Synthesis, Antimicrobial Screening, Homology Modeling, and Molecular Docking Studies of a New Series of Schiff Base Derivatives as Prospective Fungal Inhibitor Candidates

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
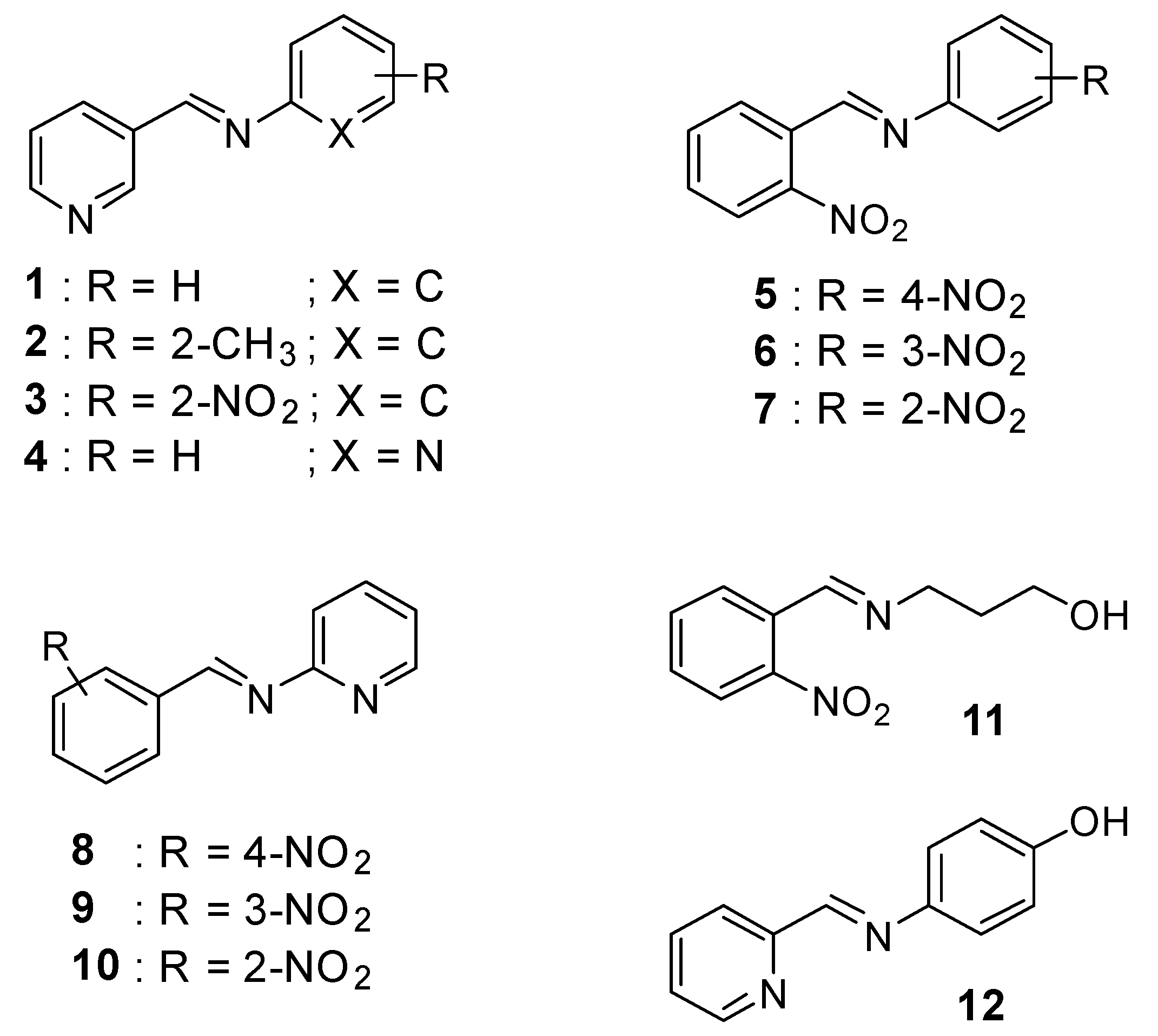
2.1. Chemistry
2.2. Biological Activity
2.3. Computational Studies
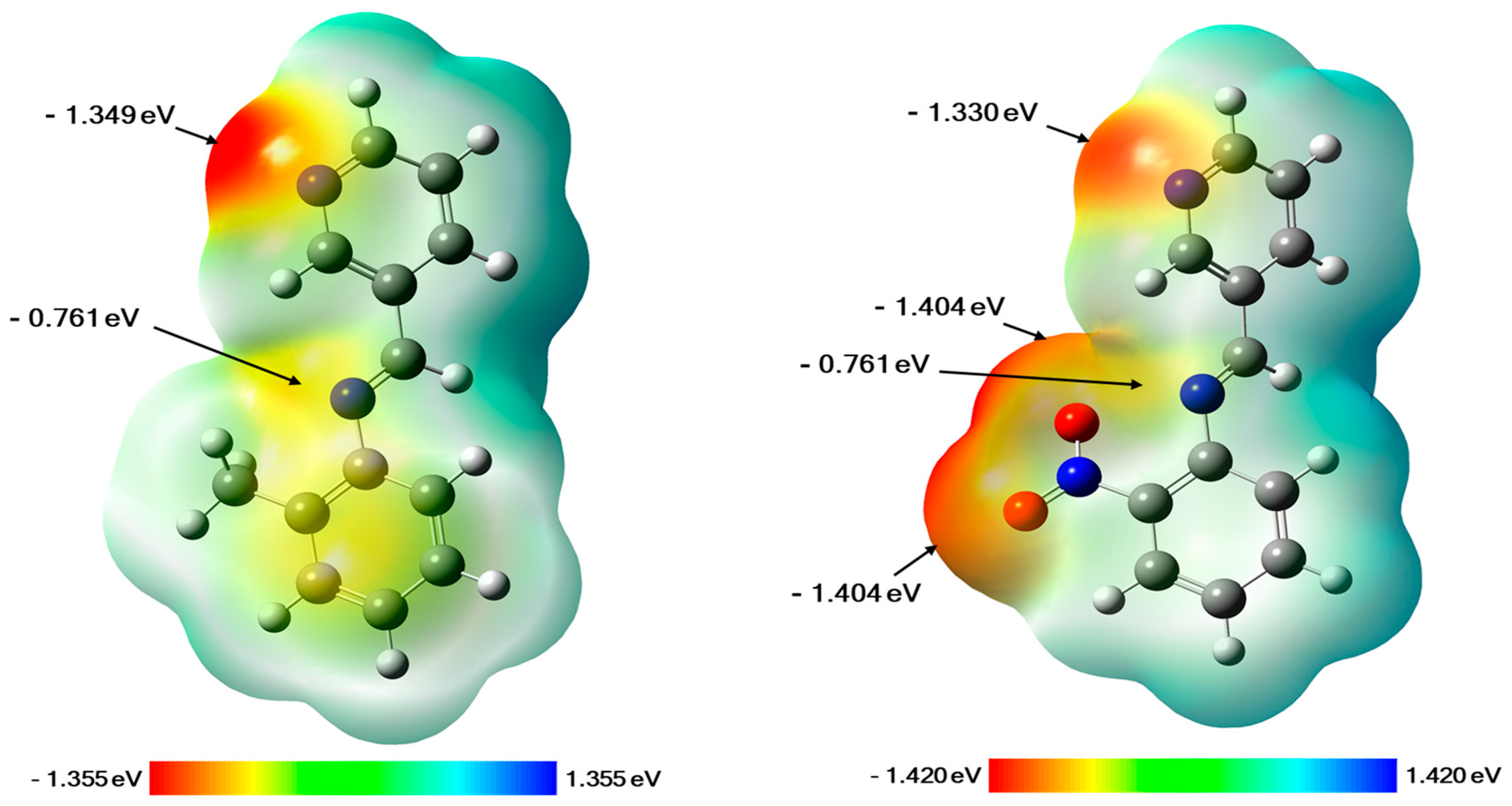
2.3.1. Molecular Electrostatic Potential (MEP) Maps
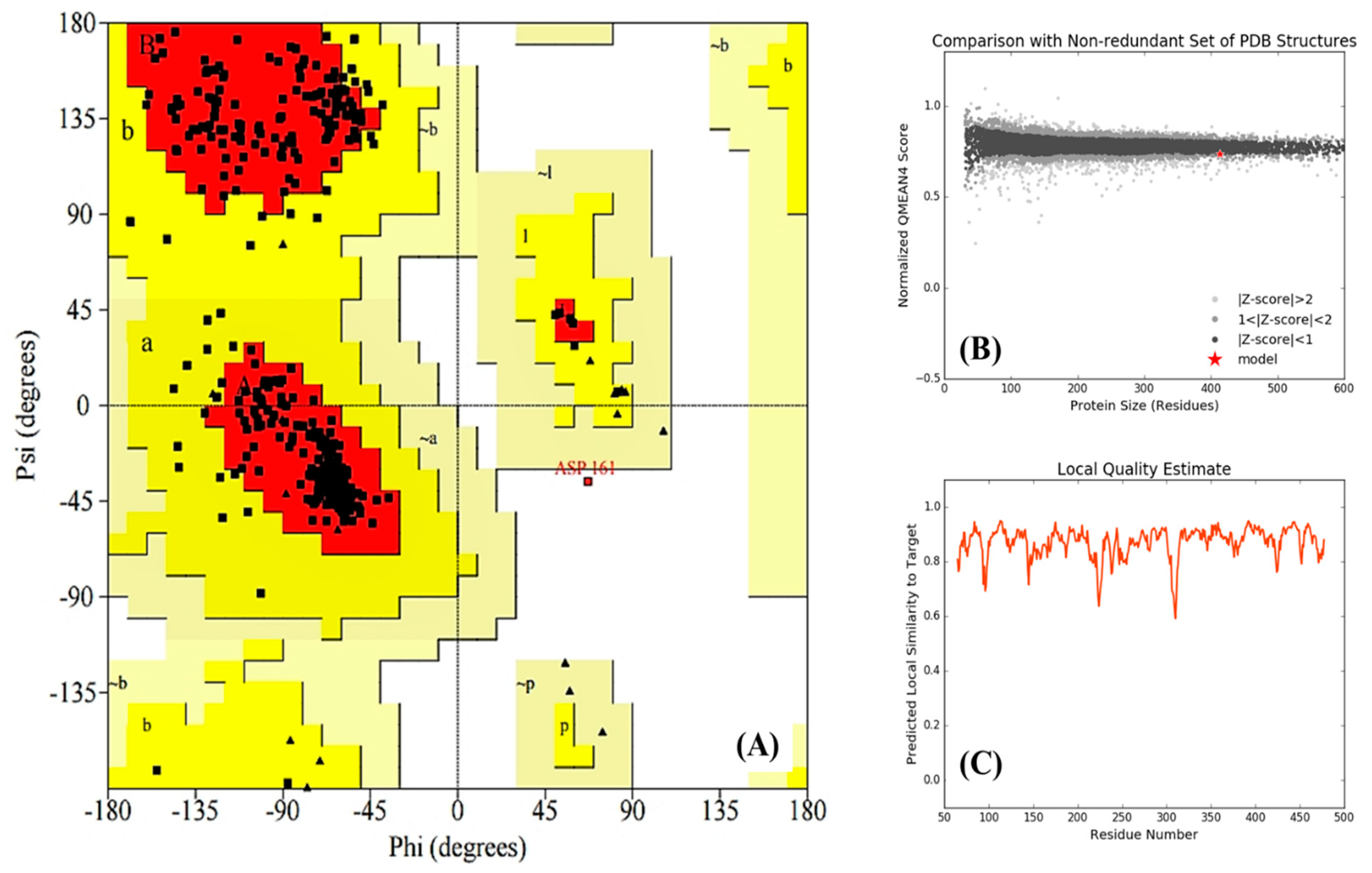
2.3.2. Homology Modeling
2.3.3. Molecular Modeling Study
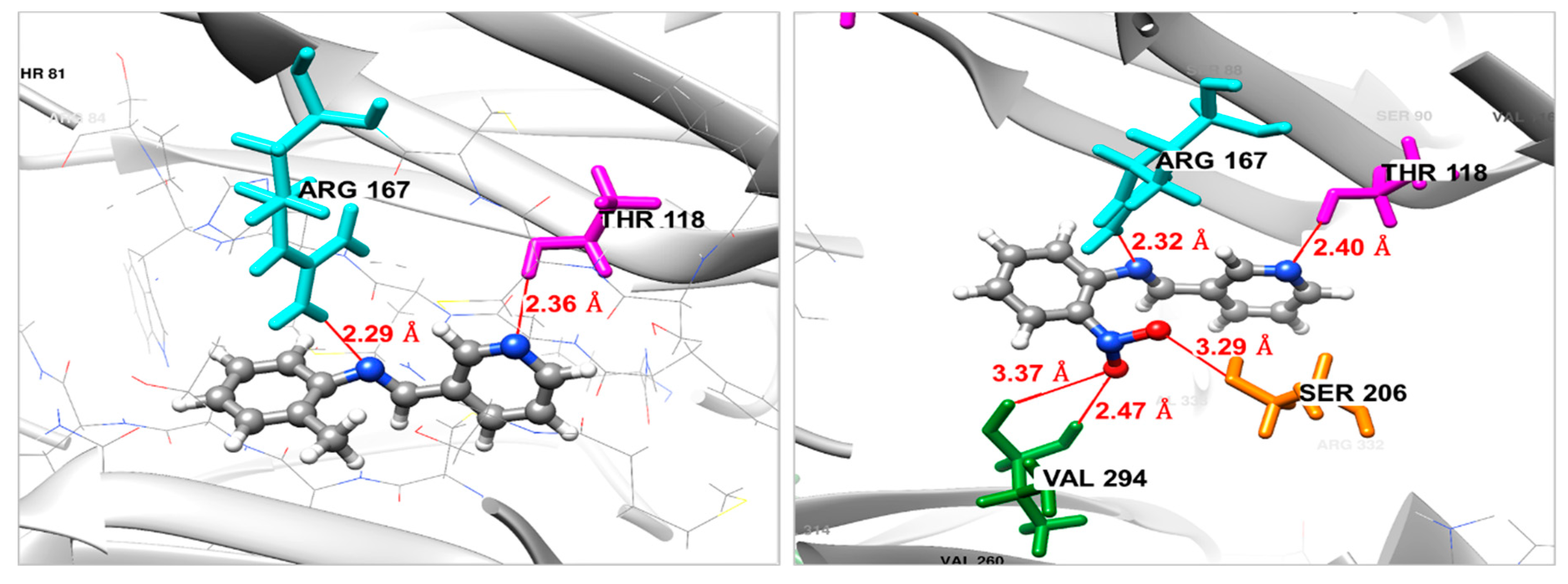
Docking against the Fgb1 Protein
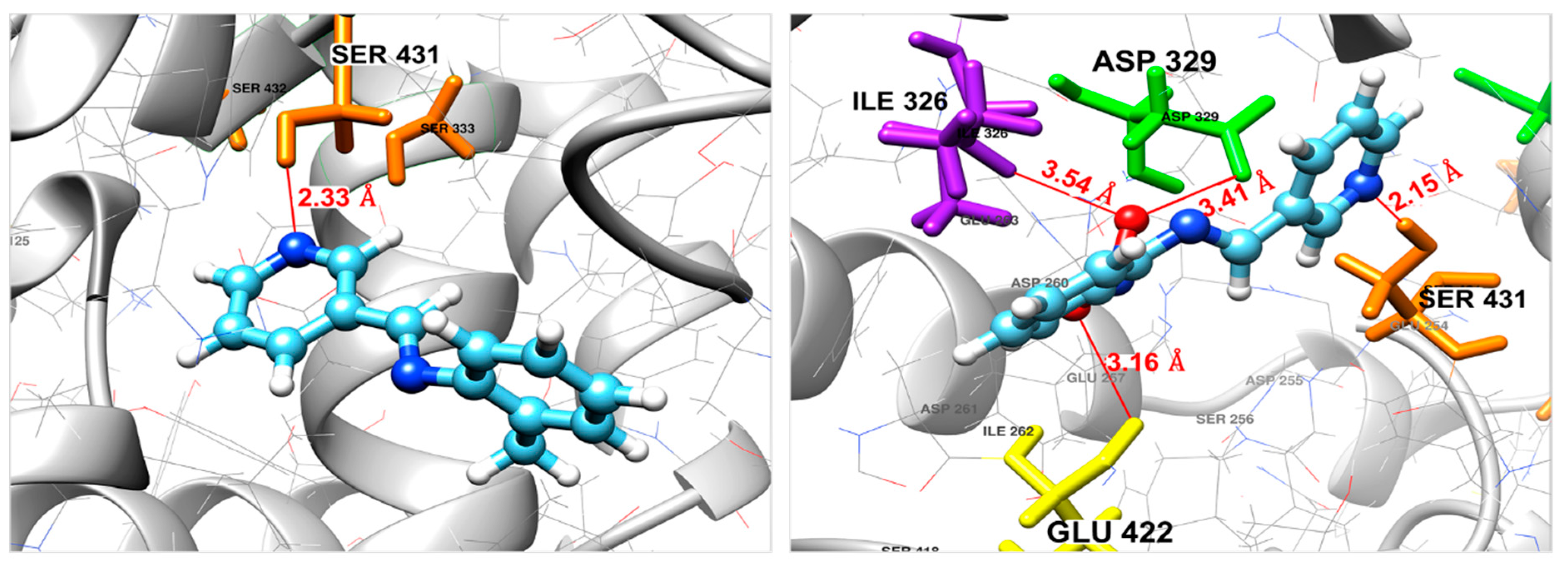
Docking Against the Fophy Protein
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Compounds 1–12
3.3. Biological Evaluation
3.3.1. In Vitro Antifungal Assay
3.3.2. Antibacterial Test
3.4. Computational Studies
3.4.1. Ligand’s Preparation
3.4.2. Homology Modeling
3.4.3. Molecular Docking
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Da Silva, C.M.; da Silva, D.L.; Modolo, L.V.; Alves, R.B.; de Resende, M.A.; Martins, C.V.B.; de Fátima, Â. Schiff bases: A short review of their antimicrobial activities. J. Adv. Res. 2011, 2, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Fekri, R.; Salehi, M.; Asadi, A.; Kubicki, M. Synthesis, characterization, anticancer and antibacterial evaluation of Schiff base ligands derived from hydrazone and their transition metal complexes. Inorg. Chim. Acta 2019, 484, 245–254. [Google Scholar] [CrossRef]
- Ferreira Mde, L.; Vasconcelos, T.R.; de Carvalho, E.M.; Lourenco, M.C.; Wardell, S.M.; Wardell, J.L.; Ferreira, V.F.; de Souza, M.V. Synthesis and antitubercular activity of novel Schiff bases derived from d-mannitol. Carbohydr. Res. 2009, 344, 2042–2047. [Google Scholar]
- Murtaza, S.; Akhtar, M.S.; Kanwal, F.; Abbas, A.; Ashiq, S.; Shamim, S. Synthesis and biological evaluation of schiff bases of 4-aminophenazone as an anti-inflammatory, analgesic and antipyretic agent. J. Saudi Chem. Soc. 2017, 21, S359–S372. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, G.G.; Mahmoud, W.H.; Diab, M.A.; El-Sonbati, A.Z.; Abbas, S.Y. Synthesis, characterization, theoretical study and biological activity of Schiff base nanomaterial analogues. J. Mol. Struct. 2019, 1181, 645–659. [Google Scholar] [CrossRef]
- Tomma, J.H.; Khazaal, M.S.; Al-Dujaili, A.H. Synthesis and characterization of novel Schiff bases containing pyrimidine unit, Arabian. J. Chem. 2014, 7, 157–163. [Google Scholar]
- Özkınalı, S.; Gür, M.; Şener, N.; Alkın, S.; Çavuş, M.S. Synthesis of new azo schiff bases of pyrazole derivatives and their spectroscopic and theoretical investigations. J. Mol. Struct. 2018, 1174, 74–83. [Google Scholar] [CrossRef]
- Rocha, M.; Di Santo, A.; Echeverría, G.A.; Piro, O.E.; Cukiernik, F.D.; Ulic, S.E.; Gil, D.M. Supramolecular self-assembly of a new multi-conformational Schiff base through hydrogen bonds: Crystal structure, spectroscopic and theoretical investigation. J. Mol. Struct. 2017, 1133, 24–36. [Google Scholar] [CrossRef]
- Purtas, F.; Sayin, K.; Ceyhan, G.; Kose, M.; Kurtoglu, M. New fluorescent azo-Schiff base Cu(II) and Zn(II) metal chelates; spectral, structural, electrochemical, photoluminescence and computational studies. J. Mol. Struct. 2017, 1137, 461–475. [Google Scholar] [CrossRef]
- Berhanu, A.L.; Gaurav, I.; Mohiuddin, I.; Malik, A.K.; Aulakh, J.S.; Kumar, V.; Kim, K.-H. A review of the applications of Schiff bases as optical chemical sensors, TrAC. Trends Anal. Chem. 2019, 116, 74–91. [Google Scholar] [CrossRef]
- Bringmann, G.; Dreyer, M.; Faber, J.H.; Dalsgaard, P.W.; Staerk, D.; Jaroszewski, J.W.; Ndangalasi, H.; Mbago, F.; Brun, R.; Christensen, S.B. Ancistrotanzanine C and related 5,1′- and 7,3′-coupled naphthylisoquinoline alkaloids from Ancistrocladus tanzaniensis. J. Nat. Prod. 2004, 67, 743–748. [Google Scholar] [CrossRef]
- de Souza, A.O.; Galetti, F.C.S.; Silva, C.L.; Bicalho, B.; Parma, M.M.; Fonseca, S.F.; Marsaioli, A.J.; Trindade, A.C.L.B.; Gil, R.P.F.; Bezerra, F.S.; et al. Antimycobacterial and cytotoxicity activity of synthetic and natural compounds. Quim. Nova 2007, 30, 1563–1566. [Google Scholar] [CrossRef]
- Yousif, E.; Majeed, A.; Al-Sammarrae, K.; Salih, N.; Salimon, J.; Abdullah, B. Metal complexes of Schiff base: Preparation, characterization and antibacterial activity. Arabian J. Chem. 2017, 10, S1639–S1644. [Google Scholar] [CrossRef] [Green Version]
- Bautista-Baños, S.; Hernández-Lauzardo, A.N.; Velázquez-del Valle, M.G.; Hernández-López, M.; Ait Barka, E.; Bosquez-Molina, E.; Wilson, C.L. Chitosan as a potential natural compound to control pre and postharvest diseases of horticultural commodities. Crop Pro. 2006, 25, 108–118. [Google Scholar] [CrossRef]
- Hassni, M.; El Hadrami, A.; El Hadrami, I.; Barka, E.A.; Daayf, F. Chitosan, Antifungal Product against “Fusarium oxysporum” f. sp. “albedinis” and Elicitor of Defence Reactions in Date Palm Roots. Phytopathol. Mediterr. 2004, 43, 195–204. [Google Scholar]
- Soundararajan, P.; Sakkiah, S.; Sivanesan, I.; Lee, K.-W.; Jeong, B.-R. Macromolecular Docking Simulation to Identify Binding Site of FGB1 for Antifungal Compounds, Bull. Korean Chem. Soc. 2011, 32, 3675–3681. [Google Scholar] [CrossRef]
- Singh, B.; Satyanarayana, T. Fungal phytases: Characteristics and amelioration of nutritional quality and growth of non-ruminants. J Anim. Physiol. Anim. Nutr. 2015, 99, 646–660. [Google Scholar] [CrossRef]
- Vats, P.; Banerjee, U.C. Production studies and catalytic properties of phytases (myo-inositolhexakisphosphate phosphohydrolases): An overview. Enzyme Microb. Technol. 2004, 35, 3–14. [Google Scholar]
- Abrigach, F.; Karzazi, Y.; Benabbes, R.; El Youbi, M.; Khoutoul, M.; Taibi, N.; Karzazi, N.; Benchat, N.; Bouakka, M.; Saalaoui, E.; et al. Synthesis, Biological Screening, POM, and 3D-QSAR Analyses of Some Novel Pyrazolic Compounds. Med. Chem. Res. 2017, 26, 1784–1795. [Google Scholar] [CrossRef]
- Panicker, C.Y.; Varghese, H.T.; Manjula, P.S.; Sarojini, B.K.; Narayana, B.; War, J.A.; Srivastava, S.K.; Van Alsenoy, C.; Al-Saadi, A.A. FT-IR, HOMO–LUMO, NBO, MEP Analysis and Molecular Docking Study of 3-methyl-4-{(E)-[4-(methylsulfanyl)-benzylidene]amino}1H-1,2,4-triazole-5(4H)-thione. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 151, 198–207. [Google Scholar] [CrossRef]
- Abrigach, F.; Rokni, Y.; Takfaoui, A.; Khoutoul, M.; Doucet, H.; Asehraou, A.; Touzani, R. In Vitro Screening, Homology Modeling and Molecular Docking Studies of Some Pyrazole and Imidazole Derivatives. Biomed. Pharmacother. 2018, 103, 653–661. [Google Scholar] [CrossRef]
- Tighadouini, S.; Radi, S.; Abrigach, F.; Benabbes, R.; Eddike, D.; Tillard, M. Novel beta-keto-enol Pyrazolic Compounds as Potent Antifungal Agents. Design, Synthesis, Crystal Structure, DFT, Homology Modeling, and Docking Studies. J. Chem. Inf. Model. 2019, 59, 1398–1409. [Google Scholar] [CrossRef]
- Neri, F.; Mari, M.; Brigati, S. Control of Penicillium Expansum by Plant Volatile Compounds. Plant Pathol. 2006, 55, 100–105. [Google Scholar] [CrossRef]
- Garrod, L.P.; O’Grady, F. Antibiotic and Chemotherapy, 3rd ed.; E. & S. Livingstone: Edinburgh, UK; London, UK, 1972. [Google Scholar]
- Clinical and Laboratory Standards Institute (CLSI), Performance Standards for Antimicrobial Susceptibility Testing; Clinical and Laboratory Standards Institute (NCCLS): Wayne, PA, USA, 2019; CLSI supplement M100 (ISBN 978-1-68440-032-4 [Print], 29th ed.; ISBN 978-1-68440-033-1 [Electronic]).
- ACD/ChemSketch, version 12.01; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2010.
- Gaussian 09, Gaussian; Inc.: Wallingford, CT, USA, 2009.
- Dassault Systèmes BIOVIA, D.S.M.E., Release 2017; Dassault Systèmes: San Diego, CA, USA, 2016.
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Schwede, T. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDB sum: Summaries and analyses of PDB structures. Nucleic Acids Res. 2001, 29, 221–222. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, A Protein-Small Molecule Docking Web Service Based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Rate of Inhibition (%) | MIC (µg·mL−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0.01 | 0.05 | 0.10 | 0.15 | 0.25 | 0.50 | 0.75 | 1.00 | 1.50 | ||
| 1 | 0 | 0 | 7 | 26 | 55 | 78 | 86 | 100 | 100 | 0.10 |
| 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 15 | 27 | 0.90 |
| 3 | 8 | 22 | 53 | 82 | 100 | 100 | 100 | 100 | 100 | 0.02 |
| 4 | 0 | 0 | 0 | 0 | 7 | 7 | 7 | 12 | 15 | 0.25 |
| 5 | 0 | 0 | 0 | 0 | 0 | 10 | 9 | 9 | 15 | 0.30 |
| 6 | 0 | 13 | 13 | 17 | 23 | 46 | 72 | 100 | 100 | 0.04 |
| 7 | 0 | 10 | 33 | 49 | 55 | 67 | 89 | 100 | 100 | 0.04 |
| 8 | 0 | 0 | 0 | 9 | 12 | 14 | 14 | 23 | 37 | 0.12 |
| 9 | 0 | 0 | 0 | 0 | 7 | 7 | 8 | 13 | 15 | 0.25 |
| 10 | 0 | 0 | 0 | 0 | 12 | 15 | 14 | 17 | 23 | 0.20 |
| 11 | 0 | 0 | 15 | 33 | 47 | 62 | 66 | 79 | 90 | 0.08 |
| 12 | 0 | 12 | 12 | 24 | 26 | 48 | 61 | 69 | 82 | 0.04 |
| (-) | - | - | - | - | - | - | - | - | - | - |
| Compound | ΔGbinding (kcal/mol) | Interaction | Bond Length (Å) |
|---|---|---|---|
| 2 | −5.84 | N(pyridine)–(H)Ser431 | 2.33 |
| 3 | −6.78 | N(pyridine)–(H)Ser431 | 2.15 |
| O(NO2)–(O)Glu422 | 3.16 | ||
| O(NO2)–(O)Asp329 | 3.41 | ||
| O(NO2)–(HN)Ile326 | 3.54 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toubi, Y.; Abrigach, F.; Radi, S.; Souna, F.; Hakkou, A.; Alsayari, A.; Bin Muhsinah, A.; Mabkhot, Y.N. Synthesis, Antimicrobial Screening, Homology Modeling, and Molecular Docking Studies of a New Series of Schiff Base Derivatives as Prospective Fungal Inhibitor Candidates. Molecules 2019, 24, 3250. https://doi.org/10.3390/molecules24183250
Toubi Y, Abrigach F, Radi S, Souna F, Hakkou A, Alsayari A, Bin Muhsinah A, Mabkhot YN. Synthesis, Antimicrobial Screening, Homology Modeling, and Molecular Docking Studies of a New Series of Schiff Base Derivatives as Prospective Fungal Inhibitor Candidates. Molecules. 2019; 24(18):3250. https://doi.org/10.3390/molecules24183250
Chicago/Turabian StyleToubi, Yahya, Farid Abrigach, Smaail Radi, Faiza Souna, Abdelkader Hakkou, Abdulrhman Alsayari, Abdullatif Bin Muhsinah, and Yahia N. Mabkhot. 2019. "Synthesis, Antimicrobial Screening, Homology Modeling, and Molecular Docking Studies of a New Series of Schiff Base Derivatives as Prospective Fungal Inhibitor Candidates" Molecules 24, no. 18: 3250. https://doi.org/10.3390/molecules24183250
APA StyleToubi, Y., Abrigach, F., Radi, S., Souna, F., Hakkou, A., Alsayari, A., Bin Muhsinah, A., & Mabkhot, Y. N. (2019). Synthesis, Antimicrobial Screening, Homology Modeling, and Molecular Docking Studies of a New Series of Schiff Base Derivatives as Prospective Fungal Inhibitor Candidates. Molecules, 24(18), 3250. https://doi.org/10.3390/molecules24183250