
Michaelis-Arbuzov-Type Reaction of 1-Imidoalkyltriarylphosphonium Salts with Selected Phosphorus Nucleophiles

 , and
, and
Abstract
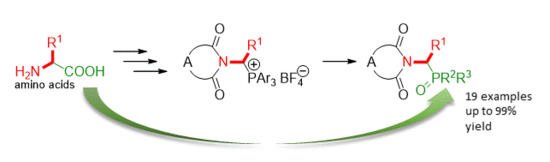
:
1. Introduction
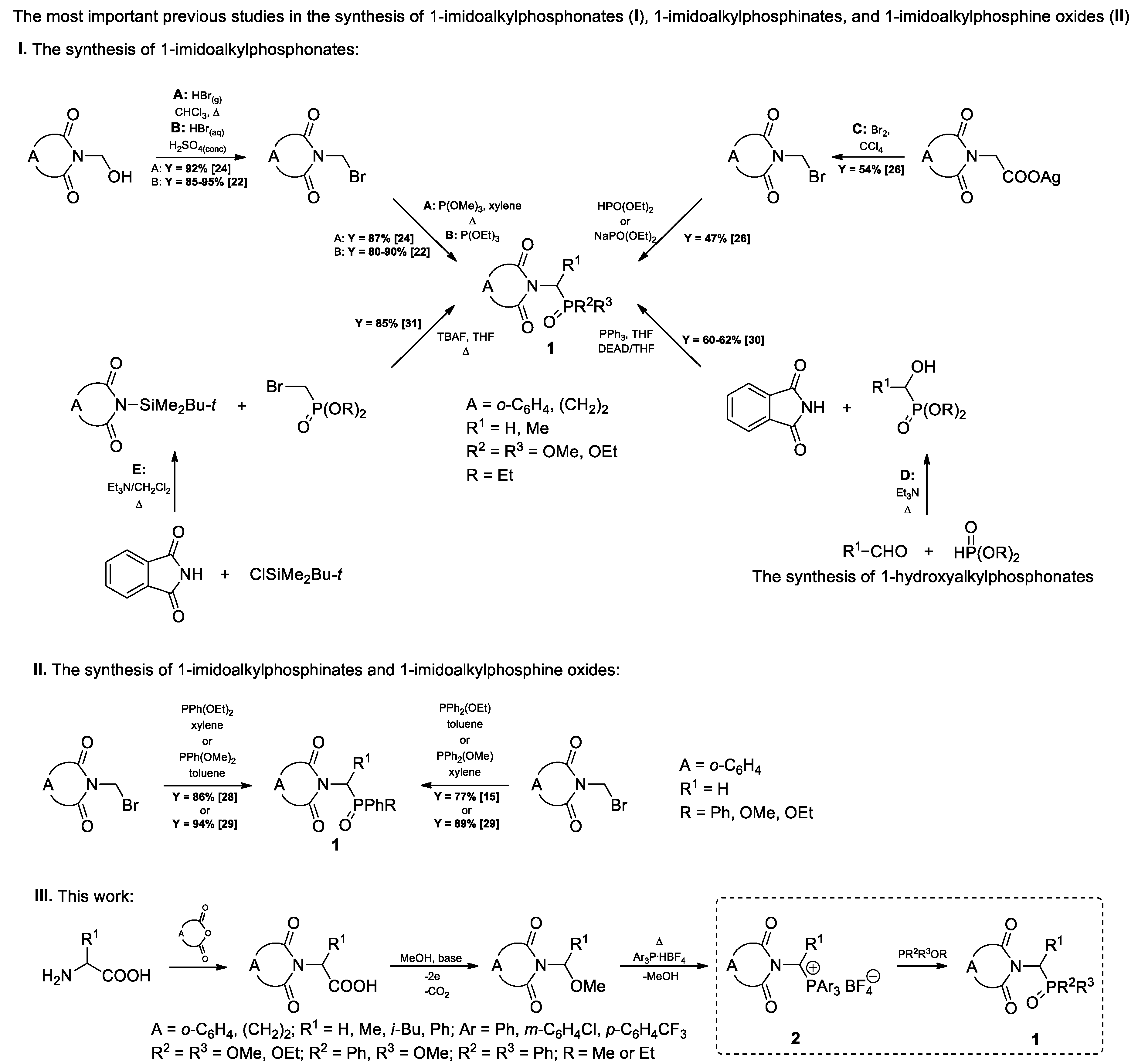
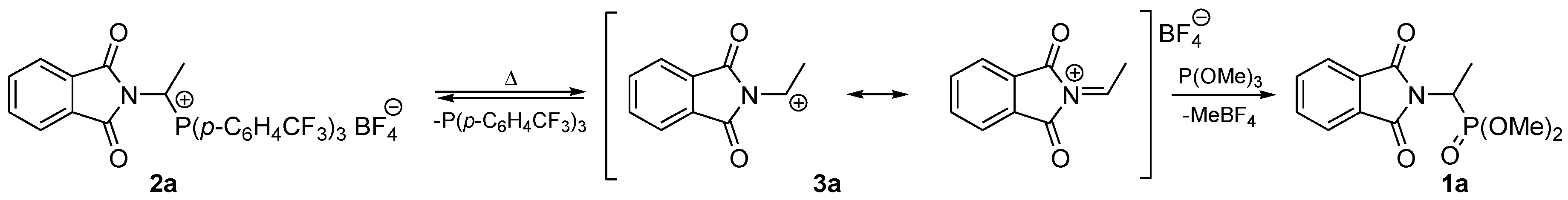
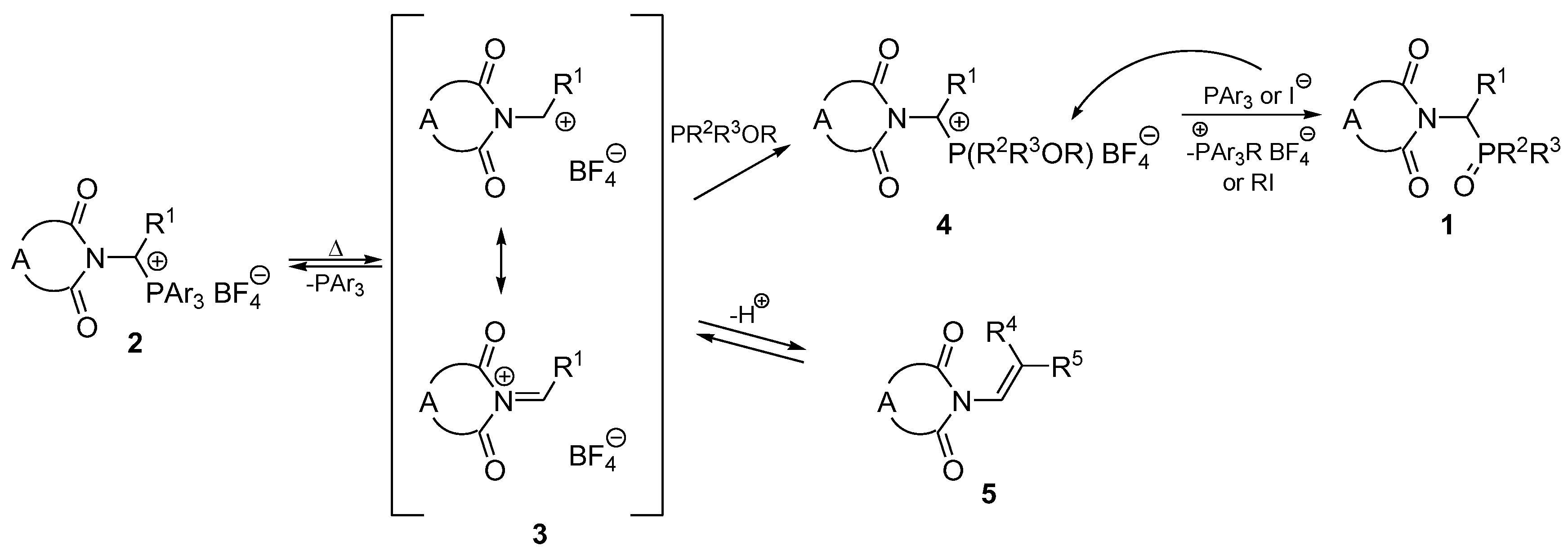
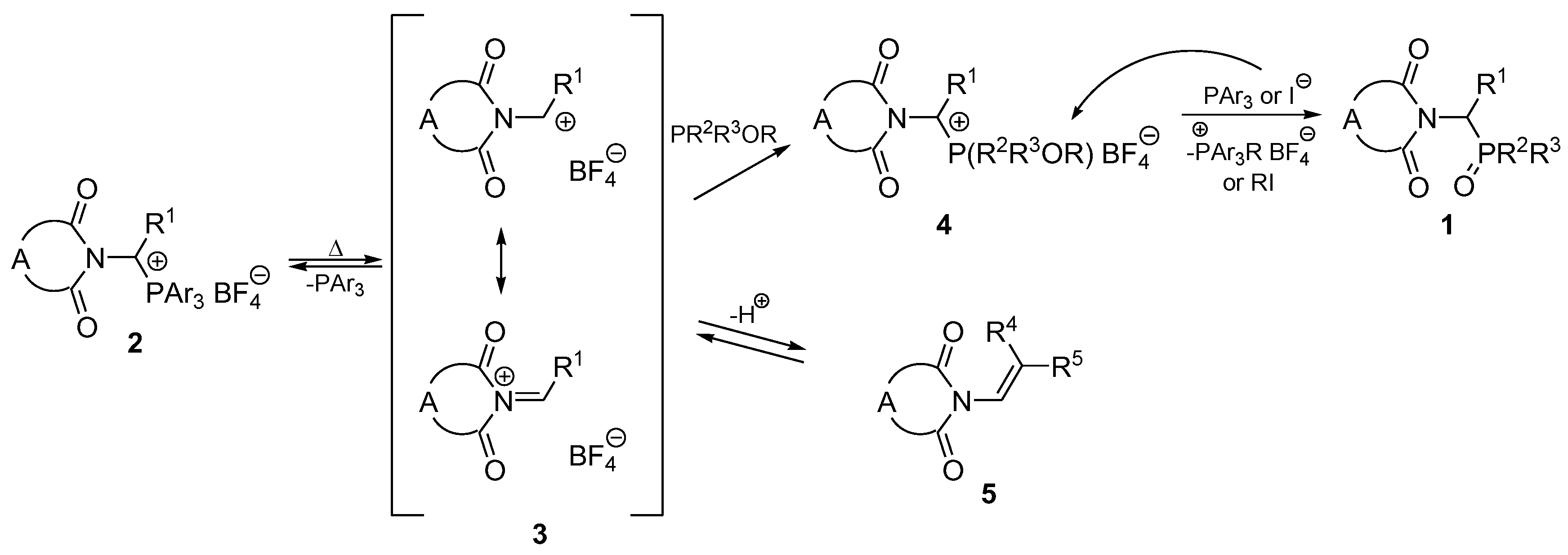
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. Synthesis
3.2.1. Substrate Synthesis
3.2.2. General Procedure for the Reaction of 1-Imidoalkyltriarylphosphonium Salts 2 with Phosphorus Nucleophiles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kukhar, V.; Hudson, H. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2000. [Google Scholar]
- Kafarski, P.; Lejczak, B. Aminophosphonic Acids of Potential Medical Importance. Curr. Med. Chem. Anti Cancer Agents 2001, 1, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, M.; Rojas-Cabrera, H.; Cativiela, C. An overview of stereoselective synthesis of α-aminophosphonic acids and derivatives. Tetrahedron 2009, 65, 17–49. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Sello, G.; Sisti, M. Aminophosphonic acids and derivatives. Synthesis and biological applications. Curr. Med. Chem. 2010, 17, 264–289. [Google Scholar] [CrossRef] [PubMed]
- Kudzin, Z.H.; Kudzin, M.H.; Drabowicz, J.; Stevens, C.V. Aminophosphonic acids-phosphorus analogues of natural Amino Acids. Part 1: Syntheses of α-aminophosphonic acids. Curr. Org. Chem. 2011, 15, 2015–2071. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable Potential of the α-aminophosphonate/phosphinate structural motif in medicinal chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef]
- Keglevich, G.; Bálint, E. The Kabachnik–Fields Reaction: Mechanism and Synthetic Use. Molecules 2012, 17, 12821–12835. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, M.; Viveros-Ceballos, J.L.; Cativiela, C.; Sayago, F.J. An update on the stereoselective synthesis of α-aminophosphonic acids and derivatives. Tetrahedron 2015, 71, 1745–1784. [Google Scholar] [CrossRef]
- Lewkowski, J.; Rodriguez Moya, M.; Chmielak, M.; Rogacz, D.; Lewicka, K.; Rychter, P. Synthesis, Spectral Characterization of Several Novel Pyrene-Derived Aminophosphonates and Their Ecotoxicological Evaluation Using Heterocypris incongruens and Vibrio fisheri Tests. Molecules 2016, 21, 936. [Google Scholar] [CrossRef]
- Cypryk, M.; Drabowicz, J.; Gostynski, B.; Kudzin, M.H.; Kudzin, Z.H.; Urbaniak, P. 1-(Acylamino)alkylphosphonic Acids—Alkaline Deacylation. Molecules 2018, 23, 859. [Google Scholar] [CrossRef]
- Huang, J.; Chen, R. An Overview of Recent Advances on the Synthesis and Biological Activity of α-Aminophosphonic Acid Derivatives. Heteroat. Chem. 2000, 11, 480–492. [Google Scholar] [CrossRef]
- Lavielle, G.; Hautefaye, P.; Schaeffer, C.; Boutin, J.A.; Cudennec, C.A.; Pierre, A. New alpha-amino phosphonic acid derivatives of vinblastine: Chemistry and antitumor activity. J. Med. Chem. 1991, 34, 1998–2003. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.-X.; Li, K.; Shi, D.-Q. A Convenient Synthesis and Herbicidal Activity of N-phosphonoalkylpyrazolo[4,3-e][1,2,4]-triazolo[1,5-d]pyrimidines. Phosphorus Sulfur Silicon Relat. Elem. 2008, 183, 3156–3165. [Google Scholar] [CrossRef]
- Malachowski, W.P.; Coward, J.K. The Chemistry of Phosphapeptides: Formation of Functionalized Phosphonochloridates under Mild Conditions and Their Reaction with Alcohols and Amines. J. Org. Chem. 1994, 59, 7616–7624. [Google Scholar] [CrossRef]
- Pradhan, B.S.; Dhokiya, K.V. Pyrrole Derivatives. U.K. Patent GB2479830, 26 October 2011. [Google Scholar]
- Fathi, R.; Huang, Q.; Syi, J.-L.; Delaney, W.; Cook, A.F. (Aminomethyl)phosphonate derivatives of oligonucleotides. Bioconjug. Chem. 1994, 5, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Nahrwold, M.; Bogner, T.; Eissler, S.; Verma, S.; Sewald, N. “Clicktophycin-52”: A Bioactive Cryptophycin-52 Triazole Analogue. Org. Lett. 2010, 12, 1064–1067. [Google Scholar] [CrossRef] [PubMed]
- Plenat, F.; Cassagne, M.; Cristau, J.H. Synthesis of new phosphorus 2,4,5-imidazolidinetriones. Tetrahedron 1995, 51, 9551–9558. [Google Scholar] [CrossRef]
- Kálmán, F.K.; Woods, M.; Caravan, P.; Jurek, P.; Spiller, M.; Tircsó, G.; Király, R.; Brülcher, E.; Sherry, A.D. Potentiometric and Relaxometric Properties of a Gadolinium-Based MRI Contrast Agent for Sensing Tissue pH. Inorg. Chem. 2007, 46, 5260–5270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leygue, N.; De Heredia, A.P.; Galaup, C.; Benoist, E.; Lamarque, L.; Picard, C. New advances in the synthesis of tripyridinophane macrocycles suitable to enhance the luminescence of Ln(III) ions in aqueous solution. Tetrahedron 2018, 74, 4272–4287. [Google Scholar] [CrossRef]
- Abdou, W.M.; Khidre, R.E. Efficient synthesis routes for various phthalimido phosphor esters as antimicrobial agents in terms of structure–activity relationship. Monatsh. Chem. 2010, 141, 214–228. [Google Scholar] [CrossRef]
- Davidsen, S.K.; Phllips, G.W.; Martin, S.F. Geminal Acylation-Alkylation at a Carbonyl Center Using Diethyl N-Benzylideneaminomethylphosphonate: 2-Methyl-2-Phenyl-4-Pentenal. Org. Synth. 1987, 65, 119–134. [Google Scholar] [CrossRef]
- Hirschmann, R.; Yager, K.M.; Taylor, C.M.; Witherington, J.; Sprengeler, P.A.; Phillips, B.W.; Moore, W.; Smith, A.B. Phosphonate Diester and Phosphonamide Synthesis. Reaction Coordinate Analysis by 31P NMR Spectroscopy: Identification of Pyrophosphonate Anhydrides and Highly Reactive Phosphonylammonium Salts. J. Am. Chem. Soc. 1997, 119, 8177–8190. [Google Scholar] [CrossRef]
- Seyferth, D.; Marmor, R.S.; Hilbert, P. Reactions of dimethylphosphono-substituted diazoalkanes. (MeO)2P(O)CR transfer to olefins and 1,3-dipolar additions of (MeO)2P(O)C(N2)R. J. Org. Chem. 1971, 36, 1379–1386. [Google Scholar] [CrossRef]
- Yamauchi, K.; Kinoshita, M.; Imoto, M. Peptides Containing Aminophosphonic Acids. II. The Synthesis of Tripeptide Analogs. Bull. Chem. Soc. Jpn. 1972, 45, 2531–2534. [Google Scholar] [CrossRef] [Green Version]
- Ösapay, G.; Szilagyi, I.; Seres, J. Conversion of amino acids and dipeptides into their phosphonic analogs: Aminoalkylphosphonic acids and peptides II. Tetrahedron 1987, 43, 2977–2983. [Google Scholar] [CrossRef]
- Elliott, R.L.; Marks, N.; Berg, M.J.; Portoghese, P.S. Synthesis and Biological Evaluation of Phosphonamidate Peptide Inhibitors of Enkephalinase and Angiotensin-Converting Enzyme. J. Med. Chem. 1985, 28, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Popoff, I.C.; Peter, B.B.; Huber, L.K. Aminoalkylphosphinic Acids. U.S. Patent 3,332,987, 25 July 1967. [Google Scholar]
- Popoff, I.C.; Huber, L.K.; Block, B.P.; Morton, P.D.; Riordan, R.P. α-Aminophosphinic Acids and α-Aminophosphine Oxides. I. Alkyl-α-aminoalkylphosphinic Acids, α-Aminoalkyl(aryl)phosphinic Acids, and α-Aminoalkyl(diaryl)phosphine Oxides. J. Org. Chem. 1963, 28, 2898–2900. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Guarneri, M.; Moroder, F.; Pollini, G.P.; Simoni, D. Synthesis of 1-Phthalimidoalkanephosphonates. Synthesis 1982, 653–655. [Google Scholar] [CrossRef]
- Chun, Y.-J.; Park, J.-N.; Oh, G.-M.; Hong, S.-I.; Kim, Y.-J. Synthesis of ω-Phthalimidoalkylphosphonates. Synthesis 1994, 909–910. [Google Scholar] [CrossRef]
- Adamek, J.; Mazurkiewicz, R.; Węgrzyk, A.; Erfurt, K. 1-Imidoalkylphosphonium salts with modulated Cα-P+ bond strength: Synthesis and application as new active α-imidoalkylating agents. Beilstein J. Org. Chem. 2017, 13, 1446–1455. [Google Scholar] [CrossRef]
- Adamek, J.; Węgrzyk, A. Catalyst-free Friedel-Crafts reaction of 1-(N-acylamino)- and 1-imidoalkyltriarylphosphonium salts with arenes. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 351–352. [Google Scholar] [CrossRef]
- Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Mazurkiewicz, R. Comparative Studies on the Amidoalkylating Properties of N-(1-Methoxyalkyl)Amides and 1-(N-Acylamino)Alkyltriphenylphosphonium Salts in the Michaelis–Arbuzov-Like Reaction: A New One-Pot Transformation of N-(1-Methoxyalkyl)Amides into Phosphonic or Phosphinic Analogs of N-Acyl-α-Amino Acids. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 967–980. [Google Scholar] [CrossRef]
- Adamek, J.; Węgrzyk, A.; Kończewicz, J.; Walczak, K.; Erfurt, K. 1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications. Molecules 2018, 23, 2453. [Google Scholar] [CrossRef] [PubMed]
- Październiok-Holewa, A.; Adamek, J.; Mazurkiewicz, R.; Zielińska, K. Amidoalkylating Properties of 1-(N-Acylamino)Alkyltriphenylphosphonium Salts. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 205–212. [Google Scholar] [CrossRef]
- Hoffmann, M.; Wasielewski, C. Amino phosphonic acids. Part VI. Diazomethane as a reagent in the synthesis of N-acylated 1-aminophosphonic acid esters. Roczniki Chem. 1976, 50, 139–146. [Google Scholar] [CrossRef]
- Yamauchi, K.; Kinoshita, M.; Imoto, M. Peptides Containing Aminophosphonic Acids. I. Reactivity of α-Aminobenzylphosphonic Acid. Bull. Chem. Soc. Jpn. 1972, 45, 2528–2531. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | 2 | A | R1 | Ar | PN a (PR2R3OR) | Solvent | Molar Ratio of 2:PN:Catalyst | T, °C | Time, h | 1 | Yield, % |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2a | o-C6H4 | Me | p-C6H4CF3 | P(OMe)3 | CHCl3 | 1:1.5:0.25 | 100 | 2 | 1a | 36 b |
| 2 | 2a | o-C6H4 | Me | p-C6H4CF3 | P(OMe)3 | CHCl3 | 1:5:0.25 | 100 | 2 | 1a | 70 b |
| 3 | 2a | o-C6H4 | Me | p-C6H4CF3 | P(OMe)3 | CHCl3 | 1:10:0.25 | 100 | 2 | 1a | 85 b |
| 4 | 2a | o-C6H4 | Me | p-C6H4CF3 | P(OMe)3 | CH3CN | 1:10:0.25 | 100 | 2 | 1a | 50 b |
| 5 | 2a | o-C6H4 | Me | p-C6H4CF3 | P(OMe)3 | C6H5Cl | 1:10:0.25 | 100 | 2 | 1a | 51 b |
| 6 | 2a | o-C6H4 | Me | p-C6H4CF3 | P(OMe)3 | CHCl3 | 1:10:0.25 | 100 | 2 | 1a | 76 c |
| 7 | 2a | o-C6H4 | Me | p-C6H4CF3 | P(OMe)3 | CHCl3 | 1:10:- | 100 | 2 | 1a | 52 c |
| 8 | 2b | o-C6H4 | Me | m-C6H4Cl | P(OMe)3 | CHCl3 | 1:10:0.25 | 120 | 2 | 1a | 70 c |
| 9 | 2c | o-C6H4 | Me | Ph | P(OMe)3 | CHCl3 | 1:10:0.25 | 150 | 0.5 | 1a | 22 c,d |
| 10 | 2d | o-C6H4 | H | m-C6H4Cl | P(OMe)3 | CHCl3 | 1:10:0.25 | 170 | 2 | 1b | 95 c |
| 11 | 2a | o-C6H4 | Me | p-C6H4CF3 | P(OEt)3 | CHCl3 | 1:10:0.25 | 100 | 2 | 1c | 90 c |
| 12 | 2a | o-C6H4 | Me | p-C6H4CF3 | P(OEt)3 | CHCl3 | 1:10:- | 100 | 2 | 1c | 86 c |
| 13 | 2e | o-C6H4 | Ph | p-C6H4CF3 | P(OEt)3 | CHCl3 | 1:10:0.25 | 80 | 0.5 | 1d | 99 c |
| 14 | 2e | o-C6H4 | Ph | p-C6H4CF3 | P(OEt)3 | CHCl3 | 1:10:- | 80 | 0.5 | 1d | 91 c |
| 15 | 2f | o-C6H4 | i-Bu | m-C6H4Cl | P(OEt)3 | CHCl3 | 1:10:0.25 | 120 | 2 | 1e | 94 c |
| 16 | 2f | o-C6H4 | i-Bu | m-C6H4Cl | P(OEt)3 | CHCl3 | 1:10:- | 120 | 2 | 1e | 83 c |
| 17 | 2g | (CH2)2 | H | p-C6H4CF3 | P(OEt)3 | CH3CN | 1:10:0.25 | 180 | 2 | 1f | nr e |
| 18 | 2h | (CH2)2 | Me | p-C6H4CF3 | P(OEt)3 | CH3CN | 1:30:0.25 | 150 | 2 | 1g | 65 c |
| 19 | 2a | o-C6H4 | Me | p-C6H4CF3 | PhP(OMe)2 | CHCl3 | 1:10:0.25 | 100 | 0.5 | 1h | 59 c,f |
| 20 | 2a | o-C6H4 | Me | p-C6H4CF3 | PhP(OMe)2 | CHCl3 | 1:10:- | 100 | 0.5 | 1h | 37 c,g |
| 21 | 2e | o-C6H4 | Ph | p-C6H4CF3 | PhP(OMe)2 | CHCl3 | 1:10:0.25 | 80 | 0.5 | 1i | 94 c,h |
| 22 | 2a | o-C6H4 | Me | p-C6H4CF3 | Ph2P(OMe) | CHCl3 | 1:10:0.25 | 100 | 0.5 | 1j | 47 c |
| 23 | 2a | o-C6H4 | Me | p-C6H4CF3 | Ph2P(OMe) | CHCl3 | 1:10:- | 100 | 0.5 | 1j | 35 c |
| 24 | 2e | o-C6H4 | Ph | p-C6H4CF3 | Ph2P(OMe) | CHCl3 | 1:10:0.25 | 80 | 0.5 | 1k | 88 c |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamek, J.; Węgrzyk-Schlieter, A.; Steć, K.; Walczak, K.; Erfurt, K. Michaelis-Arbuzov-Type Reaction of 1-Imidoalkyltriarylphosphonium Salts with Selected Phosphorus Nucleophiles. Molecules 2019, 24, 3405. https://doi.org/10.3390/molecules24183405
Adamek J, Węgrzyk-Schlieter A, Steć K, Walczak K, Erfurt K. Michaelis-Arbuzov-Type Reaction of 1-Imidoalkyltriarylphosphonium Salts with Selected Phosphorus Nucleophiles. Molecules. 2019; 24(18):3405. https://doi.org/10.3390/molecules24183405
Chicago/Turabian StyleAdamek, Jakub, Anna Węgrzyk-Schlieter, Klaudia Steć, Krzysztof Walczak, and Karol Erfurt. 2019. "Michaelis-Arbuzov-Type Reaction of 1-Imidoalkyltriarylphosphonium Salts with Selected Phosphorus Nucleophiles" Molecules 24, no. 18: 3405. https://doi.org/10.3390/molecules24183405