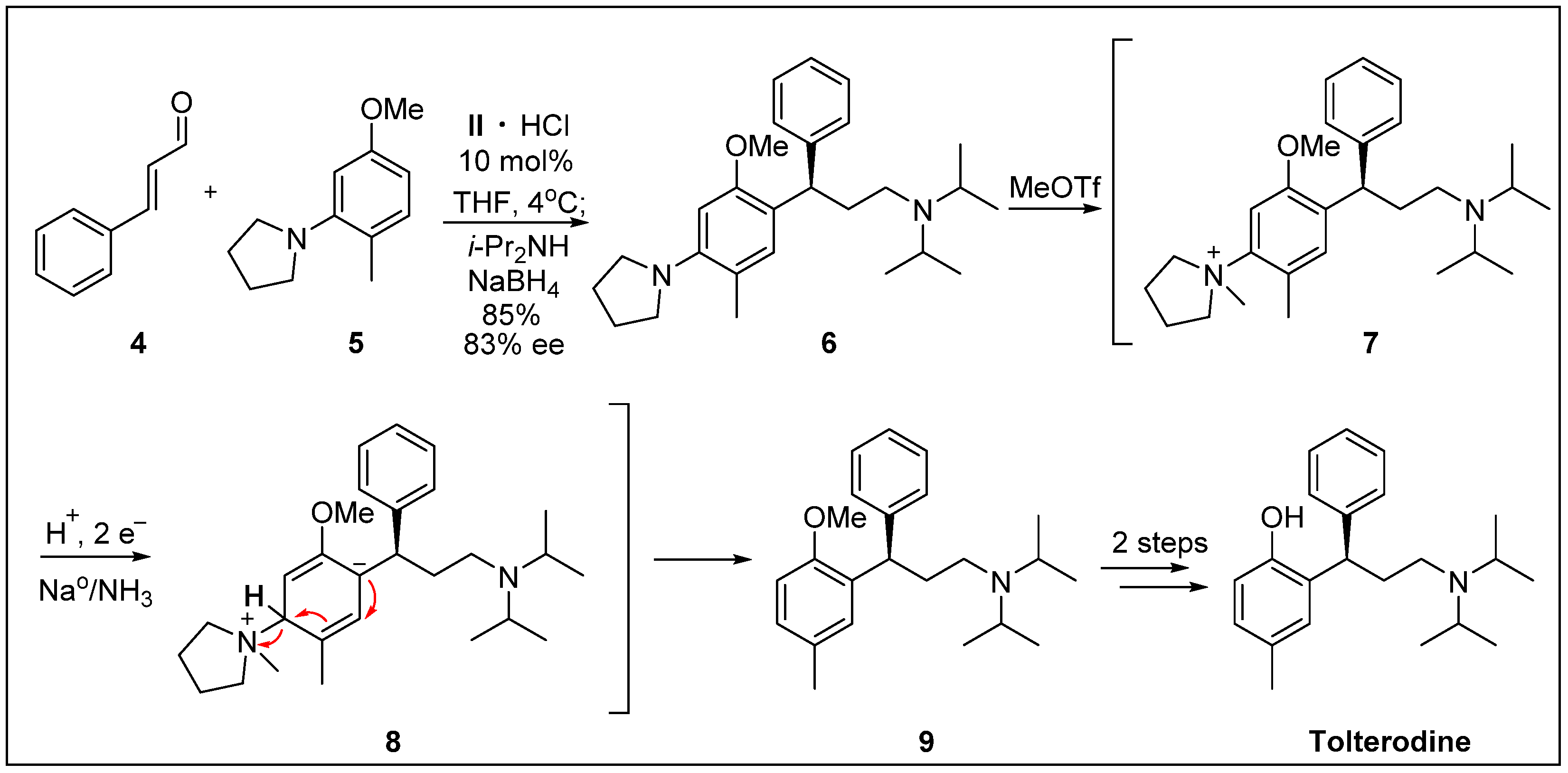
- Paras, N.A.; Simmons, B.; MacMillan, D.W. A process for the rapid removal of dialkylamino-substituents from aromatic rings. Application to the expedient synthesis of (R)-tolterodine. Tetrahedron 2009, 65, 3232–3238. [Google Scholar] [CrossRef]
- De Castro, K.A.; Ko, J.; Park, D.; Park, S.; Rhee, H. Reduction of ethyl benzoylacetate and selective protection of 2-(3-hydroxy-1-phenylpropyl)-4-methylphenol: A new and facile synthesis of tolterodine. Org. Process. Res. Dev. 2007, 11, 918–921. [Google Scholar] [CrossRef]
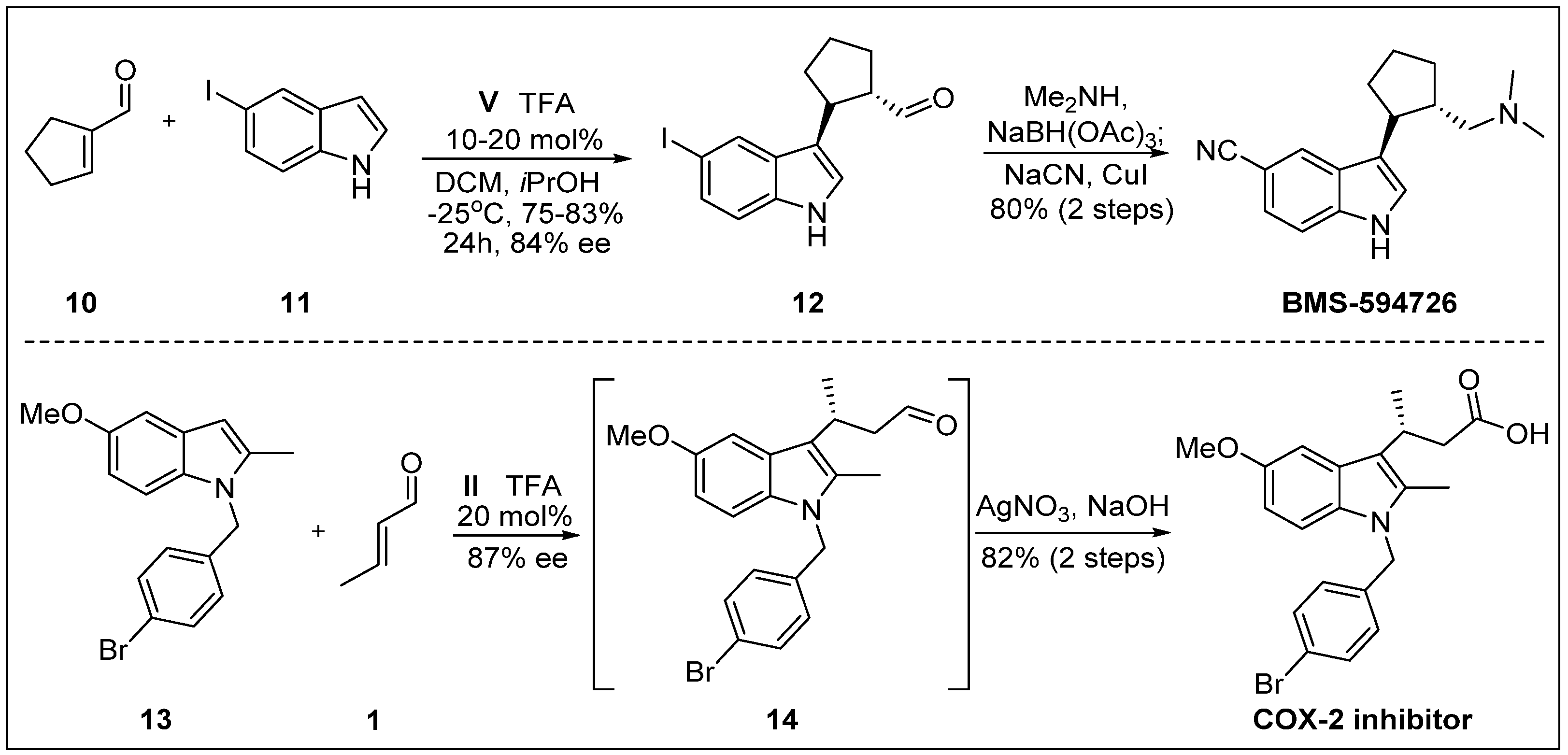
- King, H.D.; Meng, Z.; Denhart, D.; Mattson, R.; Kimura, R.; Wu, D.; Gao, Q.; Macor, J.E. Enantioselective synthesis of a highly potent selective serotonin reuptake inhibitor. An application of imidazolidinone catalysis to the alkylation of indoles with an α, β-disubstituted α, β-unsaturated aldehyde. Org. Lett. 2005, 7, 3437–3440. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.F.; MacMillan, D.W. Enantioselective organocatalytic indole alkylations. Design of a new and highly effective chiral amine for iminium catalysis. J. Am. Chem. Soc. 2002, 124, 1172–1173. [Google Scholar] [CrossRef] [PubMed]
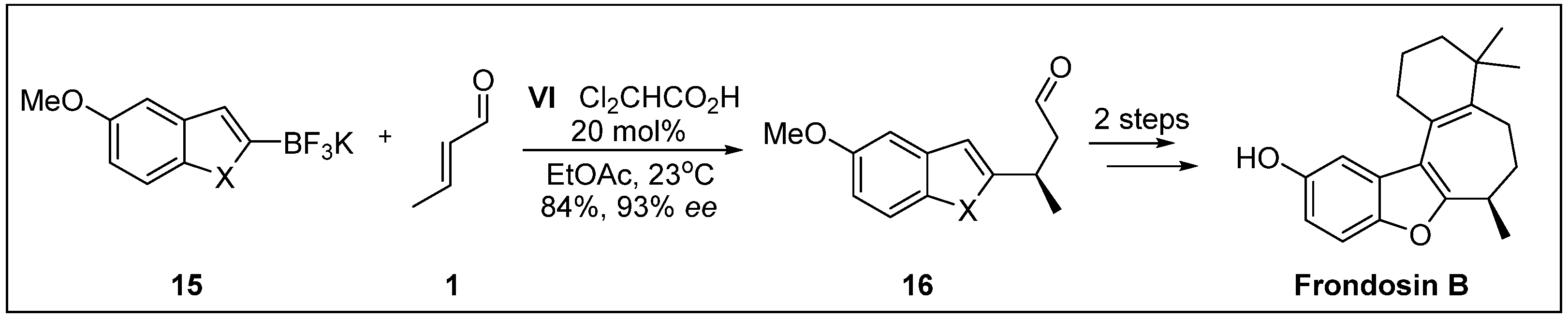
- Reiter, M.; Torssell, S.; Lee, S.; MacMillan, D.W.C. The organocatalytic three-step total synthesis of (+)- frondosin B. Chem. Sci. 2010, 1, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.D.; Freyer, A.J.; Killmer, L.; Offen, P.; Carte, B.; Jurewicz, A.J.; Johnson, R.K. Frondosins, five new sesquiterpene hydroquinone derivatives with novel skeletons from the sponge Dysidea frondosa: Inhibitors of interleukin-8 receptors. Tetrahedron 1997, 53, 5047–5060. [Google Scholar] [CrossRef]
- Hallock, Y.F.; Cardellina, J.H.; Boyd, M.R. (-)-Frondosins A and D, HIV-inhibitory sesquiterpene hydroquinone derivatives from Euryspongia sp. Nat. Prod. Lett. 1998, 11, 153–160. [Google Scholar] [CrossRef]
- Lee, S.; MacMillan, D.W. Organocatalytic vinyl and Friedel−Crafts alkylations with trifluoroborate salts. J. Am. Chem. Soc. 2007, 129, 15438–15439. [Google Scholar] [CrossRef]
- Oblak, E.Z.; VanHeyst, M.D.; Li, J.; Wiemer, A.J.; Wright, D.L. Cyclopropene Cycloadditions with Annulated Furans: Total synthesis of (+)- and (-)-frondosin B and (+)-frondosin A. J. Am. Chem. Soc. 2014, 136, 4309–4315. [Google Scholar] [CrossRef]
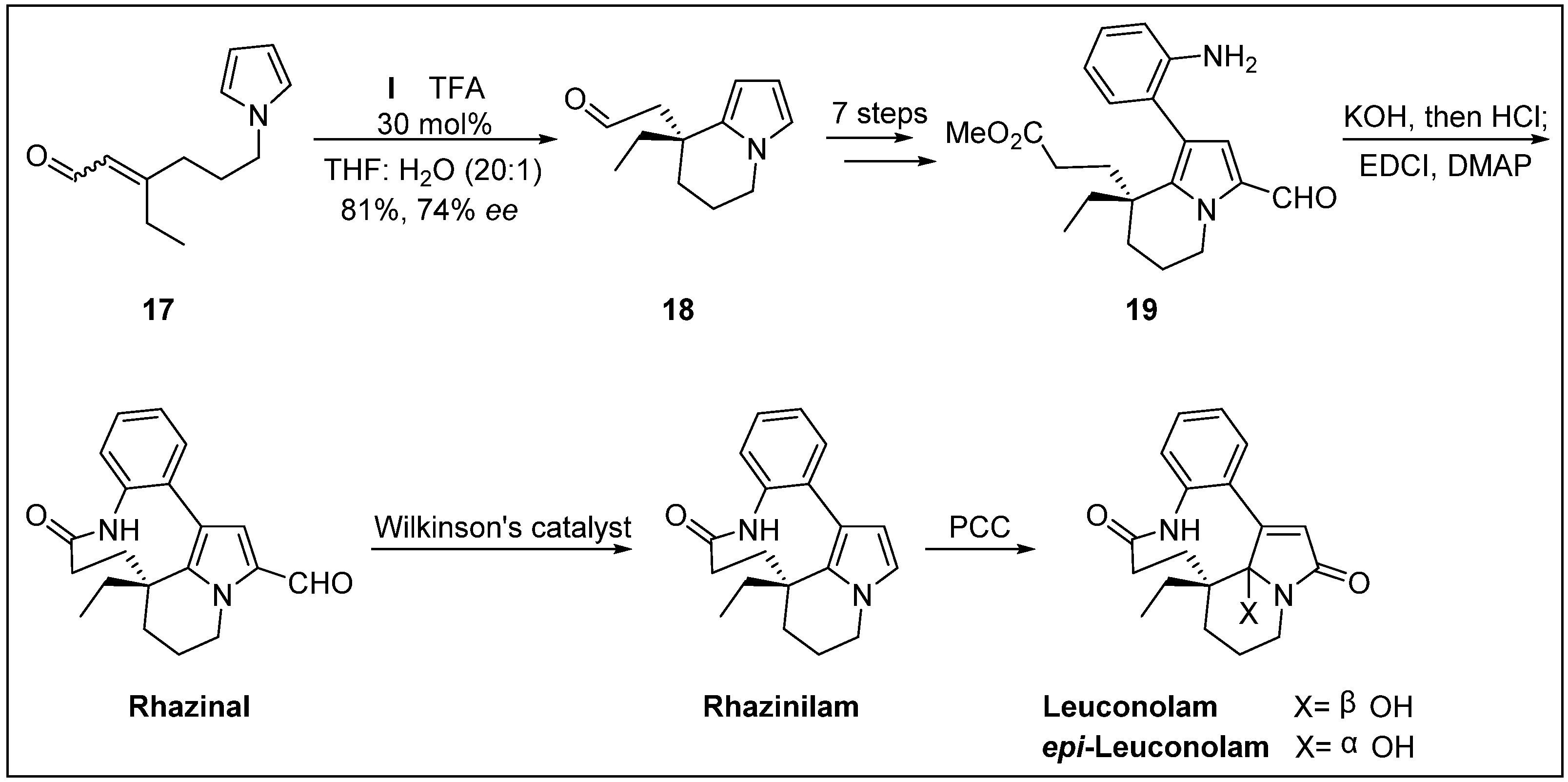
- Banwell, M.G.; Beck, D.A.; Willis, A.C. Enantioselective total syntheses of the alkaloids (-)-rhazinal,(-)-rhazinilam,(-)-leuconolam and (+)-epi-leuconolam. Arkivoc 2006, 3, 163–174. [Google Scholar]
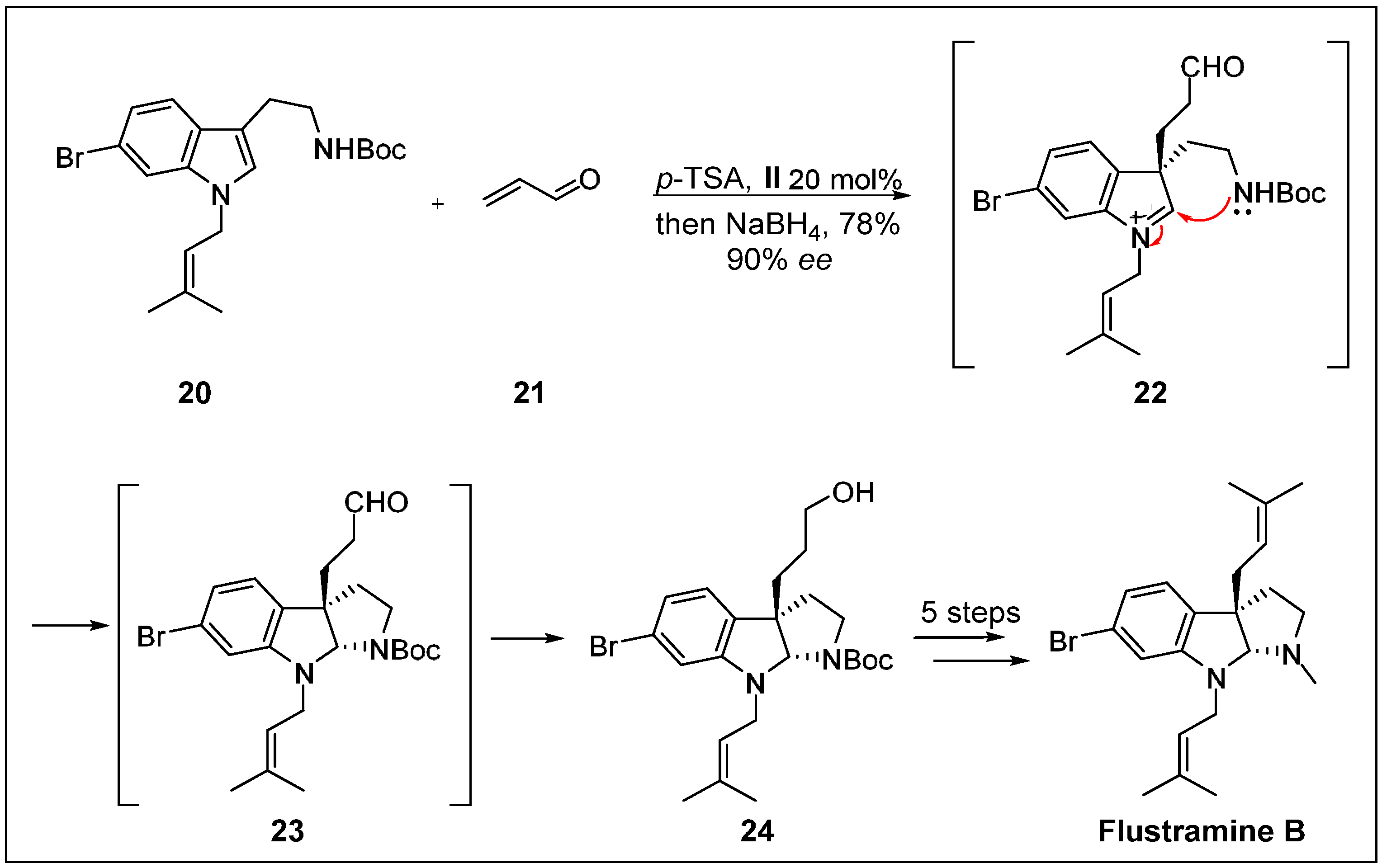
- Austin, J.F.; Kim, S.-G.; Sinz, C.J.; Xiao, W.-J.; MacMillan, D.W. Enantioselective organocatalytic construction of pyrroloindolines by a cascade addition–cyclization strategy: Synthesis of (–)-flustramine B. P. Natl. Acad. Sci. USA 2004, 101, 5482–5487. [Google Scholar] [CrossRef]
- Carlé, J.S.; Christophersen, C. Bromo-substituted physostigmine alkaloids from a marine bryozoa Flustra foliacea. J. Am. Chem. Soc. 1979, 101, 4012–4013. [Google Scholar] [CrossRef]
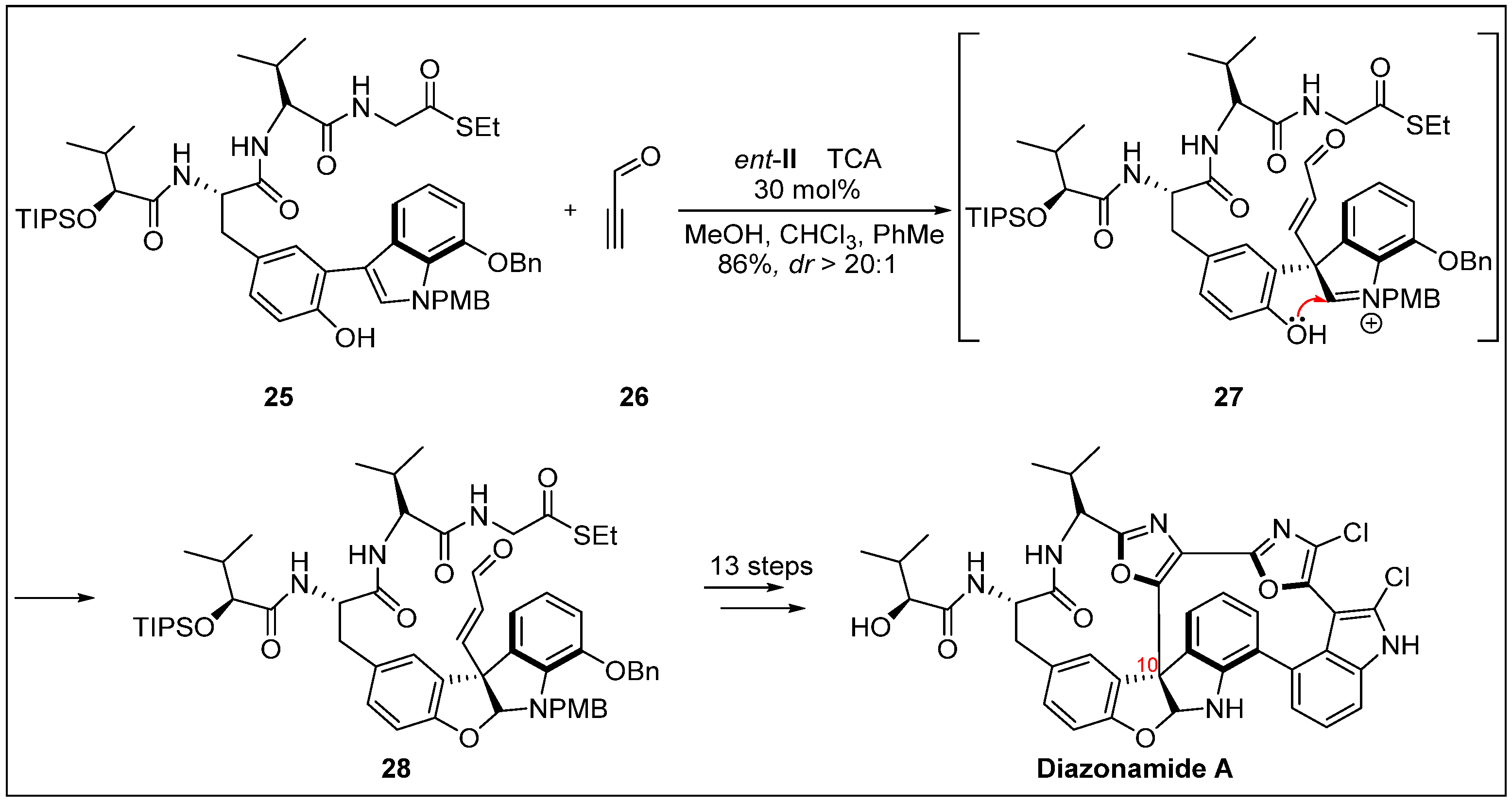
- Knowles, R.R.; Carpenter, J.; Blakey, S.B.; Kayano, A.; Mangion, I.K.; Sinz, C.J.; MacMillan, D.W. Total synthesis of diazonamide A. Chem. Sci. 2011, 2011, 308–311. [Google Scholar] [CrossRef]
- Lindquist, N.; Fenical, W.; Van Duyne, G.D.; Clardy, J. Isolation and structure determination of diazonamides A and B, unusual cytotoxic metabolites from the marine ascidian Diazona chinensis. J. Am. Chem. Soc. 1991, 113, 2303–2304. [Google Scholar] [CrossRef]
- Cruz-Monserrate, Z.; Vervoort, H.C.; Bai, R.; Newman, D.J.; Howell, S.B.; Los, G.; Mullaney, J.T.; Williams, M.D.; Pettit, G.R.; Fenical, W. Diazonamide A and a synthetic structural analog: Disruptive effects on mitosis and cellular microtubules and analysis of their interactions with tubulin. Mol. Pharm. 2003, 63, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xi, G.; Wu, Q.; Liu, W.; Ma, J.; Wang, C. Organocatalytic asymmetric Michael additions. Chin. J. Org. Chem. 2009, 29, 1018–1038. [Google Scholar]
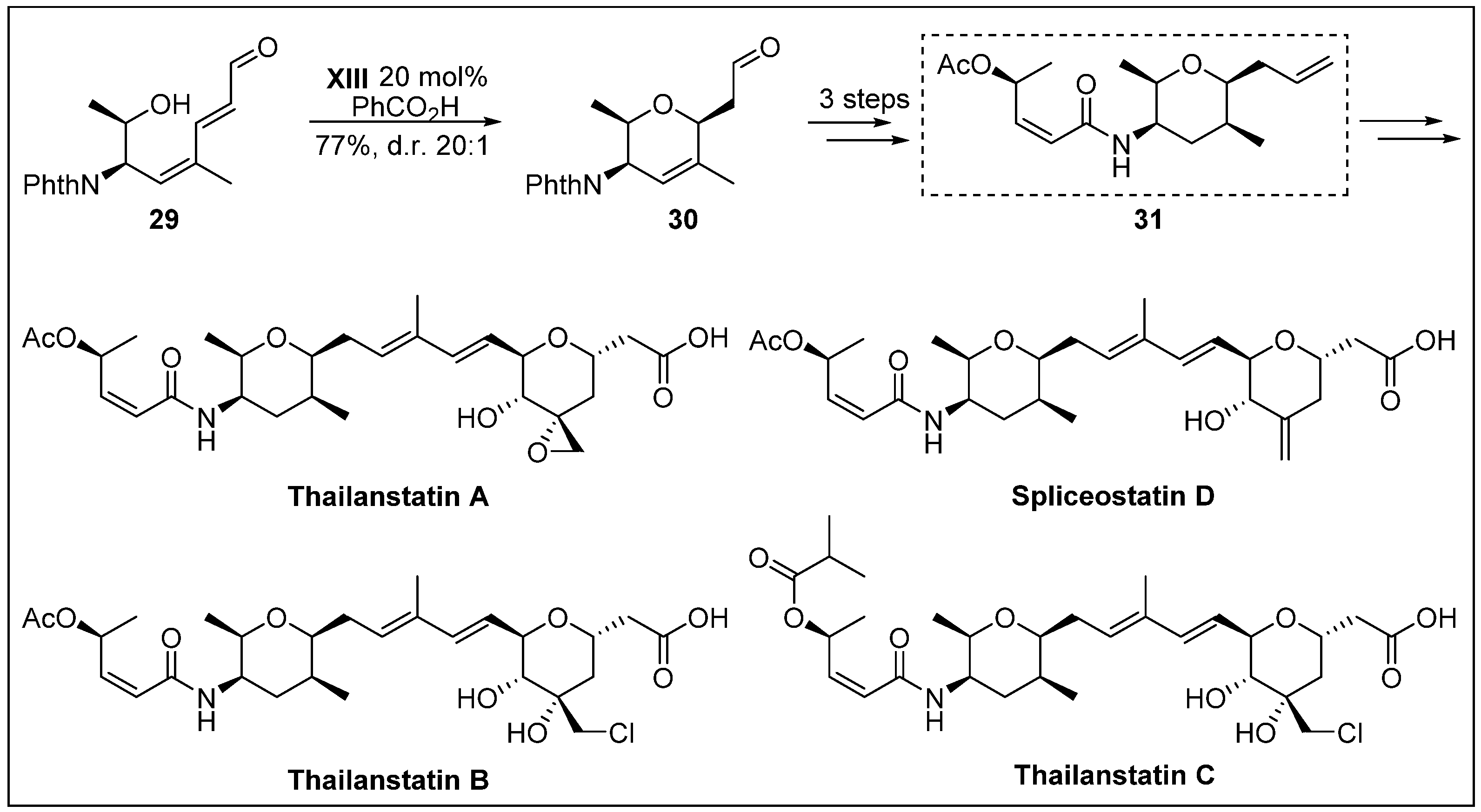
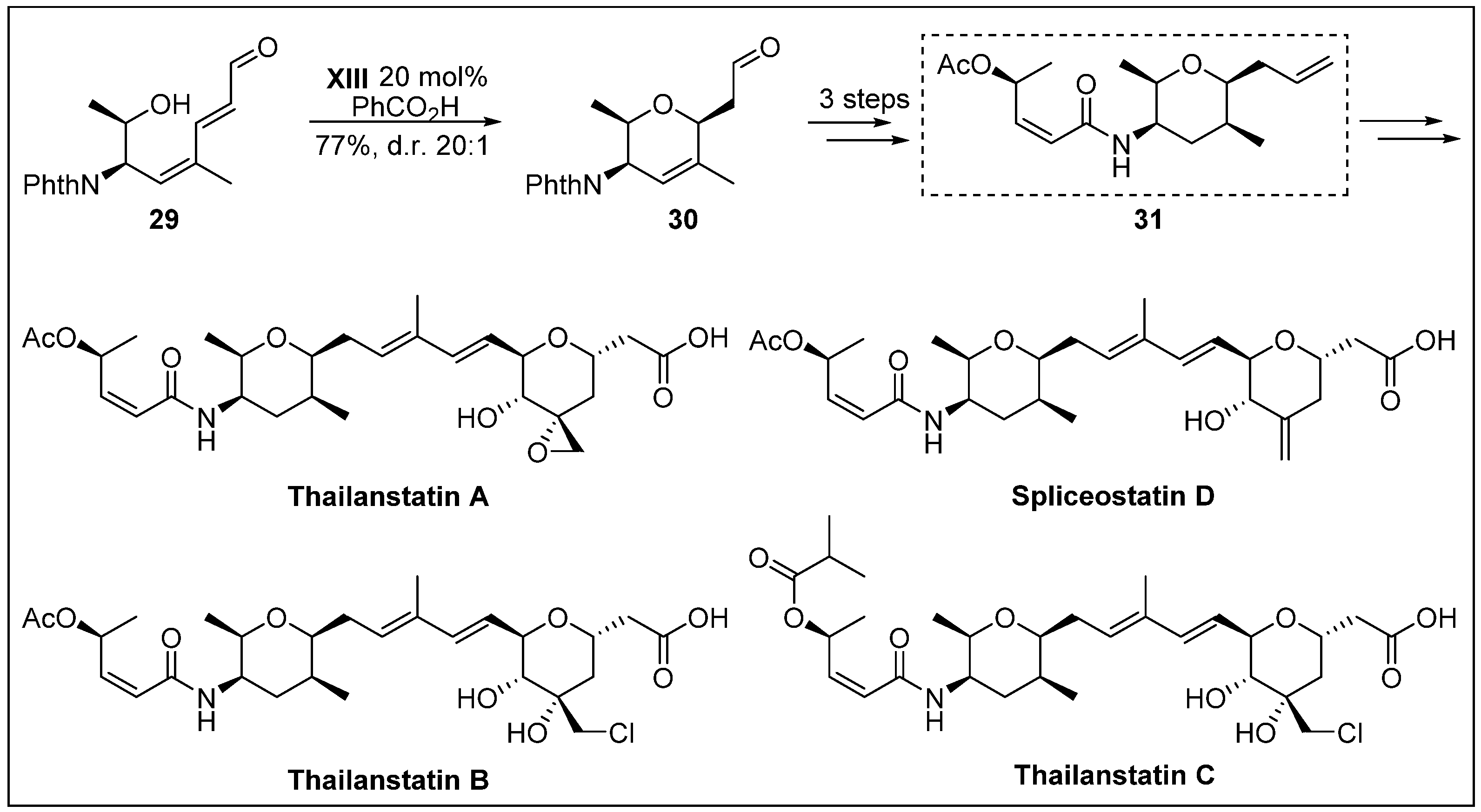
- Nicolaou, K.C.; Rhoades, D.; Lamani, M.; Pattanayak, M.R.; Kumar, S.M. Total synthesis of thailanstatin A. J. Am. Chem. Soc. 2016, 138, 7532–7535. [Google Scholar] [CrossRef]
- Liu, X.; Biswas, S.; Berg, M.G.; Antapli, C.M.; Xie, F.; Wang, Q.; Tang, M.-C.; Tang, G.-L.; Zhang, L.; Dreyfuss, G.; et al. Genomics-guided discovery of thailanstatins A, B, and C as pre-mRNA splicing inhibitors and antiproliferative agents from Burkholderia thailandensis MSMB43. J. Nat. Prod. 2013, 76, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Rhoades, D.; Kumar, S.M. Total syntheses of thailanstatins A–C, spliceostatin D, and analogues thereof. stereodivergent synthesis of tetrasubstituted dihydro-and tetrahydropyrans and design, synthesis, biological evaluation, and discovery of potent antitumor agents. J. Am. Chem. Soc. 2018, 140, 8303–8320. [Google Scholar] [CrossRef]
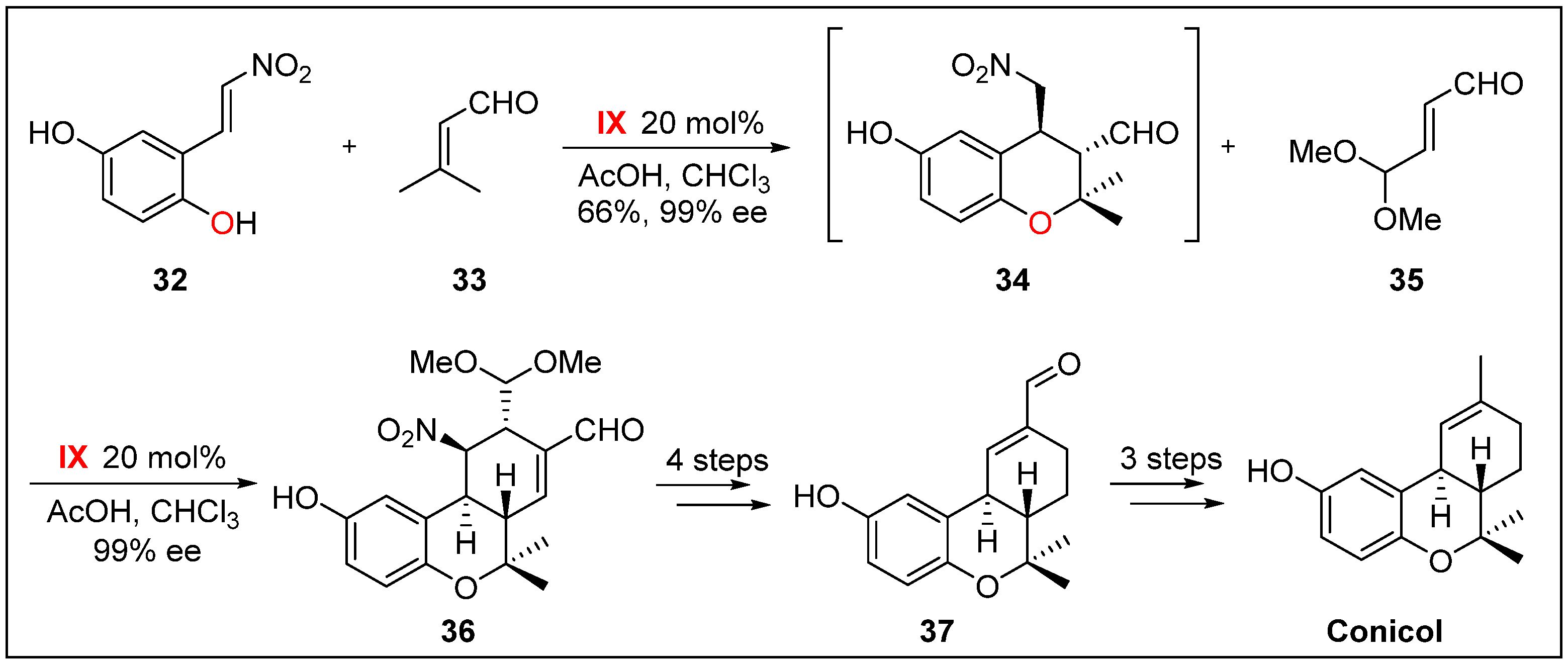
- Hong, B.-C.; Kotame, P.; Tsai, C.-W.; Liao, J.-H. Enantioselective total synthesis of (+)-conicol via cascade three-component organocatalysis. Org. Lett. 2010, 12, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Garrido, L.; Zubía, E.; Ortega, M.J.; Salvá, J. New meroterpenoids from the ascidian Aplidium conicum. J. Nat. Prod. 2002, 65, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
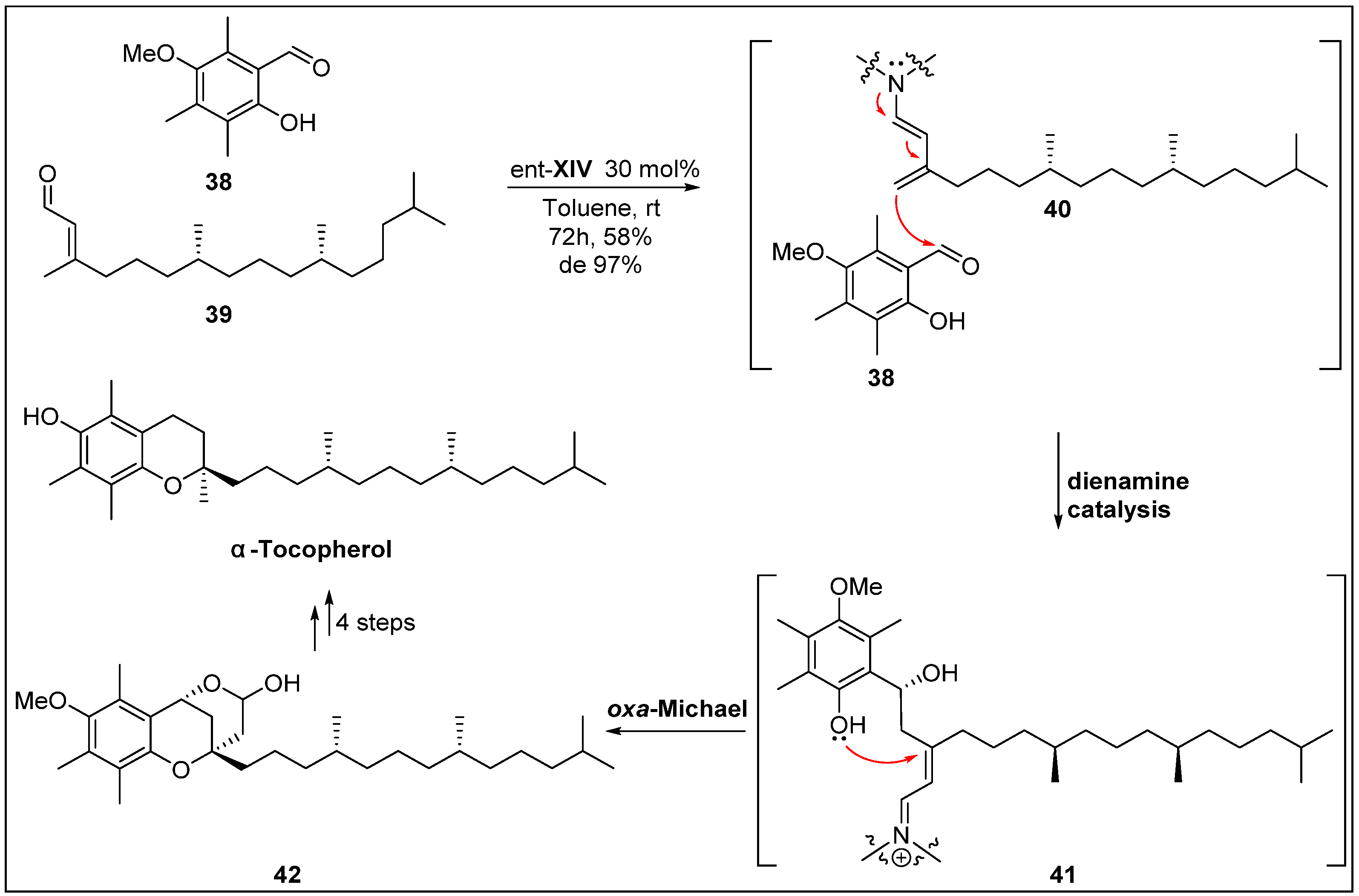
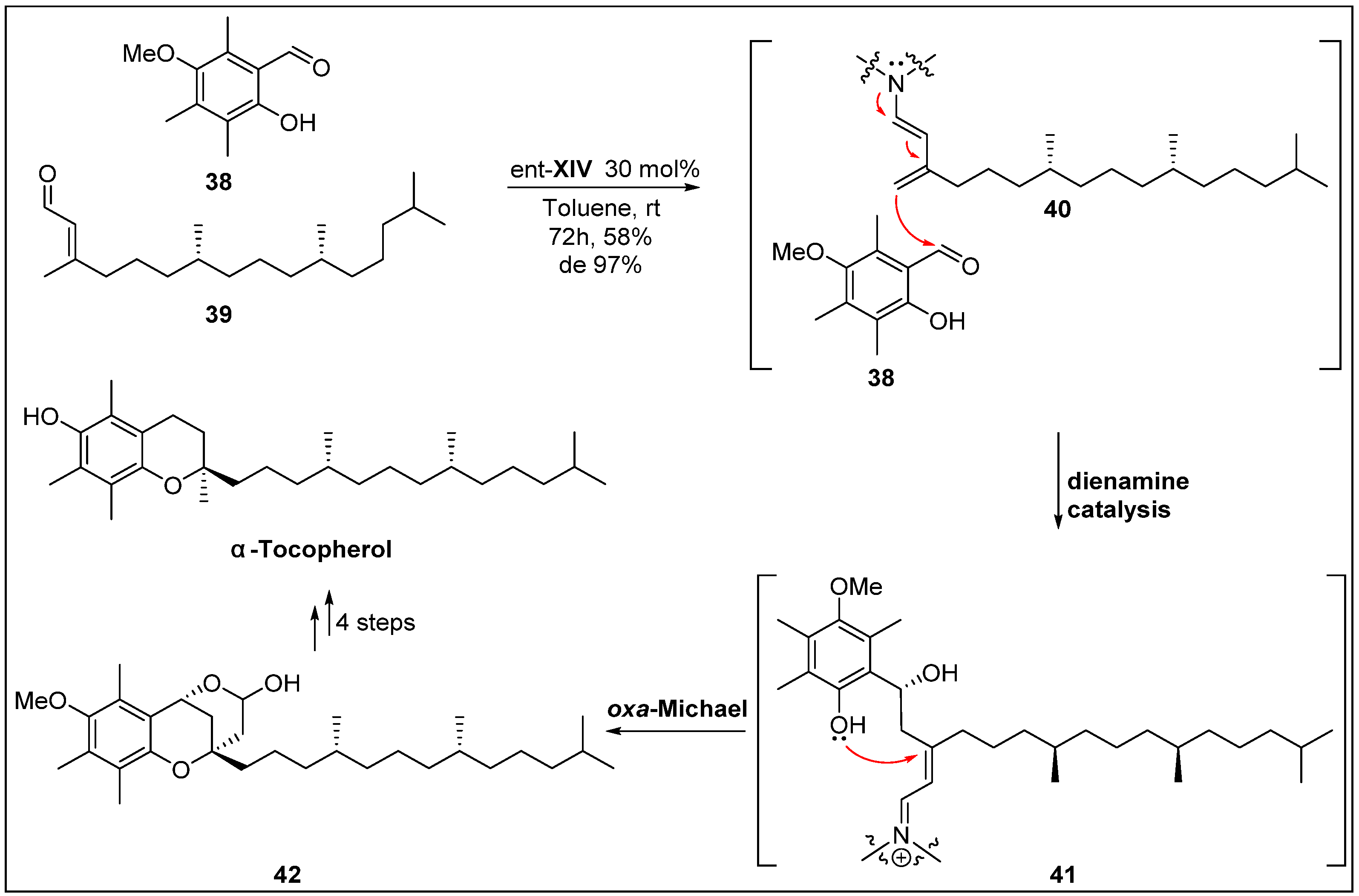
- Liu, K.; Chougnet, A.; Woggon, W.D. A short route to α-tocopherol. Angew. Chem. Int. Ed. 2008, 47, 5827–5829. [Google Scholar] [CrossRef]
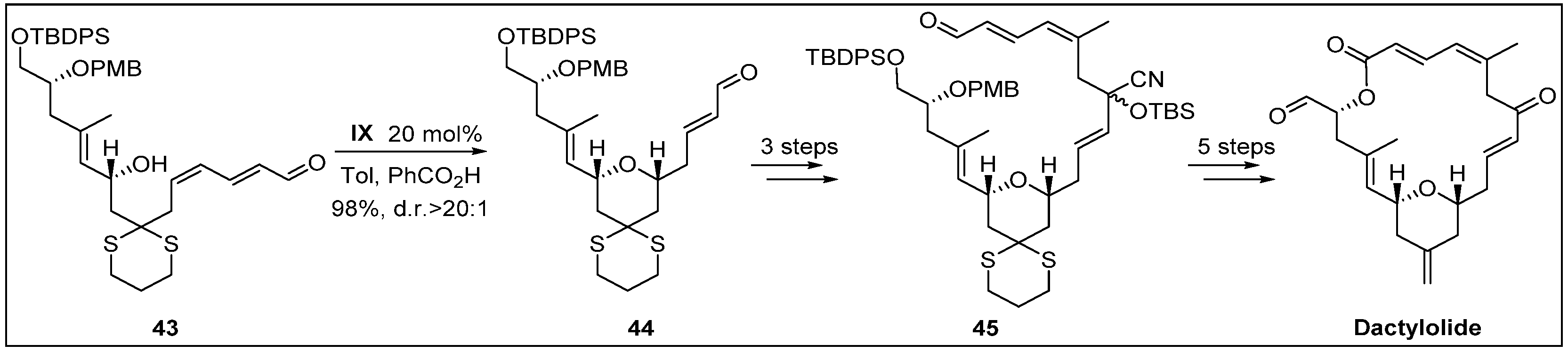
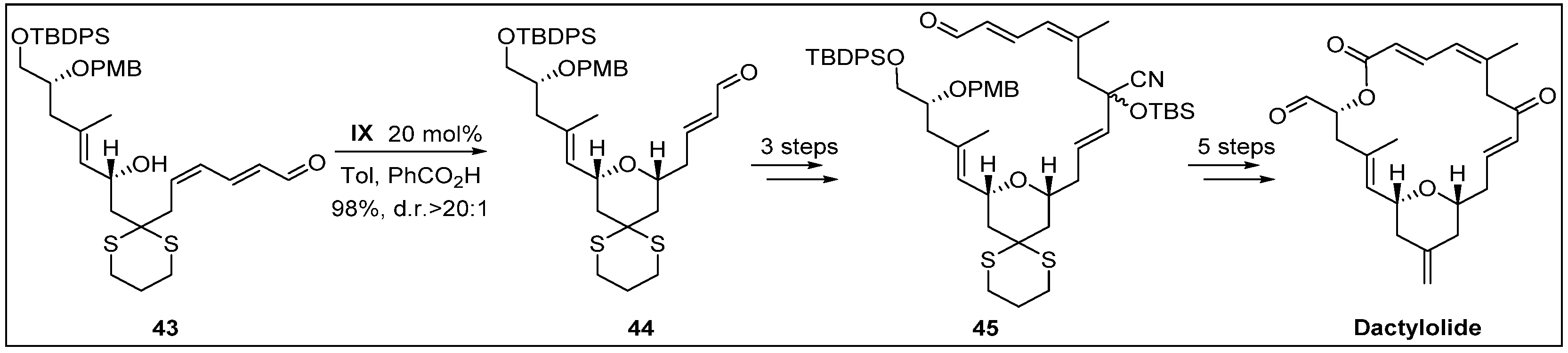
- Lee, K.; Kim, H.; Hong, J. N-Heterocyclic carbene catalyzed oxidative macrolactonization: Total synthesis of (+)-dactylolide. Angew. Chem. Int. Ed. 2012, 51, 5735–5738. [Google Scholar] [CrossRef] [PubMed]
- Cutignano, A.; Bruno, I.; Bifulco, G.; Casapullo, A.; Debitus, C.; Gomez-Paloma, L.; Riccio, R. Dactylolide, a new cytotoxic macrolide from the Vanuatu sponge Dactylospongia sp. Eur. J. Org. Chem. 2001, 2001, 775–778. [Google Scholar] [CrossRef]
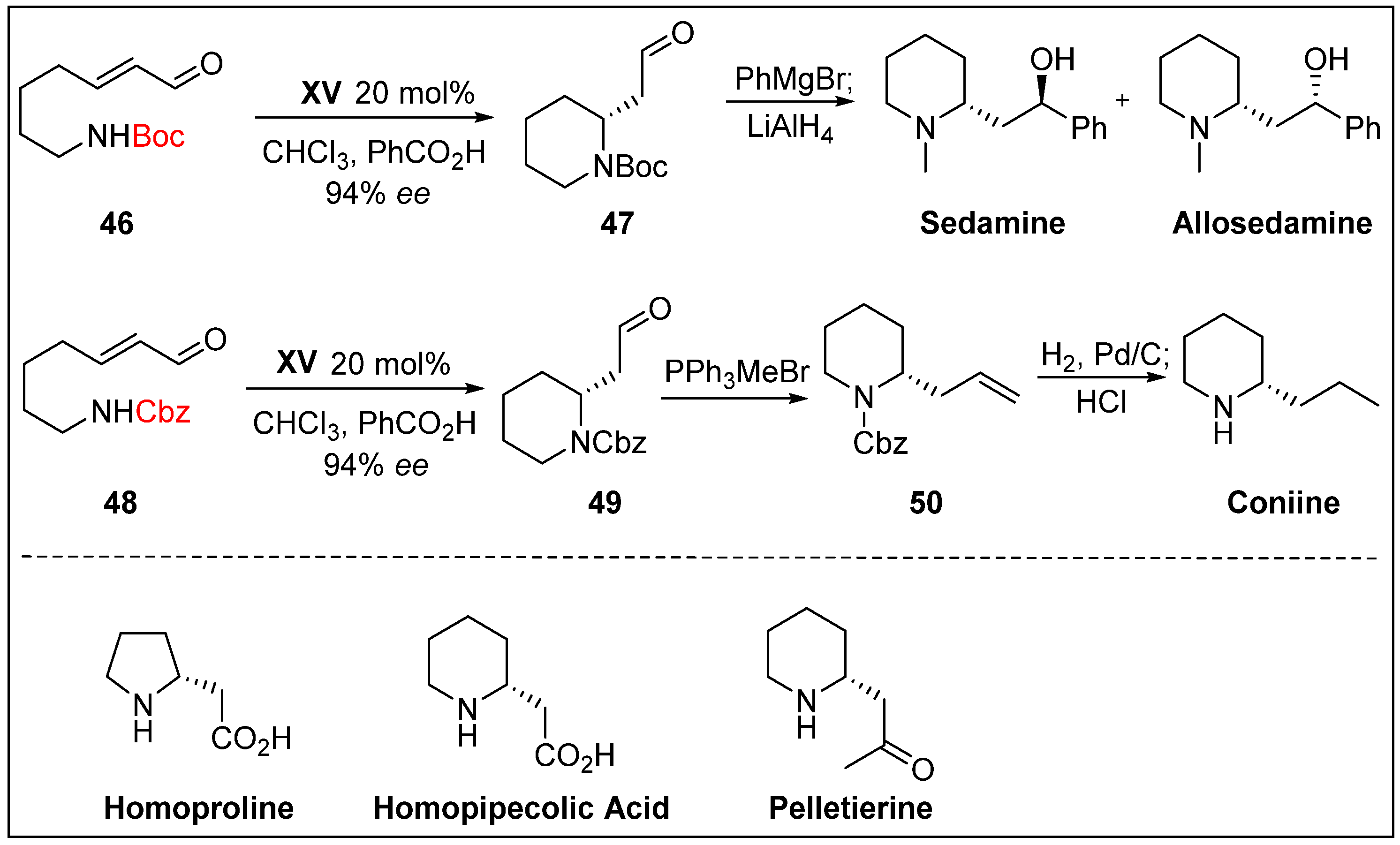
- Fustero, S.; Jiménez, D.; Moscardó, J.; Catalán, S.; del Pozo, C. Enantioselective organocatalytic intramolecular aza-Michael reaction: A concise synthesis of (+)-sedamine,(+)-allosedamine, and (+)-coniine. Org. Lett. 2007, 9, 5283–5286. [Google Scholar] [CrossRef]
- Carlson, E.C.; Rathbone, L.K.; Yang, H.; Collett, N.D.; Carter, R.G. Improved protocol for asymmetric, intramolecular heteroatom michael addition using organocatalysis: Enantioselective syntheses of homoproline, pelletierine, and homopipecolic acid. J. Org. Chem. 2008, 73, 5155–5158. [Google Scholar] [CrossRef]
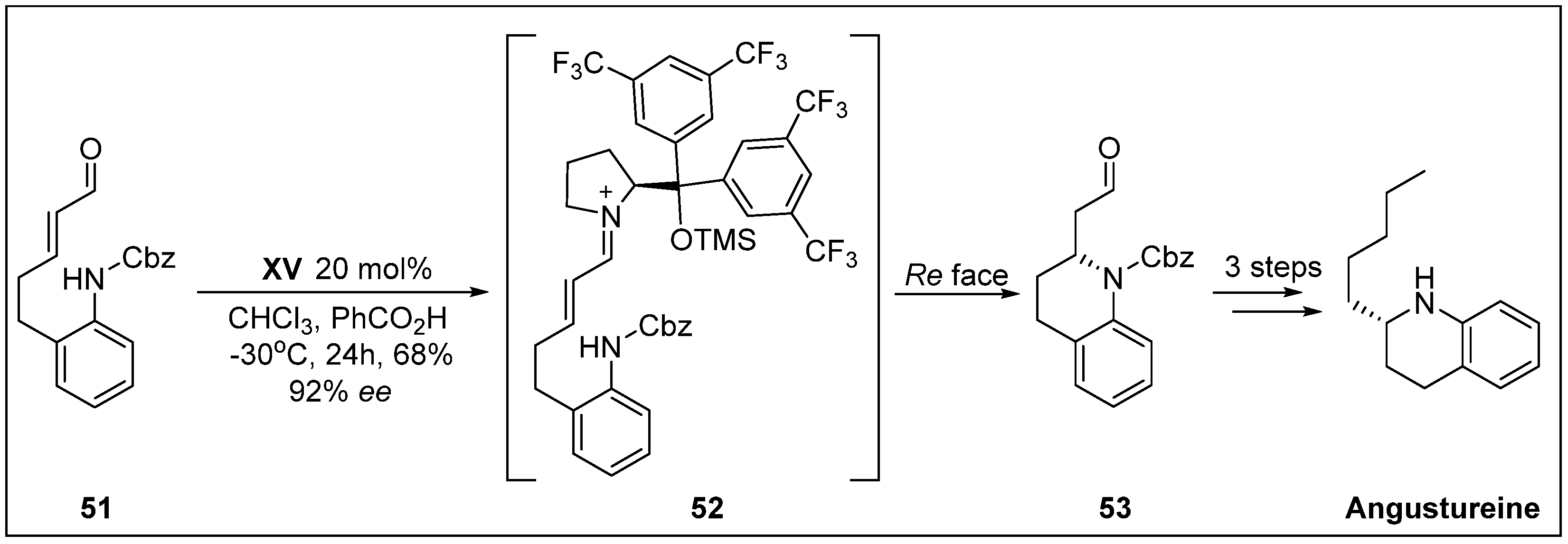
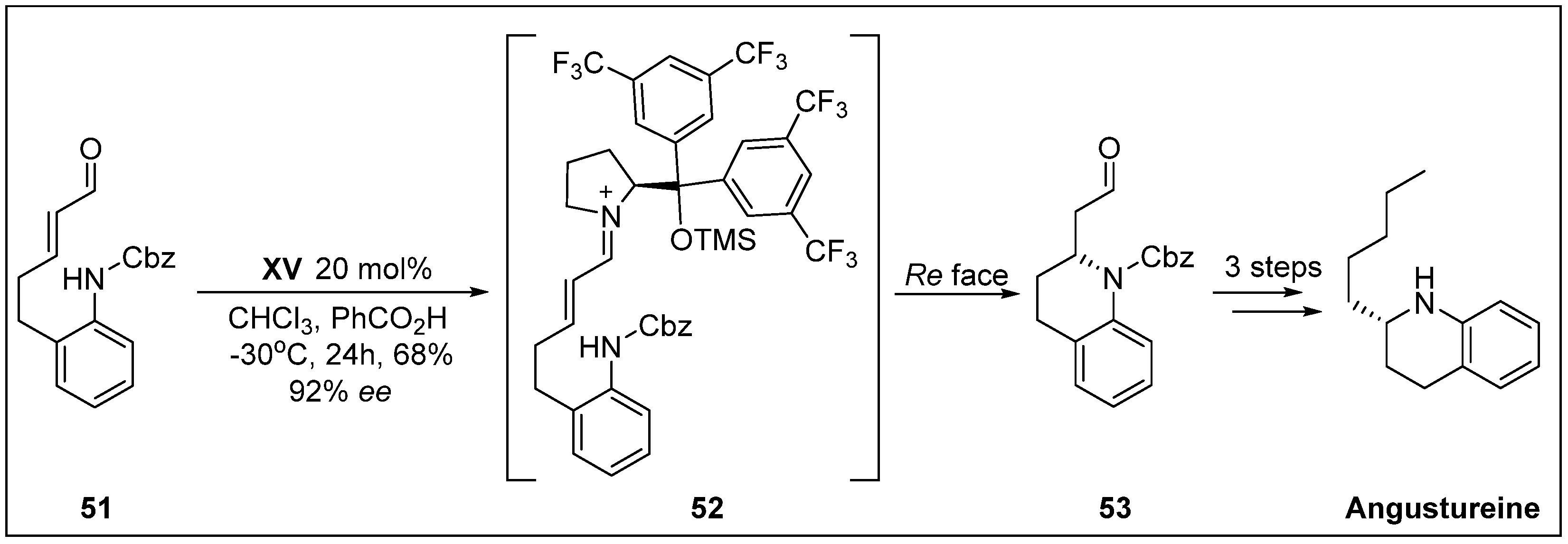
- Fustero, S.; Moscardo, J.; Jimenez, D.; Pérez-Carrión, M.D.; Sánchez-Roselló, M.; del Pozo, C. Organocatalytic approach to benzofused nitrogen-containing heterocycles: Enantioselective total synthesis of (+)-angustureine. Chem-Eur. J. 2008, 14, 9868–9872. [Google Scholar] [CrossRef] [PubMed]
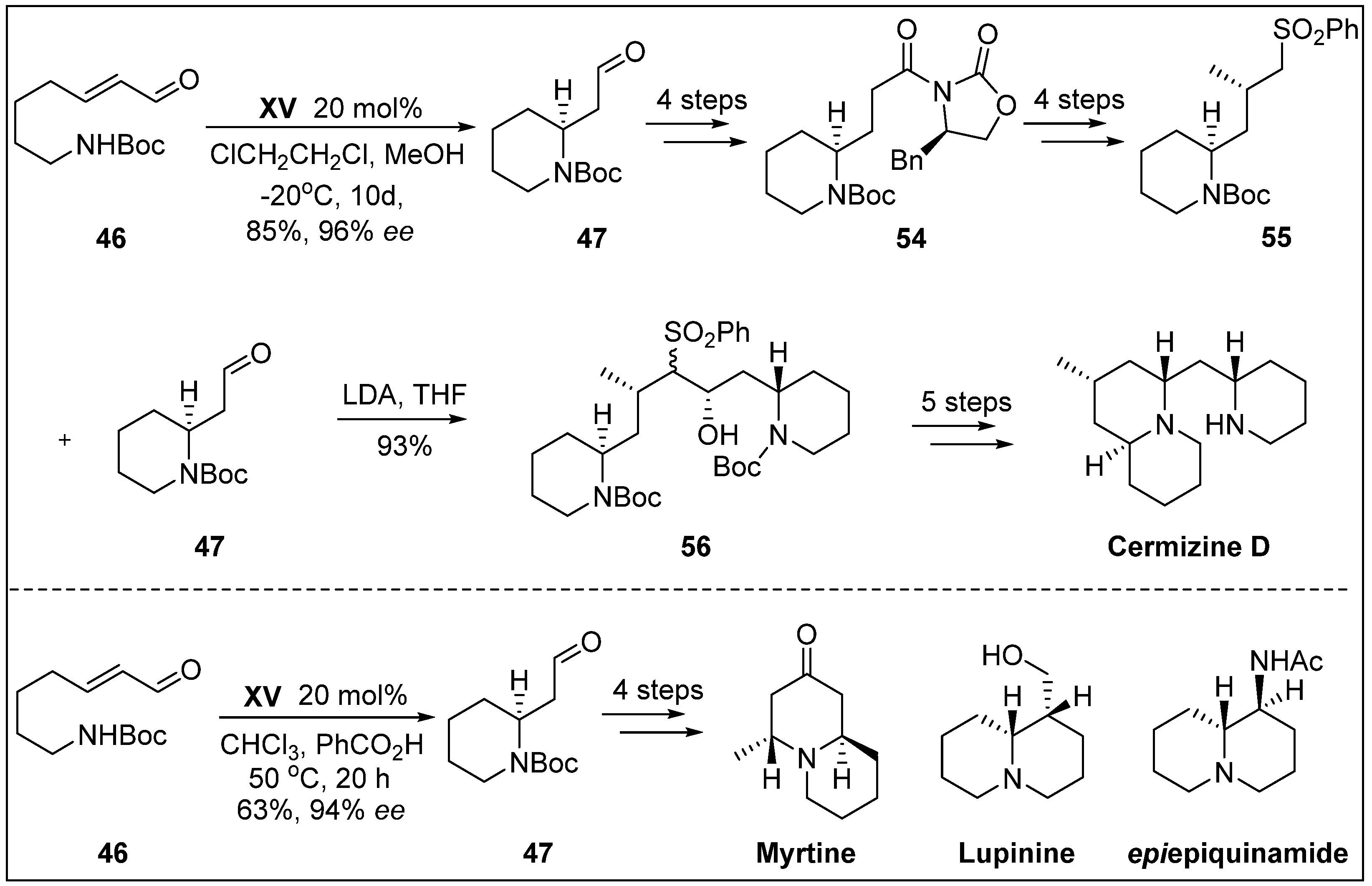
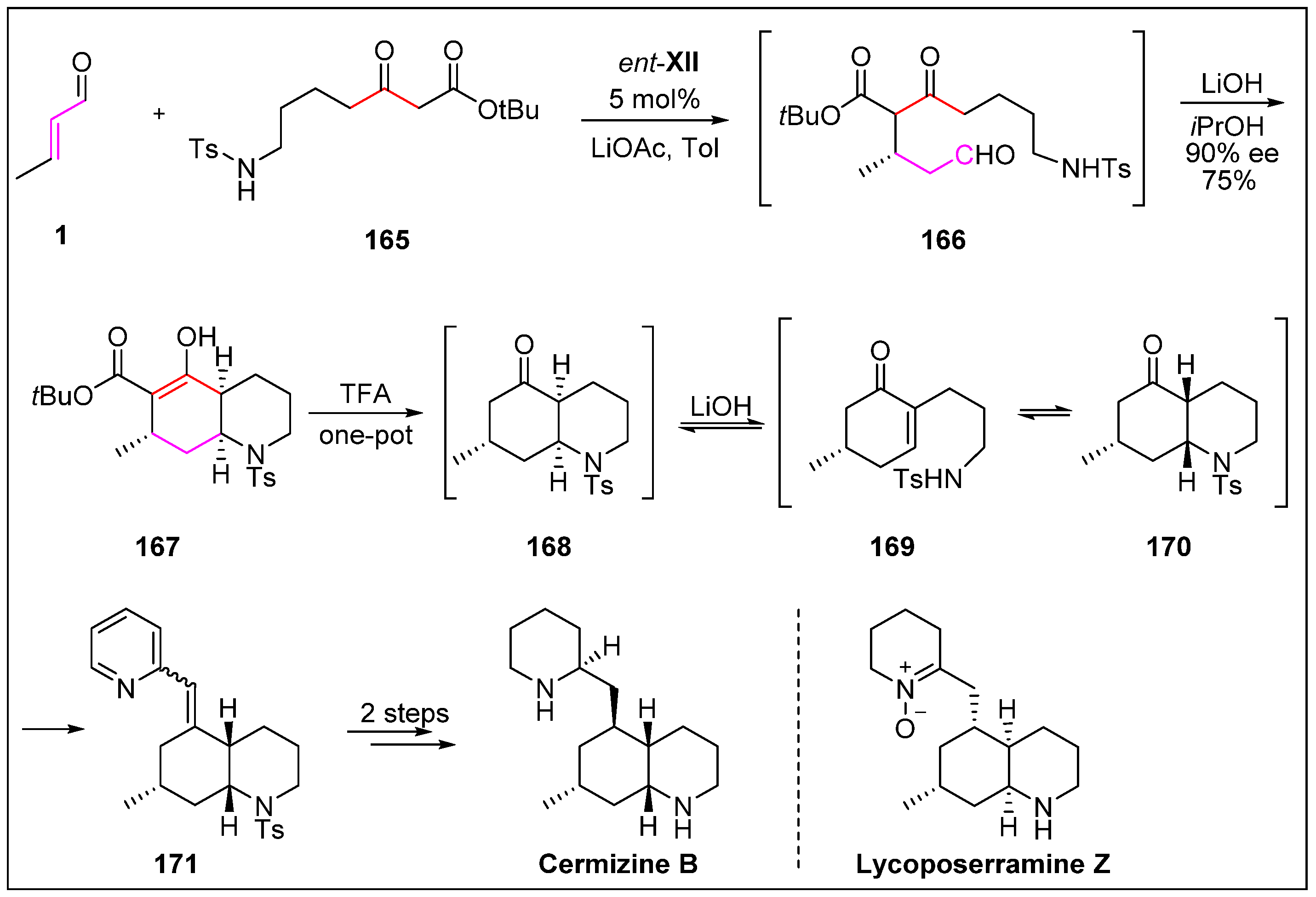
- Veerasamy, N.; Carlson, E.C.; Carter, R.G. Expedient enantioselective synthesis of Cermizine, D. Org. Lett. 2012, 14, 1596–1599. [Google Scholar] [CrossRef]
- Morita, H.; Hirasawa, Y.; Shinzato, T.; Kobayashi, J. New phlegmarane-type, cernuane-type, and quinolizidine alkaloids from two species of Lycopodium. Tetrahedron 2004, 60, 7015–7023. [Google Scholar] [CrossRef]
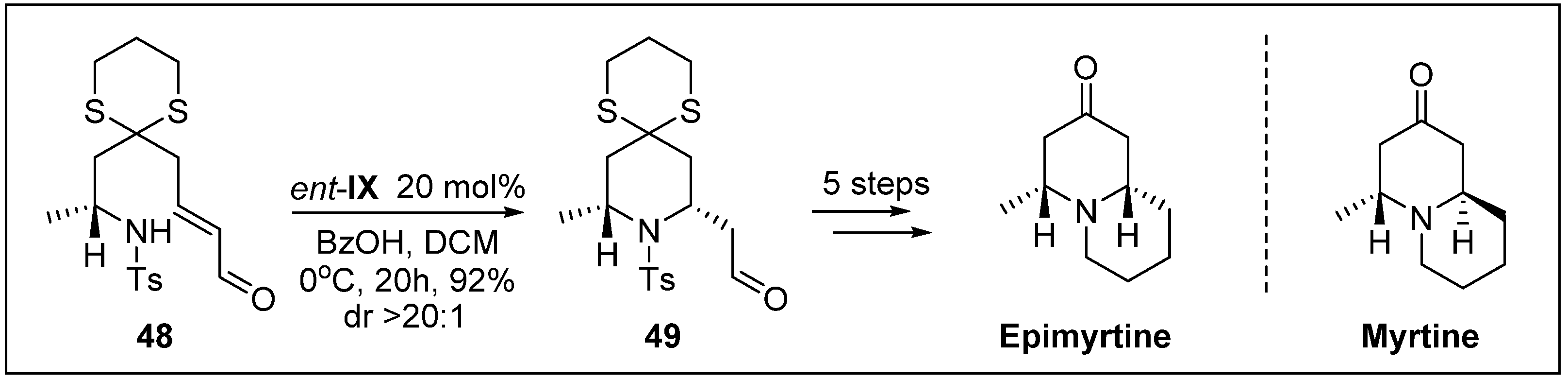
- Fustero, S.; Moscardo, J.; Sanchez-Rosello, M.; Flores, S.; Guerola, M.; del Pozo, C. Organocatalytic enantioselective synthesis of quinolizidine alkaloids (+)-myrtine, (-)-lupinine, and (+)-epiepiquinamide. Tetrahedron 2011, 67, 7412–7417. [Google Scholar] [CrossRef]
- Ying, Y.; Kim, H.; Hong, J. Stereoselective synthesis of 2, 6-cis-and 2, 6-trans-piperidines through organocatalytic aza-Michael reactions: A facile synthesis of (+)-myrtine and (−)-epimyrtine. Org. Lett. 2011, 13, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Slosse, P.; Hootele, C. Myrtine and epimyrtine, quinolizidine alkaloids from Vaccinium myrtillus. Tetrahedron 1981, 37, 4287–4294. [Google Scholar] [CrossRef]
- Trinh, T.T.H.; Nguyen, K.H.; de Aguiar Amaral, P.; Gouault, N. Total synthesis of (−)-epimyrtine by a gold-catalyzed hydroamination approach. BeilsteinJ. Org. Chem. 2013, 9, 2042–2047. [Google Scholar] [CrossRef] [PubMed]
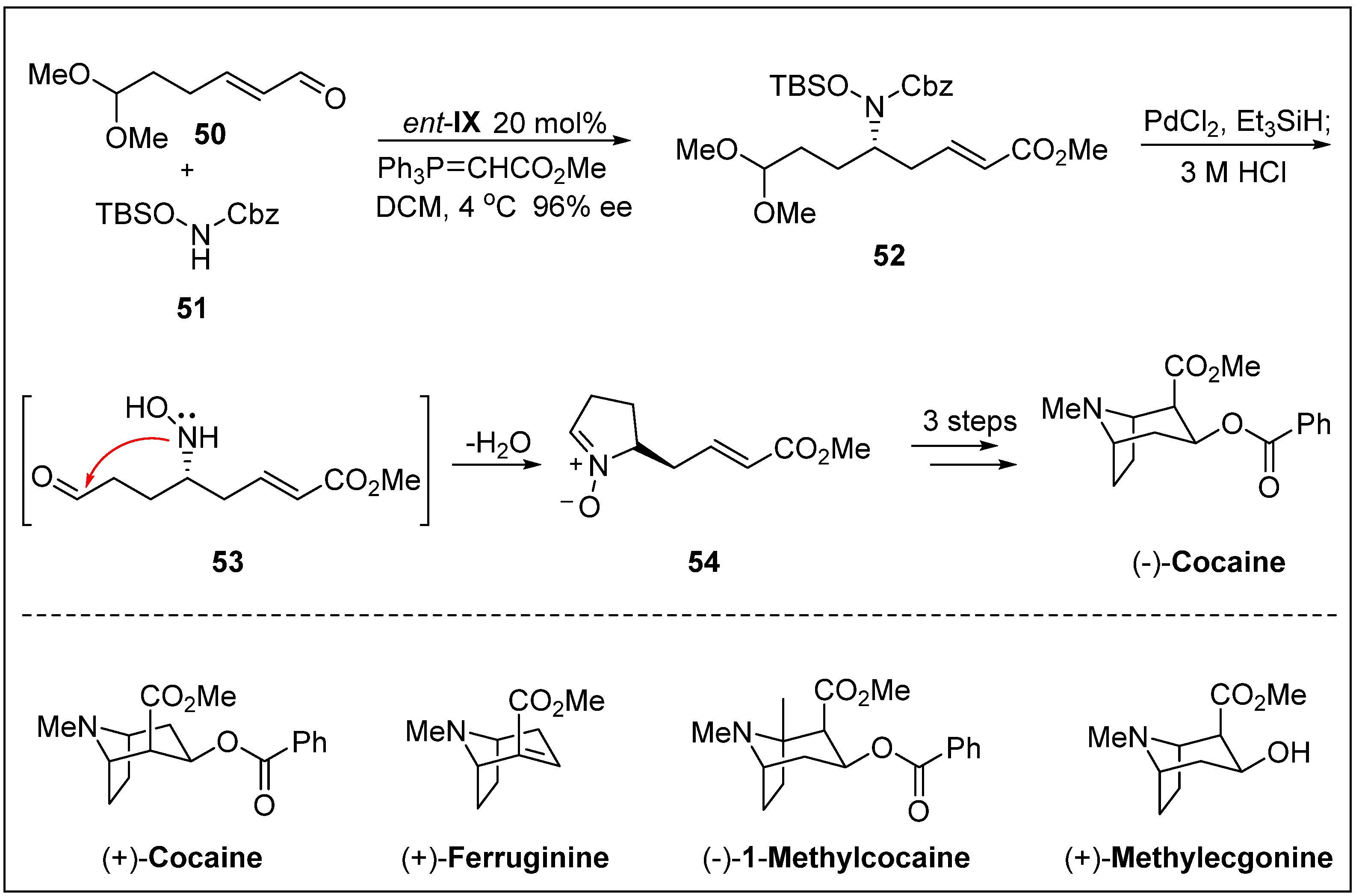
- Córdova, A.; Lin, S.; Tseggai, A. Concise catalytic asymmetric total synthesis of biologically active tropane alkaloids. Adv. Synth. Catal. 2012, 354, 1363–1372. [Google Scholar] [CrossRef]
- Koob, G.F.; Bloom, F.E. Cellular and molecular mechanisms of drug dependence. Science 1988, 242, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.P.; Goodwin, N.C.; MacMillan, D.W. The first enantioselective organocatalytic Mukaiyama-Michael reaction: A direct method for the synthesis of enantioenriched γ-butenolide architecture. J. Am. Chem. Soc. 2003, 125, 1192–1194. [Google Scholar] [CrossRef] [PubMed]
- Simmons, B.; Walji, A.M.; MacMillan, D.W. Cycle-specific organocascade catalysis: Application to olefin hydroamination, hydro-oxidation, and amino-oxidation, and to natural product synthesis. Angew. Chem. Int. Ed. 2009, 48, 4349–4353. [Google Scholar] [CrossRef] [PubMed]
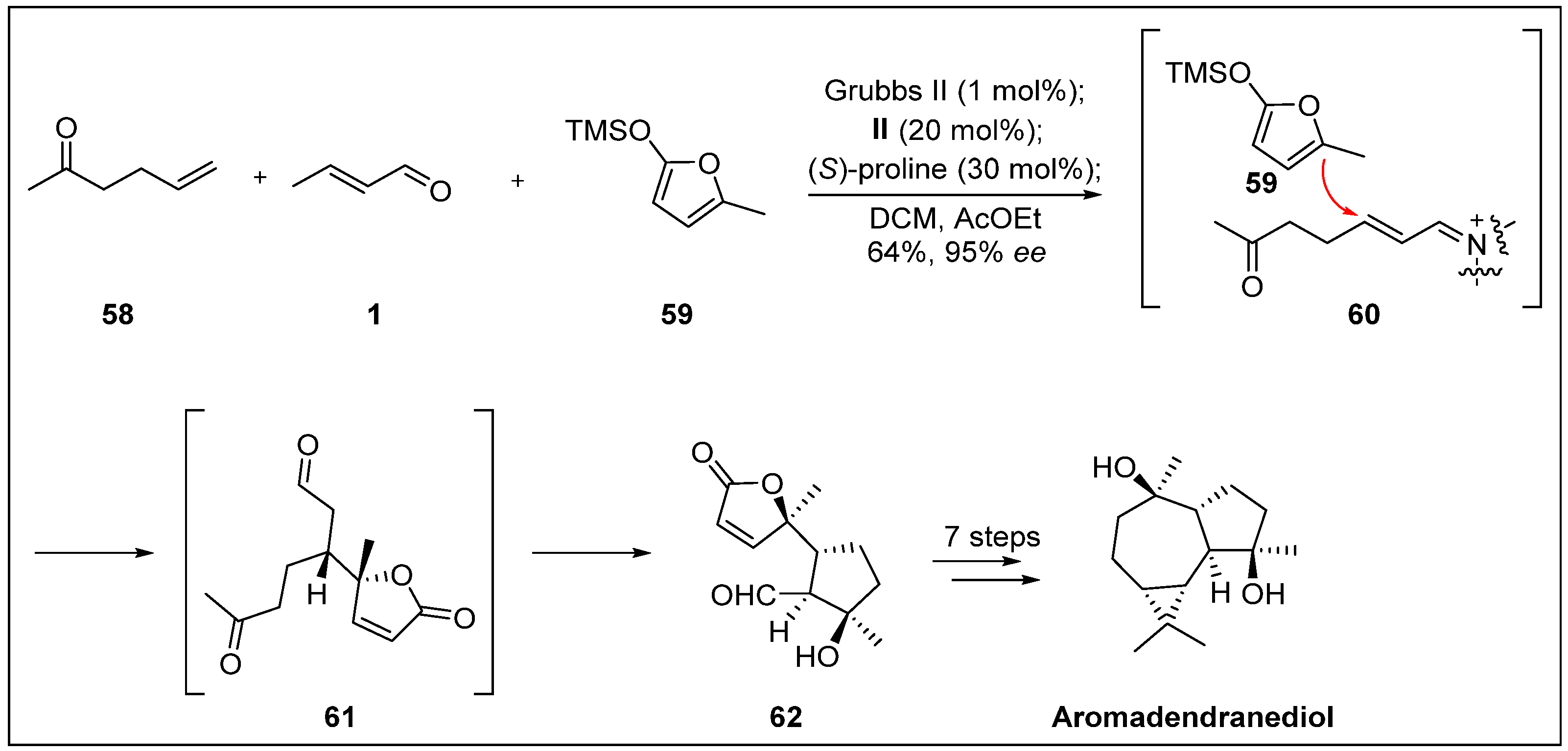
- Beechan, C.M.; Djerassi, C.; Eggert, H. Terpenoids-LXXIV: The sesquiterpenes from the soft coral sinularia mayi. Tetrahedron 1978, 34, 2503–2508. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, K.; Peuronen, A.; Rissanen, K.; Enders, D.; Tang, Y. Enantioselective total syntheses of (+)-hippolachnin A,(+)-gracilioether A,(−)-gracilioether E, and (−)-gracilioether F. J. Am. Chem. Soc. 2018, 140, 1937–1944. [Google Scholar] [CrossRef] [PubMed]
- Festa, C.; D’Amore, C.; Renga, B.; Lauro, G.; Marino, S.; D’Auria, M.; Bifulco, G.; Zampella, A.; Fiorucci, S. Oxygenated polyketides from Plakinastrella mamillaris as a new chemotype of PXR agonists. Mar. Drugs 2013, 11, 2314–2327. [Google Scholar] [CrossRef]
- Piao, S.-J.; Song, Y.-L.; Jiao, W.-H.; Yang, F.; Liu, X.-F.; Chen, W.-S.; Han, B.-N.; Lin, H.-W. Hippolachnin A, a new antifungal polyketide from the South China sea sponge Hippospongia lachne. Org. Lett. 2013, 15, 3526–3529. [Google Scholar] [CrossRef]
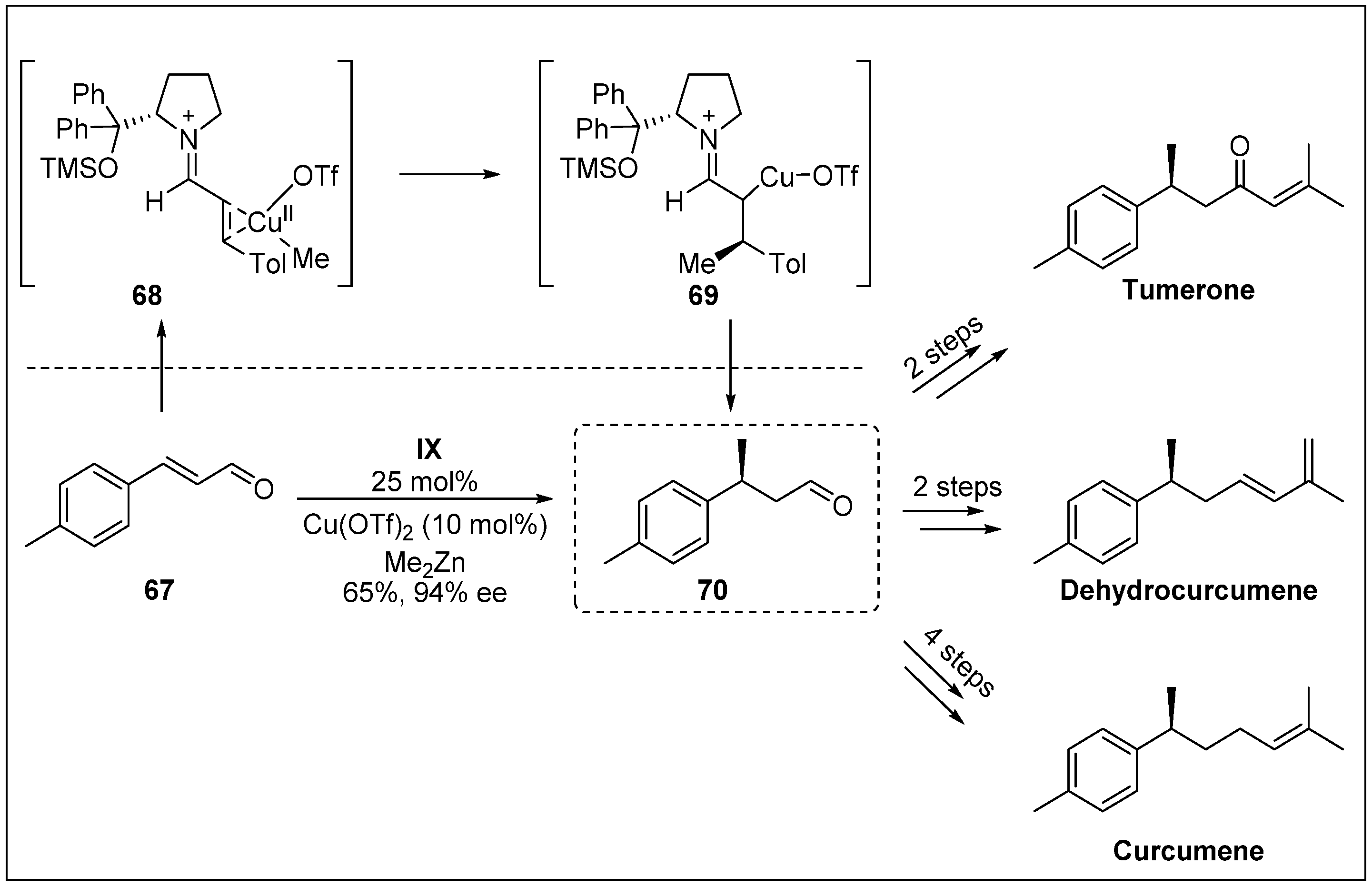
- Afewerki, S.; Breistein, P.; Pirttilä, K.; Deiana, L.; Dziedzic, P.; Ibrahem, I.; Córdova, A. Catalytic enantioselective β-alkylation of α, β-unsaturated aldehydes by combination of transition-metal-and aminocatalysis: Total synthesis of bisabolane sesquiterpenes. Chem-Eur. J. 2011, 17, 8784–8788. [Google Scholar] [CrossRef]
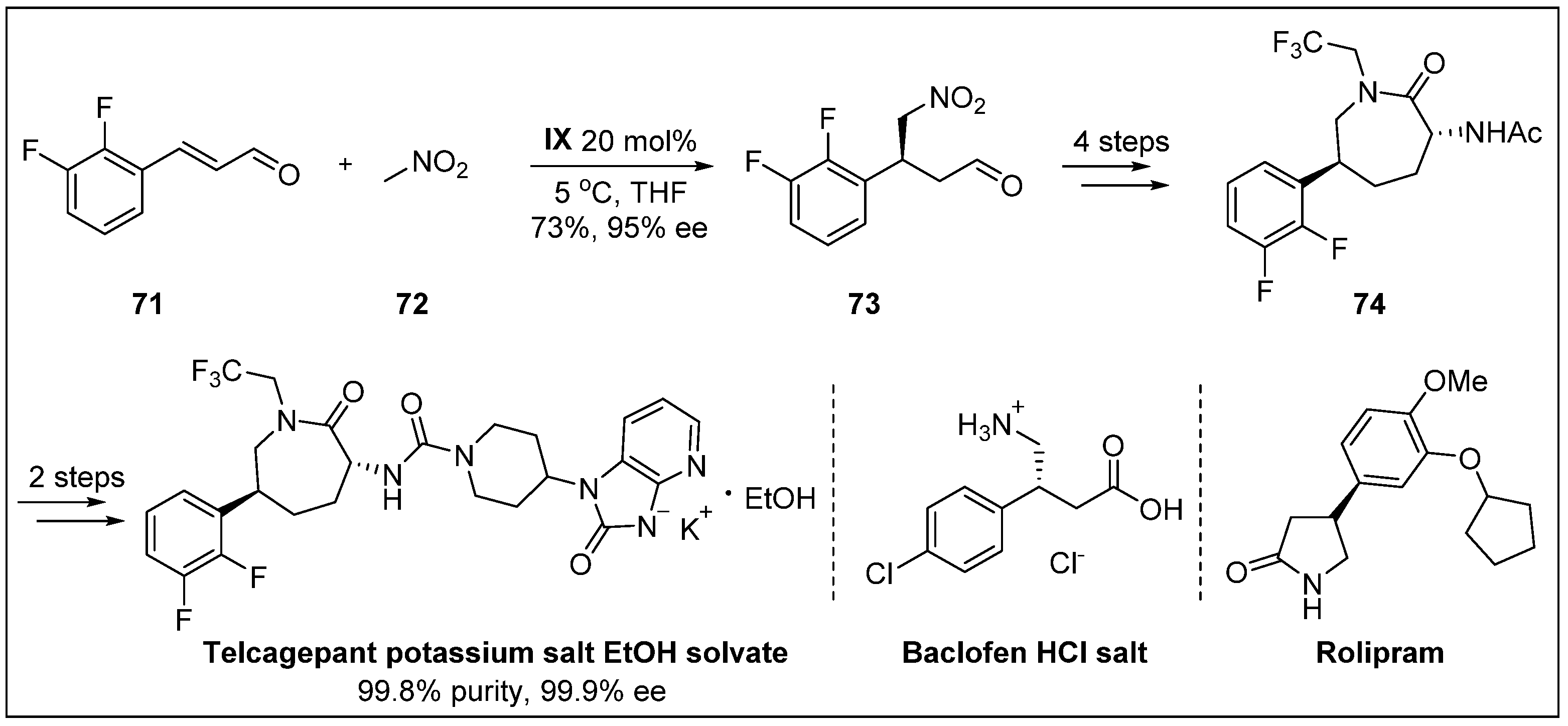
- Xu, F.; Zacuto, M.; Yoshikawa, N.; Desmond, R.; Hoerrner, S.; Itoh, T.; Journet, M.; Humphrey, G.R.; Cowden, C.; Strotman, N. Asymmetric synthesis of telcagepant, a CGRP receptor antagonist for the treatment of migraine. J. Org. Chem. 2010, 75, 7829–7841. [Google Scholar] [CrossRef] [PubMed]
- Paone, D.V.; Shaw, A.W.; Nguyen, D.N.; Burgey, C.S.; Deng, J.Z.; Kane, S.A.; Koblan, K.S.; Salvatore, C.A.; Mosser, S.D.; Johnston, V.K. Potent, orally bioavailable calcitonin gene-related peptide receptor antagonists for the treatment of migraine: Discovery of N-[(3 R, 6 S)-6-(2, 3-difluorophenyl)-2-oxo-1-(2, 2, 2-trifluoroethyl) azepan-3-yl]-4-(2-oxo-2, 3-dihydro-1 H-imidazo [4, 5-b] pyridin-1-yl) piperidine-1-carboxamide (MK-0974). J. Med. Chem. 2007, 50, 5564–5567. [Google Scholar] [PubMed]
- Zu, L.; Xie, H.; Li, H.; Wang, J.; Wang, W. Highly enantioselective organocatalytic conjugate addition of nitromethane to α, β-unsaturated aldehydes: Three-step synthesis of optically active baclofen. Adv. Synth. Catal. 2007, 349, 2660–2664. [Google Scholar] [CrossRef]
- Palomo, C.; Landa, A.; Mielgo, A.; Oiarbide, M.; Puente, A.; Vera, S. Water-compatible iminium activation: Organocatalytic Michael reactions of carbon-centered nucleophiles with enals. Angew. Chem. Int. Ed. 2007, 46, 8431–8435. [Google Scholar] [CrossRef] [PubMed]
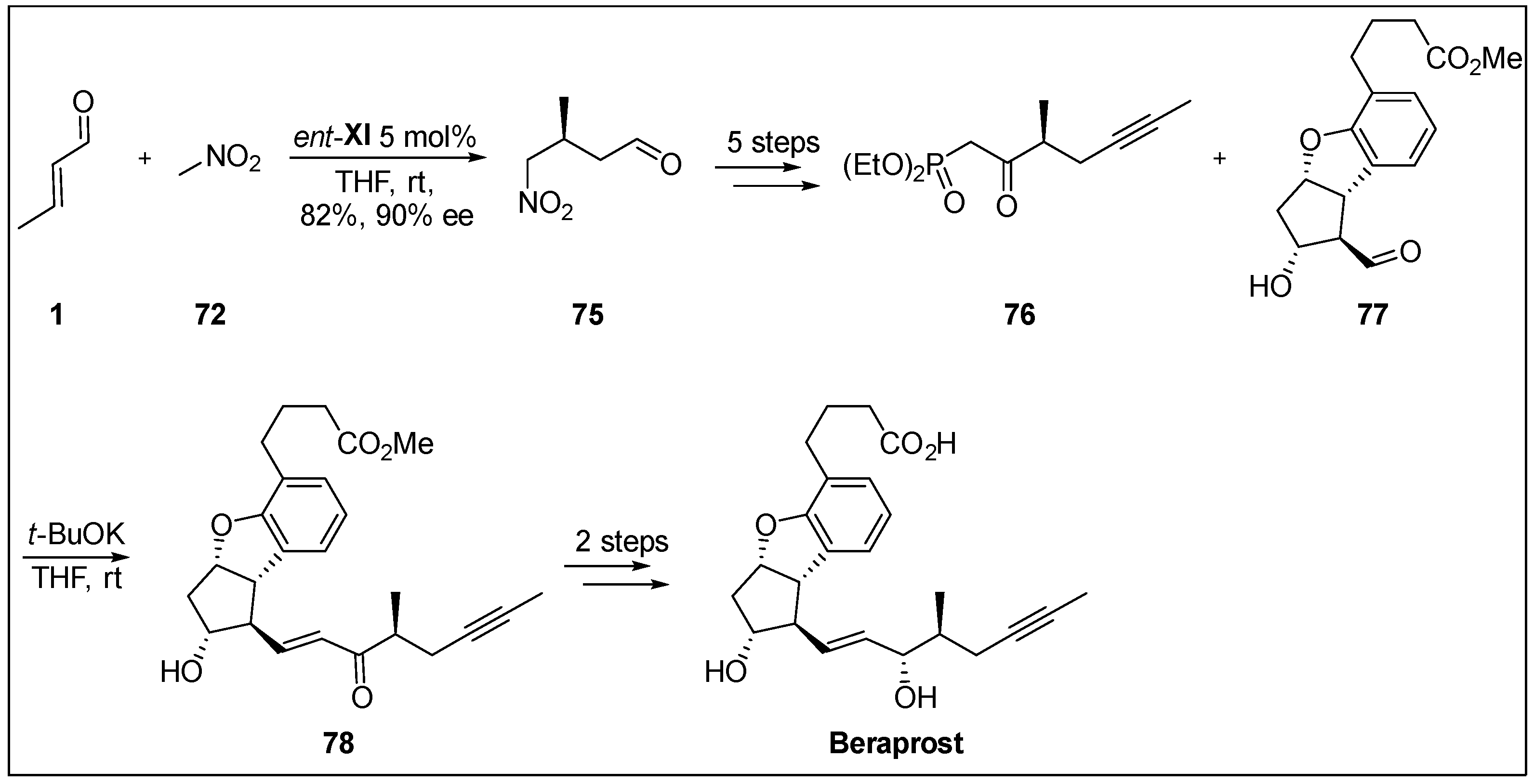
- Umemiya, S.; Sakamoto, D.; Kawauchi, G.; Hayashi, Y. Enantioselective total synthesis of beraprost using organocatalyst. Org. Lett. 2017, 19, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Wakita, H.; Yoshiwara, H.; Kitano, Y.; Nishiyama, H.; Nagase, H. Preparative resolution of 2-methyl-4-hexynic acid for the synthesis of optically active m-phenylene PGI2 derivatives and determination of their absolute configuration. Tetrahedron Asymmetry 2000, 11, 2981–2989. [Google Scholar] [CrossRef]
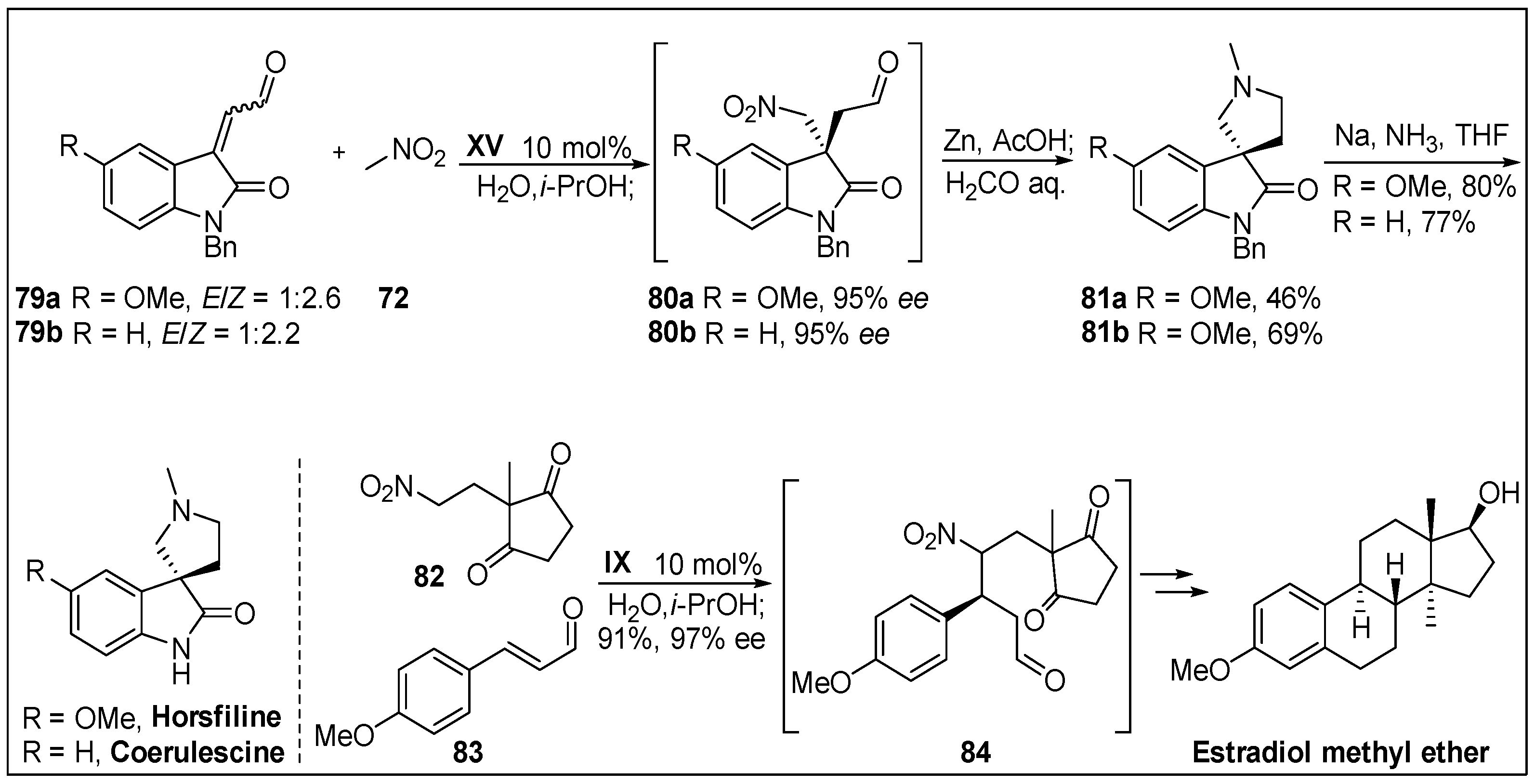
- Mukaiyama, T.; Ogata, K.; Sato, I.; Hayashi, Y. Asymmetric organocatalyzed Michael addition of nitromethane to a 2-oxoindoline-3-ylidene acetaldehyde and the three one-pot sequential synthesis of (−)-horsfiline and (−)-coerulescine. Chem. Eur. J. 2014, 20, 13583–13588. [Google Scholar] [CrossRef] [PubMed]
- Anderton, N.; Cockrum, P.A.; Colegate, S.M.; Edgar, J.A.; Flower, K.; Vit, I.; Willing, R.I. Oxindoles from Phalaris coerulescens. Phytochemistry 1998, 48, 437–439. [Google Scholar] [CrossRef]
- Jossang, A.; Jossang, P.; Hadi, H.A.; Sevenet, T.; Bodo, B. Horsfiline, an oxindole alkaloid from Horsfieldia superba. J. Org. Chem. 1991, 56, 6527–6530. [Google Scholar] [CrossRef]
- Hayashi, Y.; Koshino, S.; Ojima, K.; Kwon, E. Pot economy in the total synthesis of estradiol methyl ether by using an organocatalyst. Angew. Chem. Int. Ed. 2017, 56, 11812–11815. [Google Scholar] [CrossRef]
- Michrowska, A.; List, B. Concise synthesis of ricciocarpin A and discovery of a more potent analogue. Nat. Chem. 2009, 1, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Wurzel, G.; Becker, H. Sesquiterpenoids from the liverwort Ricciocarpos natans. Phytochemistry 1990, 29, 2565–2568. [Google Scholar]
- Wurzel, G.; Becker, H.; Eicher, T.; Tiefensee, K. Molluscicidal properties of constituents from the liverwort Ricciocarpos natans and of synthetic lunularic acid derivatives. Planta Med. 1990, 56, 444–445. [Google Scholar] [PubMed]
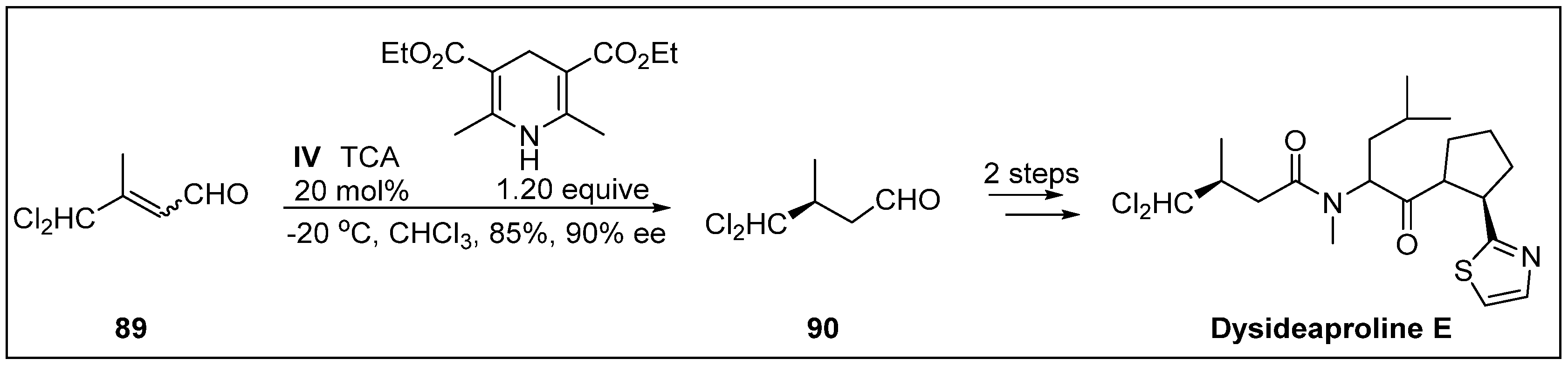
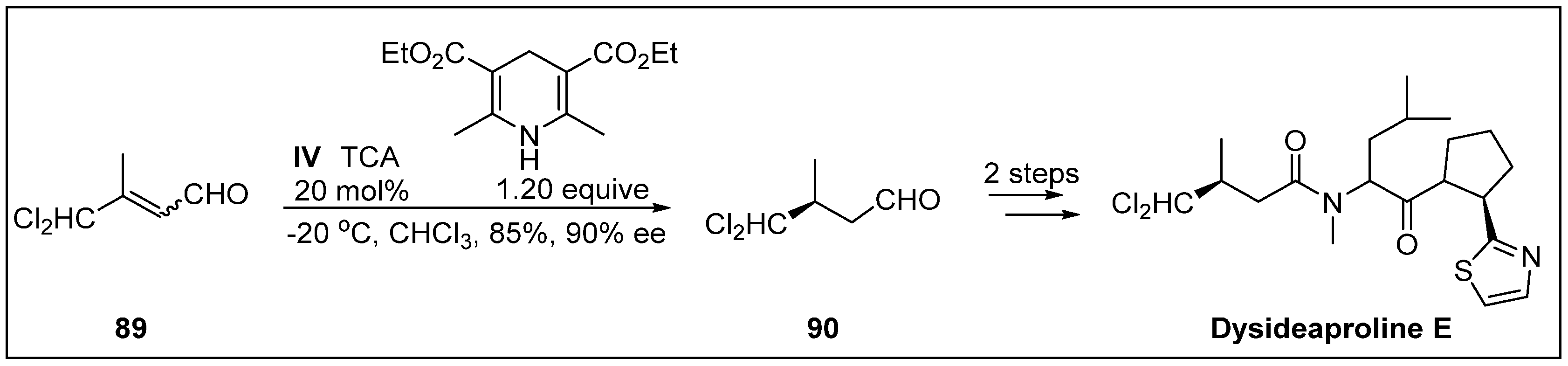
- Owusu-Ansah, E.; Durow, A.C.; Harding, J.R.; Jordan, A.C.; O’Connell, S.J.; Willis, C.L. Synthesis of dysideaproline E using organocatalysis. Org. Biomol. Chem. 2011, 9, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, G.G.; Goetz, G.H.; Luesch, H.; Yang, S.; Likos, J. Dysideaprolines A−F and barbaleucamides A−B, novel polychlorinated compounds from a Dysidea Species. J. Nat. Prod. 2001, 64, 1133–1138. [Google Scholar] [CrossRef]
- Pellissier, H. Asymmetric organocatalytic cycloadditions. Tetrahedron 2012, 68, 2197–2232. [Google Scholar] [CrossRef]
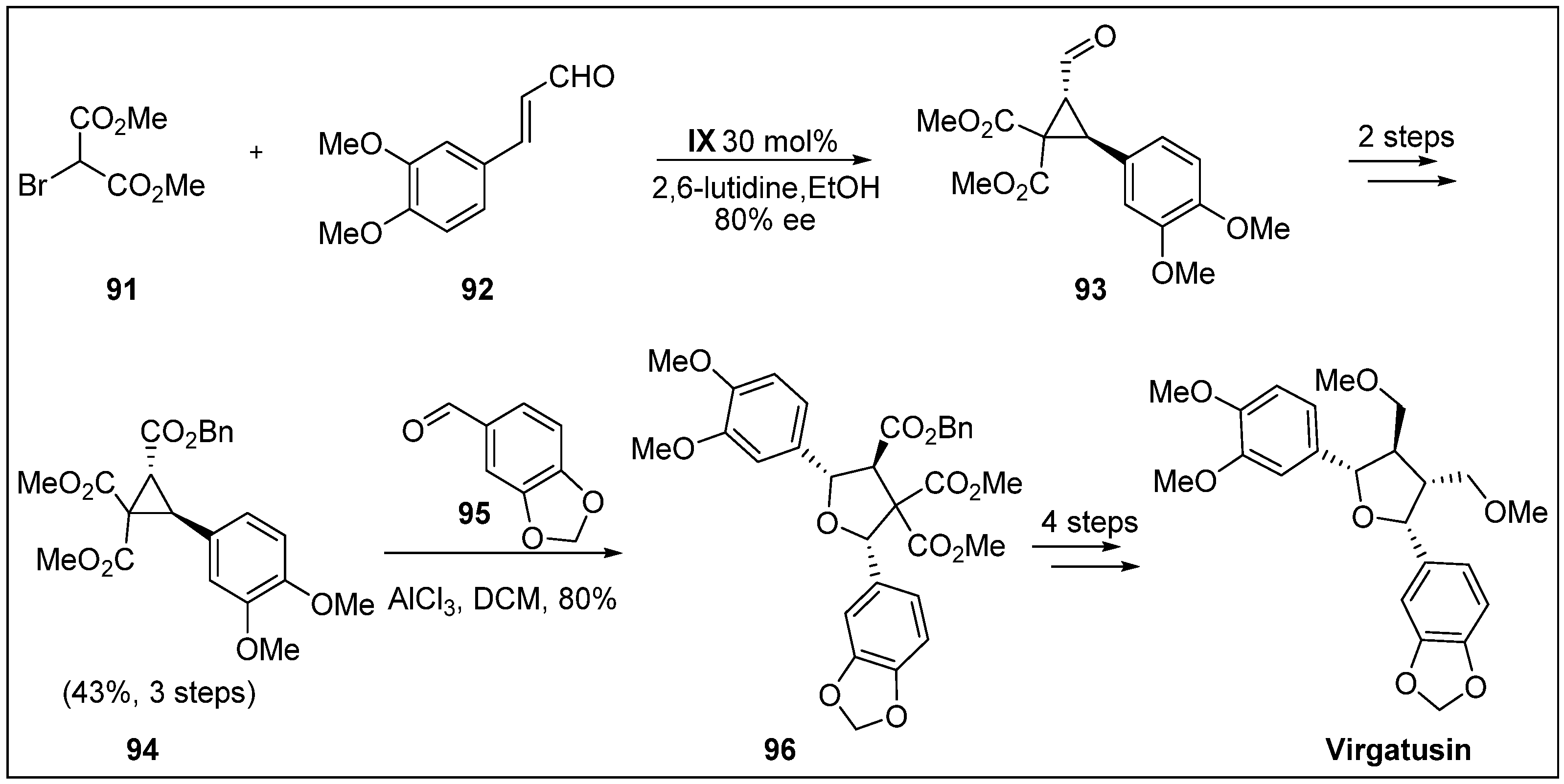
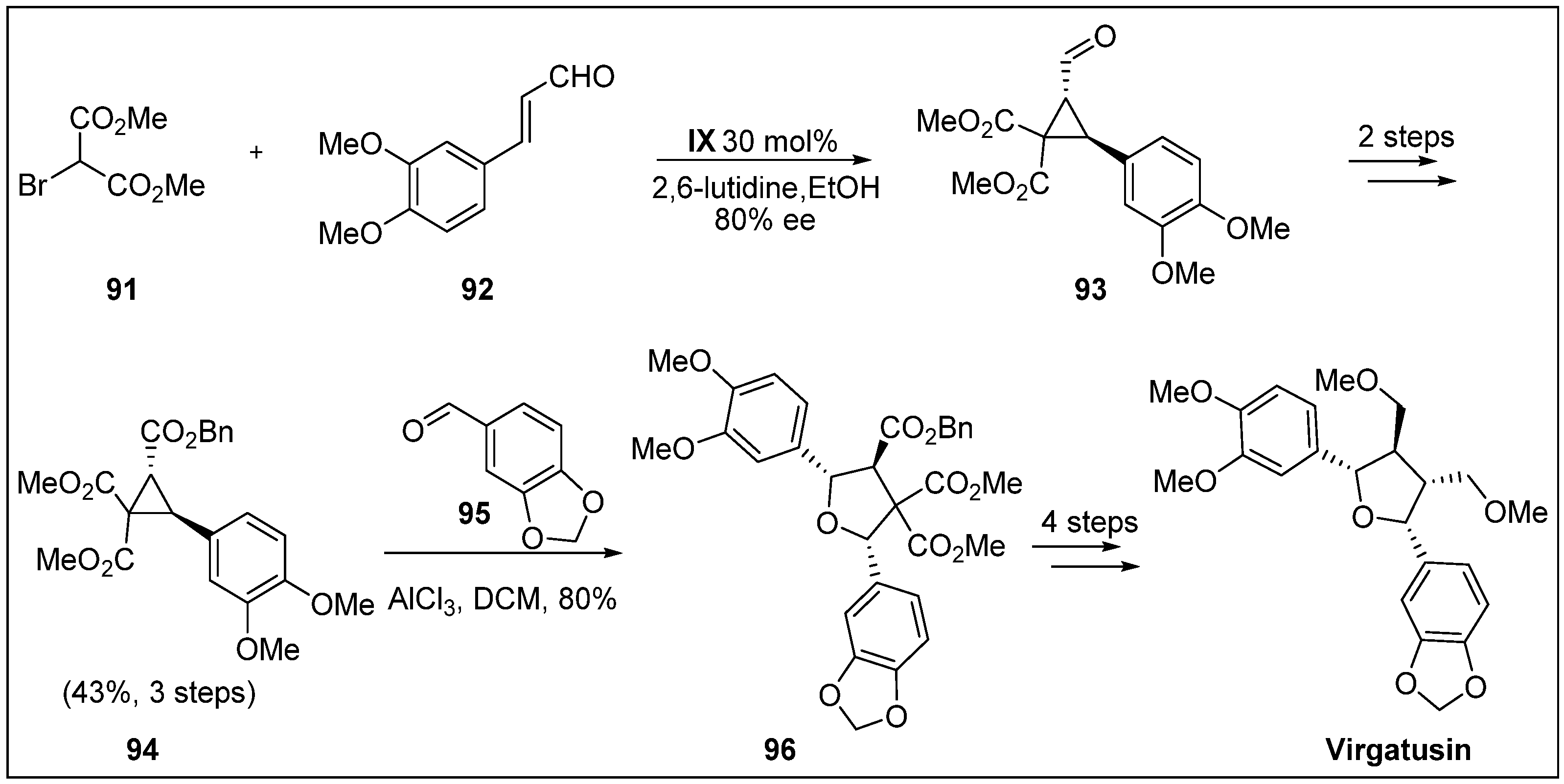
- Sanders, S.D.; Ruiz-Olalla, A.; Johnson, J.S. Total synthesis of (+)-virgatusin via AlCl3-catalyzed [3+2] cycloaddition. Chem. Commun. 2009, 5135–5137. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-L.; Chen, C.-C.; Hsu, F.-L.; Chen, C.-F. A new lignan from Phyllanthus virgatus. J. Nat. Prod. 1996, 59, 520–521. [Google Scholar] [CrossRef]
- Akiyama, K.; Yamauchi, S.; Nakato, T.; Maruyama, M.; Sugahara, T.; Kishida, T. Antifungal activity of tetra-substituted tetrahydrofuran lignan, (−)-virgatusin, and its structure-activity relationship. Biosci. Biotechnol. Biochem. 2007, 71, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
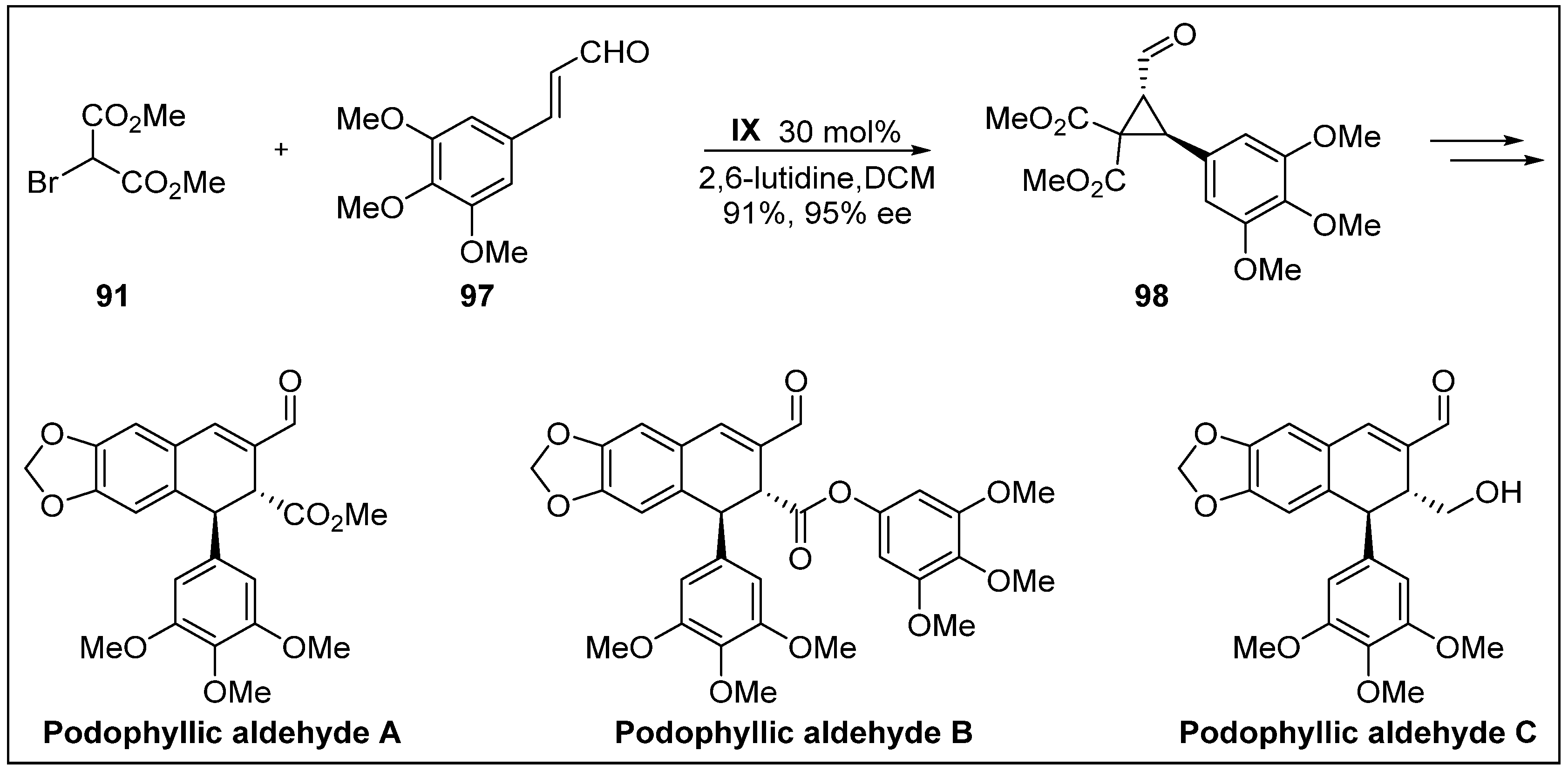
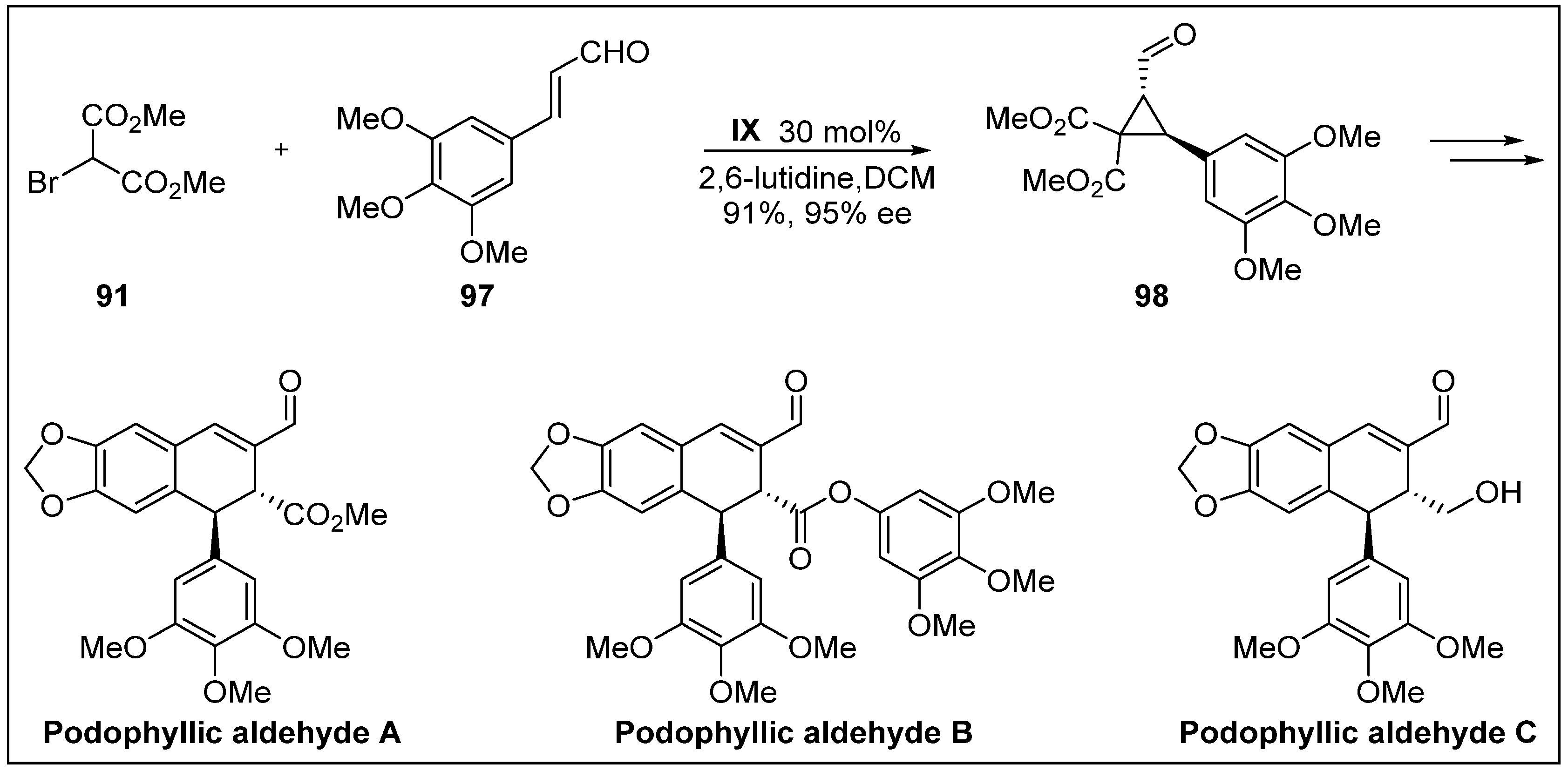
- Ito, J.; Sakuma, D.; Nishii, Y. First total synthesis of (+)-podophyllic aldehydes. Chem. Lett. 2014, 44, 297–299. [Google Scholar] [CrossRef]
- Castro, M.Á.; Miguel del Corral, J.M.; García, P.A.; Rojo, M.V.; de la Iglesia-Vicente, J.; Mollinedo, F.; Cuevas, C.; San Feliciano, A. Synthesis and biological evaluation of new podophyllic aldehyde derivatives with cytotoxic and apoptosis-inducing activities. J. Med. Chem. 2010, 53, 983–993. [Google Scholar] [CrossRef] [PubMed]
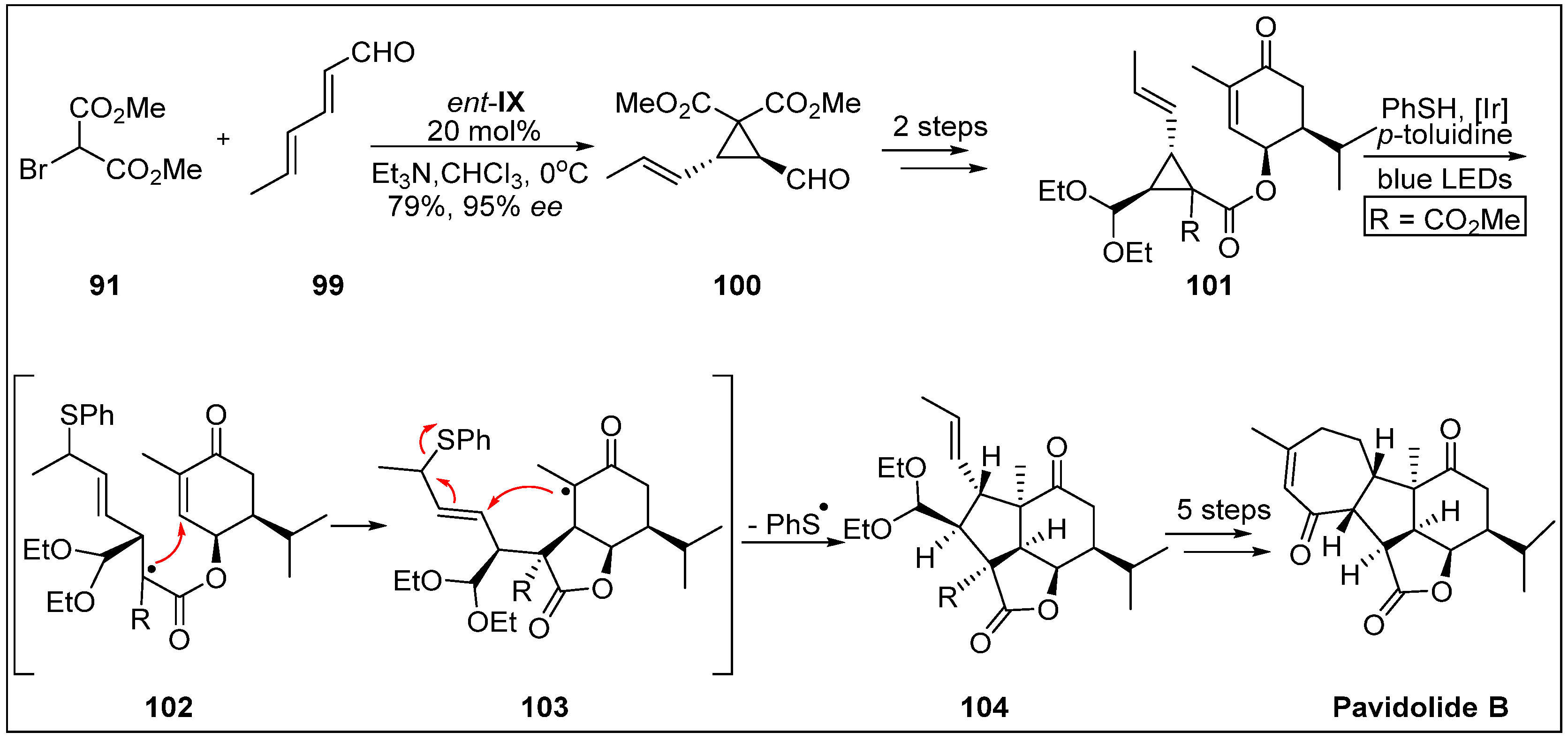
- Shen, S.; Zhu, H.; Chen, D.; Liu, D.; van Ofwegen, L.; Proksch, P.; Lin, W. Pavidolides A–E, new cembranoids from the soft coral Sinularia pavida. Tetrahedron Lett. 2012, 53, 5759–5762. [Google Scholar] [CrossRef]
- Zhang, P.-P.; Yan, Z.-M.; Li, Y.-H.; Gong, J.-X.; Yang, Z. Enantioselective total synthesis of (−)-pavidolide B. J. Am. Chem. Soc. 2017, 139, 13989–13992. [Google Scholar] [CrossRef] [PubMed]
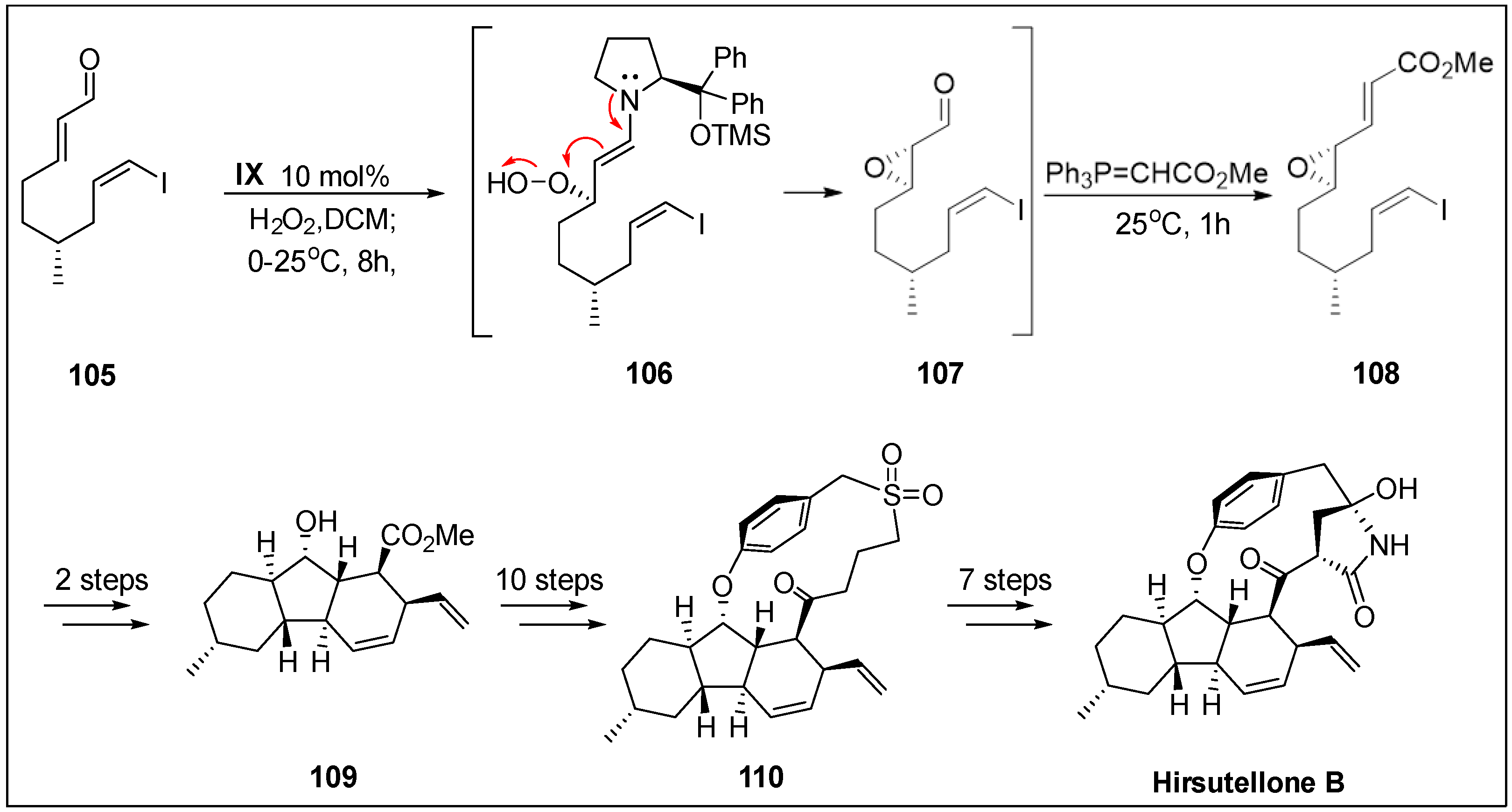
- Nicolaou, K.C.; Sarlah, D.; Wu, T.R.; Zhan, W. Total synthesis of hirsutellone B. Angew. Chem. Int. Ed. 2009, 48, 6870–6874. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Rugseree, N.; Maithip, P.; Kongsaeree, P.; Prabpai, S.; Thebtaranonth, Y. Hirsutellones A–E, antimycobacterial alkaloids from the insect pathogenic fungus Hirsutella nivea BCC 2594. Tetrahedron 2005, 61, 5577–5583. [Google Scholar] [CrossRef]
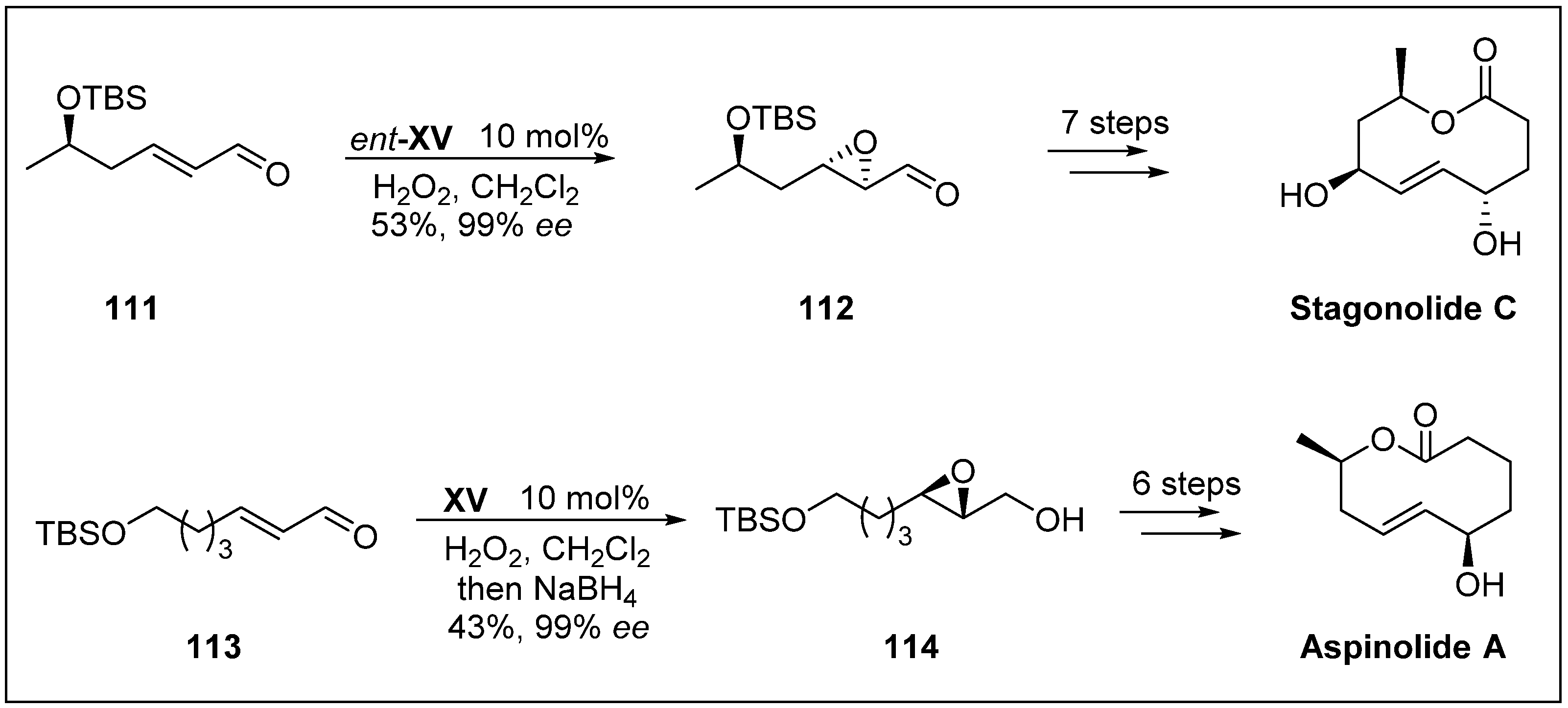
- Yuzikhin, O.; Mitina, G.; Berestetskiy, A. Herbicidal potential of stagonolide, a new phytotoxic nonenolide from Stagonospora cirsii. J. Agr. Food Chem. 2007, 55, 7707–7711. [Google Scholar] [CrossRef]
- Fuchser, J.; Zeeck, A. Secondary metabolites by chemical screening.34. Aspinolides and aspinonene/aspyrone co-metabolites, new pentaketides produced by Aspergillus ochraceus. Liebigs Ann. Recl. 1997, 1997, 87–95. [Google Scholar] [CrossRef]
- Shelke, A.M.; Rawat, V.; Suryavanshi, G.; Sudalai, A. Asymmetric synthesis of (+)-stagonolide C and (−)-aspinolide A via organocatalysis. Tetrahedron Asymmetry 2012, 23, 1534–1541. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Cai, Q.; Qin, B.; Petersen, M.T.; Mikkelsen, R.J.; Heretsch, P. Total synthesis of trioxacarcin DC-45-A2. Angew. Chem. Int. Ed. 2015, 54, 3074–3078. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Cai, Q.; Sun, H.; Qin, B.; Zhu, S. Total synthesis of trioxacarcins DC-45-A1, A, D, C, and C7 ″-epi-C and full structural assignment of trioxacarcin C. J. Am. Chem. Soc. 2016, 138, 3118–3124. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Chen, P.; Zhu, S.; Cai, Q.; Erande, R.D.; Li, R.; Sun, H.; Pulukuri, K.K.; Rigol, S.; Aujay, M. Streamlined total synthesis of trioxacarcins and its application to the design, synthesis, and biological evaluation of analogues thereof. Discovery of simpler designed and potent trioxacarcin analogues. J. Am. Chem. Soc. 2017, 139, 15467–15478. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, M.; Kuwahara, S. Total synthesis of bacilosarcins A and B. Angew. Chem. Int. Ed. 2009, 48, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Azumi, M.; Ogawa, K.-i.; Fujita, T.; Takeshita, M.; Yoshida, R.; Furumai, T.; Igarashi, Y. Bacilosarcins A and B, novel bioactive isocoumarins with unusual heterocyclic cores from the marine-derived bacterium Bacillus subtilis. Tetrahedron 2008, 64, 6420–6425. [Google Scholar] [CrossRef]
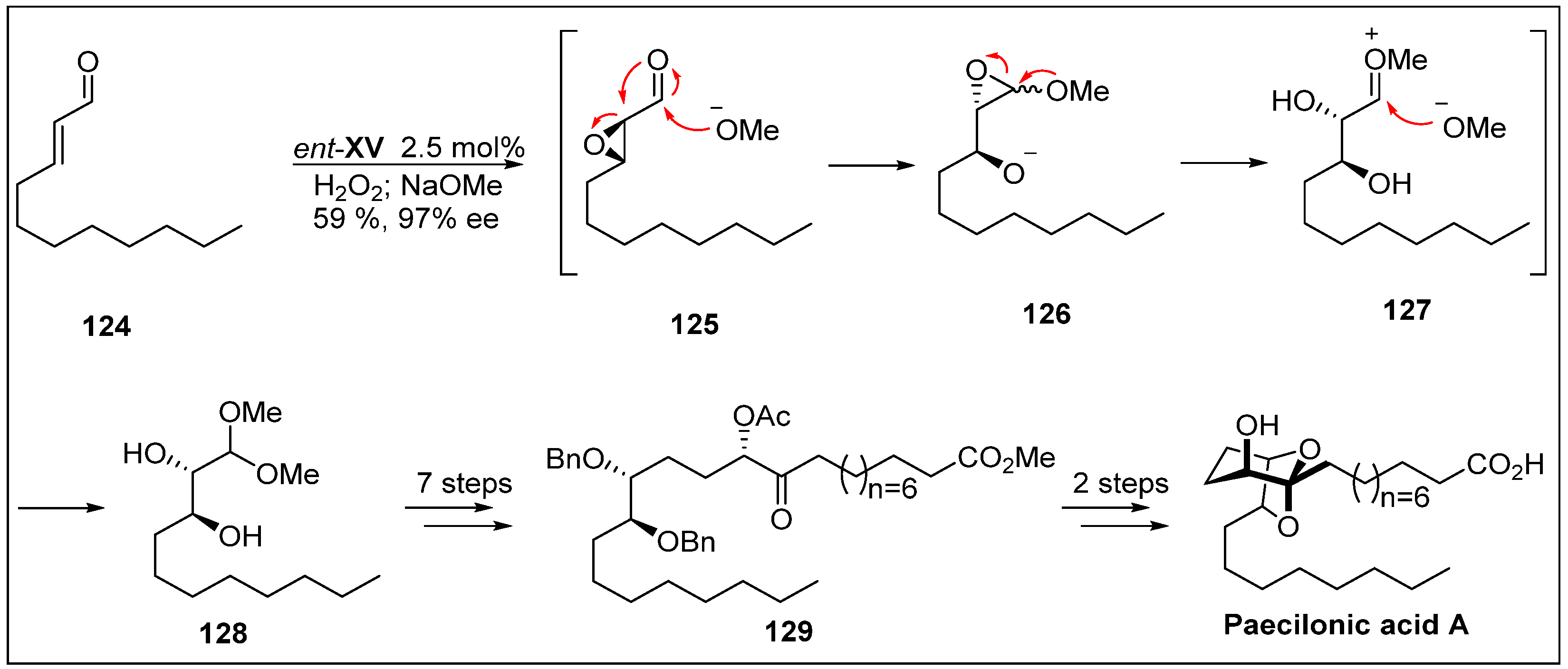
- Sommer, H.; Hamilton, J.Y.; Fürstner, A. A method for the late-stage formation of ketones, acyloins, and aldols from alkenylstannanes: Application to the total synthesis of paecilonic acid A. Angew. Chem. Int. Ed. 2017, 56, 6161–6165. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hong, J.; Yin, J.; Liu, J.; Liu, Y.; Choi, J.S.; Jung, J.H. Paecilonic acids A and B, bicyclic fatty acids from the jellyfish-derived fungus Paecilomyces variotii J08NF-1. Bioorg. Med. Chem. Lett. 2016, 26, 2220–2223. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, L.; Jiang, H.; Dickmeiss, G.; Gschwend, B.; Hansen, S.G.; Jorgensen, K.A. Asymmetric formal trans-dihydroxylation and trans-aminohydroxylation of α,β-unsaturated aldehydes via an organocatalytic reaction cascade. J. Am. Chem. Soc. 2010, 132, 9188–9196. [Google Scholar] [CrossRef] [PubMed]
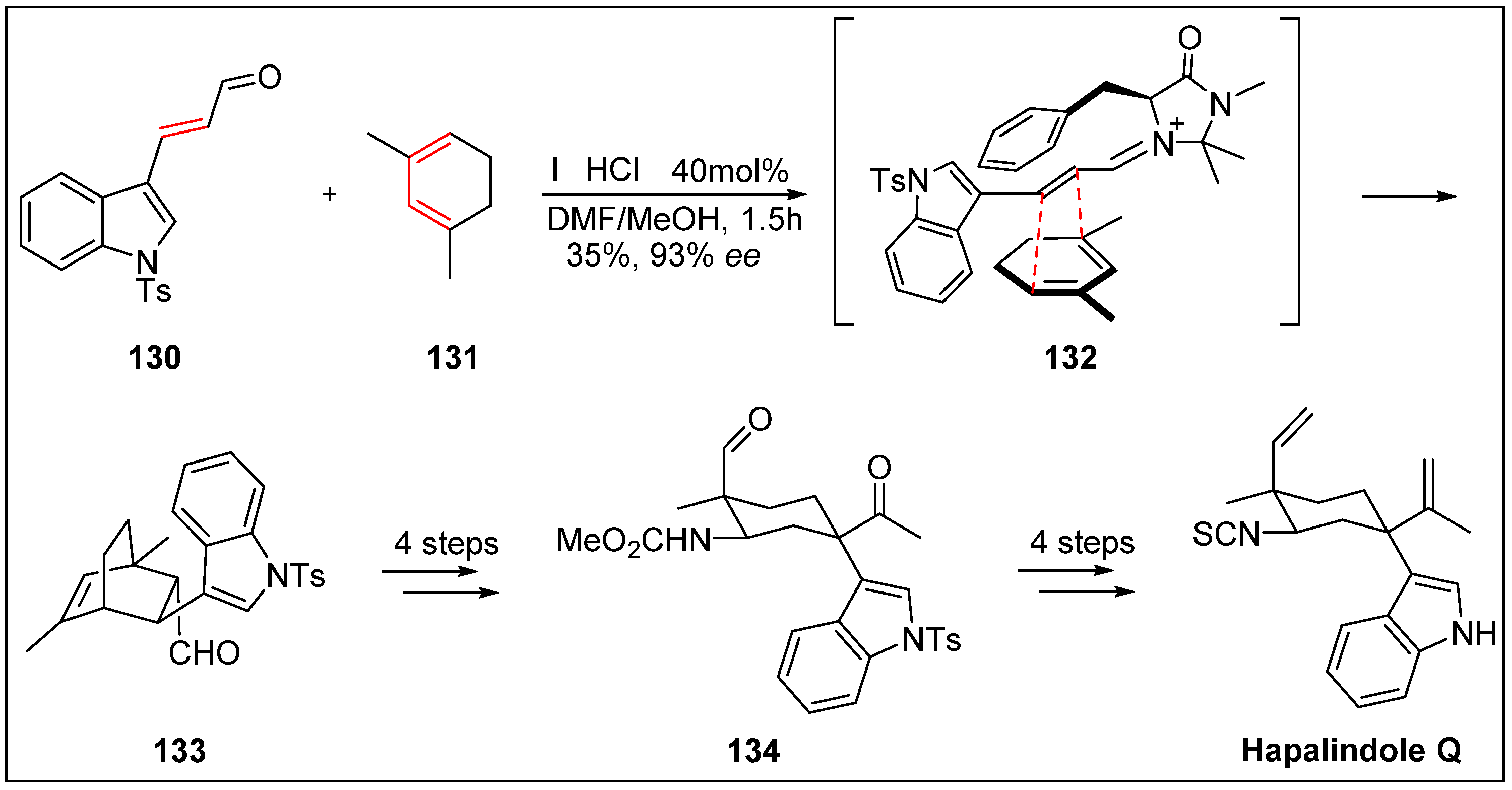
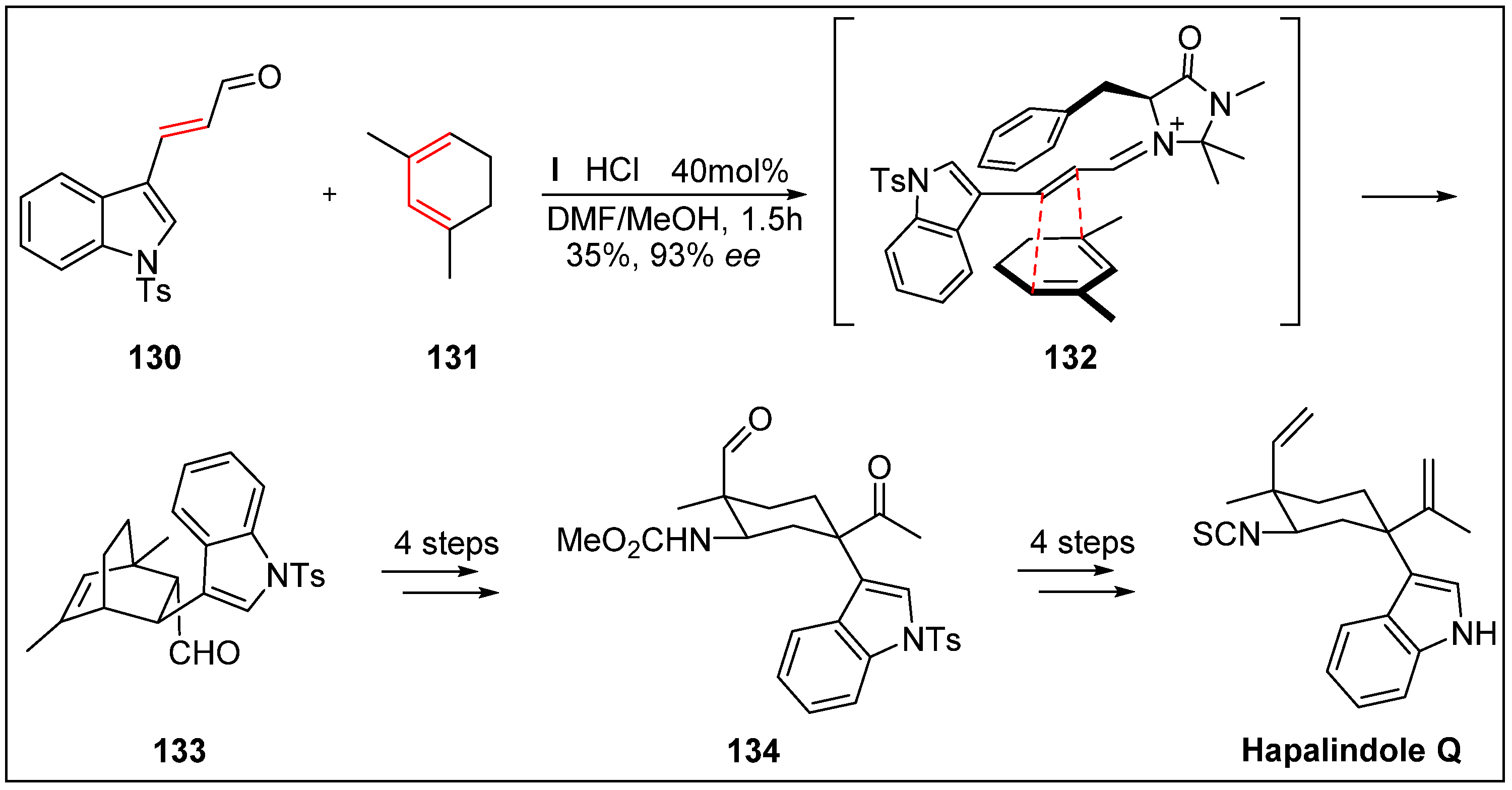
- Kinsman, A.C.; Kerr, M.A. The total synthesis of (+)-hapalindole Q by an organomediated Diels−Alder reaction. J. Am. Chem. Soc. 2003, 125, 14120–14125. [Google Scholar] [CrossRef]
- Moore, R.E.; Cheuk, C.; Yang, X.Q.G.; Patterson, G.M.; Bonjouklian, R.; Smitka, T.A.; Mynderse, J.S.; Foster, R.S.; Jones, N.D.; Swartzendruber, J.K. Hapalindoles, antibacterial and antimycotic alkaloids from the cyanophyte Hapalosiphon fontinalis. J. Org. Chem. 2002, 52, 1036–1043. [Google Scholar] [CrossRef]
- Doan, N.T.; Stewart, P.R.; Smith, G.D. Inhibition of bacterial RNA polymerase by the cyanobacterial metabolites 12-epi-hapalindole E isonitrile and calothrixin A. Fems Microbiol. Lett. 2001, 196, 135–139. [Google Scholar] [CrossRef]
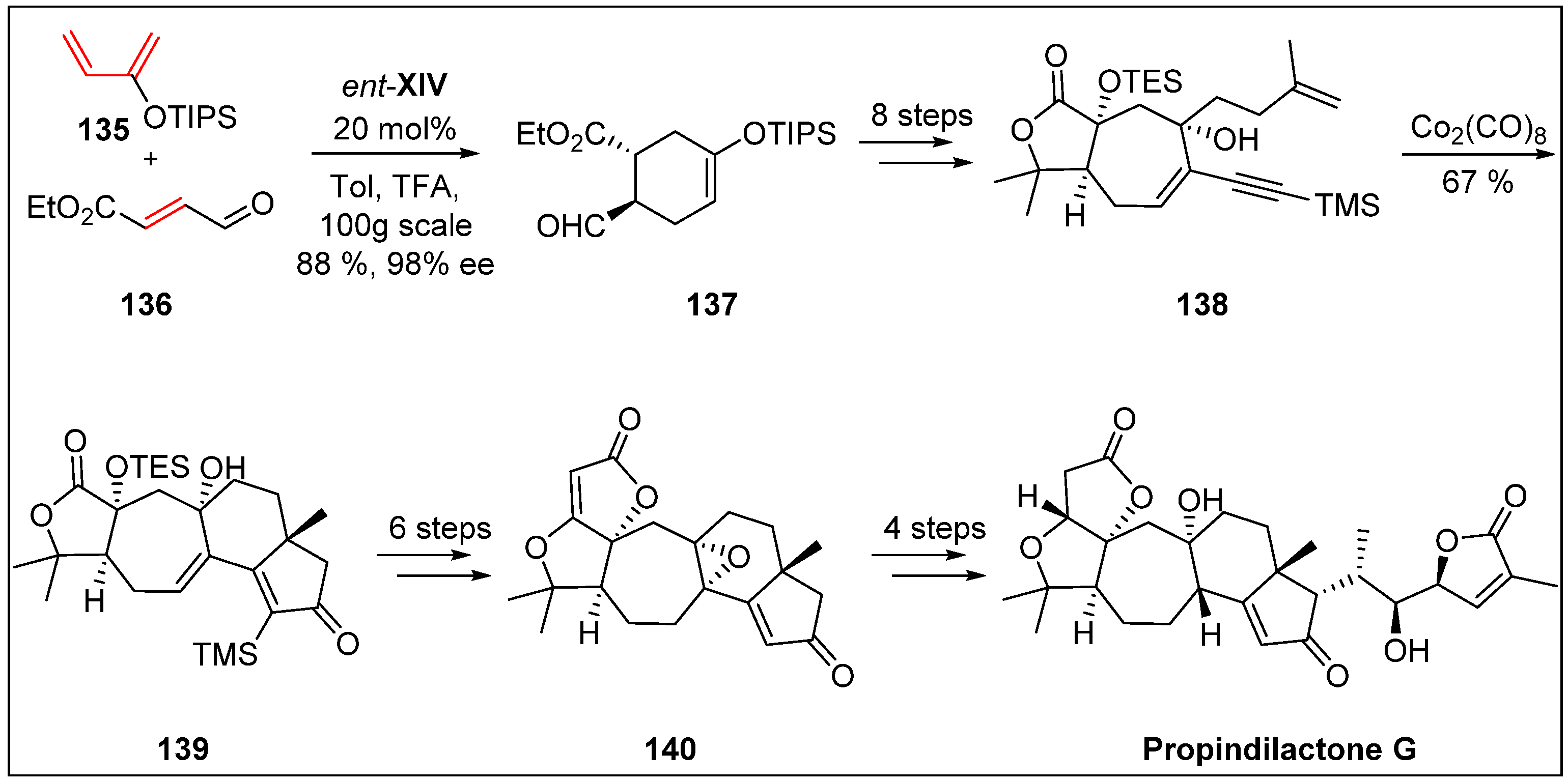
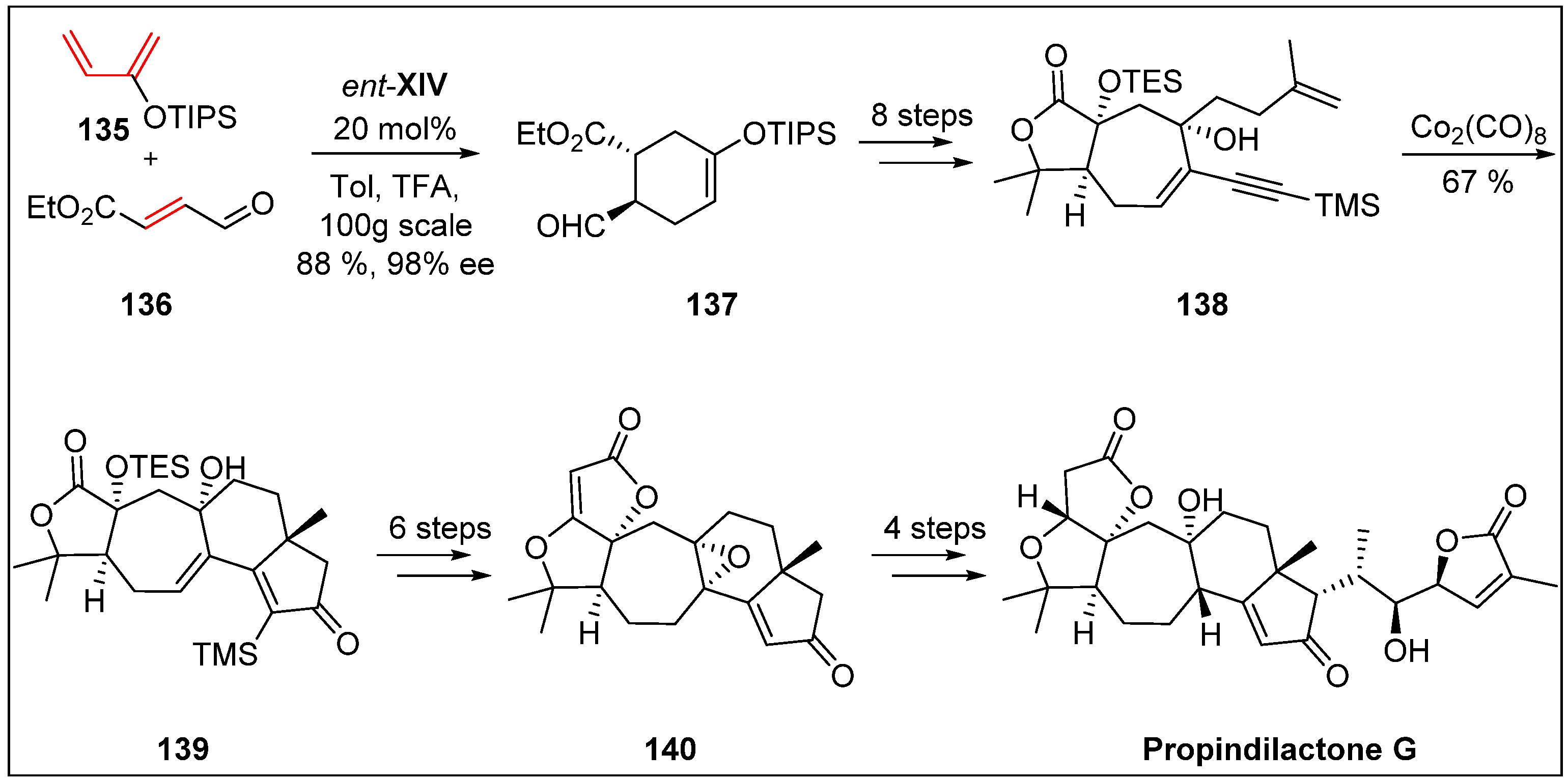
- You, L.; Liang, X.-T.; Xu, L.-M.; Wang, Y.-F.; Zhang, J.-J.; Su, Q.; Li, Y.-H.; Zhang, B.; Yang, S.-L.; Chen, J.-H. Asymmetric total synthesis of Propindilactone, G. J. Am. Chem. Soc. 2015, 137, 10120–10123. [Google Scholar] [CrossRef]
- Lei, C.; Huang, S.-X.; Chen, J.-J.; Yang, L.-B.; Xiao, W.-L.; Chang, Y.; Lu, Y.; Huang, H.; Pu, J.-X.; Sun, H.-D. Propindilactones E−J, Schiartane Nortriterpenoids from Schisandra propinqua var. propinqua. J. Nat. Prod. 2008, 71, 1228–1232. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.M.; Jen, W.S.; MacMillan, D.W. Enantioselective organocatalytic intramolecular Diels−Alder reactions. The asymmetric synthesis of solanapyrone D. J. Am. Chem. Soc. 2005, 127, 11616–11617. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, H.; Yokota, T.; Ichihara, A.; Sakamura, S. Structure and absolute configuration of solanapyrone D: A new clue to the occurrence of biological Diels–Alder reactions. J. Chem. Soc. Chem. Commun. 1989, 1284–1285. [Google Scholar] [CrossRef]
- Hagiwara, H.; Kobayashi, K.; Miya, S.; Hoshi, T.; Suzuki, T.; Ando, M.; Okamoto, T.; Kobayashi, M.; Yamamoto, I.; Ohtsubo, S.; et al. First total syntheses of the phytotoxins solanapyrones D and E via the domino Michael protocol. J. Org. Chem. 2002, 67, 5969–5976. [Google Scholar] [CrossRef] [PubMed]
- Lambert, T.H.; Danishefsky, S.J. Total synthesis of UCS1025A. J. Am. Chem. Soc. 2006, 128, 426–427. [Google Scholar] [CrossRef] [PubMed]
- Kirst, H.A.; Michel, K.H.; Martin, J.W.; Creemer, L.C.; Chio, E.H.; Yao, R.C.; Nakatsukasa, W.M.; Boeck, L.; Occolowitz, J.L.; Paschal, J.W.; et al. A83543A-D, unique fermentation-derived tetracyclic macrolides. Tetrahedron Lett. 1991, 32, 4839–4842. [Google Scholar] [CrossRef]
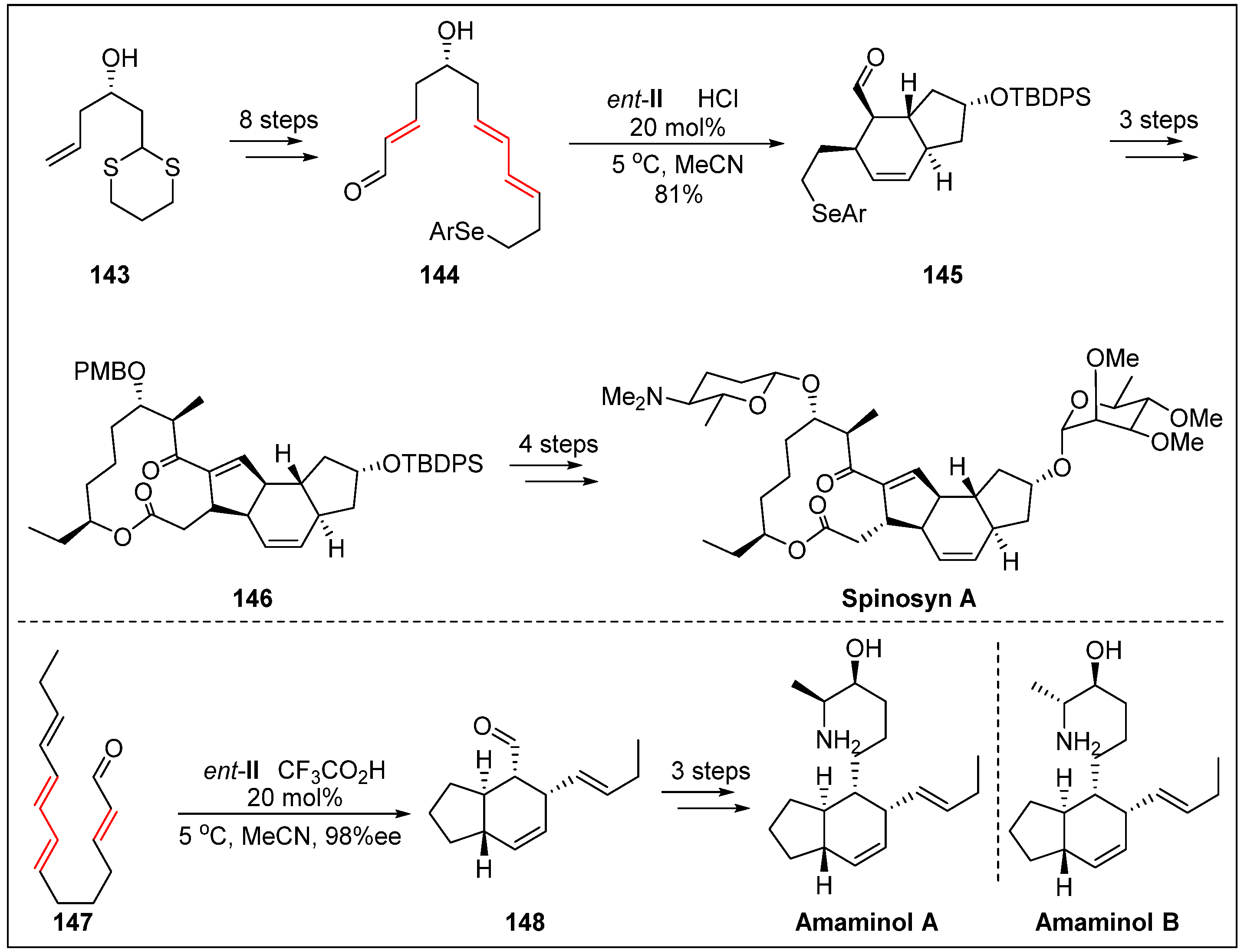
- Bai, Y.; Shen, X.; Li, Y.; Dai, M. Total synthesis of (-)-spinosyn A via carbonylative macrolactonization. J. Am. Chem. Soc. 2016, 138, 10838–10841. [Google Scholar] [CrossRef]
- Kumpulainen, E.T.; Koskinen, A.M.; Rissanen, K. Total synthesis of amaminol A: Establishment of the absolute stereochemistry. Org. Lett. 2007, 9, 5043–5045. [Google Scholar] [CrossRef]
- Jacobs, W.C.; Christmann, M. Synthesis of amaminol B. Synlett. 2008, 2008, 247–251. [Google Scholar]
- Horning, B.D.; MacMillan, D.W. Nine-step enantioselective total synthesis of (−)-vincorine. J. Am. Chem. Soc. 2013, 135, 6442–6445. [Google Scholar] [CrossRef] [PubMed]
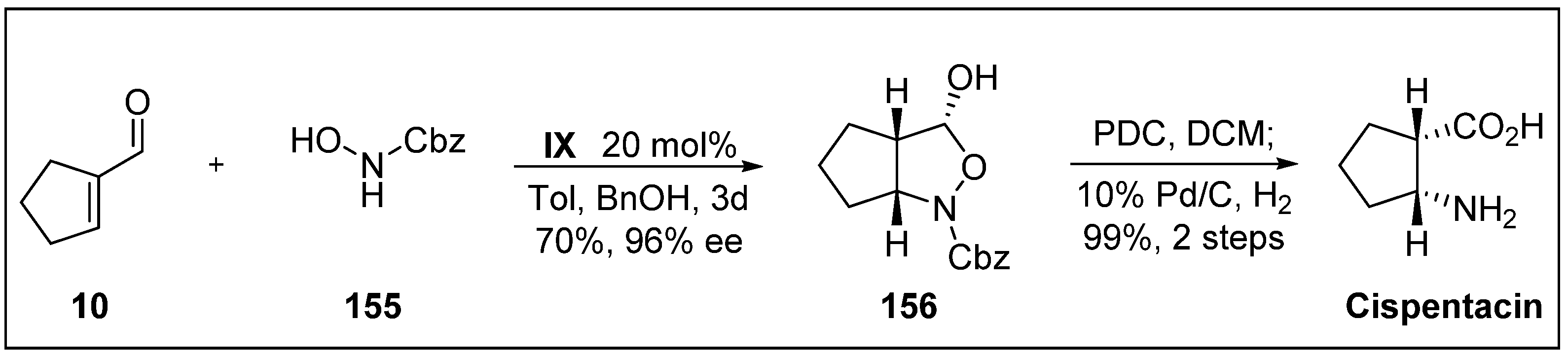
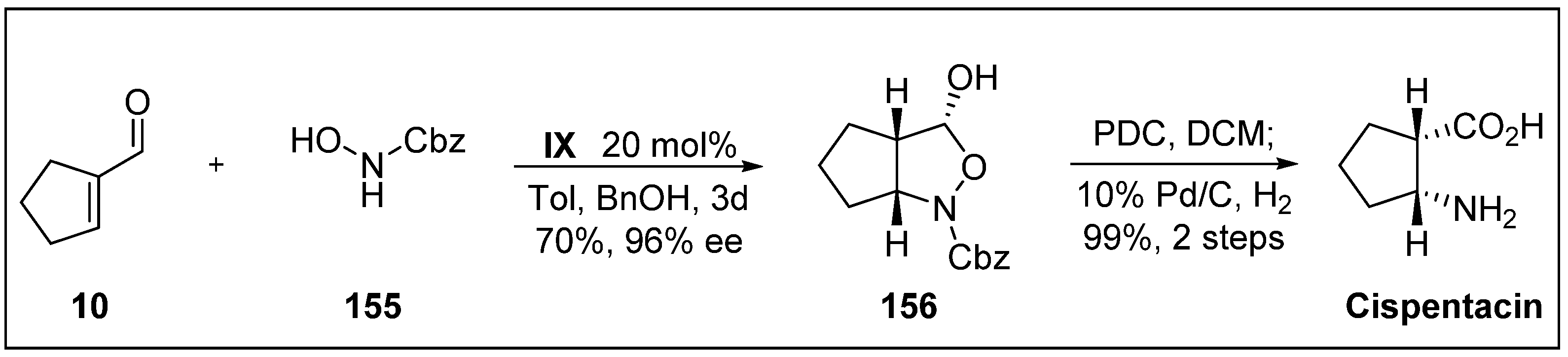
- Pou, A.; Moyano, A. Stereoselective organocatalytic approach to α, β-disubstituted-β-amino Acids: A short enantioselective synthesis of cispentacin. Eur. J. Org. Chem. 2013, 2013, 3103–3111. [Google Scholar] [CrossRef]
- Konishi, M.; Nishio, M.; Saitoh, K.; MlyakI, T.; Oki, T.; Kawaguchi, H. Cispentacin, a new antifungal antibiotic. J. Antibiot. 1989, 42, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
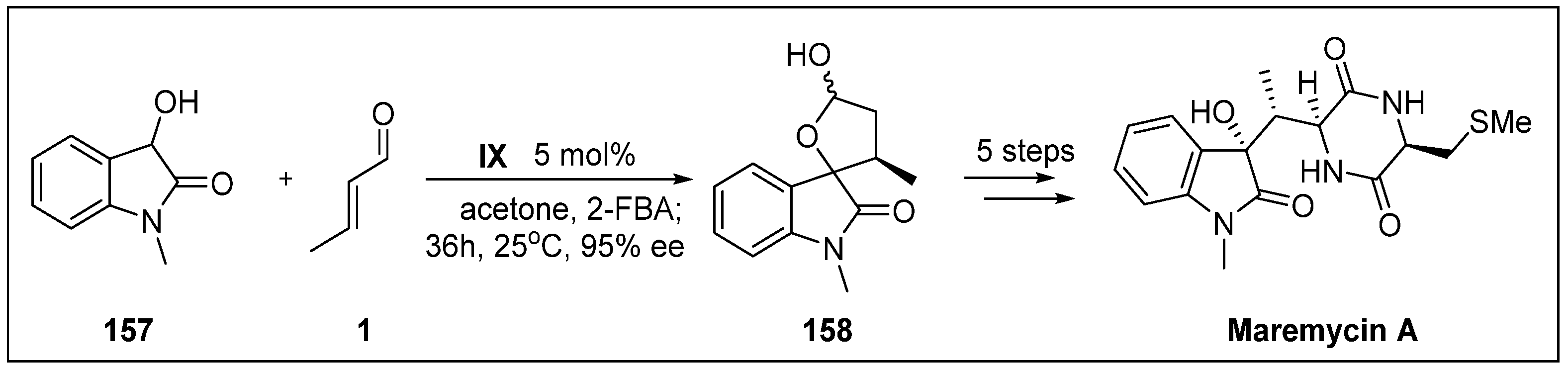
- Bergonzini, G.; Melchiorre, P. Dioxindole in asymmetric catalytic synthesis: Routes to enantioenriched 3-substituted 3-hydroxyoxindoles and the preparation of maremycin A. Angew. Chem. Int. Ed. 2012, 51, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Balk-Bindseil, W.; Helmke, E.; Weyland, H.; Laatsch, H. Marine bacteria, VIII. Maremycin A and B, new diketopiperazines from a marine Streptomyces sp. Liebigs Ann. 1995, 1995, 1291–1294. [Google Scholar] [CrossRef]
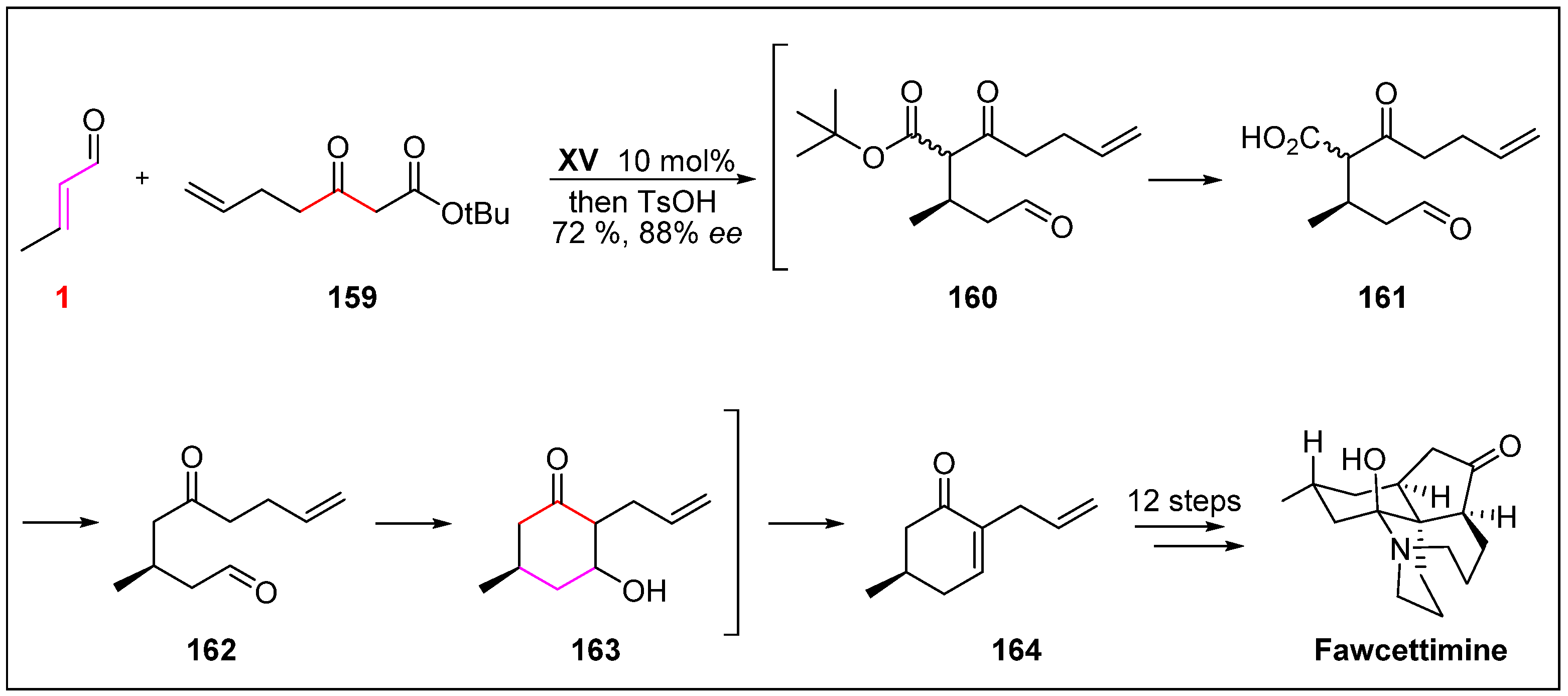
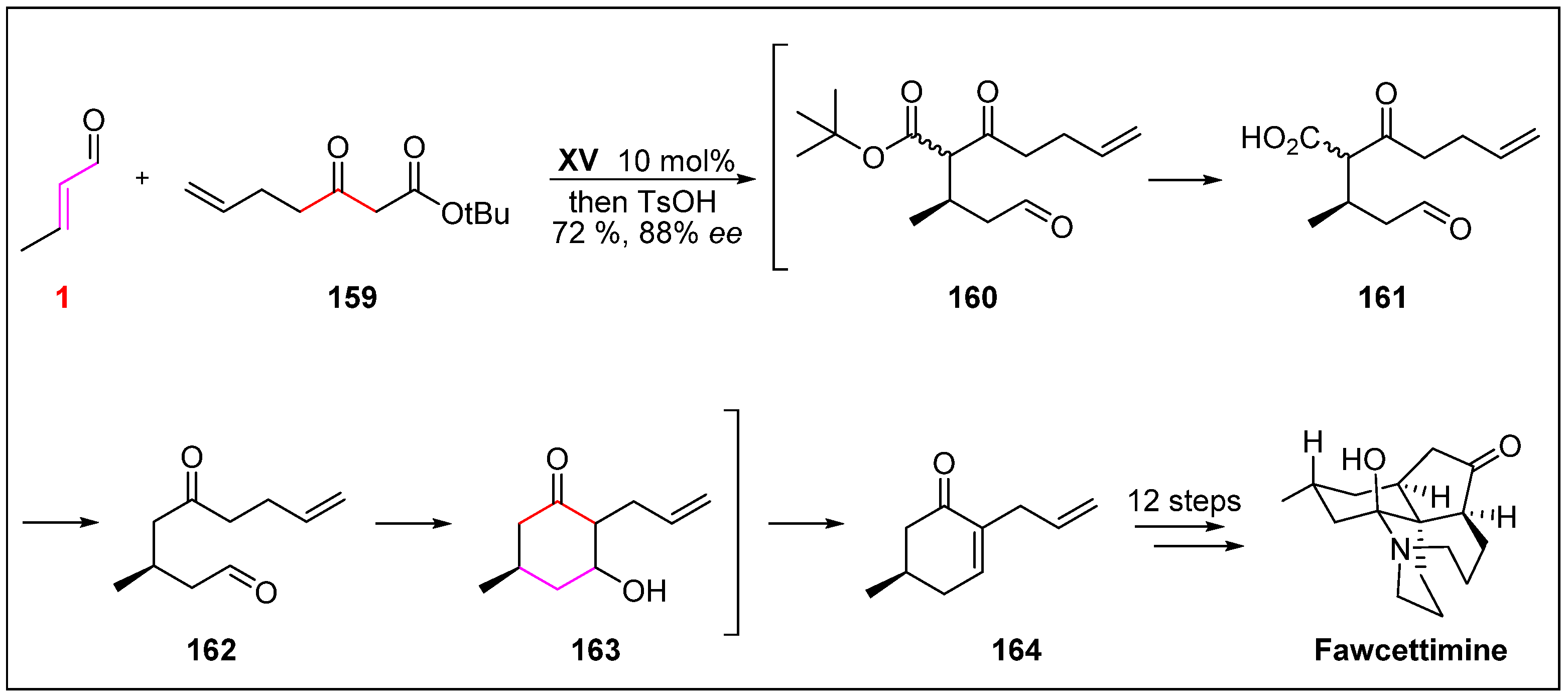
- Linghu, X.; Kennedy-Smith, J.J.; Toste, F.D. Total synthesis of (+)-fawcettimine. Angew. Chem. Int. Ed. 2007, 46, 7671–7673. [Google Scholar] [CrossRef] [PubMed]
- Burnell, R.H. Lycopodium alkaloids. 1. Extraction of alkaloids from Lycopodium fawcettii, lloyd and underwood. J. Chem. Soc. 1959, 41, 3091–3093. [Google Scholar]
- Carlone, A.; Marigo, M.; North, C.; Landa, A.; Jorgensen, K.A. A simple asymmetric organocatalytic approach to optically active cyclohexenones. Chem. Commun. 2006, 4928–4930. [Google Scholar] [CrossRef] [PubMed]
- Kocienski, P. Synthesis of (+)-fawcettimine. Synfacts 2008, 2, 124. [Google Scholar]
- Bradshaw, B.; Luque-Corredera, C.; Bonjoch, J. A gram-scale route to phlegmarine alkaloids: Rapid total synthesis of (−)-cermizine B. Chem. Commun. 2014, 50, 7099–7102. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, B.; Luque-Corredera, C.; Bonjoch, J. cis-Decahydroquinolines via asymmetric organocatalysis: Application to the total synthesis of lycoposerramine Z. Org. Lett. 2012, 15, 326–329. [Google Scholar] [CrossRef] [PubMed]
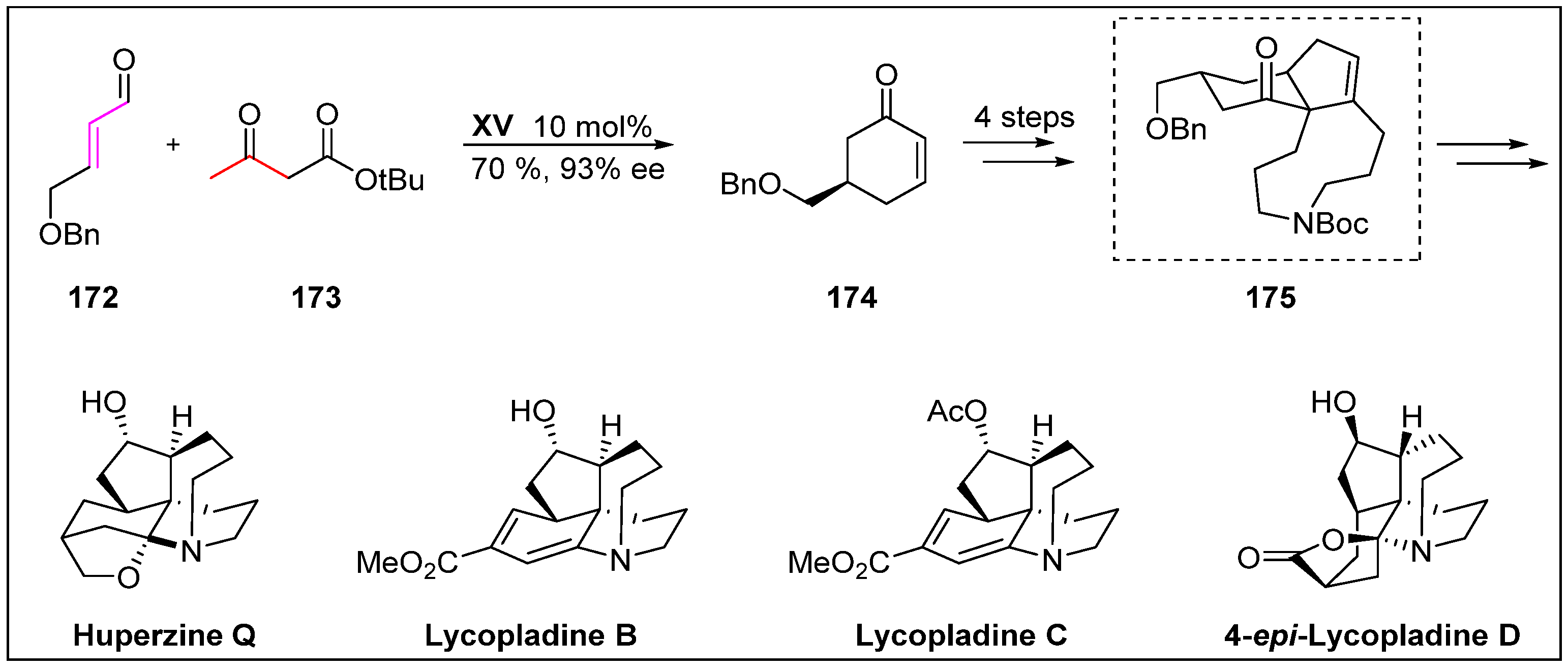
- Hong, B.; Hu, D.; Wu, J.; Zhang, J.; Li, H.; Pan, Y.; Lei, X. Divergent total syntheses of (−)-huperzine Q,(+)-lycopladine B,(+)-lycopladine C, and (−)-4-epi-lycopladine D. Chem. Asian. J. 2017, 12, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
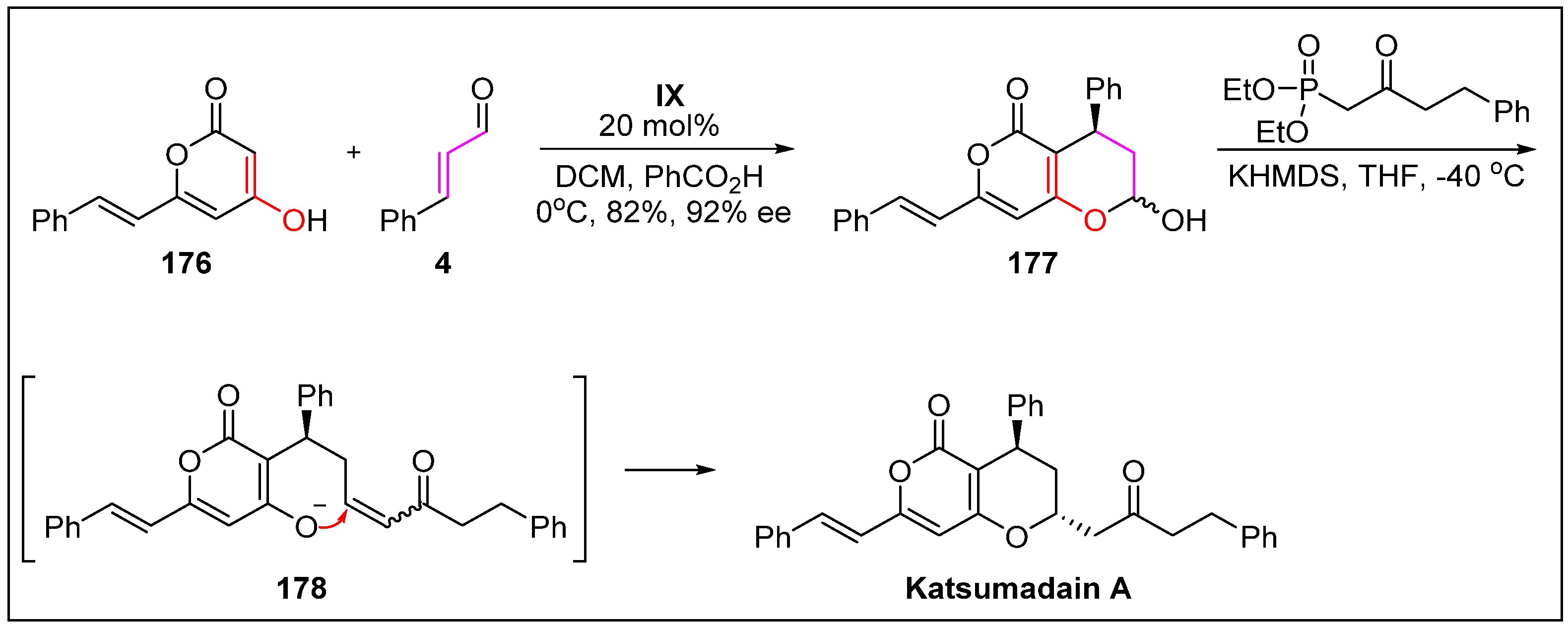
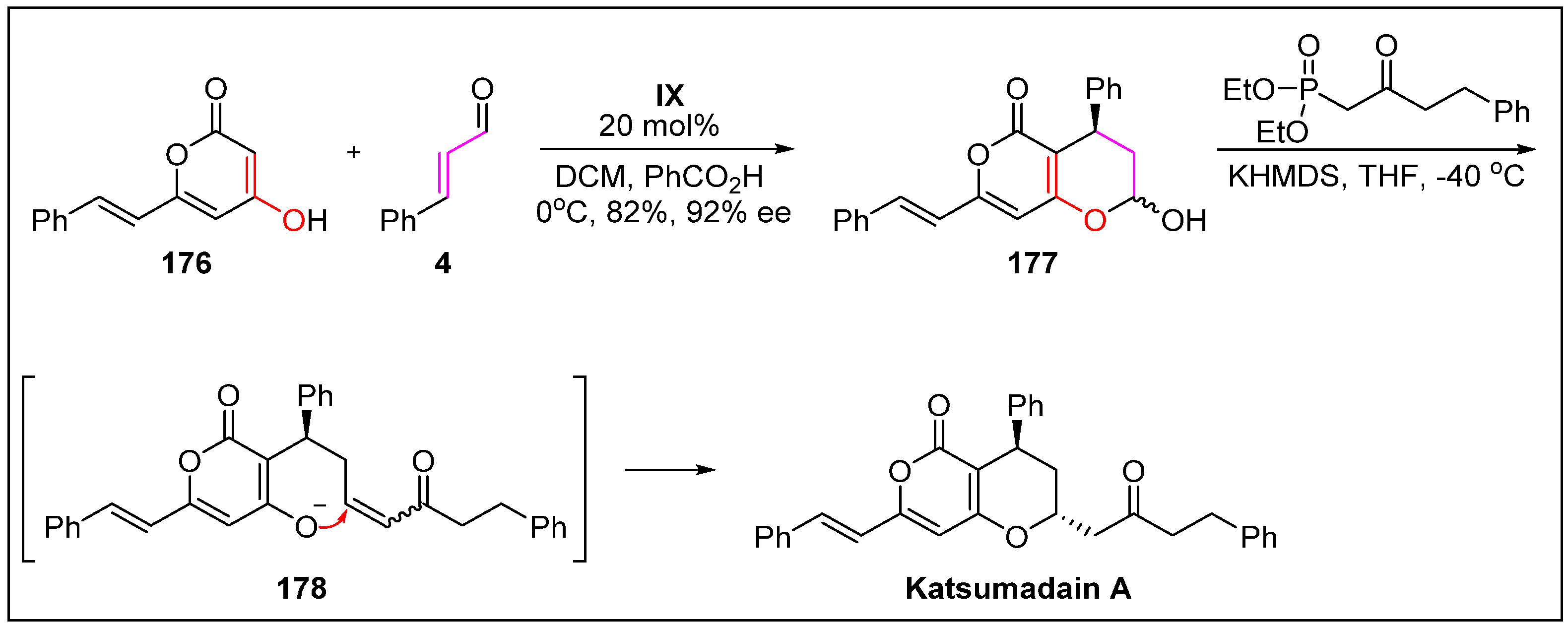
- Wang, Y.; Bao, R.; Huang, S.; Tang, Y. Bioinspired total synthesis of katsumadain A by organocatalytic enantioselective 1, 4-conjugate addition. Beil. J. Org. Chem. 2013, 9, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kinoshita, K.; Koyama, K.; Takahashi, K.; Tai, T.; Nunoura, Y.; Watanabe, K. Two novel anti-emetic principles of Alpinia katsumadai. J. Nat. Prod. 1999, 62, 1672–1674. [Google Scholar] [CrossRef] [PubMed]
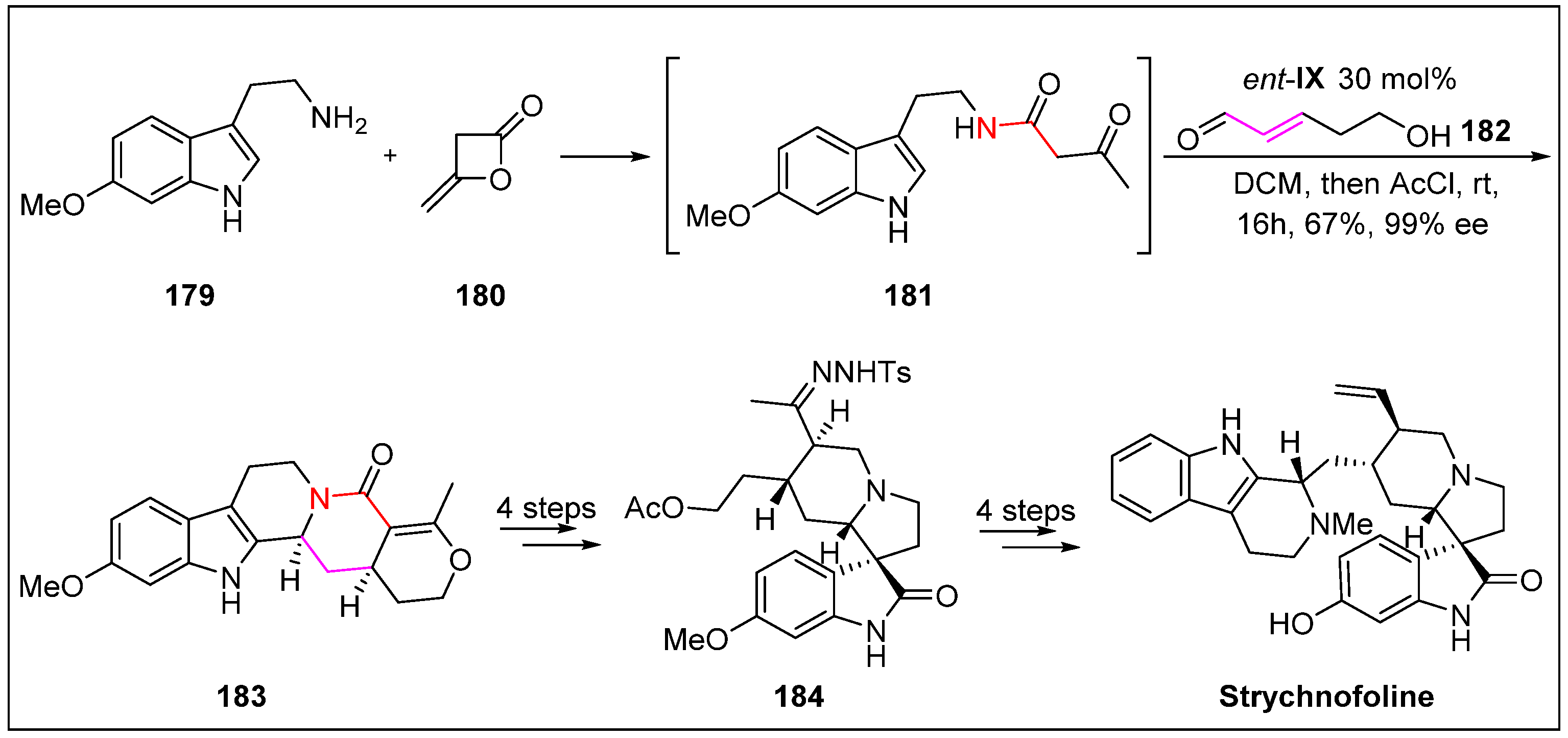
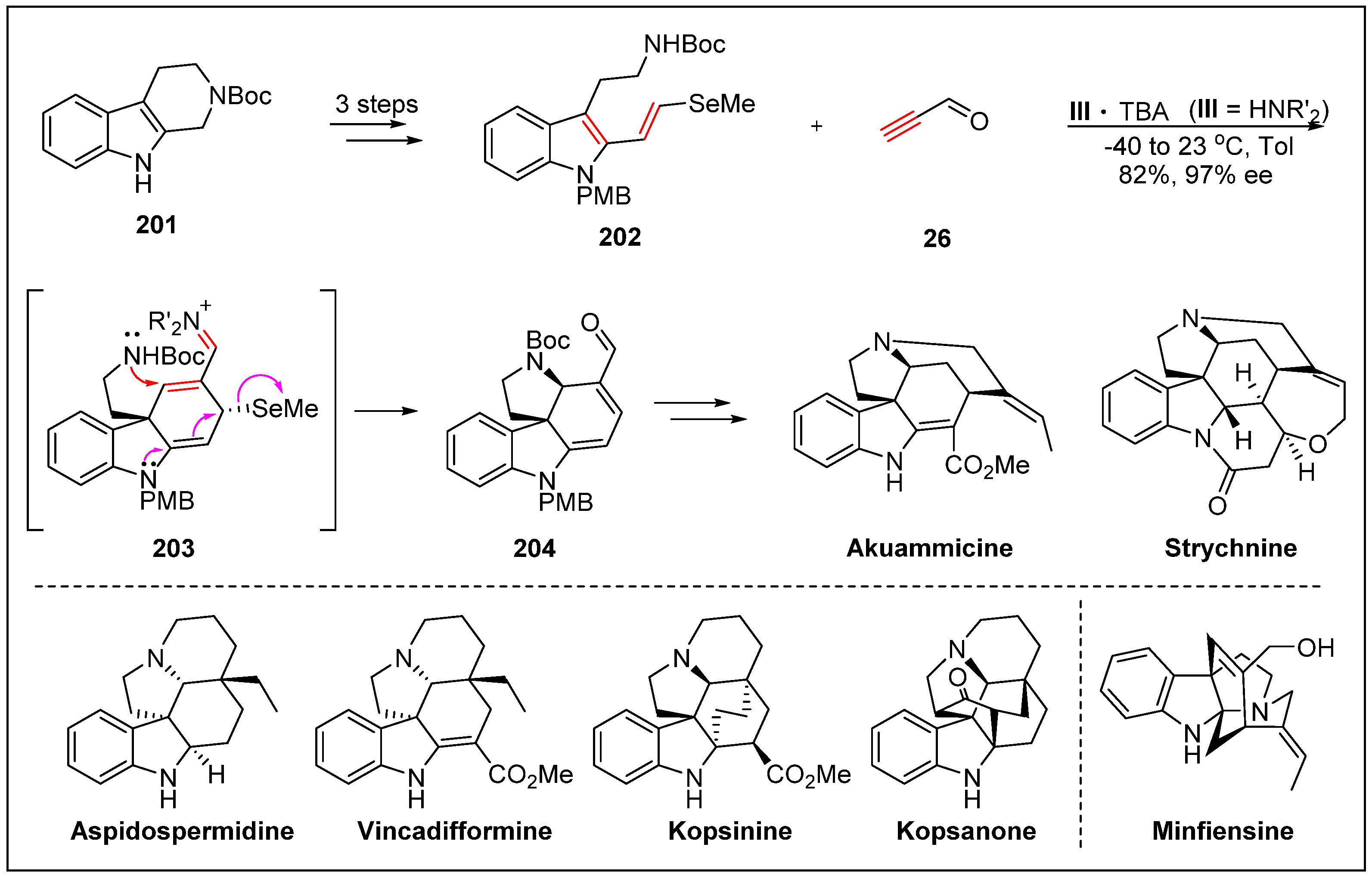
- Yu, Q.; Guo, P.; Jian, J.; Chen, Y.; Xu, J. Nine-step total synthesis of (−)-Strychnofoline. Chem. Commun. 2018, 54, 1125–1128. [Google Scholar] [CrossRef] [PubMed]
- Bassleer, R.; Depauw-Gillet, M.-C.; Massart, B.; Marnette, J.; Wiliquet, P.; Caprasse, M.; Angenot, L. Effects of three alkaloids isolated from Strychnos usambarensis on cancer cells in culture (author’s transl). Planta Med. 1982, 45, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Angenot, L. Nouveaux alcaloïdes oxindoliques du Strychnos usambarensis GILG. Plantes Med. Et Phytother. 1978, 12, 123–129. [Google Scholar]
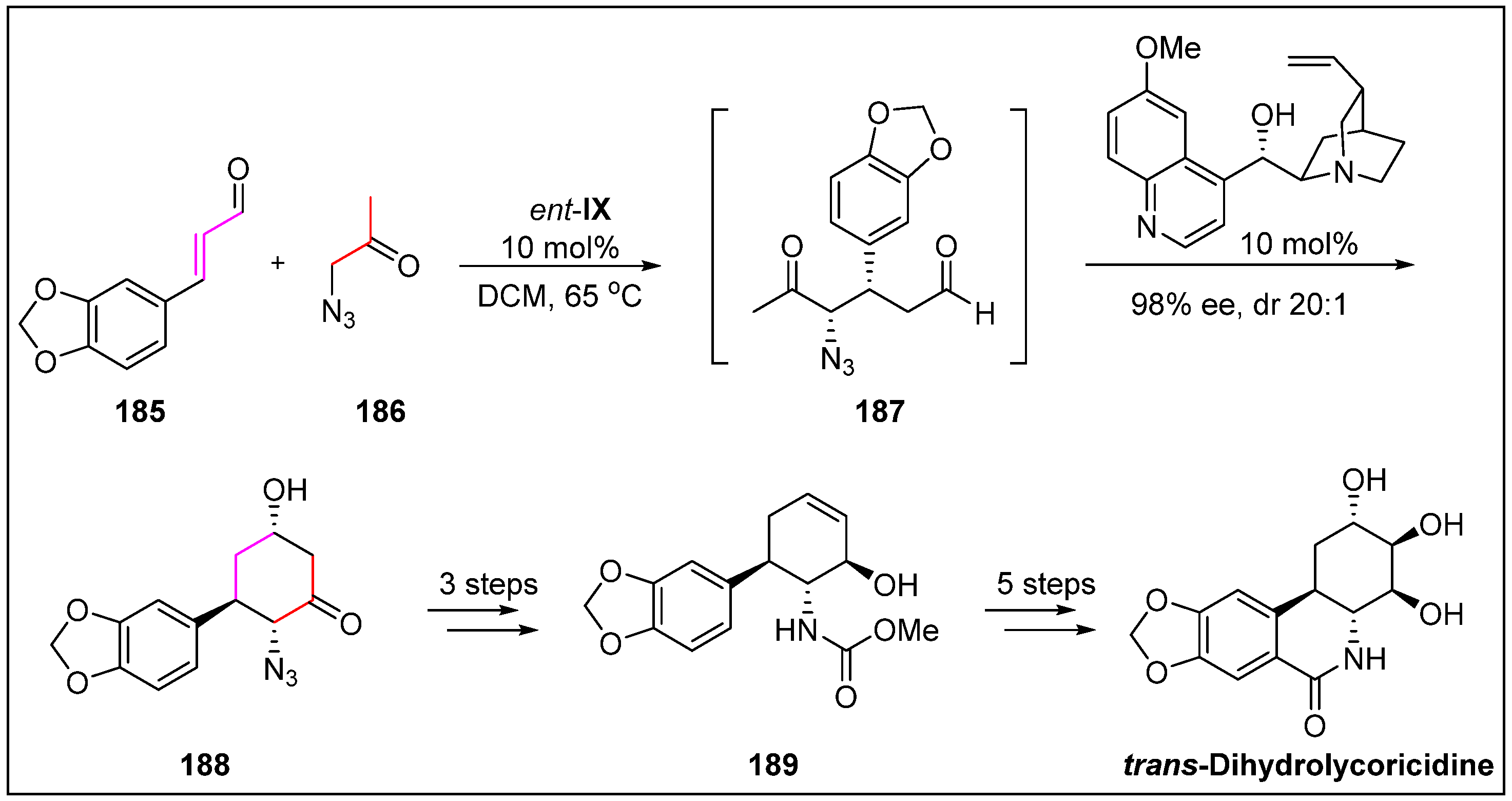
- McNulty, J.; Zepeda-Velázquez, C. Enantioselective organocatalytic Michael/aldol sequence: Anticancer natural product (+)-trans-dihydrolycoricidine. Angew. Chem. Int. Ed. 2014, 53, 8450–8454. [Google Scholar] [CrossRef]
- Pettit, G.R.; Pettit III, G.R.; Backhaus, R.A.; Boyd, M.R.; Meerow, A.W. Antineoplastic agents, 256. Cell growth inhibitory isocarbostyrils from Hymenocallis. J. Nat. Prod. 1993, 56, 1682–1687. [Google Scholar] [CrossRef]
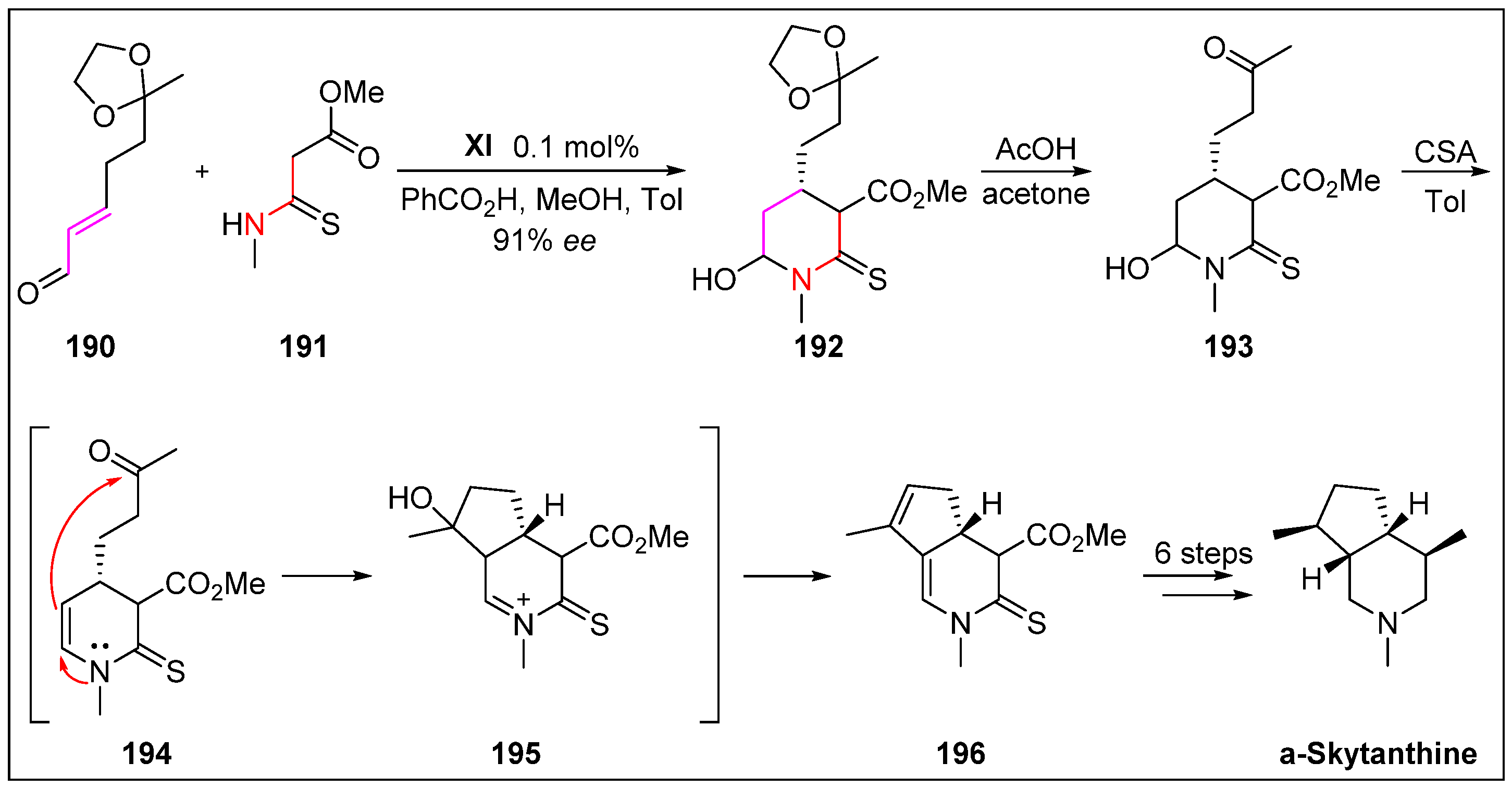
- Shiomi, S.; Sugahara, E.; Ishikawa, H. Efficient organocatalytic construction of C4-alkyl substituted piperidines and their application to the synthesis of (+)-α-skytanthine. Chem. Eur. J. 2015, 21, 14758–14763. [Google Scholar] [CrossRef]
- Djerassi, C.; Kutney, J.; Shamma, M. Alkaloid Studies-XXXII: Studies on skytanthus acutus meyen. The structure of the monoterpenoid alkaloid skytanthine. Tetrahedron 1962, 18, 183–188. [Google Scholar] [CrossRef]
- Gatti, G.; Marotta, M. Preliminary research on the pharmacologic and psychopharmacologic effect of skytanthine, an alkaloid of Skytanthus acutus Meyen. Ann. Dell’istituto Super. Di Sanita 1966, 2, 29. [Google Scholar]
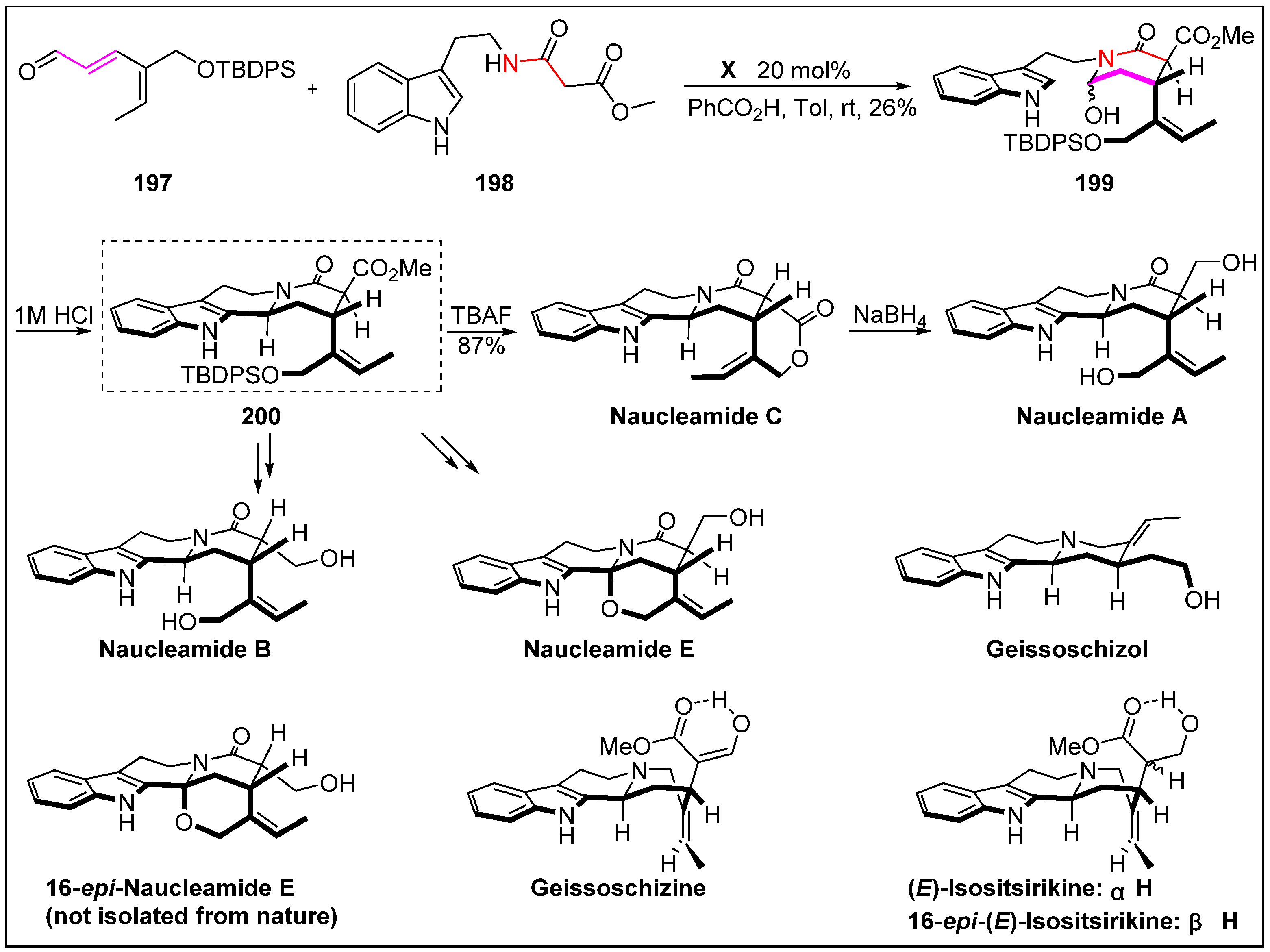
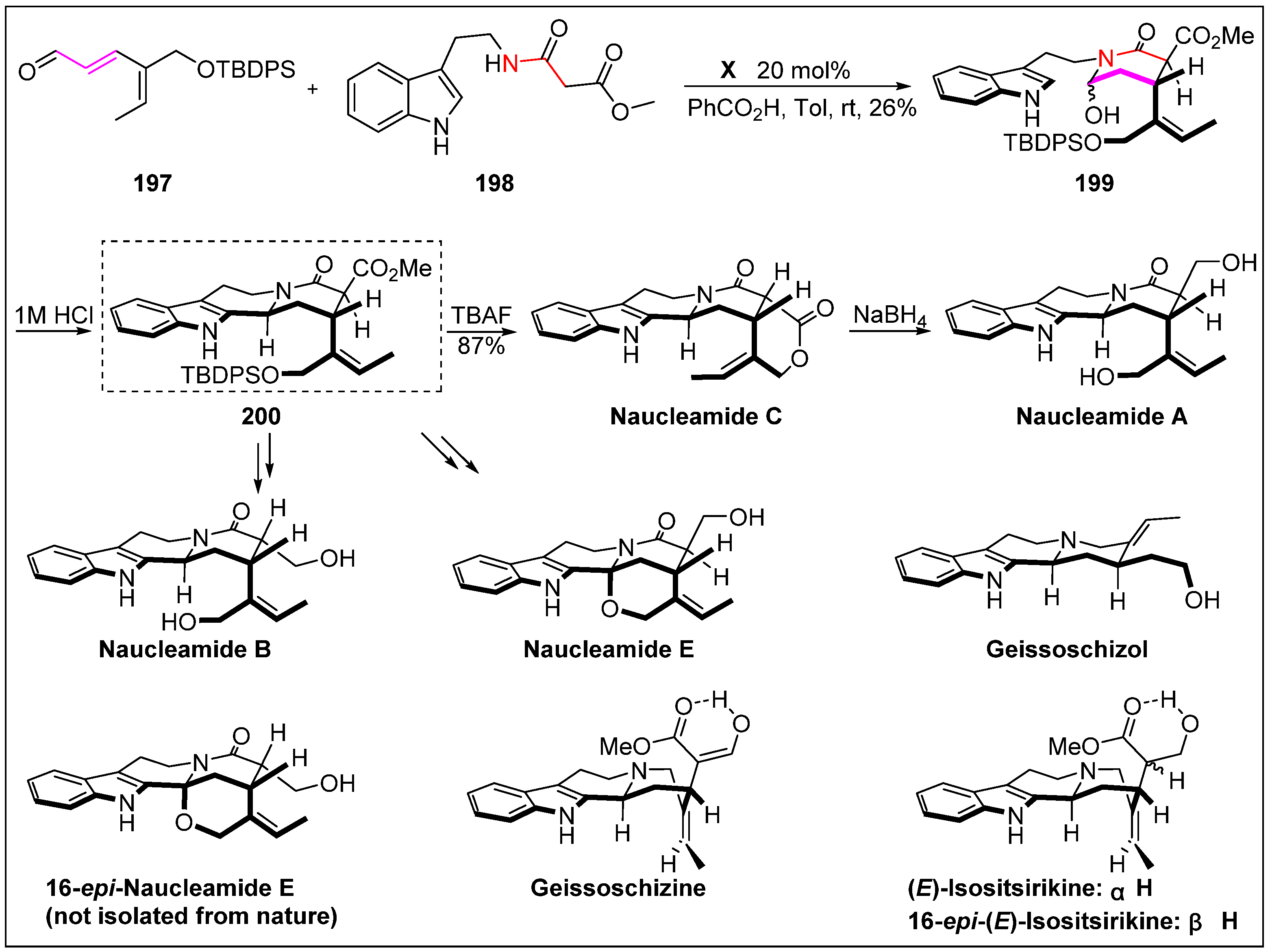
- Li, L.; Aibibula, P.; Jia, Q.; Jia, Y. Total syntheses of naucleamides A-C and E, geissoschizine, geissoschizol, (E)-isositsirikine, and 16-epi-(E)-isositsirikine. Org. Lett. 2017, 19, 2642–2645. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, H.; Xu, P.-F. Asymmetric catalytic cascade reactions for constructing diverse scaffolds and complex molecules. Acc. Chem. Res. 2015, 48, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
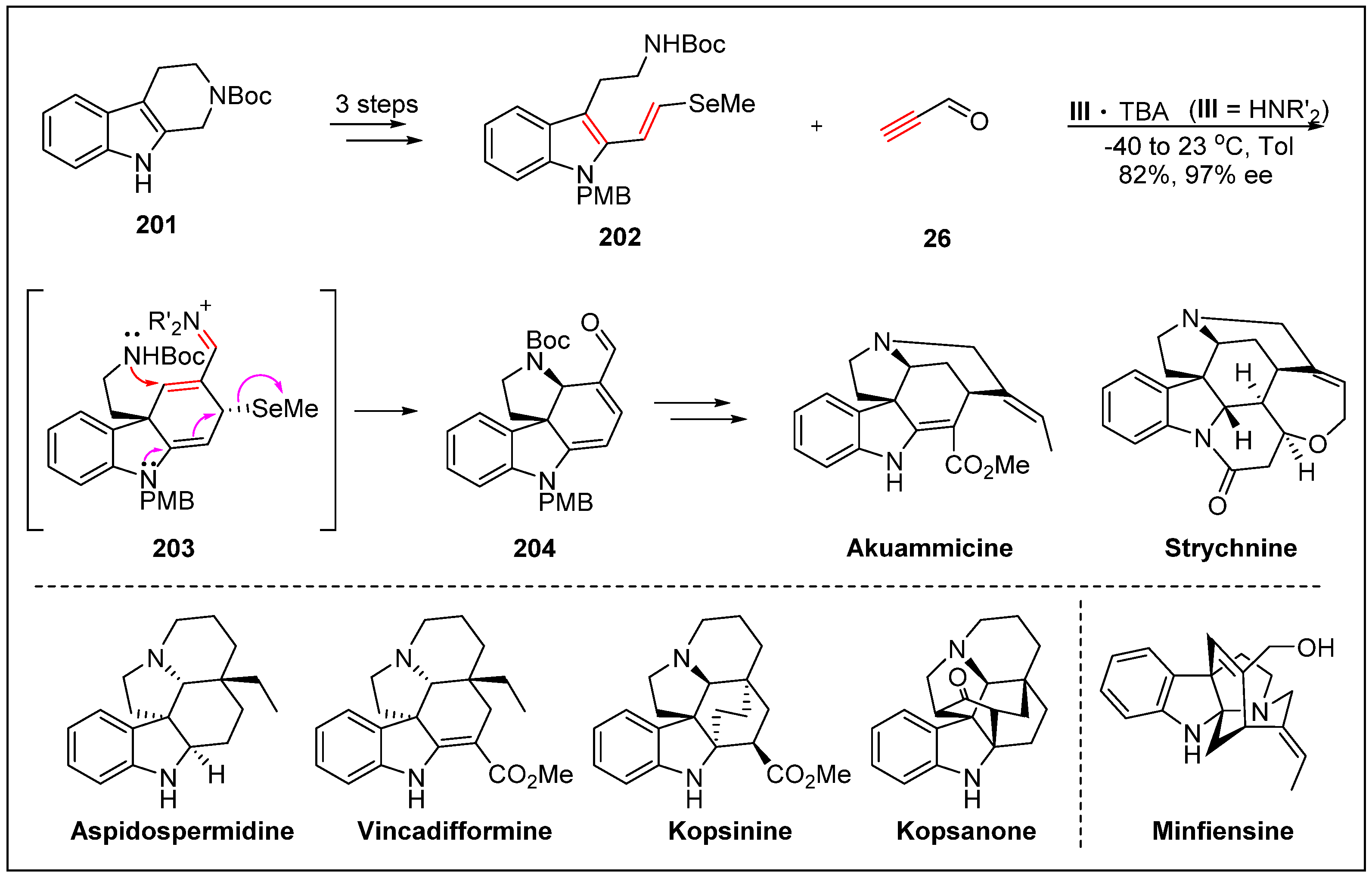
- Jones, S.B.; Simmons, B.; Mastracchio, A.; MacMillan, D.W. Collective synthesis of natural products by means of organocascade catalysis. Nature 2011, 475, 183. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.B.; Simmons, B.; MacMillan, D.W. Nine-step enantioselective total synthesis of (+)-minfiensine. J. Am. Chem. Soc. 2009, 131, 13606–13607. [Google Scholar] [CrossRef] [PubMed]
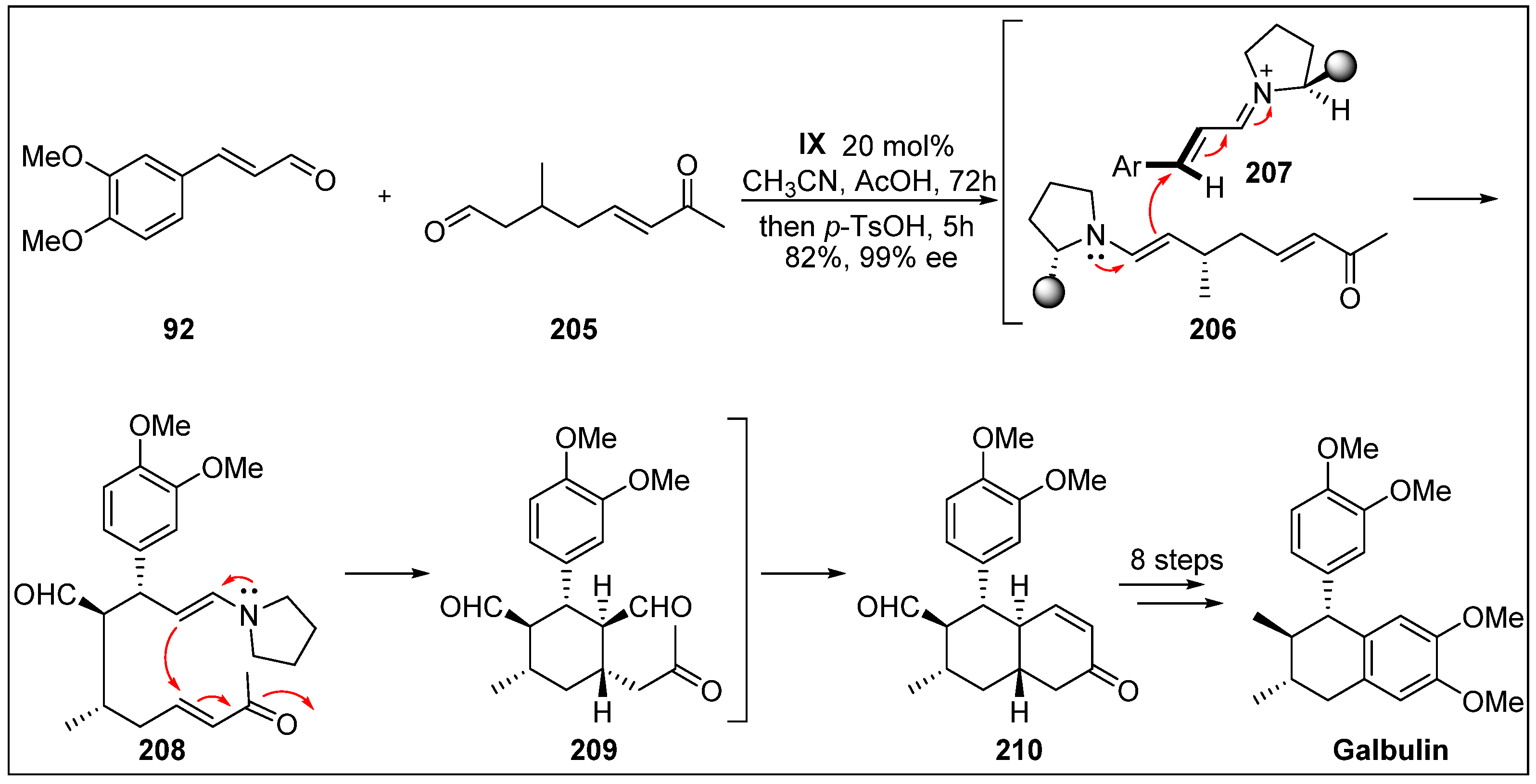
- Hong, B.-C.; Hsu, C.-S.; Lee, G.-H. Enantioselective total synthesis of (+)-galbulin via organocatalytic domino Michael–Michael–aldol condensation. Chem. Commun. 2012, 48, 2385–2387. [Google Scholar] [CrossRef] [PubMed]
- Hughes, G.; Ritchie, E. The chemical constituents of Himantandra species. I. The Lignins of Himantandra baccata Bail. and H. belgraveana F. Muell. Aust. J.Chem. 1954, 7, 104–112. [Google Scholar] [CrossRef]
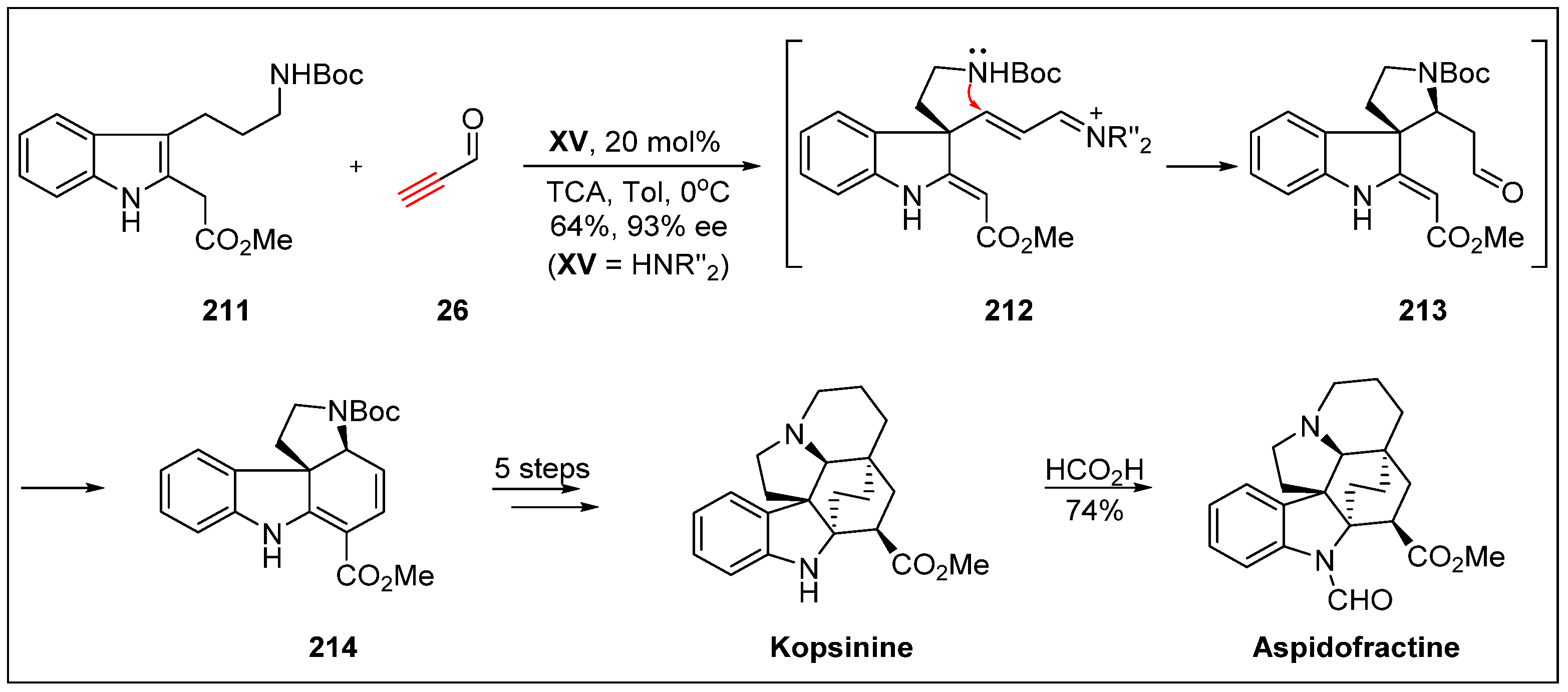
- Wu, X.; Huang, J.; Guo, B.; Zhao, L.; Liu, Y.; Chen, J.; Cao, W. Enantioselective Michael/aza-Michael/cyclization organocascade to tetracyclic spiroindolines: Concise total synthesis of kopsinine and aspidofractine. Adv. Synth. Catal. 2014, 356, 3377–3382. [Google Scholar] [CrossRef]
- Varea, T.; Kan, C.; Remy, F.; Sevenet, T.; Quirion, J.; Husson, H.-P.; Hadi, H. Indole alkaloids from Kopsia teoi. J. Nat. Prod. 1993, 56, 2166–2169. [Google Scholar] [CrossRef]
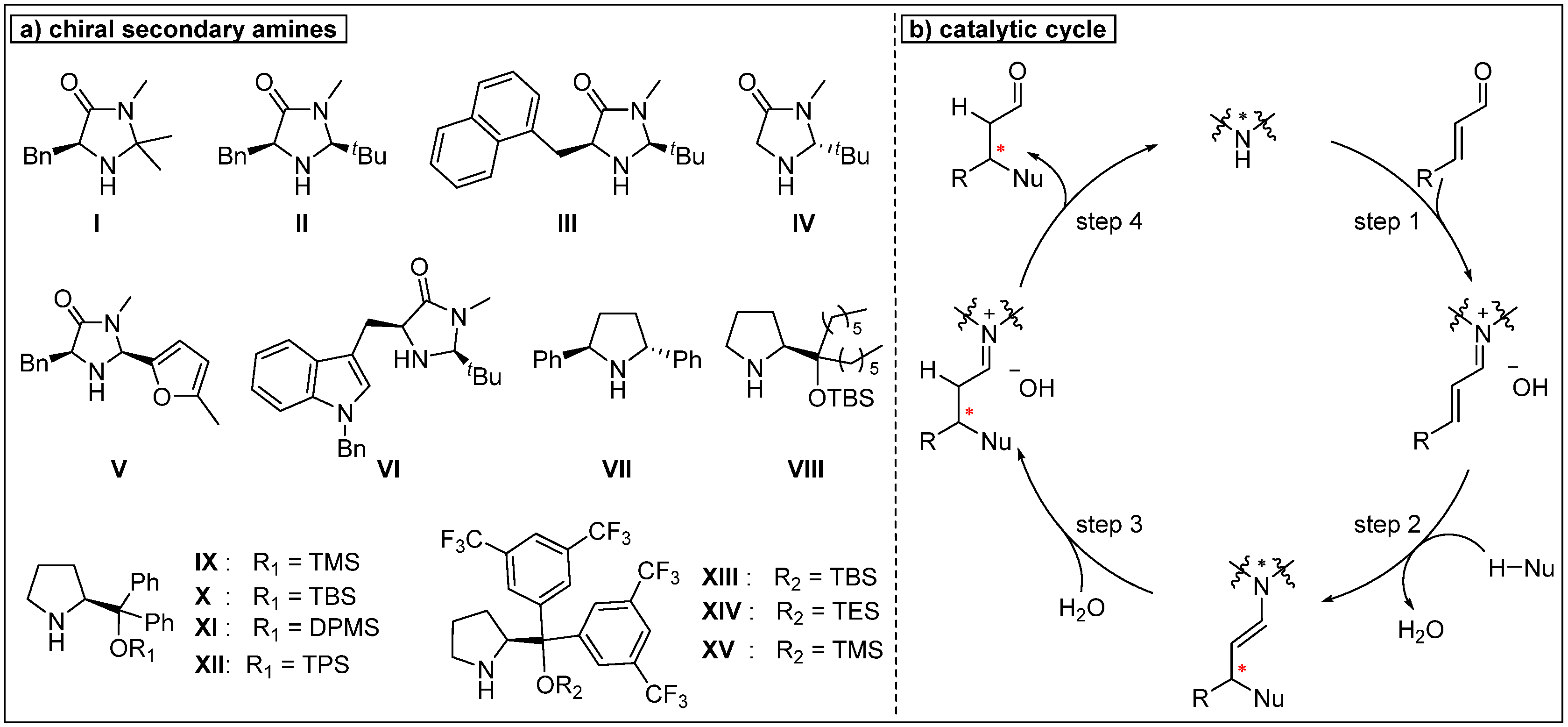













© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z. Advances in the Asymmetric Total Synthesis of Natural Products Using Chiral Secondary Amine Catalyzed Reactions of α,β-Unsaturated Aldehydes. Molecules 2019, 24, 3412. https://doi.org/10.3390/molecules24183412
Wang Z. Advances in the Asymmetric Total Synthesis of Natural Products Using Chiral Secondary Amine Catalyzed Reactions of α,β-Unsaturated Aldehydes. Molecules. 2019; 24(18):3412. https://doi.org/10.3390/molecules24183412
Chicago/Turabian StyleWang, Zhonglei. 2019. "Advances in the Asymmetric Total Synthesis of Natural Products Using Chiral Secondary Amine Catalyzed Reactions of α,β-Unsaturated Aldehydes" Molecules 24, no. 18: 3412. https://doi.org/10.3390/molecules24183412
APA StyleWang, Z. (2019). Advances in the Asymmetric Total Synthesis of Natural Products Using Chiral Secondary Amine Catalyzed Reactions of α,β-Unsaturated Aldehydes. Molecules, 24(18), 3412. https://doi.org/10.3390/molecules24183412